

Abstract
Receptor interacting protein kinase 1 (RIPK1) is an enzyme acting downstream of tumor necrosis factor alpha to control cell survival and death. RIPK1 expression has been reported to cause drug resistance in cancer cells, but so far, no published studies have investigated the role of RIPK1 in vitamin D signaling. In the present study, we investigated whether RIPK1 plays any roles in 1,25-dihydroxyvitamin D3 (1,25D3)-induced growth suppression. In our studies, RIPK1 decreased the transcriptional activity of vitamin D receptor (VDR) in luciferase reporter assays independent of its kinase activity, suggesting a negative role of RIPK1 in 1,25D3 action. RIPK1 also formed a complex with VDR, and deletion analyses mapped the RIPK1 binding region to the C-terminal ligand-binding domain of the VDR. Subcellular fractionation analyses indicated that RIPK1 increased VDR retention in the cytoplasm, which may account for its inhibition of VDR transcriptional activity. Consistent with the reporter analyses, 1,25D3-induced growth suppression was more pronounced in RIPK1-null MEFs and RIPK1-knockdown ovarian cancer cells than in control cells. Our studies have defined RIPK1 as a VDR repressor, projecting RIPK1 depletion as a potential strategy to increase the potency of 1,25D3 and its analogs for cancer intervention.
Keywords: Apoptosis, Cancer, Necroptosis, Nuclear Receptor, Receptor Interacting Protein Kinase 1, Vitamin D, Vitamin D receptor
1. Introduction
The seco-steroid hormone 1,25-dihydroxyvitamin D3 (1,25D3) and its receptor, vitamin D receptor (VDR), have long been known for their roles in calcium homeostasis and bone health. However, the widespread expression of VDR across different tissues implicates its importance in other biological processes [1]. Approximately 3% of human genes are directly or indirectly regulated by the vitamin D endocrine system [2]. Vitamin D deficiency has been consistently correlated with a number of chronic diseases including cancer; 1,25D3 and its synthetic analogs have been projected to be promising agents for cancer intervention [3–6]. Our published studies in ovarian cancer have shown that 1,25D3 inhibits cancer growth by arresting cells at specific cell cycle checkpoints [7–9]. Further analyses have demonstrated that 1,25D3 induces microRNA target genes to decrease telomerase expression for cell death induction [10, 11] and to inhibit leptin and estrogen-induced tumor growth [12], suggesting that 1,25D3 has an important role in cutting down obesity-associated cancer risks in women [13]. More recent studies have shown that ovarian cancer invasion is suppressed by 1,25D3 through the cooperative actions of epithelial and stromal VDR [14]. Overall, these studies project VDR as a new therapeutic target for ovarian cancer intervention. However, not all ovarian cancer cells responded to 1,25D3 for growth suppression in our studies, including some cells expressing VDR at levels comparable to those of sensitive cells. The analyses suggest the existence of additional molecular determinants for 1,25D3 sensitivity besides VDR expression.
Receptor interacting protein kinase 1 (RIPK1) is a member of a family of seven protein kinases that share a homologous kinase domain (KD) [15]. In addition to its N-terminal KD, RIPK1 contains a unique death domain (DD) at its C-terminus and an intermediate domain (ID) harboring a RIP homotypic interaction motif (RHIM) that is shared by another family member, RIPK3 [15]. Due to the selective utilization of its adaptor function and enzymatic activities, RIPK1 is emerging as an important determinant of cell fate and can mediate either cell death or survival in response to diverse cellular signals [16, 17]. In response to certain necroptotic stimuli, RIPK1 is known to work through RIPK3 and mixed lineage kinase domain-like (MLKL) to signal cell death in a kinase-dependent manner [18, 19]. On the other hand, RIPK1 is also known to be recruited, together with other proteins, to form a pro-survival protein complex by active TNFα receptor 1. The complex formation results in K63 and linear ubiquitination of RIPK1 and subsequent recruitment of the TAB2/TAB3/TAK1 complex and NEMO, leading to NF-κB activation and cell survival [15–17, 20, 21]. In contrast to its pro-death activity, RIPK1’s cell survival function is attributed largely to its adaptor function independent of the kinase activity, which contributes to its oncogenic activities and causes chemo-resistance in cancer cells [22–25]. Nonetheless, little is known about the role of RIPK1 in 1,25D3 signaling.
In the present study, we have examined the role of RIPK1 mediated by 1,25D3 and reported that RIPK1 is a transcriptional inhibitor of VDR and decreases 1,25D3-induced growth suppression. Interestingly, VDR inhibition occurs independently of RIPK1 kinase activity and is likely mediated through a RIPK1-VDR protein interaction that increases the cytoplasmic retention of the VDR. Our studies project RIPK1 depletion as a potential strategy to increase the potency of 1,25D3 and its analogs in cancer intervention.
2. Material and Methods
2.1 Reagents and antibodies
Anti-Flag antibody (F7425), anti-Flag M2 affinity gel (A2220), fetal bovine serum (FBS) (12306C), MG132 (C2211), myelin basic protein (MBP) (M1891), and protease inhibitor cocktail (11836170001) were purchased from Sigma-Aldrich (St. Louis, MO). Anti-HA antibody (PRB-101P) was from Covance (BioLegend, San Diego, CA). Anti-VDR (C-20 and D-6), anti-HSP60 (H-1, sc-13115), anti-α-actinin (H-2), and anti-β-actin (AC-15) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-RIPK1 antibody (BD 610458) was from BD Bioscience (San Jose, CA). Anti-PARP (9532), anti-NCAM (3606s), anti-DUSP10 (3483s), and anti-G6PD (8866s) antibodies were from Cell Signaling (Danvers, MA). Luciferase substrates were from Promega (Madison, WI). The ECL Western blotting substrates were from Thermo Scientific (Waltham, MA). Trizol (15596026) was purchased from Life Technologies (Carlsbad, CA). Complementary DNA synthesis kit (170-8891) and iTaq Universal SYBR Green Supermix (172-5121) were from Bio-Rad (Hercules, CA). Ni-NTA (635659) was purchased from Clontech (Mountain View, CA).γ-32p-ATP (BLU002A250UC) was from PerkinElmer (Waltham, MA). Penicillin and streptomycin (30-002-CI) were from Corning (Tewksbury, MA). Lipofectamine 2000 (11668-019) and trypsin (25200-056) were from Life Technologies (Grand Island, NY). 1,25D3 (calcitriol) and necrostatin-1 (Nec-1) was purchased from Calbiochem (La Jolla, CA). 1,25D3 was reconstituted in 100% ethanol (EtOH) and Nec-1 in DMSO. Both stock solutions were stored at −20 °C. Handling of 1,25D3 and Nec-1 was performed under indirect lighting.
2.2 Plasmids and cell lines
Control and RIPK1 shRNA plasmids (pRS-MIG-HRS and pRS-MIG-RIP193, respectively) [26] were kind gifts from Dr. Martin Leverkus of University of Heidelberg. The RIPK1 K45M mutant vector [27] was kindly provided by Dr. Junying Yuan of Harvard University. pCMX-Flag-RXRα was provided by Dr. Wilson H. Miller Jr. of McGill University [28, 29]. The p23 VDR reporter containing rat 24-hydroxylase promoter in pMAMMneoLuc [30] was provided by Dr. H. F. Deluca of University of Wisconsin-Madison. VDRE2TKLUC reporter was constructed by inserting two copies of a vitamin D response element (VDRE) in front of a minimal thymidine kinase (TK) promoter followed by firefly luciferase cDNA. GFP-VDR was generated by inserting human VDR cDNA in frame to the N-terminus of green fluorescence protein in pEGFP-N1 vector. His-tagged VDR in pET28a was generated as described [31]. HA-RIPK1, HA-VDR, Flag-RIPK1, and Flag-VDR plasmids were generated by cloning full-length RIPK1 and VDR cDNA into HA- and Flag-tagged pcDNA3.1 and pCMV vectors, respectively. Deletion constructs of VDR and RIPK1 were generated by PCR and cloned into Flag-tagged pcDNA3.1 and HA-tagged pCMV vectors, respectively.
293T and L929 cells were purchased from American type culture collection (ATCC) (Manassas, VA). RIPK1 wild type and null mouse embryonic fibroblasts (MEFs) [32] were kindly provided by Dr. Junying Yuan of Harvard University. PE01 cells [33] were kindly provided by Dr. Toshiyasu Taniguchi of Fred Hutchinson Cancer Research Center. OVCAR3 and PE01 cells were maintained in RPMI medium containing 2 mmol/L L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS. All other cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/ml streptomycin, and 10% FBS.
For stable knockdown of RIPK1, PE01 and OVCAR3 cells were transfected with pRS-MIG-HRS control or RS-MIG-RIPK193 plasmid. Forty-eight hours post-transfections, stable clones were selected in media containing 2 μg/mL puromycin. RIPK1 knockdown in stable clones was verified by Western blot analyses. All cells were maintained in a 37°C humidified incubator with 5% CO2.
2.3 Transfection and reporter assays
2×105 293T cells were plated in 12-well plates and transfected with plasmids the next day as indicated in the figures. Twenty-four hours post-transfections, cells were treated with either EtOH or 10−8 M 1,25D3 for an additional 24 hours. Luciferase activity was measured and normalized with cognate β-galactosidase activity as previously described [34]. Each data point is expressed as mean ± S.D of parallel analyses in triplicates (n=3).
2.4 In vitro immunocomplex kinase assays
His-VDR proteins were expressed in E. coli and purified using Ni-NTA agarose beads. Flag-RIPK1 was transfected into 293T cells and immunoprecipitated with Flag affinity beads. The kinase assays were performed by incubating 1 μg His-VDR or myelin basic protein (MBP) (M1891, Sigma) with 5 μCi of γ-32P-ATP in kinase buffer (10 mM Tris, pH7.4, 10 μM of ATP, 150 mM Nacl, 10 mM MgCl2, and 0.5 mM dithiothreitol) for 30 minutes at 30 °C. The reactions were stopped by adding 5× SDS sample loading buffer and heating for 5 minutes at 100 °C. The samples were subjected to SDS-PAGE and phosphorylation status was visualized by autoradiography.
2.5 Immunological analyses
For co-immunoprecipitations, cells were lysed in buffer containing 20 mM Tris-HCL (pH 7.5), 150 mM NaCl, 1% (v/v) NP-40, 1 mM PMSF, and protease inhibitor cocktail. After two consecutive 6-second sonications separated by a brief cooling, cells were kept on ice for 10 minutes before centrifugation. For co-immunoprecipitations of transfected proteins with tags, cellular extracts were incubated overnight at 4°C with M2 antibodies conjugated with beads. For co-precipitations of endogenous VDR and RIPK1, cellular extracts were incubated overnight at 4°C with anti-VDR antibody followed by a 4-hour incubation with protein G beads. After incubations, the beads were washed five times with lysis buffer and precipitated proteins detected by Western blot analyses.
For Western blot, immune precipitates or cellular extracts containing equal amounts of protein (20–40 μg) were separated in a 10% SDS-PAGE, transferred onto nitrocellulose membranes, and probed with cognate antibodies. ECL substrates were used for protein detections.
2.6 Cytoplasmic and nuclear fractionation
For cytoplasmic protein extraction, cells were washed and scraped with ice-cold phosphate buffered saline (PBS). After pelleting, cells were re-suspended in lysis buffer (10 mM Hepes, pH7.9; 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA) and kept on ice for 15 minutes. 10% NP-40 was added and followed by centrifugation for 1 minute at 14,000 rpm. The supernatant was collected as cytoplasmic protein. The pellet was washed with ice-cold PBS, re-suspended in nuclear extraction buffer (20 mM Hepes, pH 7.9; 400 mM NaCl, 1 mM EDTA, 1 mM EGTA), and kept on ice for 30 minutes with 3- to 5-second vortexing at 5-minute intervals. After centrifugation at 14,000 rpm for 5 minutes, the supernatant was collected as nuclear extracts.
2.7 Methylthiazol tetrazolium (MTT) assays
Cell growth was quantified using MTT assays. Cells were plated in 96-well plates and treated with either EtOH or 1,25D3 for 6 days. MTT assays were performed as described [35]. In brief, MTT was added to wells at a final concentration of 0.5 mg/mL and incubated for 3 hours. The media were removed after the incubation, and 200 μl of DMSO was added to each of the wells. The absorption at 595 nm was measured in a MRX microplate reader (DYNEX Technologies, Chantilly, VA). Each data point is expressed as mean ± S.D of 6 samples analyzed in parallel (n=6).
2.8 Real-time RT-PCR
To quantify the expression of CYP24A1 mRNA and its induction by 1,25D3, cells were treated with EtOH or 10−7 M 1,25D3 for 24 hours. Total RNA was isolated using Trizol (Life Technologies, Grand Island, NY) according to manufacturer’s protocols. cDNA was prepared in a 20 μl reaction from 1 μg of total RNA by cDNA iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA). Real-time PCR reactions were run in triplicates (n=3) in the iCycler iQ™ real-time PCR detection system (Bio-Rad, Hercules, CA) in a 20 μl reaction mixture containing 2 μl of the reverse transcription product, 0.75 μl of 10 μM stocks of forward and reverse primers [36], and 10 μl of iTaq Universal SYBR Green Supermix. Reactions were run with the following parameters: 95°C for 3 minutes followed by 40 cycles of 95°C for 10 seconds, 55°C for 1 minute and GAPDH was run as a control for CYP24A1 expression. ΔCt values and fold of induction were calculated as described previously [12]. Each data point represents three independent analyses (n=3) presented as mean ± SEM. *p<0.05, ***<0.0005, ****p<0.0001.
2.9 Statistical Analyses
Significant analyses were performed with the Student t-test. All values are presented as mean ± standard deviation (SD) unless otherwise specified. p <0.05 is considered to be statistically significant.
3. Results
3.1 RIPK1 inhibits VDR transcriptional activity independent of its kinase activity
To test the effect of RIPK1 on VDR, HEK293T cells were transfected with plasmids expressing human VDR and RIPK1 together with a luciferase reporter construct in which rat CYP24 promoter containing two VDREs were placed in front of the luciferase cDNA in pMAMMneoLuc [30]. Transfected cells were treated with a vehicle or 10−8 M 1,25D3, and the transcriptional activity of VDR was assessed by the CYP24A1 luciferase activity as an outcome. 1,25D3 stimulated luciferase activity but the co-expression of RIPK1 substantially reduced its ability to do so (Fig. 1A, bar graphs). Since RIPK1 actually increased the levels of VDR protein (Fig. 1A, lower panels), it is thus apparent that RIPK1 decreased the transcriptional activity of VDR per molecule. Subsequent analyses with a VDRE2TKLUC reporter containing two copies of synthetic VDRE in front of the thymidine kinase promoter and luciferase cDNA obtained similar results (Fig. 1B), showing that the inhibition of VDR activity by RIPK1 is not limited to CYP24A1 promoter.
Figure 1. RIPK1 decreases VDR activity in a kinase-independent manner.

A. 293T cells were transfected with 200 ng of p23 VDR reporter plasmid, 20 ng of pCMVβgal, 50 ng of Flag-VDR, and indicated amounts of Flag-RIPK1. The next day, cells were treated with either EtOH or 1,25D3 for 24 hours. B. 293T cells were transfected and treated as in panel A except VDRE2TKLUC was used as the reporter. C. 293T cells were transfected as in panel A but either with wild-type (RIPK1-WT) or kinase-dead mutant (RIPK1-K45M) RIPK1 and treated with 1,25D3 and/or RIPK1 inhibitor Nec-1 (20 μM). Luciferase activity was determined and normalized with cognate β-gal activity. Each data point was analyzed in triplicates (n-3) and reproduced two times. Western blot analyses were performed with antibodies as indicated.
Since RIPK1 is a protein kinase, we next sought to determine whether the enzymatic activity of RIPK1 is required for the inhibition of VDR. As shown in Fig. 1C, the kinase-dead K45M RIPK1 mutant inhibited 1,25D3-induced VDR activity to the same extent as wild-type RIPK1, and the addition of RIPK1 kinase inhibitor Nec-1 did not relieve the inhibition. In the absence of ectopic RIPK1, Nec-1 increased the transcriptional activity of VDR (Fig. 1B, lower panels), raising the possibility that the inhibitor may exert a positive effect on VDR through non-specific effects independent of RIPK1 [37]. In our studies, Nec-1 increased the expression levels of both wild-type RIPK1 and the K45M mutant, and both wild-type RIPK1 and the kinase-dead mutant increased the levels of VDR protein expression. The increased expression of RIPK1 and VDR by Nec-1 and RIPK1, respectively, was consistently observed in our studies.
Overall, the studies suggest that RIPK1 inhibits the transcriptional activity of VDR independently of its kinase activity. Consistent with this conclusion, RIPK1 did not phosphorylate recombinant VDR proteins (Fig 2A) in in vitro immunocomplex kinase assays though myelin basic protein was phosphorylated in parallel reactions (Fig. 2B).
Figure 2. RIPK1 did not phosphorylate VDR in in vitro immunocomplex kinase assays.
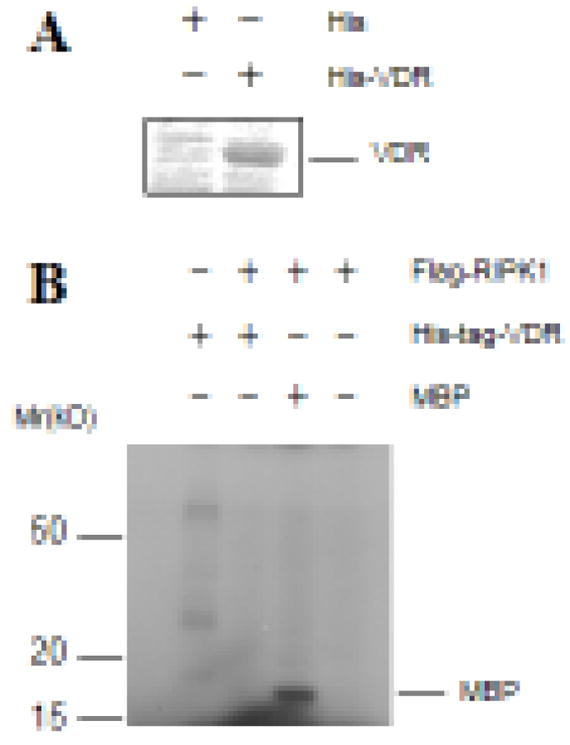
A. His-tagged VDR proteins were expressed in bacteria, purified using nickel beads, and stained with Coomassie blue. B. Flag-RIPK1 was transfected into 293T cells and immunoprecipitated with anti-Flag antibodies. In vitro immunocomplex kinase assays were performed with purified His-VDR protein as a substrate. Myelin basic protein (MBP) was included as a positive control.
3.2 RIPK1 forms a protein complex with VDR
As mentioned before, RIPK1 can perform its biological functions through its enzymatic activities or as an adaptor. The evidence that RIPK1’s ability to inhibit VDR activity is independent of its kinase activity prompted us to ask whether RIPK1 forms a complex with VDR. HEK293T cells were transfected with plasmids encoding tagged RIPK1, VDR, or both, and co-precipitation experiments performed. M2 anti-Flag beads co-precipitated HA-RIPK1 with Flag-VDR (Fig. 3A) and HA-VDR with Flag-RIPK1 (Fig. 3B) from cells expressing both proteins but not either protein alone. Thus, RIPK1 associates with VDR in transfected cells. The binding of RIPK1 to VDR was detected both in the absence and presence of 1,25D3 but was reduced by the hormone treatment (Fig. 3C). This behavior is similar to that of a typical transcriptional co-repressor for nuclear receptors [38, 39]. Moreover, the complex formation also occurred between endogenous VDR and RIPK1 in L929 mouse fibrosarcoma cells (Fig. 3D), ruling out the possibility that the complex formation might be an artifact of ectopic overexpression.
Figure 3. RIPK1 forms a protein complex with VDR, which is decreased by 1,25D3 treatments.

A – C. 293T cells were transfected with 1.5 μg of tagged VDR and RIPK1 as indicated and treated with (Panel C) EtOH or 10−7 M 1,25D3 for 24 hours. Cellular lysates were immunoprecipitated with anti-Flag antibody conjugated beads. Western blot analyses were performed with indicated antibodies. D. Whole cell lysates of L929 cells were immunoprecipitated with anti-VDR antibody followed by Western blot analyses with anti-VDR and RIPK1 antibodies as indicated.
3.3 The complex formation between RIPK1 and VDR involves the kinase domain of RIPK1 and the ligand-binding domain (LBD) of VDR
To define the site in VDR that mediates RIPK1 binding, VDR deletion constructs (Fig. 4A) were generated, and their ability to bind to RIPK1 was assessed by co-immunoprecipitation analyses. As shown in Fig. 4B, the LBD fragment of VDR was able to form a complex with RIPK1 just like the full-length receptor whereas deletion of the LBD from VDR destroyed the receptor’s ability to form such a complex, defining the LBD as the site for RIPK1 binding. The VDR fragment deleted of the activation function 2 (AF2) region retained the ability to associate with RIPK1 (Fig. 4B), revealing expendability of the AF2 region for binding.
Figure 4. RIPK1 binds to the C-terminal LBD of VDR.

A. Schematic representation of the domain structure of VDR and its deletion constructs. Amino acid residue numbers are shown at the top of the graphs. ΔC: C-terminal LBD deleted (aa.1-203); ΔN: N-terminal DNA binding domain and hinge region deleted (aa.204-427); ΔAF-2: Helix-12 deleted (aa.1-408). B. 293T cells were transfected with 1.5 μg of HA-RIPK1 together with 1.5 μg of Flag-tagged full-length (FL) VDR or its deletion constructs as indicated. Cellular extracts were subjected to co-immunoprecipitation analyses with antibodies as indicated.
Using a similar approach, the regions in RIPK1 required for VDR interaction were also defined by deletion analyses. A set of RIPK1 deletion constructs were generated (Fig. 5A) and their ability to form complexes with VDR was determined in co-precipitation analyses. As shown in Fig. 5B, neither the kinase nor the intermediate domain alone was co-precipitated with VDR whereas the fragment containing both regions was co-precipitated. The analyses suggest that the kinase or intermediate domain alone is not sufficient; both are required for binding to VDR. The deletion of C-terminal death domain and the region between RHIM and the death domain did not affect the VDR binding, showing that these regions are expendable.
Figure 5. Multiple regions of RIPK1 mediate its binding to VDR.

A. Schematic representation of the domain structure of RIPK1 and its deletion constructs. Amino acid residue numbers are shown at the top of the graphs. KD: kinase domain (aa.1-289); ID: Intermediate domain (aa.290-583); IDs: Smaller ID (aa.290-547); ΔKD: Kinase deleted (aa.290-583). B. 293T cells were transfected with 1.5 μg of Flag-VDR together with 1.5 μg of full-length (FL) or deletion constructs of HA-RIPK1 as indicated. Cellular extracts were subjected to co-immunoprecipitation analyses with antibodies as indicated.
Overall, the binding analyses suggest that RIPK1 employs multiple domains to interact with the LBD of VDR. Although the ability of RIPK1 to repress the transcriptional activity of VDR is independent of its enzymatic activity, the kinase domain is physically involved in the complex formation.
3.4 RIPK1 increases VDR retention in the cytoplasm
Besides binding to 1,25D3, the LBD of VDR is also known to be involved in heterodimer formation with RXR. In an effort to understand the mechanisms underlying VDR repression by RIPK1, we first analyzed the ability of VDR to form a heterodimer with RXR in the presence of or absence of ectopic RIPK1 expression. As shown in Fig. 6, similar amounts of RXRα were co-precipitated together with VDR from cells transfected with control vector and RIPK1, ruling out the disruption of receptor dimer formation as the mechanism for VDR repression by RIPK1. We next assessed the effect of RIPK1 on VDR cellular localization. As shown in Fig. 7, the VDR protein ectopically expressed in 293T cells was mainly localized to the nucleus but the co-transfection of RIPK1 increased the cytoplasmic VDR signal. Due to the fact that ectopic RIPK1 increased the overall levels of VDR expression, the impact of ectopic RIPK1 on the nuclear VDR was not obvious from the Western blots (Fig. 7A). However, after quantification with ImageJ and normalization with cognate whole cell signals, it became clear that the increased VDR retention in the cytoplasm was accompanied by a decrease in the nuclear VDR (Fig. 7B). The data argue that the increased cytoplasmic retention is a possible mechanism underlying the VDR repression by RIPK1.
Figure 6. RIPK1 does not alter the ability of VDR to form dimer with RXR.
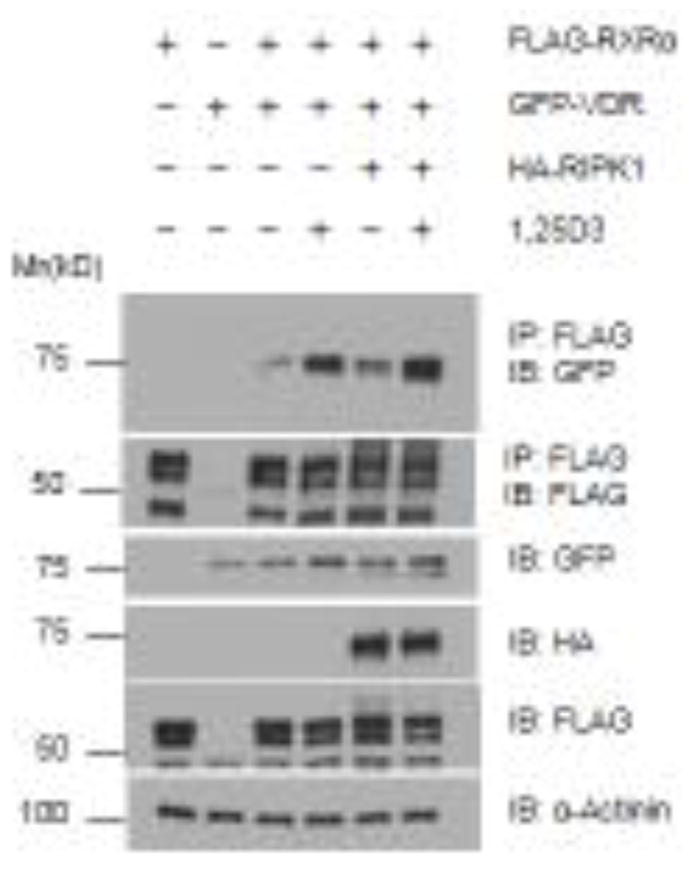
293T cells were transfected with 1.5 μg of tagged vectors expressing VDR, RXRα, and RIPK1 as indicated and treated with EtOH or 10−7 M 1,25D3 for 24 hours. Cellular lysates were immunoprecipitated with anti-Flag antibody conjugated beads. Western blot analyses were performed with indicated antibodies.
Figure 7. RIPK1 increases VDR cytoplasmic localization.

293T cells were transfected with Flag-VDR and Flag-RIPK1 constructs. Thirty-six hours post-transfections, cells were treated with EtOH or 10−7 M 1,25D3 for 24 hours. Whole cell, cytosolic, and nuclear extracts were prepared and subjected to Western blot analyses with indicated antibodies. Anti-HSP60 and PARP-1 Western blots were included as loading controls for cytoplasmic and nuclear proteins, respectively. The intensity of Western blot bands was quantified by ImageJ (NIH). VDR signals were first normalized with cognate loading controls. Cytoplasmic and nuclear VDR were further normalized with corresponding whole-cell VDR and plotted as relative VDR signals. Bar graphs represent four independent experiments (n=4) and error bars have been calculated as mean ± standard error of the mean (SEM). *p<0.05, **<0.005.
3.5 Depletion of RIPK1 expression increases sensitivity of cells to 1,25D3-induced growth suppression and target gene expression
So far, we have shown that ectopic RIPK1 represses VDR by binding to and holding the receptor in the cytoplasm. If this is true, depletion of RIPK1 expression should potentiate 1,25D3 signaling and increase the sensitivity of cells to the growth-suppressive effect of the hormone. Indeed, in comparison to wild-type MEFs, RIPK1-null MEFs exhibited a significant increase in their sensitivity to 1,25D3-induced growth suppression at all concentrations tested (Figs. 8A and 8B).
Figure 8. RIPK1 depletion sensitizes cells to 1,25D3.

A. RIPK1 expression in wild-type (RIPK1+/+) and RIPK1-null (RIPK1−/−) MEFs was verified by Western blot analyses. B. MEFs were plated in 96-well plates and treated with either EtOH or indicated concentrations of 1,25D3 for 6 days. MTT assay was performed. Six samples were analyzed in parallel for each data point (n=6) and the experiment was repeated twice. Percentages of cell growth were first calculated by subtracting MTT values at day zero from those in day 6 followed by dividing with day zero values. Percentages of growth suppression by 1,25D3 were calculated by dividing percentages of cell growth in 1,25D3-treated groups with those of the EtOH controls. **p<0.005; ***p<0.001, ****p<0.0001. C. RIPK1 and VDR expression was determined by Western blot analyses in an OVCAR3 cell pool stably expressing control or RIPK1 shRNA. D. The OVCAR3 pools were treated with EtOH or 1,25D3 for 6 days and the percentage of growth suppression was measured in MTT assays as in Panel A. E. RIPK1 and VDR expression was determined by Western blot analyses in PE01 cell clones stably expressing control or RIPK1 shRNA. F. PE01 clones were treated with EtOH or 1,25D3 for 6 days and the percentage of growth suppression was determined in MTT assays as in Panel A.
Consistent with the data from RIPK1-null MEFs, RIPK1 knockdown by stable shRNA expression in OVCAR3 (Figs. 8C and 8D) and PE01 (Figs. 8E and 8F) ovarian cancer cells significantly increased their sensitivity to 1,25D3-induced suppression. Real-time PCR and Western blot analyses showed that RIPK1 knockdown also potentiated the ability of 1,25D3 to induce CYP24A1 mRNA expression (Fig. 9A) and the expression of dual specificity phosphatase 10 (DUSP10), neural cell adhesion molecule 1 (NCAM1), and glucose-6-phosphate dehydrogenase (G6PD) proteins (Fig. 9B), respectively. All these genes have been identified as 1,25D3 target genes in ovarian cancer cells in published micro-array analyses [40]. The ability of RIPK1 depletion to potentiate both the growth response to 1,25D3 and the ability of the hormone to induce target gene expression was observed in multiple cells types with a stable shRNA pool (Figs. 8D and 9) and two independent clones (Figs. 8F and 9), ruling out the potential artifact of clonal effects and stable selection with antibiotics.
Figure 9. RIPK1 depletion increases the ability of 1,25D3 to induce target gene expression.

A. Cells were treated with EtOH or 10−7 M 1,25D3 for 24 hours. Total RNA was extracted and the expression of CYP24A1 determined by real-time RT-PCR and normalized to GAPDH. The expressions are presented as fold of induction over the vehicle control. Each data point represents three independent analyses (n=3). Student’s t-test was used for statistical analyses. *p<0.05, ***<0.0005, ****p<0.0001. B. Cells were treated with EtOH or 10−7 M 1,25D3 for 6 days. Western blot analyses were performed with antibodies as indicated. The signals of vitamin D target genes were quantified by ImageJ (NIH) and normalized with cognate signals of α-actinin as loading controls. The numbers under the blots indicated the fold of induction over vehicle control.
Overall, the growth and target gene expression analyses in RIPK1-depleted cells support the conclusion from transfection studies that RIPK1 is a VDR repressor. The studies argue that RIPK1 depletion may represent an effective strategy for increasing the potency of 1,25D3 and its analogs in cancer intervention.
4. Discussion
RIPK1 is well established as an essential molecule involved in mediating cell death and survival downstream of death receptors. In this study, we have defined a novel function for RIPK1 in nuclear receptor signaling by identifying it as a VDR repressor. Reporter analyses have first shown that RIPK1 inhibits the transcriptional activity of VDR independently of its kinase activity. Subsequent co-precipitation analyses have revealed that RIPK1 forms a complex with VDR, and deletion analyses defined the interaction interface to be the LBD of the receptor. Interestingly, the kinase domain of RIPK1 appears to be required for VDR binding even though its kinase activity is expendable for VDR repression. While the molecular mechanisms underlying VDR repression by RIPK1 remain largely to be defined, our studies suggest that increased cytoplasmic retention of VDR may be one of the underlying mechanisms. More importantly, both RIPK1 knockout and knockdown increased the sensitivity of cells to 1,25D3-induced growth suppression as well as the ability to induce target gene expression, demonstrating the significance and clinical relevance of the knowledge gained in our mechanistic studies. Overall, our studies suggest that the depletion of RIPK1 may be an effective way to increase the sensitivity of cancers to 1,25D3 and its analogs.
While the data in Figs. 1A and 1B clearly show a dose-dependent inhibition of VDR activity by RIPK1, it may appear odd that in Fig. 1C, Nec-1 significantly increased RIPK1 levels without further increasing the degree of VDR repression by RIPK1. There are two possible explanations for this. First, the amount of RIPK1 vector used for transfections in Fig. 1C was high and had already reduced VDR activity almost to its basal levels, thus leaving little room for further reduction. Second and more importantly, Nec-1 by itself increased VDR activity in the absence of ectopic RIPK1 expression (Fig. 1C), which may counteract its indirect effect on VDR through increased RIPK1 expression. Although the exact reasons that Nec-1 increased VDR activity in the absence of ectopic RIPK1 expression remains to be determined, it is most likely an off-target effect independent of RIPK1 kinase activity. Although Nec-1 has been perceived as a highly specific RIPK1 inhibitor, RIPK1-independent effects have been reported [37].
Our studies have clearly demonstrated that VDR repression by RIPK1 occurs independently of its kinase activity and is mostly likely mediated through its adaptor function as a VDR-interacting protein. Although our data suggest the increased cytoplasmic retention of VDR by RIPK1 binding as a mechanism underlying VDR repression, it should be noted that the nuclear localization signals had been identified as being in the DNA-binding domain and hinge region of VDR [41, 42] whereas the RIPK1 binding site was mapped to the LBD (Fig. 4). On the other hand, nuclear exporting signals had been located to the LBD of VDR [43]. It is thus perceivable that RIPK1 might promote VDR nuclear export. Alternatively, since our mapping analyses suggest that multiple regions of RIPK1 are involved in its binding to VDR, it is possible that, after the primary contact with the LBD is made, RIPK1 may interrupt the interaction between VDR and nuclear pore structures through steric hindrance by secondary contacts with other regions in VDR.
Since ectopic RIPK1 expression in 293T cells increased VDR cytoplasmic localization and decreased VDR activity, cytoplasmic localization may represent a mechanism underlying 1,25D3 resistance in cancer cells that overexpress RIPK1. However, it is important to point out that increased cytoplasmic localization may not be the only mechanism by which RIPK1 inhibits VDR activity. Besides hormone binding, the LBD is also known to be involved in mediating interactions between VDR and cofactors. Thus, RIPK1 binding may also cause VDR suppression by interfering with the recruitment of transcriptional coactivators. As shown in Figs. 8 and 9, RIPK1 knockdown in 1,25D3-sensitive OVCAR3 and PE01 cells increased 1,25D3-induced growth suppression as well as target gene expression. However, our cellular fractionation studies have revealed a predominantly nuclear VDR distribution in these cells; RIPK1 knockdown did not cause detectable changes in the subcellular distribution of VDR. Differing from ectopic RIPK1 in 293T cells, significant amounts of endogenous RIPK1 were detected in the nucleus of OVCAR3 and PE01 cells as it has been reported for MEFs and Hela cells [44]. It is certainly feasible for RIPK1 to bind to VDR in the nucleus in these cells to directly repress its activity by acting as a transcriptional co-repressor.
A large amount of epidemical and molecular studies have shown that vitamin D reduces cancer risks and that 1,25D3 analogs are promising agents for cancer treatments. However, the potential of 1,25D3 and its analogs in cancer intervention has yet to be realized in clinics. A majority of the published studies about vitamin D and cancer has been dedicated to pathways through which the hormone suppresses cancer growth and invasion; limited studies have been done to define the molecules and pathways that suppress VDR and cause resistance of cancer cells to 1,25D3 and its analogs [45]. RIPK1 is overexpressed in melanoma cells and causes resistance of multiple cancers to chemotherapeutic drugs [22–25, 46]. The present studies suggest that the expression of RIPK1 may also cause resistance to 1,25D3-induced growth suppression and identify RIPK1 as a molecular target for increasing the therapeutic potential of 1,25D3 analogs for cancer. The fact that VDR suppression is not mediated by the kinase activity of RIPK1 makes it more challenging to target RIPK1 for cancer intervention. However, technological advancements in small interference RNA and gene editing tools have made it possible to develop strategies for targeted depletion of RIPK1 in cancer. Alternatively, small molecules that interfere with the interaction between RIPK1 and VDR can also be developed and used together with 1,25D3 analogs for combinational cancer therapy.
Highlights.
RIPK1 forms a complex with VDR in vitro and in vivo
RIPK1 inhibits the transcriptional activity of VDR
RIPK1 depletion increases 1α,25-dihydroxyvitamin D3-induced growth suppression
Acknowledgments
The authors would like to thank Dr. Martin Leverkus of Heidelberg University for control and RIPK1 shRNA vectors, Dr. Junying Yuan of Harvard University for RIPK1+/+ and RIPK1−/− MEFs and RIPK1 kinase-dead mutant construct RIPK1 K45M, Dr. Toshiyasu Taniguchi of Fred Hutchinson Cancer Research Center for PE01 cells, and Dr. H. F. Deluca of University of Wisconsin-Madison for the p23 reporter plasmid Dr. Wilson H. Miller Jr. of McGill University for the pCMX-Flag-RXRα. The authors would also like to thank Catherine R. Bai for proofreading the manuscript. The studies were supported by Public Health Service grants CA111334 (to Bai) and CA164147 (to Zhang) from the National Institutes of Health and a project support as part of a program development grant from Ovarian Cancer Research Fund (to Bai).
Abbreviations
- 1,25D3
1α, 25-dihydroxyvitamin D3
- RIPK1
Receptor Interacting Protein Kinase 1
- VDR
Vitamin D receptor
Footnotes
Disclosure of potential conflicts of interest
The authors have no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hobaus J, et al. Role of calcium, vitamin D, and the extrarenal vitamin D hydroxylases in carcinogenesis. Anticancer Agents Med Chem. 2013;13(1):20–35. [PMC free article] [PubMed] [Google Scholar]
- 2.Bouillon R, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29(6):726–76. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welsh J. Vitamin D and prevention of breast cancer. Acta Pharmacol Sin. 2007;28(9):1373–82. doi: 10.1111/j.1745-7254.2007.00700.x. [DOI] [PubMed] [Google Scholar]
- 4.Welsh J. Vitamin D and cancer: integration of cellular biology, molecular mechanisms and animal models. Scand J Clin Lab Invest Suppl. 2012;243:103–11. doi: 10.3109/00365513.2012.682870. [DOI] [PubMed] [Google Scholar]
- 5.Feldman D, et al. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer. 2014;14(5):342–57. doi: 10.1038/nrc3691. [DOI] [PubMed] [Google Scholar]
- 6.Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7(9):684–700. doi: 10.1038/nrc2196. [DOI] [PubMed] [Google Scholar]
- 7.Jiang F, et al. G2/M arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through the induction of GADD45 via an exonic enhancer. J Biol Chem. 2003;278(48):48030–40. doi: 10.1074/jbc.M308430200. [DOI] [PubMed] [Google Scholar]
- 8.Li P, et al. p27(Kip1) stabilization and G(1) arrest by 1,25-dihydroxyvitamin D(3) in ovarian cancer cells mediated through down-regulation of cyclin E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2 ubiquitin ligase. J Biol Chem. 2004;279(24):25260–7. doi: 10.1074/jbc.M311052200. [DOI] [PubMed] [Google Scholar]
- 9.Shen Z, et al. The coupling of epidermal growth factor receptor down regulation by 1alpha,25-dihydroxyvitamin D3 to the hormone-induced cell cycle arrest at the G1-S checkpoint in ovarian cancer cells. Mol Cell Endocrinol. 2011;338(1–2):58–67. doi: 10.1016/j.mce.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang F, et al. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down-regulation of telomerase. J Biol Chem. 2004;279(51):53213–21. doi: 10.1074/jbc.M410395200. [DOI] [PubMed] [Google Scholar]
- 11.Kasiappan R, et al. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J Biol Chem. 2012;287(49):41297–309. doi: 10.1074/jbc.M112.407189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasiappan R, et al. Vitamin D suppresses leptin stimulation of cancer growth through microRNA. Cancer Res. 2014;74(21):6194–204. doi: 10.1158/0008-5472.CAN-14-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swami S, et al. Vitamin D mitigates the adverse effects of obesity on breast cancer in mice. Endocr Relat Cancer. 2016;23(4):251–64. doi: 10.1530/ERC-15-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lungchukiet P, et al. Suppression of epithelial ovarian cancer invasion into the omentum by 1alpha,25-dihydroxyvitamin D3 and its receptor. J Steroid Biochem Mol Biol. 2015;148:138–47. doi: 10.1016/j.jsbmb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7(4):243–9. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229–32. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–36. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 18.Zhou W, Yuan J. SnapShot: Necroptosis. Cell. 2014;158(2):464–464 e1. doi: 10.1016/j.cell.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 19.Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 20.O’Donnell MA, et al. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol. 2007;17(5):418–24. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ea CK, et al. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22(2):245–57. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 22.Liu XY, et al. RIP1 Kinase Is an Oncogenic Driver in Melanoma. Cancer Res. 2015;75(8):1736–48. doi: 10.1158/0008-5472.CAN-14-2199. [DOI] [PubMed] [Google Scholar]
- 23.Rosebeck S, et al. The API2-MALT1 fusion exploits TNFR pathway-associated RIP1 ubiquitination to promote oncogenic NF-kappaB signaling. Oncogene. 2014;33(19):2520–30. doi: 10.1038/onc.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Q, et al. Retaining MKP1 expression and attenuating JNK-mediated apoptosis by RIP1 for cisplatin resistance through miR-940 inhibition. Oncotarget. 2014;5(5):1304–14. doi: 10.18632/oncotarget.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, et al. Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway. J Biol Chem. 2014;289(9):5654–63. doi: 10.1074/jbc.M113.526152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geserick P, et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol. 2009;187(7):1037–54. doi: 10.1083/jcb.200904158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Degterev A, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiraki T, et al. Activation of orphan nuclear constitutive androstane receptor requires subnuclear targeting by peroxisome proliferator-activated receptor gamma coactivator-1 alpha. A possible link between xenobiotic response and nutritional state. J Biol Chem. 2003;278(13):11344–50. doi: 10.1074/jbc.M212859200. [DOI] [PubMed] [Google Scholar]
- 29.Mann KK, et al. Arsenic trioxide inhibits nuclear receptor function via SEK1/JNK-mediated RXRalpha phosphorylation. J Clin Invest. 2005;115(10):2924–33. doi: 10.1172/JCI23628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbour NC, et al. A highly sensitive method for large-scale measurements of 1,25-dihydroxyvitamin D. Anal Biochem. 1998;255(1):148–54. doi: 10.1006/abio.1997.2439. [DOI] [PubMed] [Google Scholar]
- 31.Vanhooke JL, et al. Molecular structure of the rat vitamin D receptor ligand binding domain complexed with 2-carbon-substituted vitamin D3 hormone analogues and a LXXLL-containing coactivator peptide. Biochemistry. 2004;43(14):4101–10. doi: 10.1021/bi036056y. [DOI] [PubMed] [Google Scholar]
- 32.Christofferson DE, et al. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012;3:e320. doi: 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakai W, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69(16):6381–6. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H, Bai W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002;22(16):5835–45. doi: 10.1128/MCB.22.16.5835-5845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 36.Lou YR, et al. 5alpha-dihydrotestosterone inhibits 1alpha,25-dihydroxyvitamin D3-induced expression of CYP24 in human prostate cancer cells. Prostate. 2005;63(3):222–30. doi: 10.1002/pros.20189. [DOI] [PubMed] [Google Scholar]
- 37.Cho Y, et al. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS One. 2011;6(8):e23209. doi: 10.1371/journal.pone.0023209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gurevich I, Flores AM, Aneskievich BJ. Corepressors of agonist-bound nuclear receptors. Toxicol Appl Pharmacol. 2007;223(3):288–98. doi: 10.1016/j.taap.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazar MA. Nuclear receptor corepressors. Nucl Recept Signal. 2003;1:e001. doi: 10.1621/nrs.01001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, et al. Suppression of death receptor-mediated apoptosis by 1,25-dihydroxyvitamin D3 revealed by microarray analysis. J Biol Chem. 2005;280(42):35458–68. doi: 10.1074/jbc.M506648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsieh JC, et al. Novel nuclear localization signal between the two DNA-binding zinc fingers in the human vitamin D receptor. J Cell Biochem. 1998;70(1):94–109. [PubMed] [Google Scholar]
- 42.Michigami T, et al. Identification of amino acid sequence in the hinge region of human vitamin D receptor that transfers a cytosolic protein to the nucleus. J Biol Chem. 1999;274(47):33531–8. doi: 10.1074/jbc.274.47.33531. [DOI] [PubMed] [Google Scholar]
- 43.Prufer K, Barsony J. Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol Endocrinol. 2002;16(8):1738–51. doi: 10.1210/me.2001-0345. [DOI] [PubMed] [Google Scholar]
- 44.Ramnarain DB, et al. RIP1 links inflammatory and growth factor signaling pathways by regulating expression of the EGFR. Cell Death Differ. 2008;15(2):344–53. doi: 10.1038/sj.cdd.4402268. [DOI] [PubMed] [Google Scholar]
- 45.Williams JD, et al. Tumor Autonomous Effects of Vitamin D Deficiency Promote Breast Cancer Metastasis. Endocrinology. 2016;157(4):1341–7. doi: 10.1210/en.2015-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luan Q, et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy. 2015;11(7):975–94. doi: 10.1080/15548627.2015.1049800. [DOI] [PMC free article] [PubMed] [Google Scholar]