

Abstract
Objectives
We have previously investigated an association between the genome copy number variation (CNV) and acetabular dysplasia (AD). Hip osteoarthritis is associated with a genetic polymorphism in the aspartic acid repeat in the N-terminal region of the asporin (ASPN) gene; therefore, the present study aimed to investigate whether the CNV of ASPN is involved in the pathogenesis of AD.
Methods
Acetabular coverage of all subjects was evaluated using radiological findings (Sharp angle, centre-edge (CE) angle, acetabular roof obliquity (ARO) angle, and minimum joint space width). Genomic DNA was extracted from peripheral blood leukocytes. Agilent’s region-targeted high-density oligonucleotide tiling microarray was used to analyse 64 female AD patients and 32 female control subjects. All statistical analyses were performed using EZR software (Fisher’s exact probability test, Pearson’s correlation test, and Student’s t-test).
Results
CNV analysis of the ASPN gene revealed a copy number loss in significantly more AD patients (9/64) than control subjects (0/32; p = 0.0212). This loss occurred within a 60 kb region on 9q22.31, which harbours the gene for ASPN. The mean radiological parameters of these AD patients were significantly worse than those of the other subjects (Sharp angle, p = 0.0056; CE angle, p = 0.0076; ARO angle, p = 0.0065), and all nine patients required operative therapy such as total hip arthroplasty or pelvic osteotomy. Moreover, six of these nine patients had a history of operative or conservative therapy for developmental dysplasia of the hip.
Conclusions
Copy number loss within the region harbouring the ASPN gene on 9q22.31 is associated with severe AD. A copy number loss in the ASPN gene region may play a role in the aetiology of severe AD.
Cite this article: T. Sekimoto, M. Ishii, M. Emi, S. Kurogi, T. Funamoto, Y. Yonezawa, T. Tajima, T. Sakamoto, H. Hamada, E. Chosa. Copy number loss in the region of the ASPN gene in patients with acetabular dysplasia: ASPN CNV in acetabular dysplasia. Bone Joint Res 2017;6:439–445. DOI: 10.1302/2046-3758.67.BJR-2016-0094.R1.
Keywords: Acetabular dysplasia, Copy number variation, ASPN gene
Article focus
An association between acetabular dysplasia (AD) and chromosomal structural rearrangements has only been investigated in our previous study.
We conducted a copy number variation (CNV) analysis of the ASPN gene region in cases with varying degrees of acetabular coverage.
We investigated the association between the CNV of the ASPN gene and AD to identify a possible genetic structural variation that confers a risk of AD.
Key messages
A significant loss of copy number was detected in the ASPN gene region in AD patients.
The ASPN gene copy number loss may be an independent risk factor for developing AD, and could be a predictive tool to identify severe AD patients.
Strengths and limitations
This is the first report on the association of the CNV of the ASPN gene region with AD.
The ASPN gene copy number loss showed significant association with severe AD.
Elucidation in different ethnicities in larger sets of patients and controls will be warranted for validation of the findings in the near future.
Introduction
Acetabular dysplasia (AD) is a major and common cause of secondary hip osteoarthritis (OA), and is a multifactorial disease that may involve both genetic and environmental factors. AD is characterised by a shallow hip socket and decreased coverage of the femoral head, which predisposes the hip joints to progressive damage of the cartilage, leading to OA. A nationwide epidemiological study of hip OA in Japan showed that the aetiology of hip OA was AD in 390 of 485 patients (80.4%), and the relative frequency of AD among hip OA patients was significantly greater in women (361; 92.6%) than in men (29; 7.4%).1
AD is associated with abnormally high stress on the anterolateral part of the acetabulum, which leads to degeneration of the articular cartilage. This can cause hip pain and dysfunction sufficient for the patient to require medical evaluation and treatment. The severity of AD ranges from barely detectable to severe hip malformation or dislocation. The pathogenesis of AD remains unclear, but it appears to be related to several different factors such as racial background, gender, and intrauterine positioning.1,2 Few studies have investigated the association between genetic variation and AD.3-5
Recently, copy number variations (CNVs) have been recognised as inter-individual genetic variations.6 CNVs represent structural rearrangements of the genome such as deletion or gain (including duplication and triplication). Sporadic diseases could be caused by different types of mutations, such as point mutations and genomic rearrangements. Several CNV abnormalities are significant factors in the aetiology of various diseases such as Charcot-Marie-Tooth disease, X-linked disease, and Smith-Magenis syndrome.7 To date, an association between CNV and AD has only been examined in our previous study.8 In this study we conducted a whole-genome CNV analysis, and identified a copy number loss within the region harbouring the Semaphorin 4D gene in 18 of 64 AD patients.
Asporin (ASPN) is an extracellular matrix protein that contains a unique aspartic acid (D) repeat in the N-terminal region.9,10 ASPN belongs to the small leucine-rich proteoglycan (SLRP) family; members of the SLRP family bind to transforming growth factor beta (TGF-β), a key growth factor in cartilage metabolism, and to other extracellular matrix molecules of cartilage, including collagens.11 ASPN mRNA is expressed abundantly in OA articular cartilage,9 and previous studies have implicated SLRP family genes in the aetiology of OA.12,13 Kizawa et al14 reported a significant association between a polymorphism in the D repeat of the gene encoding ASPN and OA in the Japanese population. However, the potential association between AD and chromosomal structural rearrangements of the ASPN gene region has not yet been studied.
In the present study, we investigated the association between CNV and AD to identify a possible gross genetic structural variation that confers a risk of AD. We conducted a CNV analysis of the ASPN gene region using genomic DNA extracted from peripheral blood of subjects with varying degrees of acetabular coverage.
Materials and Methods
Sample size calculation
A sample size calculation was performed using EZR software version 1.27 (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria). More precisely, it is a modified version of R commander designed to add statistical functions frequently used in biostatistics.15 The sample size calculation was based on a type I error probability of 0.05 (two-sided), 70% power, an assumed ASPN copy number loss rate of 0.3 for AD, and a minimal detectable ASPN CNV of 0.05. The study was powered to detect a change in the anticipated ASPN copy number loss rate of 0.3 for AD, as estimated on the basis of previous reports for CNV on sporadic diseases.8,16-18 According to the sample size calculation for this case-control design study, the minimum sample sizes required were 58 evaluable patients and 29 controls.
Study subjects and radiographs
We recruited 64 consecutive unrelated Japanese female patients diagnosed with AD and 32 healthy female volunteers from our hospital during the same period. A total of 96 subjects living in our city or neighbouring areas were studied. All participants gave written informed consent for the genetic analysis, and this study was approved by the medical ethics committees of our research facility.
In this study, all subjects were Japanese females who underwent clinical, radiological, and blood examinations by orthopaedic specialists. To exclude other diseases such as inflammatory arthritis (rheumatoid, polyarthritic, or autoimmune disease), post-traumatic or post-septic arthritis, and a definitely positive Mendelian family history, all subjects completed a standard questionnaire regarding their medical history. All AD cases were sporadic in nature. Case status was determined according to the examination data and symptoms. Some individuals were undergoing medical treatment and/or had received operative therapy for AD (Fig. 1b). The control group had no signs or symptoms of arthritis or joint disease (pain, swelling, tenderness, or restriction of movement). No subjects dropped out during the process of the study. The respective mean ages of the 64 AD patients and 32 control subjects were 51.3 years (sd 15.2, 21 to 75) and 35.5 years (sd 9.5, 29 to 68).

Anteroposterior radiographs of acetabular dysplasia (AD) pre- and post-operatively, and a control hip: a) case 61: AD hip of a 42-year-old female; Sharp angle 56.3°; centre-edge (CE) angle 3.5°; acetabular roof obliquity (ARO) angle 29.6°; minimum joint space width (MJSW) 3.7 mm; developmental dyaplasia of the hip, positive; b) case 61: post-periacetabular osteotomy; c) control 26: normal hip of a 30-year-old female; Sharp angle 38.6°; CE angle 28.8°; ARO angle 9.9°; MJSW 3.3 mm.
The evaluation of acetabular coverage was based on radiological findings. Radiographs were taken with a tube-to-film distance of 100 cm. The pelvises were in a supine position with the frontal anatomical plane parallel to the film plate. The central beam was directed at the midpoint between the pubic symphysis and a horizontal line connecting bilateral anterior superior iliac spines. All radiographs fulfilled the criteria for correct pelvic positioning regarding the axial and transverse pelvic rotation. The Sharp angle was defined as the angle formed between a line from the inferior pelvic teardrop to the superior lateral edge of the acetabulum and a line horizontal from the inferior pelvic teardrop.19 The centre-edge (CE) angle was defined as the angle formed by a vertical line and a line connecting the femoral head centre with the lateral edge of the acetabulum.20 The acetabular roof obliquity (ARO) angle was defined as the angle formed by a horizontal line and a line connecting the medial point of the sclerotic zone with the lateral edge of the acetabulum.21 The minimum joint space width (MJSW) was defined as the shortest distance between the femoral head margin and the acetabulum in the weight-bearing zone, as defined by the medial and lateral aspects of the acetabular sourcil.22 The diagnosis of AD was based on the presence of one of the following radiological findings: Sharp angle ⩾ 42°, CE angle ⩽ 25°, or ARO ⩾ 15° on at least one side (Fig. 1a). The radiological parameters in each case were independently measured twice by two authors (TS and EC). We could not evaluate the CE angle and MJSW in case 39 as it had a bilaterally high congenital dislocation of the hip.
We analysed the high-density oligonucleotide array using the genomic DNA extracted from peripheral blood leukocytes of the 64 female AD patients, and also that of the 32 female control subjects with Sharp angle < 42°, CE angle > 25°, and ARO < 15° on both sides (Fig. 1c).
High-density custom-made oligonucleotide tiling microarray
Agilent’s high-density oligonucleotide tiling microarray assay (Agilent Technologies, Santa Clara, California) was performed on a custom-designed microarray that targeted the 60 kb ASPN gene region on 9q22.31 (Chr9:94 250 000 to 94 310 000; NCBI Build 36.1, hg18), as described previously.23 Data analysis of the microarray experiments was conducted using the Aberration Detection Method-2 statistical algorithm (Agilent Technologies), based on the combined log2 ratios at a threshold of five, as was undertaken in our previous study.8 The data were centralised, and calls with average log2 ratios < 0.15 were filtered to exclude false positives. Abnormal copy number, losses and gains, in a complex multi-copy variable region by high-density tiling array, were identified by deviation of probe log2 ratios that exceeded a given threshold of 1.5 sd from the mean probe ratio.24,25
Statistical analysis
All numerical values are expressed as the mean and sd. Statistical analyses of the clinical data were performed with Fisher’s exact probability test, Pearson’s correlation test, and Student’s t-test. Values of p < 0.05 were considered statistically significant. All statistical analyses were performed using EZR software version 1.27 (Saitama Medical Center, Jichi Medical University, Saitama, Japan).
Results
Radiological parameters of the acetabular dysplasia patients and control subjects
The mean radiological parameters on the worst side of the AD patients and the control subjects are shown in Table I. These radiological parameters correlated significantly with each other (Table II).
Table I.
Mean radiological parameters in the acetabular dysplasia and control groups
| Radiological parameter | AD group (n = 64) | Control group (n = 32) | p-value (t-test) |
|---|---|---|---|
| Sharp angle (°) | 51.9 (3.5; 44.5 to 60.3) | 39.5 (2.0; 34.0 to 41.6) | 7.58E-33 |
| CE angle (°) | 3.1 (11.9; -24.5 to 23.9)* | 32.9 (4.1; 26.1 to 42.8) | 4.52E-24 |
| ARO angle (°) | 29.7 (8.0; 7.7 to 51.5) | 4.5 (4.0; -3.9 to 12.0) | 6.87E-30 |
| MJSW (mm) | 2.5 (1.8; 0 to 7.3)* | 3.9 (0.6; 3.3 to 5.5) | 8.12E-5 |
case 39 had a bilaterally high congenital dislocation of the hip and was excluded
Values on the worst side are presented as mean (sd; range)
AD, acetabular dysplasia; CE, centre edge; ARO, acetabular roof obliquity; MJSW, minimum joint space width
Table II.
Correlation coefficient between radiological parameters in all subjects
| Radiological parameter (n = 95)* | Correlation coefficient | p-value (Pearson’s correlation) |
|---|---|---|
| Sharp angle to CE angle | -0.810 | 3.13E-23 |
| Sharp angle to ARO angle | 0.867 | 6.76E-30 |
| Sharp angle to MJSW | -0.300 | 0.003 |
| CE angle to ARO angle | -0.893 | 5.43E-34 |
| CE angle to MJSW | 0.434 | 1.12E-5 |
| ARO angle to MJSW | -0.387 | 0.0001 |
case 39 had a bilaterally high congenital dislocation of the hip and was excluded
CE, centre edge; ARO, acetabular roof obliquity; MJSW, minimum joint space width
High-density tiling microarray analysis of the acetabular dysplasia patients and control subjects
We analysed the extent of the copy number change along the 60 kb genomic region of 9q22.31 harbouring the ASPN gene using a custom-made high-density oligonucleotide tiling microarray. A loss of copy number was found in nine of the 64 AD patients, but in none of the 32 control subjects (9/64 vs 0/32, p = 0.0212, Fisher’s exact probability test). Figure 2 shows the detailed structure of the segmental loss in nine AD patients and two controls along the 60 kb region (AD patients: Figs 2a to 2i; control subjects: Figs 2j and 2k). Figure 3 shows a schematic of the extent of the loss of copy number in the ASPN gene region in all nine of the AD patients who exhibited loss. In cases 4, 20, 39, 55, and 61, the range of the loss covered the upper side of the ASPN gene, including the ASPN D repeat region in exon 2; in the other cases, the range covered the whole ASPN gene (Fig. 3).
Fig. 2.
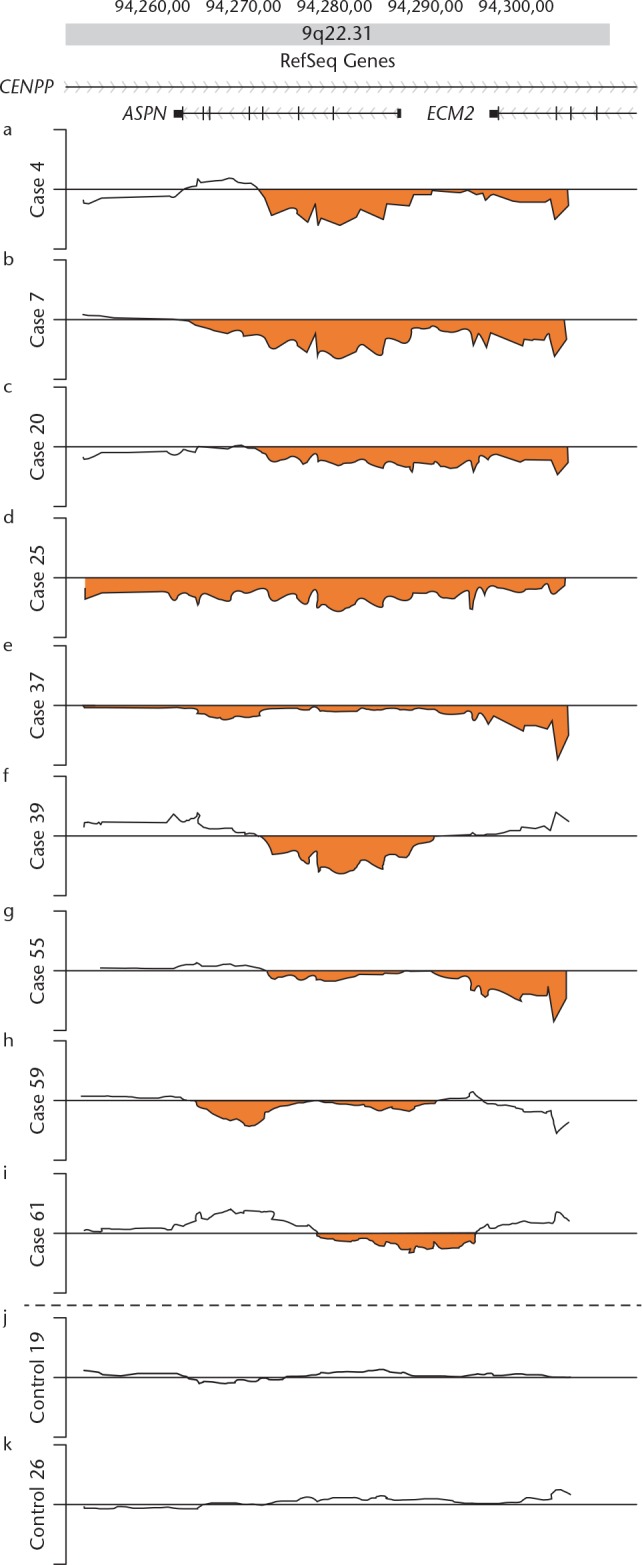
Fine structures of the loss of copy number in the 60 kb 9q22.31 region harbouring the ASPN gene resolved by Agilent high-density tiling microarray. Log2 ratio (y-axis) was plotted using the moving mean along the genome position (x-axis). Plots for the nine affected acetabular dysplasia patients (cases 4, 7, 20, 25, 37, 39, 55, 59, and 61) are shown in parallel with plots of two control subjects (controls 19 and 26). The dark line represents a copy number plot along the genome for each subject between genomic positions 94 250 000 (left) and 94 310 000 (right). The orange areas indicate ‘‘loss of copy number’’. The top map shows the positions of putative genes in the region.35 CENPP, centromere protein p; ASPN, asporin; ECM2, extracellular matrix protein 2.
Fig. 3.

Extent of the loss of copy number in the 60 kb region around the ASPN gene on 9q22.31 in the nine affected acetabular dysplasia patients. The horizontal bars represent the extent of the loss region in each case between the genomic positions 94 250 000 (left) and 94 310 000 (right). The top map shows the positions of putative genes in the region.35 CENPP, centromere protein p; ASPN, asporin; ECM2, extracellular matrix protein 2.
Characteristics of the acetabular dysplasia patients with positive copy number loss in the ASPN gene
The Sharp angle, CE angle, and ARO angle in the AD patients with loss of copy number and in the other 87 subjects with no loss in the ASPN gene are shown in Figure 4. The Sharp angle in the nine AD patients with loss was 53.6° (sd 3.6°), which was significantly larger than that in the 87 subjects that had no loss (47.2°, sd 6.6°; p = 0.0056, Student’s t-test). The CE angle in the eight AD patients with loss was -2.3° (sd 12.9°), which was significantly smaller than that in the 87 subjects with no loss (14.6°, sd 17.0°; p = 0.0076, t-test). The ARO angle in the nine AD patients with loss was 33.1° (sd 6.6°), which was significantly larger than that in the 87 subjects with no loss (20.1°, sd 13.8°; p = 0.0065, t-test). The MJSW in the eight AD patients with loss of copy number was 2.0 mm (sd 1.8 mm), which was not significantly different from that in the 87 patients with no loss (3.1 mm, sd 1.6 mm; p = 0.067, t-test). These nine AD cases all needed operative therapy, either pelvic osteotomy or total hip arthroplasty (Fig. 1b, Table III). Six of the nine AD cases had history of operative or conservative therapy for developmental dysplasia of the hip (DDH) (Table III).
Fig. 4.

Sharp angle, centre-edge (CE) angle, and acetabular roof obliquity (ARO) angle in acetabular dysplasia patients with positive loss of copy number in the ASPN gene compared with those in the 87 subjects with no loss in the ASPN gene. The vertical bar represents the extent of the radiological angle in each group. The orange round points indicate the mean radiological parameters, and the bars indicate the standard deviation.
Table III.
Characteristics of the nine acetabular dysplasia patients with positive loss of copy number in the ASPN gene
| Case | Age | Sharp angle | CE angle | ARO angle | MJSW | DDH | Surgical plan |
|---|---|---|---|---|---|---|---|
| yrs | rt/lt (°) | rt/lt (°) | rt/lt (°) | rt/lt (mm) | rt/lt | ||
| Case 4 | 59 | 50.0 / 49.8 | 14.1/15.2 | 28.5/23.4 | 2.4/3.5 | + | PO / PO |
| Case 7 | 51 | 49.1 / 45.1 | 8.7/19.1 | 30.1/15.2 | 1.9/4.5 | – | PO / – |
| Case 20 | 22 | 53.1 / 51.6 | 6.2/14.9 | 31.0/20.9 | 3.9/4.8 | – | PO / PO |
| Case 25 | 47 | 60.3 / 56.2 | -14.4/8.5 | 45.4/31.1 | 0/0 | + | THA / THA |
| Case 37 | 26 | 51.1 / 50.4 | 0/5.5 | 24.6/22.5 | 0/3.2 | – | THA / PO |
| Case 39* | 67 | 52.6 / 55.3 | –/– | 30.5/31.3 | –/– | + | THA / THA |
| Case 55 | 69 | 51.7 / 51.2 | -19.5/-7.4 | 40.7/37.5 | 0/3.0 | + | THA / PO |
| Case 59 | 45 | 52.3 / 55.4 | -5.4/-16.9 | 35.7/36.9 | 4.5/3.7 | + | PO / PO |
| Case 61 | 42 | 56.3 / 52.7 | 3.5/9.5 | 29.6/22.2 | 3.7/3.5 | + | PO / PO |
Case 39 had a bilaterally high congenital dislocation of the hip
CE, centre edge; ARO, acetabular roof obliquity; MJSW, minimum joint space width; DDH, developmental dysplasia of the hip; rt, right side; lt, left side; PO, pelvic osteotomy; THA, total hip arthroplasty
Discussion
A nationwide epidemiological study of hip OA in Japan showed that the relative frequency of AD among 485 hip OA patients was 361 (92.6%) in women and 29 (7.4%) in men of 390 AD patients,1 which indicates that the aetiology of AD may differ between genders. Therefore, in the present study, we investigated the CNV of the ASPN gene only in female patients. Our data suggest that Sharp angle, CE angle, ARO angle, and MJSW are significantly worse in AD patients (Table I). All the radiological parameters of the hip in 95 subjects significantly correlated with each parameter (Table II). Sharp angle, CE angle, and ARO angle showed a strong correlation either positively or inversely with each other, which is in accordance with previous studies.26,27 This means that each parameter could predict the other parameters, and suggests that Sharp angle, CE angle, and ARO angle are complementary for defining hip diseases such as AD and pincer-type femoroacetabular impingement.
Kizawa et al14 previously identified ASPN as a susceptibility gene for knee and hip OA in the Japanese population. They detected seven alleles of the ASPN D repeat, containing D12 to D18 residues, and found that allele D14 was significantly more common in patients with OA, and allele D13 was significantly less common in OA patients compared with controls.14 In the present study, there was a significant loss of copy number in the ASPN gene, including the ASPN D repeat region, of nine AD patients. Moreover, significantly worse radiological parameters were shown in the AD patients (Fig. 4). All nine cases needed operative therapy, and six of the nine cases had a history of therapy for DDH (Table III), indicating that loss of copy number within the region harbouring the ASPN gene on 9q22.31 was associated with severe AD. This is the first report to demonstrate the association of the CNV of ASPN with AD.
CNVs or structural variations, such as a deletion or a gain of a genomic region, are increasingly being recognised as important inter-individual genetic variations across the human genome. Each variation could range in size from about 1000 nucleotide bases to several megabases.7 As with other types of genetic variation, CNVs are significant contributors to human genetic disease and susceptibility. CNVs account for more nucleotide variation between two individuals than single-nucleotide polymorphisms.28,29 The Agilent high-density custom-made array is a high-resolution array based on the comparative genomic hybridisation method, with 60 mer probes synthesised in situ as oligonucleotide arrays, at a spacing of 100 bp to 500 bp.8,17,30 In the present study, the CNV region analysed on 9q22.31 and its flanking region contained the coding regions of the ASPN gene and the extracellular matrix protein 2 (ECM2) gene (Figs 2 and 3). This region contains multiple low-copy repeats in a small region of the genome, such as the ASPN D, which account for the instability of this region and trigger a copy number loss by an unequal crossing-over or end-joining event.31,32 These characteristics suggest the possible involvement of a de novo occurrence of a loss of copy number in AD patients, and have mechanistic implications related to the sporadic nature of the disease. Although CNV was detected in the ECM2 gene in six patients (Figs 2 and 3), there have been no previous reports on skeletal phenotypes for the ECM2 gene. ECM2 is mainly expressed in adipose tissue as well as the mammary glands, ovaries, and uterus.33 Moreover, ECM2 is related to B lymphopoiesis.34 Hence, we consider that the ECM2 gene would not be a suitable candidate for risk susceptibility to skeletal disorders such as AD. On the other hand, gene(s) in this region may predispose to AD; of these genes, ASPN is considered a strong candidate based on previous reports.9-14 We speculated that the ASPN gene in this region may be involved in the pathogenesis of AD through gene functional impairments caused by loss of copy number. Our findings indicate that decreased ASPN expression may lead to susceptibility to AD.
In conclusion, our study provides evidence for loss of genome copy number encompassing the ASPN gene region in nine AD patients. This ASPN loss may be an independent risk factor for severe AD of the hip. We analysed the AD case population using a high-density custom CNV array. As AD case populations were not large and the significance was not so high, more long-term studies should be conducted with larger sample numbers, including both genders in different ethnic groups for validation of the findings in the near future. Recent advances in DNA sequencing technology have further enabled the identification of CNVs by next-generation sequencing. Further studies on the potential relevance of ASPN function to AD will contribute not only to the determination of the pathogenesis of hip OA, but also to the identification of a novel therapeutic target for the disease. The CNV of ASPN could be a predictive tool to identify severe AD patients, and potentially delay or prevent the onset of end-stage OA of the hip by screening patients at a young age. Further study is required, however, in future.
Footnotes
Author Contributions: T. Sekimoto: Study design, Data collection and analysis, Writing the paper.
M. Ishii: Data collection and analysis, Editorial contribution.
M. Emi: Study design, Data collection and analysis, Editorial contribution.
S. Kurogi: Data collection.
T. Funamoto: Data collection.
Y. Yonezawa: Statistical analysis.
T. Tajima: Data collection.
T. Sakamoto: Data collection.
H. Hamada: Data collection.
E. Chosa: Editorial contribution.
Associated Societies: Japanese Orthopaedic Association, Japanese Hip Society.
Conflicts of Interest Statement: None declared
Funding Statement
This study was supported by a Grant-in-Aid from the Hip Joint Foundation of Japan.
We thank W. Souma, I. Tsuchimochi, Y. Ozawa, A. Saito, and M. Nagata for their technical support in the laboratory. We are also grateful to H. Sato, N. Ito, H. Iijima, R. Matoba, and K. Matsubara at the DNA Chip Research Institute for their scientific advice.
References
- 1. Jingushi S, Ohfuji S, Sofue M, et al. Multiinstitutional epidemiological study regarding osteoarthritis of the hip in Japan. J Orthop Sci 2010;15:626-631. [DOI] [PubMed] [Google Scholar]
- 2. Pollet V, Percy V, Prior HJ. Relative risk and incidence for developmental dysplasia of the hip. J Pediatr 2017;181:202-207. [DOI] [PubMed] [Google Scholar]
- 3. Sekimoto T, Kurogi S, Funamoto T, et al. Possible association of single nucleotide polymorphisms in the 3’ untranslated region of HOXB9 with acetabular overcoverage. Bone Joint Res 2015;4:50-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mabuchi A, Nakamura S, Takatori Y, Ikegawa S. Familial osteoarthritis of the hip joint associated with acetabular dysplasia maps to chromosome 13q. Am J Hum Genet 2006;79:163-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dai J, Shi D, Zhu P, et al. Association of a single nucleotide polymorphism in growth differentiate factor 5 with congenital dysplasia of the hip: a case-control study. Arthritis Res Ther 2008;10:R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature 2006;444:444-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet 2007;39(Suppl):S43-S47. [DOI] [PubMed] [Google Scholar]
- 8. Sekimoto T, Ishii M, Emi M, et al. Segmental copy number loss in the region of Semaphorin 4D gene in patients with acetabular dysplasia. J Orthop Res 2013;31:957-961. [DOI] [PubMed] [Google Scholar]
- 9. Lorenzo P, Aspberg A, Onnerfjord P, et al. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem 2001;276:12201-12211. [DOI] [PubMed] [Google Scholar]
- 10. Henry SP, Takanosu M, Boyd TC, et al. Expression pattern and gene characterization of asporin. a newly discovered member of the leucine-rich repeat protein family. J Biol Chem 2001;276:12212-12221. [DOI] [PubMed] [Google Scholar]
- 11. Ameye L, Young MF. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology 2002;12:107R-116R. [DOI] [PubMed] [Google Scholar]
- 12. Gill MR, Oldberg A, Reinholt FP. Fibromodulin-null murine knee joints display increased incidences of osteoarthritis and alterations in tissue biochemistry. Osteoarthritis Cartilage 2002;10:751-757. [DOI] [PubMed] [Google Scholar]
- 13. Ameye L, Aria D, Jepsen K, et al. Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis. FASEB J 2002;16:673-680. [DOI] [PubMed] [Google Scholar]
- 14. Kizawa H, Kou I, Iida A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet 2005;37:138-144. [DOI] [PubMed] [Google Scholar]
- 15. Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant 2013;48:452-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blauw HM, Veldink JH, van Es MA, et al. Copy-number variation in sporadic amyotrophic lateral sclerosis: a genome-wide screen. Lancet Neurol 2008;7:319-326. [DOI] [PubMed] [Google Scholar]
- 17. Kato T, Emi M, Sato H, et al. Segmental copy-number gain within the region of isopentenyl diphosphate isomerase genes in sporadic amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2010;402:438-442. [DOI] [PubMed] [Google Scholar]
- 18. Sasaki H, Emi M, Iijima H, et al. Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Mol Brain 2011;4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharp IK. Acetabular dysplasia: the acetabular angle. J Bone Joint Surg [Br] 1961;43-B:268-272. [Google Scholar]
- 20. Wiberg G. Studies on dysplastic acetabula and congenital subluxation of the hip joint: with special reference to the complication of osteoarthritis. Acta Chir Scand 1939;83(Suppl 58):28-38. [Google Scholar]
- 21. Massie WK, Howorth MB. Congenital dislocation of the hip. Part I. Method of grading results. J Bone Joint Surg [Am] 1950;32-A:519-531. [PubMed] [Google Scholar]
- 22. Terjesen T, Gunderson RB. Radiographic evaluation of osteoarthritis of the hip: an inter-observer study of 61 hips treated for late-detected developmental hip dislocation. Acta Orthop 2012;83:185-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Smith AJ, Tsalenko A, Sampas N, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet 2007;16:2783-2794. [DOI] [PubMed] [Google Scholar]
- 24. Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 2006;38:1038-1042. [DOI] [PubMed] [Google Scholar]
- 25. Lupski JR. Genomic disorders ten years on. Genome Med 2009;1:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Umer M, Thambyah A, Tan WT, Das De S. Acetabular morphometry for determining hip dysplasia in the Singaporean population. J Orthop Surg (Hong Kong). 2006;14:27-31. [DOI] [PubMed] [Google Scholar]
- 27. Park JM, Im GI. The correlations of the radiological parameters of hip dysplasia and proximal femoral deformity in clinically normal hips of a Korean population. Clin Orthop Surg 2011;3:121-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conrad DF, Pinto D, Redon R, et al. ; Wellcome Trust Case Control Consortium. Origins and functional impact of copy number variation in the human genome. Nature 2010;464:704-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park H, Kim JI, Ju YS, et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet 2010;42:400-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kudo H, Emi M, Ishigaki Y, et al. Frequent loss of genome gap region in 4p16.3 subtelomere in early-onset type 2 diabetes mellitus. Exp Diabetes Res 2011. (Epub). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Itsara A, Cooper GM, Baker C, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet 2009;84:148-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharp AJ, Locke DP, McGrath SD, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 2005;77:78-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishiu J, Tanaka T, Nakamura Y. Identification of a novel gene (ECM2) encoding a putative extracellular matrix protein expressed predominantly in adipose and female-specific tissues and its chromosomal localization to 9q22.3. Genomics 1998;52:378-381. [DOI] [PubMed] [Google Scholar]
- 34. Oritani K, Kanakura Y, Aoyama K, et al. Matrix glycoprotein SC1/ECM2 augments B lymphopoiesis. Blood 1997;90:3404–3413. [PubMed] [Google Scholar]
- 35. No authors listed. The UCSC Genome Browser Gateway. https://genome.ucsc.edu/cgi-bin/hgGateway (date last accessed 15 May 2017).