

Abstract
Estrogens and androgens influence the growth and maintenance of the mammalian skeleton and are responsible for its sexual dimorphism. Estrogen deficiency at menopause or loss of both estrogens and androgens in elderly men contribute to the development of osteoporosis, one of the most common and impactful metabolic diseases of old age. In the last 20 years, basic and clinical research advances, genetic insights from humans and rodents, and newer imaging technologies have changed considerably the landscape of our understanding of bone biology as well as the relationship between sex steroids and the physiology and pathophysiology of bone metabolism. Together with the appreciation of the side effects of estrogen-related therapies on breast cancer and cardiovascular diseases, these advances have also drastically altered the treatment of osteoporosis. In this article, we provide a comprehensive review of the molecular and cellular mechanisms of action of estrogens and androgens on bone, their influences on skeletal homeostasis during growth and adulthood, the pathogenetic mechanisms of the adverse effects of their deficiency on the female and male skeleton, as well as the role of natural and synthetic estrogenic or androgenic compounds in the pharmacotherapy of osteoporosis. We highlight latest advances on the crosstalk between hormonal and mechanical signals, the relevance of the antioxidant properties of estrogens and androgens, the difference of their cellular targets in different bone envelopes, the role of estrogen deficiency in male osteoporosis, and the contribution of estrogen or androgen deficiency to the monomorphic effects of aging on skeletal involution.
I. INTRODUCTION
Estrogens and androgens promote the acquisition of bone mass during puberty and help to maintain it thereafter. A decline of estrogen levels in females at menopause or estrogens and androgens in males later in life leads to loss of bone mass and strength and contributes to the development of osteoporosis, one of the most common metabolic disorders of old age (254, 333, 520).
In this review, we provide a comprehensive treatise of the role of estrogens and androgens in bone physiology and pathophysiology. After an introduction of the fundamental principles of bone biology for the nonexperts in the subject, we will review the molecular mechanisms of action and specific effects of estrogens and androgens on bone cells and other cell types and organs relevant to skeletal homeostasis, the effects of these two hormones on the skeleton during growth and adulthood, the pathological mechanisms causing the adverse effects of estrogen or androgen deficiency on skeletal homeostasis in either sex, and the pharmacology of natural and synthetic estrogenic or androgenic compounds used for the treatment of osteoporosis. Specific emphasis will be placed on relatively newer advances, including the crosstalk with mechanical loading, the antioxidant properties of estrogens and androgens, the difference of their cellular targets in different bone compartments, the contribution of estrogen deficiency to osteoporosis in males, and finally the contribution of estrogen or androgen deficiency to the effects of old age on the skeleton.
A. The Structure and Function of Bone
The skeleton is one of the most structurally complex and heterogeneous tissues in mammals. In adult humans it comprises a total of 206 bones of widely varying shapes and sizes. It is subdivided into an axial component, which includes the skull, spine, sternum, and the ribs, and an appendicular component that comprises long bones like the femur and radius. Its main functions are protection of internal organs and provision of levers for muscles during locomotion. Additionally, the skeleton houses the bone marrow, provides a critical niche for hematopoiesis, and serves as reservoir for calcium, phosphate, and carbonate.
Bone comprises specialized cell types and mineralized, as well as nonmineralized connective tissue matrix called osteoid, which form cortical and cancellous (also known as trabecular) structures (Figure 1). It also contains spaces that include the bone marrow cavity and vascular canals as well as lacunae and canaliculi that surround the bodies and dendritic processes of cells embedded in the mineralized matrix. Cortical bone is relatively solid and compact and represents ∼80% of the skeleton. It comprises the shafts of the long bones (e.g., femur and tibia), the shell of the vertebrae, and the surfaces of flat bones like the cranium or the pelvis. In higher mammals, cortical bone at the microscopic level is made by cylindrical concentric layers of lamellae that are crossed perpendicularly in the middle by canals in which lie the blood vessels. This organizational system is known as Haversian or osteonal. Cancellous bone has a honey comb-like appearance and consists of interconnected plates and strands. Cancellous bone is found mainly inside the ends of long bones and flat bones and is surrounded by the bone marrow. The ratio of surface to volume is much higher in cancellous than cortical bone, although this may change eventually in old age due to extensive cancellous bone loss and increasing cortical porosity (452).
FIGURE 1.

Micro-CT images of 6- and 24-month-old murine femurs depicting the cortical and cancellous envelopes. Higher resolutions of the distal epiphyses, the areas contained in the red boxes, are provided next to the images of the whole femurs. Please note the thinning of the cortex, the virtual disappearance of the cancellous bone, and the extensive cortical porosity in the 24-month-old femur as compared with the 6-month-old femur.
B. The Executive Cells of Bone
Bone is formed and removed by two highly specialized and terminally differentiated bone cell types: osteoblasts, which are responsible for the deposition of new bone matrix and its mineralization, and osteoclasts, which are uniquely capable of resorbing the mineralized matrix (324). Osteoblasts are the progeny of the mesenchymal cell lineage, whereas osteoclasts derive from hematopoietic precursors (314, 494) (Figure 2).
FIGURE 2.

Schematic representation of the remodeling process and the effects of estrogens and androgens. Osteoclasts and osteoblasts are derived from hematopoietic and mesenchymal precursors, respectively. During bone remodeling, bone matrix excavated by osteoclasts is replaced with new matrix produced by osteoblasts. Both estrogens and androgens influence the generation and lifespan of osteoclasts and osteoblasts, as well as the lifespan of osteocytes. Negative and positive effects of sex steroids on the generation and survival of the cells are depicted, by bookends and arrowheads.
The differentiation of pluripotent mesenchymal progenitors to osteoblasts is dependent on the sequential expression of two key transcription factors: the Runt domain-containing RUNX2 and OSX1, which contains three C2H2-type zinc-fingers (314). Wnts, a family of glycoproteins, engage membrane-associated Frizzled receptors and their co-receptors low-density lipoprotein receptor-related protein 5 (LRP5) or LRP6 in RUNX2/OSX 1 expressing cells and activate intracellular pathways, some of which are dependent on β-catenin (32, 314). This event promotes the replication and progression of Runx2/Osx1 cells to osteoblasts, and it is indispensable for the generation of osteoblasts. Additionally, canonical Wnt/β-catenin signaling helps to maintain a population of undifferentiated, proliferating progenitor mesenchymal stem cell (MSC), whereas noncanonical Wnts facilitate osteogenic differentiation (56). Simultaneously, canonical Wnt signaling suppresses the commitment of the pluripotent MSCs to the chondrogenic and adipogenic lineage (431). Wnts also prolong the lifespan of osteoblasts by preventing their apoptosis by both β-catenin-dependent and independent pathways (12). Genetic evidence from both humans and mice during the last few years has established that by virtue of its ability to control the supply of osteoblasts, β-catenin-dependent WNT signaling is the master regulator of bone mass accrual during postnatal life (32).
The generation of osteoclasts from progenitors of the monocyte/macrophage lineage is a multistep process (494). Initially, bone marrow macrophages differentiate into tartrate-resistant acid phosphatase (TRAP)-positive preosteoclasts. These cells then fuse with each other to form multinucleated osteoclasts. In humans, the number of nuclei per osteoclast ranges from 2 to 8 under physiological conditions. Binding of the macrophage colony-stimulating factor (M-CSF) to its receptor c-Fms and of the receptor activator of nuclear factor kappa B (NF-κB) ligand (RANKL also known as TNFSF11) to its receptor RANK are the two necessary and sufficient signals for the generation of osteoclasts (433). M-CSF promotes the proliferation and survival of osteoclast precursors via the activation of several kinases, including Src, PLC-γ, PI(3)K, Akt, and Erk (134, 189, 455). RANKL induces the association of RANK with TRAF6, which activates NF-κB and MAPKs (Erk, JNK, and p38). These kinases, in turn, induce the transcription of the nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATc1), the master transcription factor for osteoclast differentiation and function (71). Osteoprotegerin (OPG) (176) is the endogenous decoy receptor of RANKL (281). Mitochondria biogenesis and intracellular H2O2 accumulation, resulting from downregulation of catalase, an H2O2-inactivating enzyme, is a critical adaptation for the differentiation and survival of osteoclasts (37). Wnt/β-catenin signaling stimulates the production and secretion of OPG and thereby decreases osteoclast differentiation. Osteoclasts remove bone mineral and the demineralized bone matrix by secreting protons and lysosomal enzymes into a sealed microenvironment formed by a “podosome belt” that tightly adheres to the bone area targeted for removal (69, 94, 128).
Both osteoblasts and osteoclasts are relatively short-lived cells. In humans, osteoclasts live between 1 and 25 days and osteoblasts live 1-200 days. After the completion of their function, all osteoclasts and the majority of osteoblasts die by apoptosis. Some of the remaining osteoblasts become flattened and cover quiescent bone surfaces; these dedifferentiated cells are called lining cells. Some other osteoblasts undergo a dramatic morphological transformation into stellate cells, called osteocytes, with an average of 50 dendritic processes per cell. Simultaneously, the cell bodies and their processes are entombed within lacunae and canaliculi formed inside the mineralized matrix (Figure 3). As they run along the interconnected canalicular system, the dendritic processes of osteocytes form gap junctions with the processes of neighboring osteocytes to form a communication network that extends all the way to the surface of bone, the cells of the bone marrow stroma, and endothelial cells residing inside sinusoids and the wall of blood vessels. The osteocyte bodies and their processes are surrounded by a gelatinous matrix that is in continuity with the peripheral circulation. This allows the transport of solutes, small molecules (e.g., steroid hormones), and proteins as large as 70 kDa, from the circulation into and through the lacunar-canalicular system (491). Osteocytes are by far the most numerous cells of bone: 10 times more than osteoblasts and 100–200 times more than osteoclasts (334). In contrast to the short-lived osteoblasts and osteoclasts, osteocytes live as long as 50 years and, in some instances, as long as the organismal life. Osteocytes can sense and respond to changes in mechanical forces. In addition, osteocytes produce RANKL, the Wnt antagonist sclerostin (32), and fibroblast growth factor 23 (FGF23), a bone-derived hormone that regulates systemic phosphate homeostasis (413).
FIGURE 3.
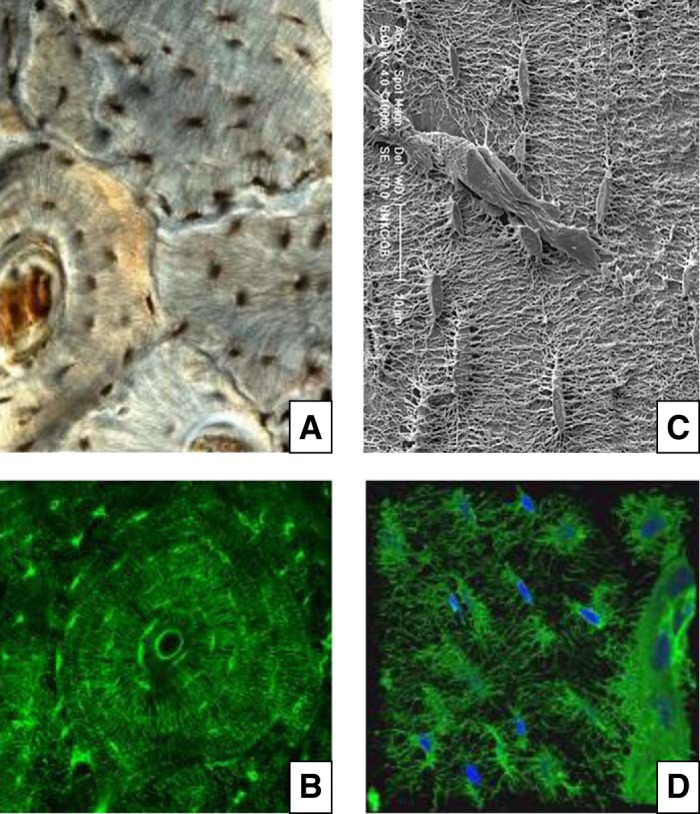
Osteocytes and their lacunar-canalicular network. A: human cortical bone section stained with India ink (courtesy of Robert S. Weinstein). B: bovine cortical bone section stained with fluorescein isothiocyanate (FITC) (from the laboratory of C. A. O'Brien). In both A and B, please note the cylindrical concentric organization of the osteocyte bodies, corresponding to the lamellar structures of osteonal bone with the blood vessel in the middle. C: electron microscopy images depicting reliefs of osteocytes and their canalicular network, following acid-etching of murine bone sections. Please note multiple attachments of the osteocyte processes to the vessels depicted in the center of the image. [From Manolagas (326).] D: confocal 3D imaging of phalloidin (green) and DAPI (blue) stained osteocytes in adult mouse long bone. Please note the intricate dendritic network and an intracortical blood vessel visible at the right of the image. [From Kamel-ElSayed et al. (241), with permission from Elsevier.]
Aberrant osteoblast and/or osteoclast number, resulting from changes in their supply as well as their lifespan, is the key pathogenic change in most acquired metabolic bone diseases, including osteoporosis (324). The effects of estrogens and androgens on bone are by and large the result of their influence on the generation and lifespan of osteoclasts, osteoblasts, and osteocytes (Figure 2).
C. Bone Modeling and Remodeling
Bone is a highly dynamic tissue that responds and adapts to changes in systemic signals, including hormones, as well as mechanical strains. During intrauterine development and postnatal growth, bones are sculpted to achieve their unique shapes and sizes. In parallel, they adapt the spatial distribution of their mineralized mass to the prevailing loads, so as to maintain optimal mechanical performance with as little weight as possible (544). This is accomplished by the resorption of bone from one site and formation in a different one. This process is termed modeling (450). During modeling, the cortical bone envelop enlarges and thickens because bone apposition at the periosteal (outer) envelope exceeds the widening of the medullary cavity by endocortical resorption. Even more remarkably, throughout life bones regenerate periodically in discrete sites via a process that is termed remodeling. In normal adult humans, the remodeling process in a particular site lasts between 6 and 9 months (324, 326). During remodeling, old, damaged, hypermineralized, or effete bone is replaced with new at the same site from which it was previously resorbed (400, 450). Additionally, remodeling contributes to extracellular calcium and mineral homeostasis. Nonetheless, not all remodeling is purposeful and targeted to replace micro damaged bone. Remodeling may also be random (also known as stochastic) with respect to localization, as for example in the setting of sex steroid deficiency (325).
In the remodeling process, teams of osteoclasts and osteoblasts assemble in distinct anatomical structures, called basic multicellular units (BMUs) (Figure 4). As the BMUs advance on trabecular, endocortical, and intracortical bone surfaces, osteoclasts, located always in the advancing front of the BMU, excavate pits. The resorption pits are subsequently filled by osteoblasts which follow in the rear of the BMU. In cortical bone, the advancing BMUs excavate and replace a tunnel; in cancellous bone they excavate and refill a trench. Capillaries are an essential component of the BMU and are most likely the route by which the hematopoietic osteoclast precursors reach the site that is targeted for remodeling (77, 402). Bone formation almost always occurs in close proximity to capillaries (73). Furthermore, perivascular cells [adventitial reticular cells (ARCs)] serve both as the mesenchymal skeletal stem cells that are pivotal for the growth and regeneration of the skeleton as well as the establishment of the hematopoietic stem cell niche (52).
FIGURE 4.
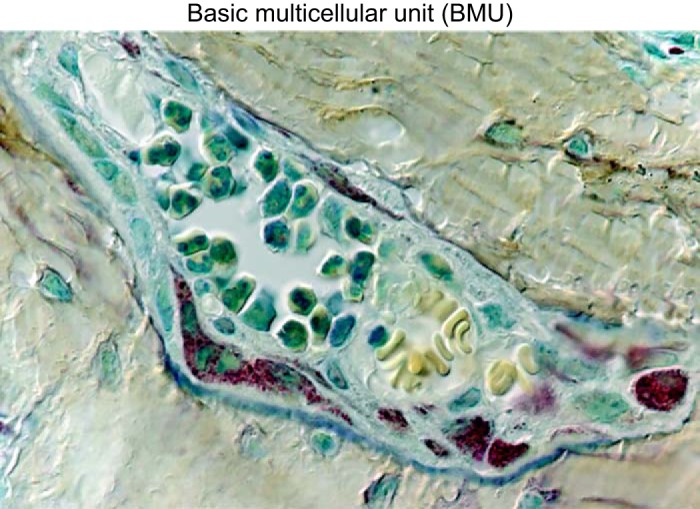
Photomicrograph of a basic multicellular unit (BMU), in a section of vertebral murine cancellous bone (×630 magnification). Please note the osteoclasts, identified by their discrete tartrate-resistant acid phosphatase-positive red granules and the osteoblasts, identified by their large nuclei with multiple nucleoli and underlying light blue osteoid. Two capillaries containing erythrocytes are also seen. Several osteocytes (blue stained cells) can be seen embedded individually within the mineralized bone (beige) surrounding the BMU. [Republished with permission of The Endocrine Society from Weinstein et al. (555); permission conveyed through Copyright Clearance Center, Inc.]
The rate of bone remodeling is rapid during growth (but much slower after puberty), and it varies widely from one bone to the other or even different anatomical localities in the same bone. For example, the mandible remodels several times faster than the ileum, whereas the ossicles in the inner ear never remodel. On average, the periodic replacement (turnover) of bone is 10% per year. This corresponds to a mean lifespan of ∼10 years and a mean age of ∼5 years; thus most of the skeleton in adult humans is no more than 10 years old (401). The rate of remodeling in different bones is determined, by and large, by the magnitude of the mechanical strains and the physical demand experienced by the particular anatomical locality (for example, the strains exerted during mastication on the mandible) and thereby the chances of micro damage. If not repaired, micro damage compromises structural integrity and eventually leads to the mechanical failure of the tissue, i.e., fracture. Loss or decrease of mechanical loading, as for example in weightlessness during space flights or immobilization, increases the rate of remodeling, but in these instances the end result is removal of bone that is likely perceived as unneeded. The latter situation is well illustrated also by the alveolar bone atrophy in edentulous mandible specimens compared with specimens with teeth (532).
In the last few years, a substantial body of evidence has revealed that remodeling is orchestrated and targeted by osteocytes to a particular site that is in need for repair (574). Osteocytes have mechanosensory properties that allow them to sense mechanical loads and effete bone and direct the homing of osteoclasts and osteoblasts to the site that is in need of remodeling (398). The central role of osteocytes in the choreography of remodeling relies on their ability to independently control bone resorption and formation, by producing rate-limiting factors for the generation of osteoclasts and osteoblasts (58, 574). Additionally osteocytes control and modify the mineralization of the matrix deposited by osteoblasts, by producing a systemic phosphate controlling hormone, FGF23 (57, 325). In agreement with this role, reduced osteocyte density in human central cancellous bone is associated with increased surface remodeling (412), which is an independent contributor to bone fragility (200). Furthermore, signals arising from apoptotic and old, or dysfunctional, osteocytes may be seminal cellular culprits in the pathogenesis of osteoporosis caused by sex steroid deficiency or old age (334, 335).
Under physiological conditions, bone resorption and formation during remodeling are linked in time and space; this spatial and temporal relationship is termed coupling. Additionally, under physiological conditions, the cavities formed by osteoclastic resorption are refilled by an equal amount of new bone formed by osteoblasts. The relative amount of bone resorbed and formed within individual BMUs is referred to as balance. Balanced remodeling depends on the adequate supply of osteoblasts at the right place and time (399). This is accomplished by the generation of stimulatory signals for osteoblast formation produced by osteoclasts and osteocytes as well as matrix-derived factors released during resorption. An increase in the rate of tissue level remodeling due to increased number of BMUs (i.e., more osteoclasts and osteoblasts), as it occurs for example in the estrogen deficient state, predisposes to trabecular penetration and increased cortical porosity; in addition, the negative balance in individual BMUs contributes to bone loss and structural change. A negative balance within an individual BMU can be caused by either too many osteoclasts relative to the need for repair, e.g., in estrogen deficiency, or too few osteoblasts relative to the need to refill the resorption cavities, e.g., aging. Over the years, several signals have been proposed to account for coupling (464). Whether defective “coupling” plays a role in these, or for that matter, in any other cause of osteoporosis, remains unknown.
D. The Contribution of Knowledge Gained From Rodent Models
Rats and mice have provided invaluable insights to our understanding of bone physiology and pathophysiology. Moreover, discoveries from rodents complemented with human studies have led to the development of both anabolic (368, 490) and anticatabolic (70, 115, 280) therapies for diseases of low bone mass (e.g., osteoporosis), including the class of selective estrogen receptor modulators (SERMs) (88, 125). As we will detail in the following sections, studies in rodents and in particular modern mouse genetics have also contributed a great deal to our understanding of the cellular and molecular mechanisms of sex steroid action on bone and the mechanisms of the adverse effects of their deficiency on skeletal homeostasis. In particular, together with clinical observations in humans, studies in rodents have established that estrogens and androgens contribute to the maintenance of bone mass during adulthood, primarily by slowing the rate of bone remodeling. Nevertheless, as we have reviewed elsewhere, rodents differ from humans in many important ways, and genetic manipulations have their inevitable limitations and need to be interpreted cautiously (333, 520). It will, therefore, be useful to point out at this time the similarities and differences between rodents and humans in the context of bone remodeling and its modulation be sex steroids.
In contrast to humans, rats and mice do not experience the abrupt loss of estrogens at menopause, nor do androgen levels seem to decrease with age in male mice (14). Nevertheless, gonadectomy of mice or rats over the age of 4–5 months, the time at which both species have achieved peak bone mass, faithfully replicates the loss of cancellous and cortical bone mass caused by estrogen or androgen deficiency in humans (197). In contrast, gonadectomy of immature mice and rats causes a failure to gain bone mass. Insufficient peak bone mass acquisition related to abnormal gonadal steroids may contribute to the risk of osteoporosis later in life (288, 453, 515). Nevertheless, findings from studies with gonadectomized growing rodents cannot be extrapolated to the effects of acquired sex steroid deficiency after menopause or in aging men, a situation in which preexisting bone is lost. This important caveat is frequently overlooked in preclinical studies, leading to inappropriate conclusions.
Old age and estrogen deficiency are the two most critical factors for the development of osteoporosis in both women and men. However, it is unknown whether the molecular changes caused by estrogen deficiency and aging are similar or distinct or how they may influence each other. Because of the abrupt decline of ovarian function at menopause in women and a slower decline of both androgen and estrogen levels in men with advancing age, the two conditions inexorably overlap making it impossible to dissect their independent contribution to the cumulative anatomic deficit. Because mice do not experience menopause, they are an invaluable model for dissecting the contribution of sex steroid deficiency versus aging per se to the involution of the skeleton. We (S. C. Manolagas and M. Almeida) have recently elucidated that both female and male mice exhibit all major features of skeletal aging, including the decline of cortical and cancellous bone mass as well as the development of cortical porosity by 18 months of age, independently of sex steroid deficiency as mice are sex steroid sufficient at the time that all these changes are already manifested (506). These discoveries from the mouse model indicate for the first time that the effects of aging, one of the most fundamental biologic processes experienced by all living organisms, and sex steroid deficiency on a mammalian skeleton are independent.
Finally, rats and mice do not normally exhibit osteonal remodeling. Therefore, particular caution must be exercised in translating results to human cortical bone. That being said, osteonal organization is likely a consequence of body size and habitual loading related to it, rather than of phylogenetic origin. In fact, histologic evidence supports the contention that rodents do, indeed, have osteonal remodeling, albeit not as well-organized as humans (197).
II. MOLECULAR MECHANISMS OF SEX STEROID ACTION ON BONE
Estrogens and androgens are derived from cholesterol and are synthesized in the gonads and the adrenals. In addition, they are locally activated or catabolized within target tissues such as bone (520). In women, estradiol (E2) is made primarily in the granulosa and theca cells of the ovarian follicles. In men, ∼15% of E2 is secreted directly from the testes, and the remaining ∼85% is derived from peripheral aromatization (170). Contrary to the abrupt decline of E2 during menopause, older men do not experience a true “andropause,” and total E2 concentrations remain above a level sufficient to maintain skeletal homeostasis (152, 168, 217). Testosterone (T), the primary circulating androgen, is made by the Leydig cells of the testicles and acts unmodified or following conversion to the more potent dihydrotestosterone (DHT). T can also be converted to E2 by the aromatase (CYP19A1) enzyme. The bioactivity of circulating estrogens and androgens is controlled by gonadotropins [follicle-stimulating hormone (FSH) and luteinizing hormone (LH)] via hypothalamic-pituitary feedback. In humans, the bioavailability of either estrogens or androgens is restricted by high-affinity binding to circulating sex hormone-binding globulin (SHBG) (293). Only 1–5% of circulating T, DHT, and E2 (the free fraction that is not bound to SHBG, albumin, or other proteins) is thought to be biologically active.
A. Receptors
As in other tissues, the effects of estrogens and androgens on bone are exerted upon binding with high affinity to the estrogen receptor (ER) α and β (also known as NR3A1 and NR3A2) and the androgen receptor (AR) (also known as NR3C4), respectively (43, 410). The genes encoding these receptors share an evolutionary conserved template with other members of the large nuclear receptor (NR) family of ligand-inducible transcription factors (146). Like other NRs, the ERs and the AR display a modular structure consisting of an amino-terminal domain (NTD), a DNA-binding domain (DBD) containing two zinc-fingers, a hinge region, and a carboxy-terminal ligand-binding domain (LBD) (105, 278). Two critical coactivator interaction surfaces called transcriptional activation functions, AF-1 and AF-2, are located in the NTD and the LBD, respectively.
All three sex steroid receptor proteins form homodimers that bind to DNA sequences called hormone response elements (EREs or AREs) (Figure 5A). EREs or AREs comprise palindromes of two hexads of nucleotide sequences separated by three base pairs. The first of the two zinc fingers makes sequence-specific contacts with the DNA, while the second is involved in receptor dimerization (105, 202). In the absence of ligand, the homodimers recruit a complex of factors (co-repressors) that repress transcription. Binding of the ligand to a hydrophobic pocket of the LBD causes conformational changes that reveal a nuclear localization signal as well as an AF-2 surface to which coactivators can bind (359, 378).
FIGURE 5.

Mechanisms of estrogen receptor action. A: classical genomic signaling in which the ligand-activated receptor dimer attaches to estrogen response elements (ERE) on DNA, and activates or represses transcription. B: ERE-independent genomic signaling pathway in which the ligand-activated receptor binds to other transcription factors (e.g., p50 and p65 subunits of NF-κB) and prevents them from binding to their response elements. C and D: nongenotropic mode of action in which the ligand-activated receptor (in the plasma membrane) activates cytoplasmic kinases which in turn cause the phosphorylation of substrate proteins and transcription factors (e.g., Elk-1 and c-jun) that positively (C) or negatively (D) regulate transcription. [From Manolagas et al. (333).]
Several proteins are capable of coactivating or co-repressing sex steroid receptors (201). The best-characterized subgroup is the p160 family, which includes the steroid receptor coactivator 1 (SRC-1) (386). SRC-1 interacts with ERα or AR via their LBD as well as their NTD (47, 102). The AR-NTD also harbors a polyglutamine stretch (CAG repeats) of variable length, which decreases AR transcriptional activity when sufficiently long (84, 223). In full agreement with its role as a coactivator, SRC-1 knockout mice of both sexes display trabecular bone loss, due to estrogen resistance in females (356) and androgen resistance in males (355, 576).
Apart from ligand-dependent transactivation, nuclear receptors may also display ligand-independent effects. At least some of the effects of ERα on bone (particularly its role in bone's response to mechanical loading) appear to be independent of estrogens (15). To date, there is no evidence for ligand-independent actions of AR on bone (529).
B. Putative Gene Targets
Estrogen- and androgen-responsive genes are well-documented in breast and prostate cancer, respectively. In contrast, direct target genes of sex steroid action on bone, defined as genes whose expression is regulated in 24 h or less by ER or AR binding to their promoter or enhancer (274), remain poorly characterized.
Based on in vitro evidence from cell lines and primary cell cultures, several genes have been proposed over the years as putative targets of the antiresorptive effect of estrogens on bone. This long list includes cytokines such as interleukin (IL)-1β, IL-6, IL-7, tumor necrosis factor (TNF)-α, M-CSF, RANKL, and OPG produced by bone marrow stromal cells, T and B lymphocytes, macrophages, and dendritic cells (332, 556). The IL-1 receptor, c-jun, c-fos, cathepsins, and TRAP expressed in osteoclasts have also been implicated (272, 323, 393, 485). Heretofore, it remains unknown whether any of these genes are direct targets of estrogen action, as evidence for DNA binding of the ERα or -β using techniques such as chromatin immunoprecipitation (ChIP) is missing.
Fasl, which encodes the proapoptotic protein Fas ligand, is directly regulated by estrogen-activated ERα signaling in osteoblasts, but not in osteoclasts, via an ERE-containing transcriptional enhancer located downstream from this gene (275). The biologic relevance of this finding, however, remains unclear as genetic evidence that will be discussed later shows that the ERα in osteoclasts, but not in osteoblasts, mediates the proapoptotic effect of estrogens on osteoclasts, and estrogens have an antiapoptotic, not proapoptotic, effect on osteoblasts (270, 340, 365). In recent studies from the authors' laboratories (M. Almeida and S. C. Manolagas), we have searched for estrogen target genes by starting with ERα-deficient cells from mice with cell-specific targeted deletions (226, 506). The advantages of this approach are that it ensures a priori that 1) putative target genes are indeed ERα sensitive and 2) are expressed in cell targets that have been functionally validated in vivo. The Fas ligand (FasL) mRNA levels were not affected by the ERα deletion in our studies. Moreover, contrary to an earlier report (365), we found that FasL plays no role in the effects of estrogen deficiency on the murine skeleton or the OVX-induced increase in osteoclast numbers (506). Instead, we found that ERα deletion in cells of the myeloid lineage (targeted by LysM-Cre) causes an increase in the expression of the calcium binding protein S100A8 (also known as MRP8, Calgranulin A, or CP-10); and silencing S100A8 greatly attenuated osteoclastogenesis and increased osteoclast apoptosis in vitro (226).
Over the years, several genes in cells of the osteoblast lineage have been proposed as targets of a putative “bone forming” effect of estrogens, based again on evidence from in vitro studies with osteoblast-like cell lines. This list includes retinoblastoma-binding protein 1 (RBBP1, a RUNX coactivator), transforming growth factor (TGF)-β-inducible early gene-1 (TIEG, a modulator of OPG), GATA4, and alkaline phosphatase (Alpl) (198, 276, 353, 358). RBBP1 and TIEG were shown to be regulated by ERα and ERβ, respectively, and this regulation was confirmed by ChIP. Other ChIP-validated ERα target genes in osteoblasts are Svep1 (175), insulin-like growth factor (IGF) binding protein 4 (127), ERα itself (282), and MMP3 (164). Finally, three gene clusters (CTSZ-SLMO2-ATP5E, TRAM2-TMEM14A, and MAP4K4), associated with risk for bone fractures in ER-positive breast cancer patients treated with aromatase inhibitors, have been shown to be regulated by estrogens in human fetal osteoblasts transfected with ERα (313), but evidence of direct ER regulation by ChIP is missing.
In our (S. C. Manolagas and M. Almeida) latest work, we have found that ERα deletion in mesenchymal/stromal cells (targeted by Prx1- or Osx1-Cre), as well as estrogen or androgen deficiency in wild-type mice causes an increase in the expression of stromal derived factor 1 (SDF1), also known as CXCL12 (226). SDF1 is a C-X-C motif containing chemoattractant with a seminal function in the BM niche (4, 130, 180, 506). The major source of SDF1 is mesenchymal progenitor-derived cells called CXCL12 abundant reticular (CAR) cells, a heterogeneous population of reticular cells closely associated with the perivascular niches in the BM. SDF1 functions as a key recruiting signal for hematopoietic cells into the bone compartment and promotes osteoclastogenesis in vitro (178, 179, 570, 579) and in vivo (267). ERα deletion in mesenchymal/stromal cells also increases the expression of the matrix metalloproteinase 13 (MMP13), which promotes osteoclast fusion independent of its enzymatic activity (158). These latest insights suggest that the protective effects of estrogens against bone resorption result not only by actions in both osteoclasts and mesenchymal/stromal cells, but also via different target genes in each cell type, S100A8 in the former and MMP13 and SDF1 in the latter.
TGF-β, cathepsin B, and TRAP have been suggested as direct target genes of the antiresorptive effects of androgens (408), but recent genetic evidence has revealed that the osteoclast AR plays no role in bone resorption (469, 504), casting doubts on the biologic significance of AR target genes in this cell type. TGF-β, IL-6, OPG, MYBL2, HOXD11, and ADCYAP1R1 have been suggested as direct targets of a bone-forming effect of androgens (209, 210, 245, 350), but once again, there has been no demonstration of AR binding to DNA by ChIP.
Finally, in addition to direct DNA binding on EREs or AREs, sex steroid receptors are able to bind chromatin and regulate transcriptional activity indirectly by tethering with other transcription factors such as Mef2 and SRF (536, 573) (Figure 5B). Through such mechanism, ERα can repress the transcription of the IL-6 gene in osteoblast lineage cells by binding to components of the NFκB complex (162). In addition, AR may form a heterodimer with RUNX2 in osteoblasts (250). The biological significance of these observations to the in vivo situation remains unknown.
C. Nonnuclear Initiated Signaling Pathways
In addition to nuclear initiated actions, estrogens or androgens bind to subsets of their cognate receptors that are localized outside the nucleus, either on the cell membrane or in the cytosol. Binding of these ligands to receptors localized on the membrane initiates signal transduction cascades that involve the production of cyclic nucleotides, calcium flux, and activation of cytoplasmic kinases. Activated kinases, in turn, phosphorylate substrate proteins and transcription factors that then modulate gene transcription (Figure 5,C and D) (190). At least in breast cancer cells, the number of genes regulated by the ERs through nonnuclear initiated actions is larger than the one regulated by the direct association of the ERs with DNA (415).
During the last 15 years, it has been shown in rodents that synthetic ligands that can selectively activate nonnuclear initiated actions of the ERα, but have minimal or no effects on the nuclear-initiated actions of this receptor, may replicate the beneficial effects of estrogens on skeletal maintenance without affecting reproductive organs, such as the uterus and the breast. These synthetic ligands have been dubbed activators of nongenomic estrogen-like signaling (ANGELS), and may represent a safer alternative to estrogen therapy in postmenopausal women (270, 271, 330, 331). The synthetic compound originally used to test this hypothesis, 4-Estren-3α-17β-diol, was shown in retrospect to bind to the androgen receptor with 30-fold lower affinity than DHT, and thereby affect reproductive organs, at least under some experimental conditions (329, 565). Albeit, studies using an isomer of this compound showed that it had minimal effects on the prostate and the seminal vesicles of rats but still prevented the loss of bone and muscle caused by orchidectomy (ORX) (396). Similarly, several other synthetic compounds that selectively activated nonnuclear initiated actions of the ERα, but had minimal or no effects on nuclear-initiated actions, were shown to have potent bone protecting effects of E2, but had minimal effects on the uterus and breast (392, 559).
More recently, the idea that the beneficial effects of estrogens on bone and other nonreproductive target tissues can be dissociated from their effects on reproductive organs by selectively activating nonnuclear initiated actions of the ERα has been tested using a cell membrane-impermeable E2 conjugate. In this compound, E2 is attached to a large, inert, positively charged nondegradable poly(amido)amine (PAMAM) dendrimer via a hydrolytically stable linkage (195). The E2-dendrimer conjugate (EDC) binds to ER with the same affinity as E2 and is very effective in stimulating nonnuclear initiated signaling (264). It cannot, however, enter the nucleus and stimulate nuclear ER target gene expression (195). With the use of this compound, it was shown that one can simulate the protective effects of E2 on the cardiovascular system in mice without affecting uterine or breast cancer growth (95).
The EDC also prevents the loss of cortical bone mass in the OVX mouse model as effectively as E2, but unlike E2, it does not prevent the loss of cancellous bone mass; on the other hand, unlike E2, EDC has no effect on the uterus in OVX mice (36). These results suggest that the protective effects of estrogens on cortical bone mass result from nonnuclear-initiated actions of the ERα and are mechanistically different from the nuclear-initiated actions of the ERα on reproductive organs. Taken together with evidence from mouse models with conditional deletion of the ERα from osteoclasts and osteoblast progenitors (discussed in the following section), the existing evidence suggests that EDC protects cortical bone mass through nonnuclear signaling on osteoblast progenitors. On the other hand, the protective effects of estrogens on cancellous bone result from nuclear-initiated actions of the ERα. In line with these conclusions, deletion of a coactivator of the nuclear actions of the ERα, SRC1, or inactivation of the AF1 domain of the ERα in mice, and thereby prevention of its' nuclear-initiated actions, abrogates the protective effect of estrogens on cancellous bone, but it has no effect on cortical bone (66, 356).
III. EFFECTS OF SEX STEROIDS ON BONE CELLS
ERα, ERβ, and AR have been detected in several cell types along the differentiation progression of mesenchymal and myeloid precursors to osteoblasts and osteoclasts (522), as well as in other cell types residing in the bone marrow or even in tissues distant from bone that may indirectly influence bone homeostasis. However, it was not until the development of mouse models with global or cell-specific deletion of these receptors that their function in bone, and by extension the role of their ligands, could be gleaned in vivo (333).
Mice with global deletion of ERα have a complex bone phenotype that is incongruent with the known effects of estrogen deficiency (463). This is because of high circulating levels of estrogens and androgens, resulting from the loss of receptor function in the pituitary gland. The skeletal phenotype of mice with global deletion of ER-β is minimal (196, 533). However, deletion of ERβ specifically in osteoblast lineage cells, in the absence of splice variants, increases trabecular bone mass in females and the differentiation of osteoblast precursor cells into osteoblasts (374). The skeleton of male mice with global deletion of the AR exhibits low bone mass and high bone turnover, consistent with the effects of androgen deficiency (249, 577).
During the last few years, the development of mouse models with targeted deletion of the ER and the AR in specific cell types using Cre/LoxP technology has provided a much clearer picture of the cellular targets of sex steroid action in vivo (333, 520). With the use of a cycling recombinase (Cre) driven by a cell-specific promoter, a genomic region flanked by LoxP sites (floxed allele, e.g., crucial exons of ERα or AR) can be selectively deleted. This can be accomplished very early in myeloid precursors of osteoclasts (LysM-Cre) or mesenchymal precursors of osteoblasts (Prx1-Cre, Osx1-Cre, Runx2-Cre), leading to deletion in all descendent cells of the lineage; or in differentiated osteoclasts (e.g., CathepsinK-Cre) and mature osteoblasts and osteocytes [2.3 kb Col1A1-Cre, osteocalcin (OCN)-Cre, and Dmp1-Cre] (Tables 1 and 2). Importantly, in a recent report the investigators have used high-resolution microscopy of bone sections and flow cytometry to carefully define the targeting specificity of the OCN-Cre and Dmp1-Cre. They found that in addition to mature osteoblasts and osteocytes, OCN-Cre and Dmp1-Cre target broader stromal cell populations including the CXCL12-abundant reticular (CAR) cells, referred to earlier in section IIB, and both venous sinusoidal and arteriolar pericytes (582).
Table 1.
Murine models of targeted deletion of ERα or -β in bone cells
| Cell Type | Mouse Model | Sex | Cancellous Bone | Cortical Bone | Age at Phenotyping, wk | Reference Nos. |
|---|---|---|---|---|---|---|
| Myeloid progenitor | LysM-Cre;ERαf/f | F | Increased osteoclast number | ND | 4, 8, 12, 18, 22, 28 | 339 |
| M | ND | ND | 12 | 504 | ||
| Osteoclast | CtsK-Cre;ERαf/f | F | Increased osteoclast number | 12, 14 | 365 | |
| M | ND | ND | 12 | |||
| Mesenchymal progenitor | Prx1-Cre;ERαf/f | F | ND | Low bone mass, reduced periosteal bone formation rate | 8, 12, 28 | 15 |
| M | ND | Low bone mass (transient) | 6, 8, 18 | |||
| Prx1-Cre;ERβf/f | F | Increased bone mass | ND | 6, 12, 16 | 374 | |
| Osteoblast progenitor | Osx1-Cre;ERαf/f | F | ND | Low bone mass | 8, 12, 24 | 15 |
| M | ND | Low bone mass (transient) | 6, 10, 26 | 504 | ||
| Osteoblasts and osteocyte | Col1-Cre;ERαf/f | F | Increased osteoblast apoptosis | ND | 4, 8, 12, 26 | 15 |
| M | ND | ND | 4, 8, 12 | 504 | ||
| OCN-Cre;ERαf/f | F | Low bone mass and decreased osteoblast number | Low bone mass | 14, 24, 48, 20 | 318, 348 | |
| 12, 18 | ||||||
| Dmp1-Cre;ERαf/f | F | None or low bone mass | ND | 12 | 268, 564 | |
| 11, 16 | ||||||
| M | None or low bone mass with reduced bone formation rate | ND | 12 |
ND, no changes detected.
Table 2.
Murine models of targeted deletion of AR in bone cells
| Cell Type | Mouse Model | Sex | Cancellous Bone | Cortical Bone | Age at Phenotyping, wk | Reference Nos. |
|---|---|---|---|---|---|---|
| Myeloid progenitor | LysM-Cre;ARf/y | M | ND | ND | 12, 26 | 504 |
| Osteoclast | CtsK-Cre;ARf/y | M | ND | ND | 12, 16 | 469 |
| Mesenchymal progenitor | Prx1-Cre;ARf/y | M | Low bone mass with increased osteoclast number | ND | 7, 16, 26 | 504 |
| F | Low bone mass | ND | 16 | |||
| Osteoblast and osteocyte | 2.3 kb Col1A1-Cre;ARf/y | M | Low bone mass | ND | 6, 12, 32 | 376 |
| OCN-Cre;ARf/y | M | Low bone mass with increased osteoclast number | None or transient low bone mass | 6, 12, 24, 12, 20, 24 | 101, 319 | |
| F | Low bone mass | ND | 12, 20, 24 | 319 | ||
| Dmp1-Cre;ARf/y | M | Low bone mass | ND | 12, 32 | 468 |
ND, no changes detected.
A. Osteoclasts
In humans and rodents alike, estrogens or androgens suppress bone resorption in trabecular and endocortical bone surfaces by decreasing osteoclast numbers. This results from attenuation of the differentiation of osteoclasts as well as shortening their life span by stimulating apoptosis. The anti-osteoclastogenic actions of sex steroids are mediated via direct effects in cells of the osteoclast lineage as well as indirectly. Mouse models with targeted deletion of ERα in osteoclasts (using CathepsinK-Cre) or the entire myeloid lineage (using LysM-Cre) exhibit increased osteoclast number in trabecular bone and are resistant to the loss of bone mass caused by ovariectomy (OVX) (339, 365). The loss of bone mass at the endocortical surfaces following OVX is unaffected in these mice, indicating that the predominant, if not exclusive, mechanism of the antiresorptive actions of estrogens on cortical bone is not the result of direct actions on osteoclasts. As we will detail in section IIIB, the antiresorptive effects of estrogens on cortical bone are instead mediated via actions on cells of the osteoblast lineage. In contrast to females, male mice with targeted ERα deletion in mature osteoclasts (using cathepsinK-Cre) exhibit no change in osteoclast numbers or cancellous bone mass, indicating that direct actions of estrogens on osteoclasts play no role in the maintenance of cancellous bone in males (365, 504).
Over the years, experimental evidence has implicated several factors as mediators of the effects of estrogens on osteoclasts (Table 1). More recently, it was shown that Wnt/β-catenin signaling is a negative regulator of osteoclastogenesis (32). In support of this evidence, mice with deletion of β-catenin in osteoclast lineage cells exhibit increased osteoclast number and decreased bone mass (7, 552). In addition, Wnt ligands like Wnt3a reduce osteoclast formation (7) and promote osteoclast apoptosis (M. Almeida, unpublished data). Estrogens act on osteoclasts to increase the expression of the Wnt co-receptor LRP5 as well as β-catenin (543). This finding raises the possibility that estrogens attenuate bone resorption by potentiating Wnt signaling directly in osteoclasts.
Heretofore, the most compelling explanation for the suppressive effects of estrogens on bone resorption, at least in cancellous bone, is that estrogens promote osteoclast apoptosis (214, 240). Nonetheless, the molecular mechanism responsible for this pro-apoptotic effect remains unknown. Some studies have suggested that estrogens promote osteoclast apoptosis by increasing the transcription of the FasL gene (365). Consistent with this contention, estrogens stimulate FasL expression in osteoclasts derived from human monocytes (543). However, this conclusion remains controversial because the stimulatory effect of estrogens on FasL expression in osteoclasts has not been observed in some other studies (275, 339, 438). Furthermore, as we discussed in the previous section, contrary to a report by others (275, 339, 365), we have found that FasL plays no role in the effects of estrogen deficiency on the murine skeleton or the OVX-induced increase in osteoclast numbers. Instead, our latest findings support the hypothesis that an increase of S100A8 expression in osteoclasts in the estrogen-deficient state is the proximal mediator of the increased resorption of cancellous bone.
Similar to the case with estrogens, in vitro evidence has suggested that T or the nonaromatizable androgen DHT act directly on osteoclast progenitors and mature osteoclasts to inhibit osteoclastogenesis and promote osteoclast apoptosis (365). However, genetic evidence from mice with osteoclast-specific AR deletion indicates that androgen signaling in osteoclasts plays no role in the antiresorptive effect of androgens on the cancellous or cortical bone compartments. Indeed, mice with targeted deletion of AR in osteoclast lineage cells exhibit no changes in osteoclast number or bone mass (469, 504) (Table 2).
In concluding this section, genetic evidence from murine models of targeted ER or AR deletion indicates that direct sex steroid actions on osteoclasts account only for the antiresorptive effect of estrogens on cancellous bone in females. As will be discussed below, the direct targets of the antiresorptive effects of androgens on cancellous bone as well as the antiresorptive effects of both estrogens and androgens on cortical bone are evidently uncommitted pluripotent mesenchymal progenitors.
B. Osteoblasts
Sex steroids help to maintain bone mass predominantly by decreasing osteoclastic bone resorption and thereby suppressing the rate of bone remodeling. Consistent with this conclusion, antiresorptive agents such as bisphosphonates are effective inhibitors of the bone loss caused by estrogen or androgen deficiency. However, because during remodeling bone formation is inexorably tied to bone resorption, loss of sex steroids is accompanied by an increase in bone formation; albeit, the remodeling balance in this setting is negative and the end result is bone loss. Estrogen or androgen replacement attenuates bone formation in the setting of sex steroid deficiency (183, 271, 524, 571). It remains unclear, however, whether the suppressive effect of sex steroids on bone formation is secondary to their antiresorptive properties as opposed to a direct inhibition on osteoblast generation. Irrespective of whether sex steroids may or may not suppress osteoblast generation, extensive evidence from studies in humans and rodents shows that either class of sex steroids attenuates osteoblast and osteocyte apoptosis, an effect that is diametrically opposite to their proapoptotic actions on osteoclasts (271, 495, 496).
The generation of mouse models with targeted ERα or AR deletion at different stages of the differentiation progression of the osteoblast lineage, including pluripotent mesenchymal osteoblast progenitors (using Prx1-Cre), bipotential osteoblast precursors (using Osx1-Cre), or mature osteoblasts and osteocytes only (using collagen1-Cre, OCN-Cre, or Dmp1-Cre) has been very informative on the role of direct estrogen or androgen signaling on this lineage. Deletion of ERα from mesenchymal progenitors or from osteoblast progenitors using Prx1- or Osx1-Cre, respectively, causes a decrease in periosteal bone apposition and cortical bone mass (15). These effects result from the potentiation of Wnt/TCF signaling by ERα and, thereby, stimulation of proliferation and differentiation of periosteal osteoblast progenitor cells. Deletion of ERα from osteoblasts and osteocytes expressing Col1a1- or Dmp1-Cre has very modest or no impact on bone mass and architecture (15, 268, 564). Albeit, deletion of ERα with OCN-Cre, which is expressed in chondrocytes and osteoblasts, causes a decrease in trabecular and cortical bone mass, as well as a decrease in osteoblast number (318, 348). The decrease in osteoblast number could be due to an increase in apoptosis, as seen in mice with deletion of ERα with Col1a1-Cre (15). Be that as it may, the conclusions of the findings from mice with ERα deletion using Dmp1-Cre and OCN-Cre need to be reassessed in view of the latest evidence that OCN-Cre and Dmp1-Cre target broader stromal cell populations than previously appreciated, discussed in section IIB (582). In any case, the evidence that the bone formation rate in cancellous and endocortical surfaces is not altered by ERα deletion in progenitors and mature osteoblasts indicates that estrogens may play no significant role in osteoblast generation (15, 348). Intriguingly, deletion of the ERβ with Prx1-Cre increases cancellous bone mass, without changes in osteoblast or osteoclast numbers or activity at 6 or 12 weeks (wk) of age, raising the possibility that the deletion affected bone development (374) (Table 1).
In males, deletion of ERα from osteoblast progenitors causes a delay in cortical bone mass accrual during puberty (15). However, in contrast to females, this effect is transient and at 3 months of age male mice have normal bone mass, suggesting that androgens acting via AR compensate for the lack of ERα. Indeed, studies with mice with global deletion of ERα, AR, or both have suggested that both receptors contribute to periosteal bone accrual in males (529). Surprisingly, deletion of AR from the entire mesenchymal lineage including progenitors, osteoblasts, and osteocytes has no effect on cortical bone mass, indicating that the actions of androgens on this compartment are indirect (101, 319, 376, 468, 504). In contrast, direct actions in cells of the osteoblast lineage are responsible for the effect of androgens on cancellous bone. Indeed, mice lacking AR in osteoblasts exhibit increased cancellous osteoclast number but no changes in osteoblasts, indicating that androgens attenuate osteoclast numbers in cancellous bone indirectly (319, 504). Nevertheless, the levels of RANKL and OPG are not altered by AR deletion in osteoblast and osteocytes (468), suggesting that other factors are responsible for the antiresorptive effects of androgens on cancellous bone. Conversely, transgenic mice overexpressing AR under the control of the 2.3-kb α1(I)-collagen promoter fragment have low bone turnover and increased cancellous bone volume (567). In female mice, deletion of the AR in mesenchymal progenitors or mature osteoblasts decreases cancellous bone mass in the femur, but this effect is less pronounced in females than males (319, 504). Consistent with the possibility that androgens promote the accrual or maintenance of bone mass in females, administration of DHT to ovariectomized mice protects against loss of bone mineral density (BMD), as determined by dual energy X-ray absorptiometry (DXA) (14, 271). The evidence for a positive skeletal effect of androgens in women is contradictory (45, 215, 337, 418) (Table 2).
Estrogens restrain the production of osteoclastogenic cytokines produced by cells of the osteoblast lineage and via this indirect mechanism inhibit osteoclast formation and bone resorption (260, 324, 328). This mechanism accounts for the protective effect of estrogens on cortical bone. This conclusion is based on the observation that mice with deletion of ERα in mesenchymal progenitors are protected from the OVX-induced increase in osteoclast numbers in the endocortical surface and the loss of cortical bone mass (15). In contrast, the OVX-induced loss of cortical bone is unaffected in mice lacking ERα in osteoblasts and osteocytes.
As discussed in section IIB, ERα deletion in mesenchymal/stromal cells increases the expression of SDF1 and MMP13, and loss of ovarian or testicular function in mice increases the levels of secreted SDF1 in the bone marrow plasma (229). Furthermore, E2 prevents the ORX-induced loss of cortical bone mass in mice. On the other hand, DHT has no effect on the ORX-induced endosteal bone resorption and loss of cortical bone mass in adult mice, but it does prevent the ORX-induced loss of cancellous bone (229, 517). These latest findings support the conclusion that estrogens protect against endocortical bone resorption in both female and male mice; likely via an ERα-mediated suppression of SDF1 in uncommitted mesenchymal progenitors. Of note, these results in male mice allow the re-interpretation of earlier studies in men (147, 255, 299). In those earlier studies, in men with suppressed sex steroid production with a GnRH agonist and selective replacement of either estrogens, T, or both, estrogens accounted for ∼70% and T for at most ∼30% of the protective effect of sex steroids on bone resorption, consistent with the fact that the skeleton is ∼80% cortical and ∼20% cancellous (255). Taken together, the evidence from humans and mice suggests that estrogens protect against endocortical resorption in both males and females, at least in part, via ERα-mediated actions (upon aromatization of androgens to estrogens in males) on mesenchymal/stromal cells. In addition, our latest findings from the mouse models with ERα deletion in specific cell types (226) support the notion that the protective effects of estrogens against the resorption of cortical bone result from the suppression MMP13 and SDF1 in mesenchymal/stromal cells. Hence, an increase in MMP13 and SDF1 expression in the estrogen deficient state may be the proximal mediator of the increased resorption of cortical bone.
In summary, the available evidence indicates that the ERα in osteoblast progenitors plays a critical role in the accrual of cortical bone mass in female mice. This effect results from an increase in bone formation at the periosteum. In addition, ERα signaling in mature osteoblasts may contribute to the maintenance of cancellous bone mass. However, the molecular details by which ERα signaling in mature osteoblasts helps to maintain cancellous bone mass remain unclear. AR signaling in osteoblasts is responsible for the protective effects of androgens on cancellous bone mass. Such signaling leads to a decrease in osteoclast numbers and bone resorption. On the other hand, the target cell(s) for the effects of AR on periosteal bone formation remains unknown. Finally, estrogens protect against endocortical resorption in both males and females, at least in part, via ERα signaling in mesenchymal/stromal cells. Elucidation of the target cells of androgen action on cortical bone and the molecular mediators of estrogen and androgen action in this compartment awaits future studies.
C. Osteocytes
ERα is required for load-induced bone formation in female mice, apparently without the need for a ligand (301). In addition, ERα mediates many of the effects of estrogen on cortical and cancellous bone remodeling as determined by germline ERα mutations. Osteocytes are thought to participate in sensing changes in load and have been shown to produce factors that control bone remodeling (118). Therefore, it is possible that ERα expression in osteocytes participates in one or both of these actions. If this were the case, then deletion of ERα specifically from osteocytes would be expected to have the following consequences. First, changes in bone formation in response to changes in skeletal load should be blunted in mice lacking ERα in osteocytes. Second, these mice should display a high remodeling phenotype mimicking that seen with estrogen deficiency.
Conditional ERα alleles in mice have been deleted at various stages of osteoblast differentiation. In each of these studies, ERα was deleted from osteocytes since Cre-mediated recombination is irreversible and osteocytes are derived from osteoblasts. However, none of the Cre driver strains used in these studies was specific for osteocytes. Nonetheless, since the Dmp1-Cre driver strain causes gene deletion primarily in mature osteoblasts and osteocytes, deletion of ERα using this model should provide the best evidence for or against a role for this receptor in osteocytes in vivo. Two such studies have been performed. The first analyzed the skeletons of female and male mice at 11 wk of age and found that female mice lacking ERα in osteocytes had normal skeletons compared with control littermates (564). In contrast, male conditional KO mice had reduced cancellous bone volume in the tibia and L5 vertebra, which was associated with a reduced bone formation rate. In the second study, which analyzed mice at 12 wk of age, female conditional KO mice had low bone mass, but there was no effect on the skeletons of male mice (268). There was not a significant reduction in bone formation rate in the conditional KO mice, but osteoblast number was reduced. Thus the two studies agree that ERa in osteocytes is important for cancellous bone mass, but they disagree about the sex of the animals in which this occurs (Table 1).
An explanation for the discrepant outcomes of the two studies is not obvious. It is important to point out that the histological changes observed in the mice with low bone mass in either study (reduced bone formation) do not resemble those seen with estrogen deficiency (elevated bone formation and resorption). Moreover, tail-suspension was performed in the second study and revealed that bone loss due to unloading still occurred in the mice lacking ERα in osteocytes (268). Overall, the results of these studies do not support the idea that ERa in osteocytes mediates the known effects of ERα on either physiological bone remodeling or in response to load.
An increase in osteocyte apoptosis during estrogen deficiency has been observed in humans, rats, and mice (270, 495, 496). Moreover, osteocyte apoptosis in estrogen-deficient mice has been spatially correlated with regions of increased bone resorption (142). Based on this evidence, it has been proposed that loss of estrogen increases osteoclast formation and bone resorption, in part, as a result of the increased osteocyte apoptosis. Although osteocyte death correlates with increases osteoclast formation in some models (492), this is not a universal finding (360). Be that as it may, if estrogen suppresses osteoclast number via control of osteocyte viability or function, such regulation must be indirect since none of the models in which ERα was deleted from osteocytes displayed increased osteoclast number or in bone resorption.
The AR has also been deleted using the Dmp1-Cre transgene, and this led to a moderate reduction of cancellous bone volume in the long bones and trabecular number in the spine of 8-month-old male mice (468). This change, however, was not yet present at 3 months of age. Although a definitive cellular mechanism could not be demonstrated in this study, the results suggest that the effects of androgens on trabecular bone mass are partly mediated by late osteoblasts and osteocytes (Table 2).
To conclude this section, the study of conditional deletion models has unexpectedly revealed that the effects of both estrogens and androgens on the cancellous and cortical bone compartments are mediated via different cell types. The antiresorptive effect of estrogens on cancellous bone in females results, by and large, from direct actions on osteoclasts, whereas the antiresorptive effects of androgens on cancellous bone in the male are exerted indirectly. In cortical bone, estrogens protect against resorption in both females and males, at least in part, via ERα-mediated actions (upon aromatization of androgens to estrogens in males) on uncommitted mesenchymal progenitors.
D. Attenuation of Reactive Oxygen Species
Mechanistic studies of the effects of sex steroid deficiency on the murine skeleton have revealed that loss of estrogens or androgens, similar to old age, leads to an increase in reactive oxygen species (ROS) in bone cells (14, 295, 326). Conversely, systemic administration of antioxidants attenuate the loss of bone mass due to sex steroid deficiency in male and female mice (14, 295). These observations have raised the possibility that an increase in ROS may be a common mechanism of the adverse effects of sex steroid deficiency and old age on bone homeostasis; and that sex steroid deficiency may accelerate the effects of aging on skeletal involution.
Generation of ROS occurs primarily in mitochondria during respiration and is caused by the escape of electrons passing through the electron transport chain (174, 371). This process generates superoxide, which is rapidly converted to H2O2, the more stable and most abundant form of ROS (31, 96, 117). High ROS levels cause damage to proteins, lipids, and DNA, leading to cell demise (194). However, at low levels ROS can also promote intracellular signaling for physiological cell functions (149, 233, 454).
In line with a pathogenetic role of oxidative stress on bone formation, ROS attenuate osteoblastogenesis via activation of FoxOs, transcription factors that promote compensatory adaptations in response to oxidative stress and growth factor deprivation (11, 13). In the osteoblast lineage, the overriding function of FoxOs is to provide an optimal balance among the maintenance of self-renewing stem cells, the replication of lineage-committed intermediates, and the survival of the terminally differentiated progeny, for the purpose of compensatory adaptations to stresses that accumulate in bone with advancing age (18, 227). Nonetheless, as is the case with several other defense responses against aging, FoxO activation can eventually aggravate the effects of aging on bone and become a culprit of involutional osteoporosis. Administration of antioxidants to ovariectomized or aged mice prevents osteoblast and osteocyte apoptosis, indicating that ROS shorten the lifespan of these cells (14, 235).
Consistent with the role of “lower levels” of ROS in signaling involved in physiological cell functions, RANKL and MCS-F, the two cytokines that are indispensible for osteoclast generation, promote mitochondria biogenesis and the accumulation of ROS in osteoclasts. These events are essential for the bone-resorbing function of osteoclasts and the bone loss caused by estrogen deficiency (37, 166, 185, 224, 302). Indeed, mice expressing mitochondria-targeted catalase, an enzyme that prevents H2O2 accumulation, in osteoclasts exhibit a decrease in osteoclast numbers and increased bone mass (37). Moreover, these mice are protected against the loss of cortical bone caused by either OVX or ORX (505); however, they undergo the same amount of cortical bone loss caused by aging as their littermate controls. These findings indicate that whereas attenuation of H2O2 generation in osteoclasts is sufficient to prevent the adverse effects of sex steroid deficiency on cortical bone, it has no impact on the effects of old age. On the other hand, mice expressing mitochondria-targeted catalase in cells of the osteoblast lineage are protected from the effects of old age on cortical bone, indicating that increased H2O2 generation with old age in cells of the mesenchymal lineage is a seminal culprit of the effects of aging on cortical bone (10). At present it remains unknown whether estrogens suppress the generation of H2O2 directly or indirectly, for example, by attenuating osteoclastogenesis via the suppression of pro-osteoclastogenic cytokines produced in cells of the osteoblast lineage, such as SDF1. The relevance of these findings in mice to humans remains unknown.
IV. EFFECTS OF SEX STEROIDS ON BONE VIA EXTRASKELETAL ACTIONS
A. B and T Lymphocytes
Lymphocytes have been proposed to contribute to the bone loss caused by sex steroid deficiency via a variety of mechanisms (211, 395). The role of T lymphocytes has been addressed primarily using various mouse strains deficient in this cell type. Specifically, it has been shown that a number of different T cell deficient mouse strains are protected from bone loss caused by OVX (395). The major mechanism is thought to be increased production of T cells that produce TNF-α (307), with a possible additional contribution of RANKL produced by T cells (497). Consistent with the latter possibility, increased production of RANKL by T cells has been observed in postmenopausal women compared with premenopausal controls (140). In contrast to these studies, others have shown that various mouse strains lacking T cells do lose cortical bone after OVX (303). More recently, the authors (S. C. Manolagas, M. Almeida, and C. A. O'Brien) and others have demonstrated that mice lacking RANKL specifically in T cells have normal bone mass and lose bone similar to control mice after OVX (366, 385). Thus it remains unclear whether T lymphocytes contribute to the bone loss caused by sex steroid deficiency.
On the other hand, numerous studies have reproducibly demonstrated that the number of B lymphocytes increases in the bone marrow of rodents after loss of either estrogen or androgens (304, 342, 354, 387, 503, 563), raising the possibility that this cell type plays an important role in the bone loss caused by sex steroid deficiency (354). This view is also supported by evidence that elevation of B cell numbers via administration of IL-7 is sufficient to increase osteoclast number and cause bone loss in mice (354). However, mature B cells are not required for bone loss induced by OVX as OVX leads to similar amounts of bone loss in mice lacking mature B cells compared with control littermates (310).
Deletion of RANKL from the entire B cell lineage prevents the increase in osteoclast number and loss of cancellous bone caused by OVX in mice (385). The amount of RANKL mRNA or protein expressed by B cells in wild-type mice does not increase, either 2 or 6 wk after OVX, supporting the idea that it is the increase in the total number of B cells expressing RANKL, rather than the expression level per cell, that stimulates osteoclastogenesis after loss of estrogen. Consistent with this, the number of bone marrow cells expressing RANKL is elevated in estrogen-deficient women, compared with women treated with estrogen for 3 wk or premenopausal women (493). However, a second study suggests that estrogen suppresses the amount of RANKL on the surface of B cells, as well as in T cells and bone marrow stromal cells, but does not change total cell number (140). The reason for the difference between the latter two studies is unknown but may be related to different durations of estrogen deficiency.
An increase in B cell number may contribute to osteoclast formation via multiple mechanisms. Perhaps the simplest scenario is that RANKL on the surface of B cells interacts with RANK on osteoclast progenitors and stimulates their differentiation into osteoclasts. However, it is also possible that B cells may be a source of cytokines other than, or in addition to, RANKL that promote osteoclast differentiation or function. Another possibility is that B lymphocytes, at specific stages of commitment, may act as a source of osteoclast progenitors, which are known to increase in the bone marrow after loss of sex steroids.
Multiple studies have shown that isolated B lymphocytes can act as osteoclast progenitors, at least in vitro (55, 247, 411, 443). These studies have used various methods to isolate cells expressing cell surface markers for B cells, such as B220 or CD19, and then exposed these cells in vitro to RANKL and M-CSF. Under these conditions, the cells that are formed are multinucleated, express TRAP and calcitonin receptor, and can resorb bone matrix. Nonetheless, some have attributed osteoclast formation in these cultures to contamination with macrophage-lineage cells (232). None of these studies has examined the ability of B cells to form osteoclast in vivo using lineage-tracing, and such studies will be required to definitively address this question.
How sex steroids control B cell number is only partially understood. Male mice with germline deletion of the AR exhibit increased B cells in the bone marrow (17, 562). Deletion of the AR gene in either osteoblast-lineage cells or in B lymphocytes also increases B cell number in the bone marrow, suggesting that androgens act both directly and indirectly on B cells to suppress their number (17, 562). Less is known about estrogen control of B lymphopoiesis. However, reconstitution studies using bone marrow from wild-type and ERα null mice suggest that, similar to androgens, estrogen acts on both hematopoietic and nonhematopoietic cells to control B cell number (203).
B. Muscle Cells
Estrogens and androgens are important for the growth and homeostasis of both bone and muscle, and a decline in the circulating levels of sex steroids leads to loss of mass and functional integrity in either tissue (86). Moreover, as it will be discussed in the following section on mechanical loading, it has been long postulated that mechanical strains exerted on bone by muscles are critical signals for bone mass accrual and strength (157). Therefore, the effects of sex steroids on bone may result in part via the effects of these hormones on muscle.
It is widely believed that in humans androgens, but not estrogens, are responsible for the beneficial effects of sex steroids on muscle. Strong support for this view has been recently provided by compelling evidence that in men with suppressed endogenous T and estrogen levels, the deficiency of androgens, but not estrogens, is responsible for the decreases in lean mass, muscle size, and strength (151, 220). Some studies in mice, on the other hand, have suggested a role of estrogens on muscle mass. Indeed, ERα as well as aromatase, but not ERβ, deletion reduces mass as well as contractile properties of some muscles in mice (74). In addition, combined ablation of AR and ERα results in a further decrease in quadriceps weight compared with AR ablation alone (83). These results, however, may have been confounded by effects of estrogens on the GH/IGF-I axis. Thus the contention for an indirect effect of estrogens on skeletal homeostasis, secondary to an anabolic effect on muscle, remains a matter of conjecture.
T increases muscle mass both in young (49) and older healthy men (51). Similarly, in postmenopausal women, which also have low androgen levels, androgens augment muscle mass (132, 213) as well as muscle protein synthesis (475). Nonetheless, while there is agreement that androgens increase muscle mass, it is not clear that T administration improves muscle strength and physical function (116, 172). Myocyte- or satellite-cell specific AR ablation reduces muscle mass or strength. Additionally, androgens may exert muscle-anabolic actions via paracrine mechanisms or actions on muscle-resident fibroblasts (136–138, 389). Importantly, muscle-specific AR ablation in mice has hitherto not been shown to affect cortical or cancellous bone (389).
In conclusion, the effects of androgens on bone are not mediated by direct actions on muscle, at least in mouse models. Whether the same is true in humans remains unknown.
C. Other Putative Targets
Estrogen deficiency causes an increase in fat mass in rodents (83, 153, 279, 344). A recent randomized trial in men also showed that estrogen deficiency promotes fat accumulation in men (87). In postmenopausal women however, it remains unclear whether body weight gain and fat accumulation are caused by estrogen deficiency (151). On the other hand, high fat mass may increase extragonadal estrogen production through aromatization in adipocytes. On the basis of these two lines of evidence, as well as higher BMD measurements in obese subjects, it has been thought in the past that overweight postmenopausal women may be protected from osteoporosis because of increased estrogen levels as well as increased mechanical strain on their skeleton. However, recent studies using quantitative computed tomography (QCT), which is less likely to be confounded by projectional artifacts, indicate that bone volumetric density, geometry, or strength-to-load ratio are not necessarily increased in obese subjects (457, 578). Additionally, higher bone mass in obese people does not necessarily translate into decreased fracture susceptibility (108, 238). BMI may also have site-specific effects, with increased hip fracture risk associated with low BMI and relatively increased risk of upper arm fractures in obesity (238). Moreover, estrogen levels are lower in obese subjects with type 2 diabetes (129).
The central nervous system (CNS) is a target organ of estrogens and androgens, and AR, ERα, and ERβ are widely expressed in the brain (173). And, central nervous pathways may control skeletal homeostasis (244). Taken together, these two lines of evidence have raised the possibility that sex steroids regulate bone mass in part via influencing neuronal pathways. In support of this notion, ERα deletion in neuronal cell progenitors using nestin-Cre led to higher bone mass because of increased accrual of bone during early growth (381). AR deletion leads to decreased spontaneous physical activity, which may explain some of the skeletal effects of androgen deficiency (388, 416).
The possibility that actions of sex steroids on the adipose tissue and CNS mediate some of their effects on bone requires further investigation.
V. DISTINCT CELLULAR TARGETS IN CANCELLOUS VERSUS CORTICAL BONE AND WHAT IT MEANS
The most unexpected discovery and perhaps the most significant insight gained from the generation and study of mouse models with cell-specific deletion of the ER and AR is that the effects of sex steroids on cancellous and cortical bone are mediated by different cell types and mechanisms (Tables 1 and 2). The implications of these new insights deserve highlighting here as they represent important conceptual advances in our understanding of the role of sex steroids in skeletal physiology and pathophysiology. Moreover, they will help the reader to better understand the pathophysiology of skeletal disorders caused by sex steroid deficiency, the subject of subsequent sections.
To recap the evidence from the cell-specific deletion models of the ER and AR, in females the osteoclast ERα mediates the protective effect of estrogens on the cancellous, but not the cortical bone compartment, which represents the majority of the skeleton (339, 365). The ERα in committed osteoblast progenitors targeted by Osterix1 (Osx1-Cre), on the other hand, promotes cortical bone accrual at the periosteum in both females and males; while the ERα in uncommitted mesenchymal progenitors targeted by Prx1-Cre mediates the protective effect of estrogens against endocortical, but not cancellous, bone resorption in females (15). As in the case of estrogens in females, the effects of androgens on the cancellous and cortical bone compartment in the male are mediated via different cell types. Nonetheless, whereas the antiresorptive effects of estrogens on cancellous bone result from direct actions on osteoclasts, the antiresorptive effects of androgens on cancellous bone are exerted indirectly (339, 365). Indeed, androgen signaling through the AR expressed in cells of the mesenchymal lineage, most likely the mature osteoblasts and osteocytes, mediates the protective effects of androgens on cancellous bone by indirectly decreasing osteoclast numbers and restraining bone resorption in this compartment (520).
Collectively, these new insights point to the inexorable conclusion that the signals of estrogens and androgens are orchestrated and fine-tuned in different anotomical sites and for different purposes. Specifically, these hormonal signals are modified and integrated at different bone sites with different environmental cues (mechanical strains or the local concentration of paracrine cytokines and growth factors).
Importantly, the same principle of signal orchestration and fine-tuning in different anatomical sites and for different purposes is becoming increasingly more clear from advances in other areas of bone biology, including the mechanisms responsible for the development and maintenance of the skeleton, topics that will be our focus in subsequent sections. Briefly here, another good example of bone site-specific signal orchestration is the evidence that RANKL is not only produced by multiple cell types but the contribution of one cell type versus another differs depending on the particular bone site and physiological versus pathological conditions (321, 377). Moreover, the production and/or release of RANKL is modulated differently at particular environmental niches by local cues, including other factors that can enhance RANKL activity (Wnt5a), mimic it (SDF1), or dampen it (OPG). In line with the general principle, osteoblast/osteocyte-specific deletion of RANKL causes a cancellous, but not cortical, phenotype (574). Similarly, Wnt5a increases bone resorption and decreases bone mass in the cancellous, but not the cortical, compartment; whereas Wnt16 increases cortical, but not cancellous, bone mass (364).
VI. CROSSTALK BETWEEN SEX STEROIDS AND MECHANICAL LOADING
Mechanical loading or lack thereof are seminal signals for the formation and removal of bone, respectively, and are critical factors for the optimal accrual of bone mass during growth and the development of osteoporosis later in life. Mechanical loading tilts the balance between bone formation and resorption in favor of the former, by stimulating bone formation and suppressing bone resorption (426, 436). Mechanical unloading has the exact opposite effects. For example, exposure to frequent high-impact loading with physical exercise promotes bone formation as evidenced by the increased bone mass in the dominant arm of tennis players (222, 239). Conversely, the unloading that results from immobilization, prolonged bed rest, or weightlessness during space flights causes precipitous and marked reduction in bone mass, associated with decreased bone formation and increased bone resorption (284, 296). Because the adaptation of bone to mechanical loading is greater during growth than during adulthood, the accrual of bone mass and size in youth has profound influence on the risk for osteoporosis and fractures later in life (60, 206, 548). The major effects of mechanical loading or unloading on the skeleton have been readily reproduced in rodent models employing tibia- or ulna-loading, tail suspension, or neurogenic paralysis of the hindlimbs (292, 345) Nevertheless, the cellular and molecular mechanism(s) mediating the effects of physical signals initiated by loading remain poorly understood.
A. Crosstalk With Estrogens
Harold Frost was the first to postulate that estrogens enhance the mechanoresponsiveness of bone, and estrogen deficiency has the opposite effect (156). He proposed that similar to disuse, changes in estrogen levels like the ones that occur with puberty or menopause affect the adaptive response of bone to mechanical loading, but only in bone adjacent to the marrow, i.e., cancellous and endocortical bone. These ideas have received considerable attention and followed up with extensive experimental work showing that the ERα, in and of itself (i.e., in its unliganded state), rather than estrogens participate in mechanosensing (41, 286). Loss of estrogens in female mice or rats has no effect on the response of cortical bone to loading (187, 566). On the other hand, female mice with global ERα deletion fail to exhibit the expected response of cortical bone to mechanical loading (301). In contrast, male mice with global ERα deletion have normal response to mechanical loading (80, 445).
Studies with mice lacking the AF1 or the AF2 domains of the ERα have further revealed that the effects on mechanical loading are dependent on the AF1 function domain of ERα, whereas the AF2 domain is dispensable (445, 566). Conversely, the effects of estrogens on bone mass maintenance require the AF2, but not the AF1 domain (66). Interestingly, global deletion of ERβ in mice enhances the response of cortical bone to mechanical loading, raising the possibility that ERα and -β may exert opposite effects in the adaptation of bone to loading (445, 447).
Several cell types of bone are capable of responding to mechanical loading (437). The earliest and most critical event in the adaptive response of bone to mechanical loading is an increase in the proliferation of mesenchymal/stromal cells (407, 439, 502). Consistent with this evidence, the ERα expressed in mesenchymal/stromal cells (expressing Osx1-Cre) is indispensable for optimal periosteal bone accrual and the periosteal response to loading (15, 228). Furthermore, osteoblastic cell proliferation in vitro in response to strain requires ERα and is associated with stimulation of IGF-I and Wnt signaling (15, 26, 121, 161, 484). Taken together, these lines of evidence raise the possibility that both mechanical forces and ERα exert their effects on bone by stimulating Wnt signaling. Consistent with this hypothesis, Wnt/β-catenin signaling in osteoblast lineage cells is a critical mediator of the bone formation induced by mechanical loading. Loading increases the expression of Wnt ligands, such as Wnt16, and reduces the expression of Sost, the gene encoding the potent Wnt antagonist sclerostin (427, 557). Moreover, mice lacking the Wnt co-receptor Lrp5 or Wnt16, or mice overexpressing Sost, exhibit reduced osteogenic response to mechanical loading (444, 446, 500, 557).
Osteocytes are traditionally thought of as the principal mechanosensing cell in bone (3, 59). However, mice with targeted deletion of ERα in mature osteoblasts and osteocytes (using Dmp1-Cre) have normal cortical bone, and the cortical bone response to mechanical loading is unaffected. This evidence argues against the possibility that osteocytes are the mediators of the osteogenic actions of estrogens or the unliganded ERα itself (268, 564).
B. Crosstalk With Androgens
In contrast to the extensive study of estrogenic effects on loading-induced bone accretion in male and female rodents, only a few studies have addressed the potential role of androgens in modulating bone's adaptive response to mechanical loading.
Contrary to the effect of ERα deletion in female mice, ORX increases the osteogenic loading response in cancellous bone of male mice (155). This counterintuitive result is consistent with the finding that genetic AR disruption enhances periosteal bone formation in response to identical strain magnitudes compared with the wild-type littermates (80). Furthermore, Both T and DHT suppress the loading-related increase in cortical thickness and periosteal bone formation, indicating a direct role for AR activation in inhibiting skeletal mechanoresponsivity (470). Similarly, T decreases the response of wild-type but not ARKO-derived primary bone cells to mechanical stimulation (80). Altogether, these lines of evidence indicate that AR activation limits bone's response to mechanical loading, at least in mice. Whether the same mechanism also occurs in humans requires further investigation.
The increase in the RANKL/Opg mRNA ratio in bone following ORX in mice is prevented by loading, indicating that in the context of androgen deficiency physical activity may ameliorate the loss of bone through an antiresorptive effect (470). Notably, forced physical exercise also ameliorates some of the musculoskeletal deficits observed in male ARKO mice (388). Mice with global AR deletion display a stronger load-induced inhibition of sclerostin (80). Similarly, in castrated mice, T stimulates sost expression and abrogates the suppressive effect of loading on sost (470). Thus AR signaling seems to suppress load-induced bone formation by inhibiting Wnt/β-catenin signaling secondary to increasing sost. A similar mechanism is observed in muscle, where AR signaling directly upregulates myostatin, a negative regulator of muscle mass, as a feedback mechanism to avoid excessive muscle growth (137).
The evidence discussed in the previous paragraph notwithstanding, the effects of androgens on sost expression are not necessarily the result of AR signaling in osteocytes. Indeed, osteocyte-specific deletion of the AR in males has no effect on the loading response (470). Therefore, the inhibition of mechanoresponsiveness may result from AR actions in other cell types, such as mesenchymal/stromal or periosteal cells.
In conclusion, mouse models with androgen deficiency or global AR deletion have decreased cortical and trabecular bone mass, but their skeletal response to mechanical loading is not impaired and may even be increased.
VII. EFFECTS OF SEX STEROIDS ON SKELETAL DEVELOPMENT AND GROWTH
Mammalian skeletons grow in three dimensions. In the long bones and the vertebrae, longitudinal growth (Z-axis) is mediated by chondrocytes at the epiphyseal growth plates, and appositional growth (X- and Y-axis), the outward bone expansion, results from osteoblastic bone formation at the periosteal surface. Simultaneously, bone is resorbed at the endosteal surface to expand the marrow cavity. All these events are sexually dimorphic and in humans contribute to sex differences in bone strength and fracture risk later in life (76, 373, 483).
A. Longitudinal Growth and Final Height
A sex difference in longitudinal bone growth in humans is universally recognized from the smallest pygmy tribes to the Neanderthals. Adult men are on average 7–8% taller than women (184, 432). In the year 2000 United States growth charts, the P50 values for women and men are 164 versus 176 cm, a difference of about two standard deviations (379). Such a difference is quite remarkable given the fact that adult human height is a highly polygenic trait associated with no less than 697 variants in recent genome-wide meta-analysis, and ESR1 (the gene for ERα) is just one of these (569). Most of the common autosomal variants appear to influence prepubertal growth. Sex differences in growth and ultimate height on the other hand are accrued during puberty. Before puberty, skeletal features show no significant sex differences (375, 451). Thus sexual dimorphism in height is likely regulated by pubertal sex steroids. Nonetheless, growth-controlling factors on the X and Y chromosomes are also known to exert a strong influence (391).
The main determinant of greater ultimate height in men is the later onset of puberty in boys compared with girls. The pubertal growth spurt starts on average 2 years later in boys, allowing more time for prepubertal growth which, at a rate of about 5.5 cm/yr, already explains an 11 cm difference (Figure 6). In other words, boys are taller than girls before their growth spurts start, with less difference in height gained during their growth spurts. Within each sex there is considerable variation in the timing of puberty, but later onset of puberty is generally associated with greater final height (53, 199). Peak height velocity is also somewhat greater in males (252, 458), but its effect on ultimate height is considerably smaller. A third possible mechanism is delayed growth plate closure in men, i.e., longer duration of the growth spurt (225).
FIGURE 6.
Mean height velocity (in cm/yr) in non-African American children according to age and sex [from the U.S. Bone Mineral Density in Childhood Study (252).] The growth spurt starts ∼2 yr later in boys (9 vs. 11 yr). The striped area under the height velocity curve indicates the effect of delayed growth spurt onset on ultimate height in boys compared with girls. Peak height velocity (PHV) is also somewhat greater in boys on average, and growth velocity may be better maintained (and growth plate closure delayed) in late puberty (225). However, the latter factors play only a minor role in the sex difference in ultimate height, as indicated by the size of the two respective gray areas under the height velocity curve. [Based on results from Kelly et al. (252), with permission from the Endocrine Society.]
In both sexes, prepubertal growth velocity is greater in the appendicular than in the axial skeleton (40, 72, 225, 451). Therefore, prepubertal children or patients with disorders of puberty exhibit so-called eunuchoid proportions (i.e., long limbs relative to the spine). At the start of puberty, truncal growth accelerates, while appendicular growth does so only minimally, before it decelerates at the end of puberty. Because of the longer duration of prepubertal growth (predominantly in the appendicular skeleton) in boys, the average man has longer legs than the average woman, but sitting height is affected less (40, 72, 225, 451).
In summary, men are on average ∼8% taller than women. This difference in adult height is mainly due to later onset of puberty in males, allowing more time for prepubertal, predominantly appendicular growth. Greater peak height velocity and longer duration of intrapubertal growth (delayed epiphyseal closure) play only a minor role (225). Notably, taller women have greater fracture risk than shorter women, possibly due to increased fall impact as well as comparatively more porous and slender bones (54). It is unknown, however, whether sex differences in height also contribute to sex differences in fracture risk.
B. Peak Bone Mass Acquisition
The sex difference in osteoporotic fracture risk can be attributed in part to differences in peak bone mass [PBM, i.e., peak bone mineral content (BMC) at the end of the skeletal maturation] and bone width. Differences in subsequent bone loss are another important contributor. Mathematical models predict that even small increases in PBM, followed by identical rates of age-related bone loss, would considerably delay and decrease fracture burden (206). However, in preclinical and clinical interventional studies aiming to increase PBM (mainly by physical exercise), BMD returns to a homeostatic set point after the intervention stops (44). It is important to note that DXA BMD is only a two-dimensional (not a volumetric) surrogate measurement of bone mass that relies on the attenuation of an X-ray beam by the mineral content (i.e., hydroxyapatite) of bone. The BMD value is derived by dividing BMC in grams by bone area in square centimeters. BMC is another important determinant of bone strength in and of itself. Structural bone adaptations, especially development of a wider cortex gained, for example, by exercising early in life, can be maintained into old age (547, 548). Conversely, studies in osteoporotic men and their sons suggest that low PBM continues to be relevant into old age (256, 288, 515). Thus it is plausible that the decreased risk of osteoporotic fractures in older men versus women is at least partly due to greater PBM development in young men, especially cortical bone expansion.
Young adult men have almost 25% greater whole body BMC compared with women (63), but such difference is not more than what can be expected for their ∼8% greater height (extrapolated to three dimensions, 3√125% ∼108%). Upon closer examination, PBM at different sites shows time- and sex-specific differences, but these rely almost entirely on greater bone area (42, 44, 205, 316). For example, BMC in one study peaked at ages 21–22 in men and ages 23–28 in women, with greater BMC at the femoral neck (+28%), ultradistal radius (+44%), and lateral spine (+12%) in men compared with women (205). Areal BMD differences are however much smaller (∼5–10%) or even absent at certain si tes like the lumbar spine, especially following adjustment for bone size (44, 63, 204). In fact, calculated volumetric BMD (vBMD) tends to be lower in men, especially at the lumbar spine (205, 541). In other words, men develop greater PBM because their bones are longer and also wider, but certainly not denser.
Later puberty is associated with greater final height, but lower PBM particularly in girls (99, 123, 230). During young adulthood, skeletal consolidation (or catch-up development) may counter some of the adverse effects of delayed puberty on PBM, but subtle deficits in cortical and trabecular bone structure may persist (60, 98–100, 123, 380).
Thus PBM advantages in men appear to be site-specific and mainly due to greater bone area. Still, because of the low resolution and the two-dimensional nature of DXA, advanced techniques like high-resolution, peripheral quantitative computed tomography (HR-pQCT) (148, 291) are necessary to determine true differences in vBMD, cortical bone area, cortical porosity, or trabecular bone volume (Figure 7). As it will be discussed below, the single most important determinant of greater bone strength and peak bone mass in young men is development of a wider bone due to greater periosteal bone expansion. Whether skeletal microarchitecture (e.g., osteocyte lacunae or vascular channel volume) or material properties are sexually dimorphic in humans remains unknown.
FIGURE 7.

Schematic representation of the differences in cortical bone structure and density for girls (G) and boys (B) across puberty [by Tanner (T) stage]. Boys acquire higher estimated bone strength (failure load) due to their greater cortical bone diameter. The medullary cavity is also wider in boys, resulting in only mildly greater cortical thickness. Volumetric bone mineral density (vBMD) is higher in girls (whither cortex). Trabecular bone volume and cortical porosity (Ct.Po) are also higher in boys (differences not depicted). [From Nishiyama et al. (375), with permission from John Wiley & Sons, Inc.]
1. Cortical bone
HR-pQCT has recently provided unprecedented understanding of sex differences in peri-pubertal bone development (148). However, some limitations should be considered. First, HR-pQCT can only be performed on the distal radius and tibia. Although microstructure at these sites predicts both vertebral and nonvertebral fractures (534), it may not be representative of the entire skeleton (in particular of the spine). Second, microporosity from osteocyte lacunae or vascular channels is below the resolution of HR-pQCT scanners; therefore, measured porosity mainly reflects larger resorption pores. Bone strength is not measured directly but estimated from finite element analysis modeling, which does not take into consideration possible variations in material properties or nanostructure. Finally, estimation of cortical thickness critically depends on the boundary between cortical and trabecular bone, which is arbitrary due to trabecularization of the endocortical surface with aging (580).
All parameters measured by HR-pQCT in the radius and tibia are different between young adult men and women, with the exception of total volumetric BMD (2). These findings clearly show that men build a structurally stronger skeleton with similarly or even slightly less mineralized material (Table 3).
Table 3.
Determinants of greater peak bone strength in men
| Men Versus Women at PBM (age 20–30 yr) | |
|---|---|
| Bone strength* | ⇈ |
| Strength-to-load ratio | ↑ |
| Cortical bone | |
| Volumetric BMD | ↓ |
| Cortical thickness | ≈ or ↑ |
| Total cross-sectional area or periosteal circumference | ⇈ |
| Endosteal (marrow) area | ↑ |
| Cortical porosity | ↑+ |
| Trabecular bone | |
| Trabecular bone volume | ⇈ |
| Trabecular number | ≈ or ↑ |
| Trabecular thickness | ↑ |
| Nanostructure | ? |
| Material properties | ? |
Relative difference in failure load (in N) estimated by micro-finite element analysis. +Although the sex difference is large in relative terms, it is still quite low in both sexes in absolute terms. ≈, Not significantly changed; ↑, slightly increased; ↓, slightly decreased; ⇈, markedly increased.
As compared with women, men have a greater cortical bone diameter due to greater periosteal apposition, placing the cortex further away from the neutral axis. Bone strength scales as the fourth power to bone diameter, independent of cortical thickness (76, 120, 193, 193, 261, 320, 375). This is the key reason why men have stronger bone. The medullary cavity is also wider in men (i.e., periosteal expansion is accompanied by endosteal resorption and net outward bone expansion), while cortical thickness is only mildly increased (Table 3 and Figure 7) (193, 261, 320). Contrary to earlier ideas that girls have more endosteal apposition than boys, endocortical resorption dominates in both sexes, but less so in females (160). Thus, even though estrogens may stimulate endosteal bone formation resulting in net endosteal contraction in some situations (489), normal pubertal bone development is characterized by diminished endosteal expansion in girls. In other words, the marrow cavity enlarges during puberty in both sexes but less so in females. The idea that estrogens shrink the bone marrow cavity in girls is most likely erroneous. Importantly, sex differences in bone structure are accrued during puberty, as evidenced by minimal or no sex differences in prepubertal children by (HR-)pQCT (375).
On the other hand, young adult men appear to have slightly but significantly lower cortical vBMD and higher intracortical porosity (76, 193, 320, 375, 542). Especially during rapid growth, a transient deficit in cortical bone thickness and increased porosity coincide with the peak incidence of fractures in childhood, typically at the radius and more frequently so in boys (265, 545). Still, the greater cortical bone diameter far outweighs the lower cortical vBMD and greater porosity, with an estimated 34–48% greater ultimate failure load in young adult men versus women (193).
2. Trabecular bone
Young men develop greater trabecular bone volume in late puberty, at least at the distal radius and tibia. This is mainly due to greater trabecular thickness, at least at the radius. Trabecular number may also be higher in men, especially at the tibia (76, 120, 261, 265, 320, 375).
At central skeletal sites, however, the situation seems to be opposite. Calculated vBMD of the lumbar spine is in fact lower in men (205, 541). QCT studies confirm that at PBM, men have wider lumbar spine and femoral neck with paradoxically lower trabecular vBMD (46, 423). This may be due to the dominant effect of cortical bone which bears most of the loads, thus obviating the need for additional trabecular support. Further studies on bone structure at central sites in different sexes and ages are needed to clarify this subtlety.
In summary, men have a biomechanically superior PBM acquisition characterized mainly by wider bones due to greater periosteal bone expansion. Trabecular bone volume in men is also greater at peripheral, but not central skeletal sites.
C. Periosteal Expansion
Periosteum is a thin layer of connective tissue that covers the external surfaces of most bones and is rich in osteogenic cells. Periosteal bone apposition is, by and large, responsible for the enlargement of bones during growth. At the completion of longitudinal growth and epiphysial closure, periosteal apposition slows precipitously (9, 452). Importantly, greater periosteal expansion during puberty accounts for the larger bones in men compared with women (423), hence the increased strength and reduced fracture risk in men (6, 390).
Sex steroids are critical contributors to periosteal bone expansion. Indeed, higher serum levels of T are associated with larger periosteal circumference in long bones of young adult men (316). However, part of the effect of androgens on periosteal expansion results from aromatization to estrogens, a conclusion strongly supported by evidence that estrogen administration to aromatase-deficient adolescents (with normal androgen levels) increases bone size due to periosteal expansion (67).
Similar to humans, estrogens and androgens promote the expansion of the periosteum in growing male rodents. Indeed, suppression of estrogen synthesis by administration of an aromatase inhibitor reduces periosteal expansion over and above the reduction caused by ORX (82). Furthermore, male mice with global AR or ERα deletion exhibit lower periosteal circumference, which is further diminished in mice lacking both of these receptors (529). Female mice lacking ERα globally similarly exhibit lower periosteal circumference (463). Mice with targeted ERα deletion in mesenchymal/stromal cells (using Osx1-Cre) have decreased periosteal bone apposition and cortical bone mass. In contrast, mice with targeted ERα deletion in osteoblasts and osteocytes have no cortical bone phenotype (15, 268, 564). This genetic evidence strongly suggests that the ERα pool in mesenchymal/stromal cells, but not in mature osteoblasts, is responsible for the effects on periosteal expansion. As discussed in section V, however, the effects of ERα on periosteal apposition are independent of the binding of the ligand.
Targeted deletion of the AR across all stages of the osteoblast differentiation progression from mesenchymal progenitors to mature osteoblasts and osteocytes (using Prx1-Cre, Col1-Cre, OCN-Cre and Dmp1-Cre) has no effect on cortical bone mass in mice, indicating that the stimulatory actions of androgens on periosteal expansion are not mediated via osteoblasts or their progenitors (101, 319, 376, 468, 504). Thus the target cell(s) of the actions of androgens on the periosteum remains unknown.
D. Epiphyseal Closure
During postnatal life, long bones grow and elongate via the process of endochrondral ossification, during which cartilage made by chondrocytes is formed and then replaced by bone (273). Puberty and the associated production of sex steroids exert major regulatory influence on this process. At the onset of puberty, the relatively low concentration of estrogens and androgens stimulate the growth spurt by increasing growth hormone (GH)/IGF-I levels. The stimulatory effects of androgens during early puberty is most likely due to modulating influences on the pattern of GH production. During the late stages of puberty, the higher estrogen concentrations, but not androgens, shut off longitudinal growth in both girls and boys by stimulating the closure of the epiphyseal growth plates, an effect mediated via direct actions of estrogens on proliferating chondrocytes (Figure 8) (65, 520).
FIGURE 8.

Proposed biphasic regulation of longitudinal bone growth and epiphyseal closure by sex steroids. Early in puberty, rising sex steroid concentrations stimulate longitudinal growth via indirect effects on growth hormone (GH) and insulin-like growth factor I (IGF-I), which both stimulate growth plate chondrocytes. In late puberty, higher estrogen concentrations exert an overruling inhibitory effect via ERα in chondrocytes. Androgens have effects mainly on GH while peripheral and central aromatization are believed to be more important for circulating IGF-I. AR is also expressed in chondrocytes, but whether this contributes to sex differences in longitudinal growth remains unclear. [Adapted from Börjesson et al. (65), with permission from John Wiley & Sons, Inc.]
Invaluable insights on the critical role of estrogens in longitudinal bone growth have been provided by the study of rare genetic disorders of estrogen action. Indeed, individuals with aromatase (CYP19) deficiency have tall stature and delayed bone age, consistent with the restraining effect of estrogens on epiphyseal closure and final height. These patients continue to grow at prepubertal rates into adulthood but lack a pubertal (mainly truncal) growth acceleration, resulting in long limbs compared with their trunk height (eunuchoid proportions; see sect. VIIA) (361, 430). Similarly, one woman and a man with loss of function ERα mutations did not experience clear pubertal growth spurt. On the other hand, their epiphyses remained open and were taller than their predicted adult height due to continued growth into adulthood (414, 473). Consistent with this, chondrocyte-specific ERα inactivation in mice did not affect the pubertal growth spurt but prevented epiphyseal fusion and growth deceleration thereafter (65). Collectively, this genetic evidence strongly supports the hypothesis of biphasic effect of estrogens on bone growth: stimulatory at low concentrations early in puberty during the growth spurt and inhibitory at the growth plate at the end of puberty. Additional support for the biphasic model of estrogen action is provided by evidence in the general population, showing that common variants in the ESR1 gene are significantly associated with adult human height (569).
In subjects with androgen insensitivity syndrome (AIS) due to inactivating AR mutations, adult height is greater than female and close to male reference values (122, 191). The taller stature compared with women is probably due to impaired epiphyseal fusion from estrogen deficiency as well as growth-stimulating factors on the Y chromosome. The lower height compared with normal male values in turn is probably due to lack of sex steroid actions on GH and IGF-I (122, 191).
Support of the biphasic model of sex steroid regulation of bone growth is also provided by both observational as well as therapeutic intervention studies. For example, E2 and IGF-I levels in girls are positively associated with tibia length early in puberty, while E2 is negatively associated with tibia length thereafter (575). High-dose estrogens, on the other hand, cause fusion of the growth plates and prevent undesirable longitudinal growth in girls. Low-dose estrogens in turn can trigger the pubertal growth spurt in girls with Turner syndrome or constitutional delay of growth and puberty (CDGP), without adverse effects on ultimate height (167, 417, 551). Conversely, aromatase inhibitors (which decrease estrogen and increase androgen concentrations) prevent growth plate closure in boys and advance bone age, with positive effects on predicted adult height (208, 440, 561).
Short courses of low-dose T or the weak, nonaromatizable androgen oxandrolone have been used in boys with CDGP to accelerate puberty and height velocity, without adversely affecting age/epiphyseal closure or ultimate height (107, 417). Likewise, oxandrolone treatment of girls with Turner syndrome receiving GH increases ultimate height by accelerating height velocity, without altering bone maturation or pubertal progression (167, 409, 434). The mechanism by which nonaromatizable androgens accelerate growth in the face of supraphysiological GH treatment remains unexplained, but an increase in IGF-I due to androgen modulation of GH patterns remains a possibility (112, 409).
E. Interaction of Sex Steroids and Growth Factors
1. Effects on longitudinal growth
GH has direct effects on chondrocytes and osteoblasts as well as indirect effects via stimulation of hepatic secretion of IGF-I and IGF-binding proteins (IGFBPs). Moreover, osteoblasts and chondrocytes produce IGFs and IGFBPs and deposit them locally within the bone matrix. Mice with genetic deficiency in GH or IGF-I action have short stature and reduced periosteal bone expansion, but normal trabecular bone mass. Similarly, humans with GH deficiency have decreased areal BMD, but volumetric density is unaffected, suggesting an adverse effect on bone size (68, 322). The role of GH in skeletal maintenance is far less clear; albeit, low circulating IGF-I levels have been associated with increased fracture risk (165, 172, 382). Moreover, recombinant IGF-I/IGFBP3 complex treatment has beneficial effects on bone density, grip strength, and functional recovery in postmenopausal women with hip fracture (62).
Puberty is accompanied by a rise in both amplitude and duration of GH secretory peaks as well as IGF-I concentrations in both sexes. However, circulating IGF-I as well as average GH concentrations are not sexually dimorphic in humans. The main sex difference is the pattern of GH spikes, with males exhibiting a more ordered secretion than females, both before and after puberty (528). Sex steroids affect GH secretion both in puberty and adulthood (104, 441). Additionally, the perinatal T peak determines male-pattern pituitary GH secretion as well as sex-specific hepatic steroid metabolism via imprinting mechanisms (234). Accordingly, androgen-resistant (testicular feminization, Tfm) rats display femalelike GH secretion (351).
Observational and experimental studies in humans support a role for both E2 and T in modulating different properties of GH spikes, while GH and estrogens codetermine circulating IGF-I levels (526–528) (Figure 8). Similarly, IGF-I is increased in estrogen-treated (362, 531) and decreased in ERαKO, ERαβKO, aromatase-KO, and aromatase inhibitor-treated male mice. These observations suggest that aromatization plays a key role in the maintenance of circulating IGF-I, probably via both GH-dependent and direct hepatic effects (82, 83, 394, 529). Aromatase inhibition may also decrease IGF-I in young men (287), while androgenic stimulation seems to have no clear effect (370). The effects of sex steroids on IGF-I in rodents are conflicting. Studies in orchidectomized mice treated with an aromatase inhibitor suggest that sex differences in body size rely on synergistic effects of both androgen actions and GH/IGF-I (82). A direct role for androgens on GH-dependent growth (independent of aromatization) is further supported by evidence that mice with conditional deletion of the AR in the nervous system (nestin-AR−/−) have decreased length and IGF-I concentrations (419). Furthermore, GH-receptor-KO (GHRKO) mice are not only small but have decreased pubertal growth (82). The Jak/STAT5b-pathway acts downstream of the GH-receptor with STAT5b-KO mice being resistant to GH pulses. These mice have female-like growth curves in both sexes (507). Still, E2 can restore defective hepatic Jak/STAT5b signaling in GHRKO mice, thereby independently stimulating IGF-I secretion (531). Furthermore, patients with Laron syndrome (congenital GH receptor deficiency with low IGF-I) do not experience a growth spurt despite a delayed but full puberty. Furthermore, a unique study in GnRH-deficient (hypogonadal, hyp) mice showed that the perinatal T surge and consequent hypothalamic and hepatic imprinting effects are required for an optimal growth spurt (462). Collectively, these data show that GH/IGF-I actions are primordial mediators of sex steroid actions on the pubertal growth spurt.
Conversely, sex differences in height do not rely entirely on GH/IGF-I. Indeed, Laron patients still display a sexually dimorphic growth during adulthood, with males having a lower height velocity but delayed growth plate closure compared with females, the latter resulting in continued growth and greater ultimate height in adult males compared with female Laron subjects (269, 289, 290). This pattern is in line with a GH-independent effect of estrogens on hepatic IGF-I secretion, which explains why girls with Laron syndrome exhibit earlier height velocity acceleration, and growth plate chondrocytes. This also explains why these girls experience earlier growth plate closure compared with men with Laron syndrome. Also in subjects with combined GH and gonadotropic deficiency, GH replacement restores prepubertal growth rates, but add-on treatment with low-dose E2 in girls or T in boys is required to initiate the growth spurt (417).
In summary, the effects of androgens and estrogens on the pubertal growth spurt are mediated via effects on the GH secretion patterns, while estrogens also have direct effects on hepatic IGF-I release (Figure 8).
2. Effects on periosteal bone expansion
Male mice have greater periosteal bone formation during early puberty, and concomitantly greater radial bone expansion due to actions of both AR and ERα (82, 83, 394). GHRKO mice, however, show no sexual dimorphism in cortical bone size. Hence, the effects of sex steroids on cortical bone expansion in male pubertal mice rely largely on GH/IGF-I signaling (81, 82, 530). Similarly, osteoblast-specific deletion of GH-receptor results not only in reduced osteoblastic sensitivity to IGF-I in vitro but also in a striking feminization of cortical bone geometry in male mice in vivo (467). Nevertheless, castration and replacement studies reveal that androgens still enhance cortical bone development in GH-deficient rats and mice (263, 530, 583) without clear IGF-I changes (82, 530). Furthermore, the perinatal T surge is not required for the effects of androgens on cortical bone expansion (462). Thus androgens regulate cortical bone expansion both directly and indirectly via circulating GH/IGF-I.
Global or osteoblast-specific ERαKO decreases periosteal bone formation and cortical bone expansion (15, 83). Conversely, OVX increases cortical bone expansion as well as the endocortical perimeter (82, 177, 263, 583). Periosteal expansion is inhibited by estrogens and stimulated by GH-signaling in females (154, 263, 583). Since the stimulatory effects of ERα on periosteal bone expansion occurs through osteoblast-lineage cells and estrogens have the opposite effect, neither of these effects is likely to involve systemic GH/IGF-I regulation.
Data on cortical bone in human growth hormone resistance remain scanty but suggest sex differences in cortical thickness (269). At least one study showed significantly greater bone diameter and cortical thickness in adult men compared with women with untreated GH deficiency (219). Therefore, the mechanistic insights from mouse models are applicable to humans, although further investigation into cortical bone geometry in humans with GH/IGF-I deficiency or resistance is needed.
Trabecular bone on the other hand is probably regulated directly by androgens and estrogens and not indirectly via GH/IGF-I. Indeed, male GHRKO mice have normal trabecular bone mass and respond normally to androgen or estrogen deficiency and replacement (530). Yet, OVX results in bone loss at trabecular sites even in GH- or IGF-I-deficient female rodents (154, 583).
In conclusion, both AR and ERα are required for optimal cortical bone expansion in males, acting largely but not exclusively via systemic GH/IGF-I signaling. In females, ERα is required for optimal periosteal expansion while estrogens maintain the endosteal perimeter and limit periosteal expansion; both of these effects are independent of the somatotropic axis.
VIII. EFFECTS OF SEX STEROIDS ON SKELETAL MAINTENANCE
The progressive increase in the mass of bone during development and growth is accomplished primarily by bone modeling. The maintenance of skeletal assets during adulthood, on the other hand, depends on the balance between bone resorption and formation during the continuous regeneration of the skeleton by remodeling. Because both osteoclasts and osteoblasts are short-lived cells, balanced remodeling in turn depends on the timely and coordinated supply of these two cell types. An oversupply of osteoclasts relative to the need for remodeling or an undersupply of osteoblasts relative to the need for cavity repair are, by and large, responsible for the pathophysiological changes of most acquired metabolic bone diseases (324, 332).
Estrogens and androgens slow the rate of bone remodeling and help to maintain a focal balance between bone resorption and formation. These effects, as it was discussed earlier, result from their ability to restrain the birth rate of both osteoclasts and osteoblasts. In addition, sex steroids influence the lifespan of both of these cells in opposite direction by exerting pro-apoptotic effects on osteoclasts and anti-apoptotic effects on mature osteoblasts. Estrogen or androgen deficiency causes loss of bone mass which is associated with an increase in the bone remodeling rate, increased osteoclast and osteoblast numbers, and increased resorption and formation, albeit unbalanced (324).
Estrogens or androgens attenuate also the apoptosis of osteocytes, and estrogen or androgen deficiency increases the prevalence of osteocyte apoptosis in both cancellous and cortical bone in animals and humans (271, 495, 496). The increased osteocyte apoptosis caused by loss of estrogens is regional, rather than uniform, and in the bone cortex the location of the apoptotic osteocytes is tightly correlated with the areas where endocortical resorption is subsequently activated (142). Furthermore, while osteocyte apoptosis triggers the bone remodeling response to microdamage, the neighboring nonapoptotic osteocytes are the major source of RANKL, and both the apoptotic and osteoclast-signaling osteocyte populations are localized in a spatially and temporally restricted pattern consistent with the targeted nature of this remodeling response (253). In addition, ablation of osteocytes in young mice recapitulates at least some of the effects of old age on bone and rapidly leads to decreased bone strength, microfractures, and osteoporosis (492). Collectively, these observations support the idea that the live neighbors of apoptotic osteocytes can sense mechanical damage and/or the death of their neighbors and become beacons for the excavation and repair of a specified area of bone and the removal of the dead cells (325). In agreement with this contention, OVX or ORX in adult mice and rats increases osteocyte apoptosis and causes an increase in RANKL levels in bone marrow plasma (309).
At the age of peak bone mass, long bones in men have greater total cross-sectional area, whereas women have a narrower endosteal area (Figure 9). This sex difference in skeletal assets accounts for greater bone strength in men, in spite of the fact that cortical thickness is similar at this stage or only mildly greater than in women, an advantage that remains present throughout the subsequent stages of life.
FIGURE 9.

Schematic representation of sex differences around the age of peak bone mass (20 yr) and subsequent lifetime changes at the endosteal and periosteal surface at the tibia [based on the longitudinal QCT findings from Lauretani et al. (294)]. The black interrupted circumferences represent the lower endosteal expansion in women and greater periosteal expansion in men, as compared with the other sex. Changes in cortical thickness are shown as bars in the inset; differences become progressively greater with age. The yellow circles represent the average degree of endosteal bone resorption (and cortical trabecularization) at the tibia in both genders (greater in women). The outer gray circles represent ongoing periosteal expansion in adulthood, which is particularly greater in men between the ages of 20 and 50. The black outer circles indicate ongoing periosteal expansion in old age, which is similar or even slightly greater in women at the tibia (but lower at the radius). [Adapted from Lauretani et al. (294), with permission from John Wiley & Sons, Inc.]
After reaching peak bone mass, periosteal apposition continues in both sexes but is greater in men. Endosteal resorption, on the other hand, is greater in women (487, 581). However, the mechanisms of cortical thinning may be not only gender specific but also site specific. In a population-based cross-sectional study, men showed smaller declines in cortical area from age 20 to 90 (−8% at the radius and −9% at the tibia) compared with women (−17 and −18%, respectively). This difference was mainly due to greater periosteal expansion at the radius in men (423). In a longitudinal QCT study at the tibia, it was found that these differences are not linear between ages 20 and 90 (294). Until age 60–70, men gained more cross-sectional area than women and continue to increase their cortical bone area. Women, however, lost cortical bone area from age 50, due to endosteal expansion. After age 70, women had a wider marrow cavity than men, and periosteal expansion approached or even slightly exceeded that of men, but it could not compensate for the increased endocortical resorption (294). Another population-based cross-sectional HR-pQCT study showed that the greater losses of cortical area and thickness at the tibia in women are mainly due to greater endocortical expansion, while at the radius the greater cortical area loss in women is due to insufficient periosteal expansion (320). The failure of periosteal expansion at the radius in women, but not men, confirms earlier findings (482, 487). Thus greater cortical bone area loss in women is due to greater endosteal resorption, but failure of periosteal bone expansion also plays an important role at the radius, but not at the tibia.
Soon after the attainment of peak bone mass, during the third decade of life in humans, the balance between bone formation and bone resorption begins to progressively tilt in favor of the latter, in both women and men (315). This change begins long before and independently of any changes in sex steroid levels. This process is slowed by the presence of sex steroids and accelerated, at least for a few years, following gonadectomy in both females and males or the abrupt decline of the circulating levels of estrogens at menopause. The rate of bone loss in women slows within 5–10 years after menopause and is followed by a slower phase of bone loss that also occurs in men. This later phase affects primarily cortical bone, and a significant portion of it is due to increased endocortical resorption and intracortical porosity (581). Similar to humans, mice experience a progressive loss of bone mass almost immediately after the attainment of peak bone mass, but unlike women, mice do not experience the equivalent of menopause (326). Nonetheless, as in postmenopausal women, the effects of OVX in adult female mice are transient and the OVX-induced increases in osteoclastogenesis and osteoblastogenesis return to baseline in less than 2 months (237).
Genetic evidence from the mouse model has also revealed that increased H2O2 generation with old age in cells of the mesenchymal lineage is a seminal culprit of the loss of cortical bone (10). In contrast, increased H2O2 generation in cells of the osteoclast lineage is the culprit of the loss of cortical bone caused by acute sex steroid deficiency, but not old age (505, 506). Hence, the cellular culprits of cortical bone loss in the two conditions may be distinct. Finally, consistent with the biologic role of osteocytes in the choreography of bone remodeling, emerging evidence indicates that signals arising from apoptotic and/or old dysfunctional osteocytes are seminal culprits in the pathogenesis of involutional, postmenopausal, steroid-, and immobilization-induced osteoporosis (335). In other words, in conditions of overwhelming stress, including age-accumulated damage, physiological mechanisms of bone repair are exaggerated and eventually become disease mechanisms.
IX. SEX STEROID DEFICIENCY AND THE DEVELOPMENT OF OSTEOPOROSIS
Osteoporosis is a skeletal disorder predisposing to fractures (1). Each year there are ∼2 million osteoporotic fractures in the United States and 3.5 million in Europe, which are associated with substantial mortality, morbidity, and economic losses (186, 207, 465).
The lifetime risk of osteoporotic fractures in high-risk Caucasian populations is ∼50% in women versus 20–25% in men (2, 311, 372). Regardless of ethnicity, hip fracture risk displays an almost constant 1:2 male-to-female ratio worldwide (243). As such, sex is a key clinical risk factor for osteoporosis, second in importance only to aging. In women, fracture incidence starts to rise from around the age of menopause, while in both sexes the risk increases exponentially from the seventh decade as a consequence of aging. This sex difference can largely be attributed to sex steroid actions, since the most recent genome-wide association meta-analysis found no significant autosomal gene-by-sex interactions (312). However, even such large meta-analysis may be underpowered to detect such interactions because of adjustment for multiple testing. It remains, therefore, possible that autosomal genes may still play an important role in skeletal sexual dimorphism (221).
A. Female Osteoporosis
An association between the decline of estrogen levels at menopause and the development of osteoporosis was first noted in 1940 by Fuller Albright in a seminal article in which he coined the term “postmenopausal osteoporosis” (8). In the following 50–60 years, attention to postmenopausal osteoporosis grew exponentially, and the condition came to be recognized as one of the most common metabolic disorders in older women. The heightened awareness and interest in postmenopausal osteoporosis owed a great deal to the development of convenient and inexpensive methods for the determination of BMD by single- and later dual X-ray absorptiometry (DXA). Simultaneously, replacement therapy with estrogens (alone or in combination with progesterone) became the mainstay of therapy for the prevention and treatment of osteoporosis. However, with the turn of the 21st century, major research advances, newer imaging technologies, including the advent of high-resolution peripheral quantitative computed tomography (HR-pQCT), as well as the startling realization of the side effects of estrogen-related therapies on breast cancer and cardiovascular diseases has changed considerably the landscape of our understanding of the relationship between menopause and osteoporosis as well as the treatment of the condition.
It is widely appreciated nowadays that osteoporosis, the Greek word for porous bones, has been erroneously used for a long time as synonymous with low BMD. The disease is bone fragility, and low BMD is just one of many other risk factors. In fact, most osteoporotic fractures occur in patients with normal BMD (27, 110, 540). Moreover, even though low BMD is predictive of fractures in untreated patients, an increase in BMD in patients that are treated with anti-osteoporosis medications accounts very little for the decrease of fracture incidence (114). Similarly, bone strength is, by and large, determined genetically, and of the genetic contribution to bone strength, very little is mediated by BMD (308, 558).
Faced with such improved understanding, in 2001 the National Institutes of Health changed the definition of osteoporosis to “a skeletal disorder characterized by compromised bone strength predisposing a person to an increased risk of fractures.” Extensive research conducted since then has made it clear that osteoporosis is a multifactorial disease of both sexes (349, 405, 535, 553, 560) in which a decline in ovarian or testicular function is only one of several other progressive and cumulative pathologies. Old age is the most critical predictor of fractures (218), the clinical manifestation of the disease. Additional non-BMD factors that determine susceptibility to fractures are small bone size (135), disrupted bone architecture (266), excessive rate of bone remodeling (200), changes in the quality of the bone matrix and the maturity of its mineral (28), delayed repair of fatigue micro-damage (79), loss of osteocyte viability with age (554), falls (242), and increased cortical porosity (581). As a result of these and several more advances in our understanding of the cellular and molecular mechanisms of osteoporosis, the “estrogen-centric” paradigm of osteoporosis as a disease largely caused by a decrease in sex hormones is slowly yielding ground to the recognition that fundamental age-related processes play a primary role in the development of osteoporosis and changes in the ovaries are contributory (326, 327, 334). In fact, osteoporosis may not be an isolated disease entity resulting from its own unique mechanisms; instead, it appears to share several pathological processes with other age-related disorders, such as atherosclerosis, myocardial hypertrophy, sarcopenia, insulin resistance, and Alzheimer's disease (327).
Bone loss in either sex begins within 10 years after the achievement of peak bone mass, which occurs during the third decade of life. This is clearly much earlier and independent of any changes in sex steroid levels, in both rodents and humans (14, 258, 373, 424). In fact, a significant portion of the loss of cancellous bone in estrogen sufficient women is age-related and estrogen-independent (424). At menopause, the loss of cancellous bone in the spine accelerates. Importantly, however, the “menopausal” bone loss is a composite caused by estrogen deficiency and aging per se, with estrogen deficiency being perhaps more critical for the loss of bone in the spine compared with the loss of bone in the hip and total body (420). Indeed, as measured prospectively by DXA, women lose ∼22% of their total body bone mineral between menopause and the age of 75. Of this amount only 7.75 percent is due to estrogen deficiency, with the remaining 13.3 percent due to aging. In the femoral neck, only 5.3 percent of the loss is due to estrogen deficiency while 14 percent is “age related” (420). Moreover, as determined by HR-pQCT, the contribution of estrogen deficiency versus aging to the loss of cancellous and cortical bone is much greater in the former compartment. Indeed, in women over the age of 65, most bone loss is cortical, not trabecular; and after the age of 80, 90% of the decline of bone mass is cortical. Notably, non-vertebral, non-hip fractures are approximately five times more common and burden healthcare resources twice as much as spine and hip fractures combined (549).
The acceleration of cancellous bone loss that ensues with estrogen deficiency results predominantly from a decrease in trabecular number caused by trabecular perforation and loss of connectivity (Figure 10). The precise mechanism of this pathological event is unclear, but histological evidence from human bone biopsies suggests that acute estrogen deficiency gives rise to “killer osteoclasts” (403) capable of complete perforation of trabeculae in the cancellous bone compartment, such that it precludes subsequent refilling of the cavities by the bone-forming osteoblasts (Figure 10). The evidence from the study of mice with cell-specific deletion of the ERα, discussed in previous sections, supports the notion that the acceleration of trabecular bone loss following menopause is due to the prolongation of osteoclast lifespan, which results from the loss of the direct pro-apoptotic effects of estrogens on osteoclasts (339, 365).
FIGURE 10.
Microphotograph of a human trabecula perforated by osteoclastic resorption. The image is taken from an iliac crest biopsy specimen. Please note that resorption from either side of the trabecula has left only a thin thread of bone. [From Weinstein (553a).]
With the use of HR-pQCT, it has been shown that in women between the ages of 50 and 80, the majority of the loss of cortical bone is largely due to increased cortical porosity (29, 30, 373, 581). Consistent with this finding, most fractures over the age of 65 occur predominantly at cortical sites (425). Importantly, the marked increase of cortical porosity with age is not captured by DXA BMD (373, 581). The age-dependent increase in cortical porosity and the decrease in cortical thickness in mice is clearly due to increased bone resorption (236). Be that as it may, this later phase of bone mass decline that affects primarily the cortical bone compartment is also associated with a decrease in osteoblast number and bone formation rate. Indeed, the most consistent histological finding in both elderly women and men with osteoporosis is decreased wall width, the hallmark of decreased osteoblast work output. This finding is in line with the idea that decreased bone formation, combined with increased resorption, is a critical pathogenetic contributor to the loss of bone due to aging per se (192, 398, 404). In support of this contention, the cellular culprits responsible for the loss of cortical bone following acute sex steroid deficiency in mice are different from those responsible for the loss of trabecular bone, and also different from the mechanisms causing loss of cortical bone with old age (333). Specifically, as was discussed earlier, H2O2 generated in the mitochondria of osteoclasts is required for the loss of cortical bone mass caused by estrogen or androgen deficiency, but not aging (505, 506). On the other hand, attenuation of H2O2 accumulation in mesenchymal/stromal cells abrogates the effects of aging on cortical bone. The dissimilarity of the effects of acute estrogen deficiency (as in menopause) and prolonged estrogen deficiency (as in elderly osteoporotic females) indicates that the effects of loss of estrogens are drastically modified and perhaps overridden by aging mechanisms intrinsic to bone.
Following menopause, periosteal bone apposition increases (6). This observation is also consistent with the evidence from the mouse model that in the presence of estrogens periosteal expansion is attenuated as a result of estrogen signaling via the ERα of osteoblast progenitors (15). Nonetheless, endosteal bone resorption in women far outpaces the effect of periosteal expansion, cancelling the benefit of the outward expansion on strength.
Finally, estrogen deficiency may contribute to the development of osteoporosis by decreasing the sensitivity of bone to mechanical loading. Support of this contention has been provided by evidence that the ERα enhances the responsiveness of bone to mechanical loading (5, 301, 566), at least in part, by potentiating the activation of the Wnt signaling (15, 26). However, the effect of ERα on mechanical loading does not require ligand binding. Therefore, one would have to postulate that estrogen deficiency decreases estrogen responsiveness, by decreasing the expression of the ERα.
In conclusion, estrogen deficiency plays an important role in the development of osteoporosis in women. Nonetheless, during the last 10 years, genetic evidence from the mouse model has provided a paradigm shift from the traditional “estrogen-centric” account of the pathogenesis of involutional osteoporosis to one in which age-related (sex monomorphic) mechanisms intrinsic to bone, including mitochondrial dysfunction, oxidative stress, FoxO activation, senescence of mesenchymal stem cells and osteocyte, and declining autophagy are protagonists, and age-related changes in other organs and tissues, such as the ovaries, are contributory (16, 227, 326, 334, 384). Similar to humans, sex steroid-sufficient female and male mice experience an age-dependent progressive decline in bone mass and strength. In mice, this is temporally associated with increased oxidative stress. Although both estrogen deficiency and aging inexorably contribute to the development of osteoporosis, the extent to which pathogenetic mechanisms resulting from the former overlap or amplify mechanisms resulting from the latter remains unknown.
B. Male Osteoporosis
In contrast to the inevitability of menopause, a true andropause does not exist. In fact, total T levels decline only slightly with age, and in the majority of elderly men, T levels are maintained above a threshold that separates normalcy from symptomatic hypogonadism (∼200–300 ng/dl or 8–11 nM/l) (48, 572). Nonetheless, old age in men is consistently associated with a mild to moderate increase in SHBG. As a result, the free (non-protein bound) or bioavailable (non-SHBG bound) concentration of either T or E2 shows a decline with age in a substantial portion of older men (48).
In the last 50 years, most, but not all, cross-sectional or longitudinal studies in men have found an association between lower serum E2 or T concentrations and low BMD or the rate of loss of BMD. Bioavailable or free E2, higher SHBG, or both of these measurements independently generally show a stronger positive association with BMD. The association between total or free T and BMD, on the other hand, is weaker, absent, or dependent on E2 (181, 257, 472). Based on these associations, it was hypothesized in the late 1990s that estrogen deficiency is the cause of not only postmenopausal osteoporosis, but also idiopathic male osteoporosis and the osteoporosis of old age (422). Specifically, it was proposed that estrogen deficiency in the elderly leads to the so-called type II or involutional osteoporosis by impairing intestinal calcium absorption and causing secondary hyperparathyroidism. With the hindsight of advances in our understanding of estrogen action on bone and the biology of skeletal aging since that time (discussed in previous as well as following sections of this article), a mechanistic link between estrogen deficiency and secondary hyperparathyroidism is no longer a plausible explanation of involutional osteoporosis. Nonetheless, in a portion of the elderly, particularly those residing in northern latitudes, estrogen deficiency can coincide with vitamin D deficiency and secondary hyperparathyroidism.
Epidemiological evidence has also suggested that low bioavailable serum E2, SHBG, or both independently, but not serum T, is associated with higher bone remodeling markers, cross-sectional and longitudinal BMD differences (20, 24, 92, 169, 256, 277, 305, 317, 486, 501, 513, 514, 568). More recently, low free E2, but not free or total T have been also associated with increased cortical porosity (518) as well as decreased cortical volumetric BMD, cortical thickness, endosteal expansion, and trabecular density (25, 259, 488, 546) in older men. In addition, cortical, but not cancellous, bone loss is accelerated in older men below a threshold around the median bio-E2 value (<40 pM for BMD at cortical sites, and <30 pM for cortical vBMD) (169, 256, 259). However, this threshold has not been confirmed in other studies.
All of these observational studies, as well as more recent ones using reliable mass spectrometry methods, as opposed to less reliable immunoassays, consistently show an association of male bone loss with bioavailable (but not total) estrogens and/or SHBG (61, 89, 212, 383, 518, 521). In the largest of these studies (MrOS), longitudinal BMD loss was associated with bioavailable E2 (bioE2) but not T; however, the combination of high SHBG, low bio-E2, and bio-T was associated with threefold faster BMD loss (89). Circulating T and E2 levels are intrinsically related because T is the substrate for E2 synthesis via aromatization. Associations with T may still reflect estrogen actions on bone, because of local aromatization within the tissue (471). Moreover, bioavailable and free sex steroid calculations are based on SHBG concentrations, making it impossible to disentangle independent contributions of each one of these measures. Albeit, it is unlikely that SHBG, in and of itself, has a direct role in skeletal homeostasis, as opposed to being just a biomarker (212). Importantly, sons of men with idiopathic osteoporosis have lower bioavailable sex steroids and low BMD, raising the possibility that the cross-sectional associations in older men could be due to the earlier effects of sex steroids on peak bone mass acquisition (288, 515).
Epidemiological studies have also shown associations between sex steroid and SHBG concentrations with fracture outcomes in older men (19, 297, 346, 347, 568). However, the value of these measurements in predicting fracture risk remains unknown, and in the larger one of these studies (MrOS), the attributable risks of low bioavailable E2, high SHBG, and low bioavailable T were 5.7, 7.7, and 1.5% (91, 519). Hence, in spite of the fact that sex steroids play an important role in skeletal homeostasis and an association between low E2 and fracture risk is plausible, the added benefit of sex steroid measurements will be modest at best. Still, the epidemiologic evidence leaves open the possibility that low sex steroid concentrations and, in particular, high SHBG are biomarkers of increased r isk rather than direct mediators of this effect (93, 212). This possibility is strengthened by the fact that incident fractures are increased in men with a combination of low sex steroid and vitamin D, but with neither in isolation (33).
The associations between sex steroid levels with BMD and fracture risk in elderly men aside, meta-analysis of genome-wide studies has shown an association between polymorphisms of ESR1 and DXA BMD as well as cortical volumetric BMD (144, 312), but not fractures in both sexes (499, 516). On the other hand, there is no association between polymorphisms of the AR with BMD, vBMD, fractures, or even frailty (406). If any, a higher number of CAG repeats in the AR (polyglutamine tracts which decrease AR activity when above a certain length) are associated with higher BMD resulting from higher T concentrations, due to hypothalamic feedback, and thus increased substrate for peripheral aromatization and estrogen actions (216, 499).
Additional support for the contention that declining E2, rather than T, levels are a culprit of the decline in BMD and strength with advancing age is, at least at first glance, provided by the two case reports of a man and woman with loss of function of ERα mutations (414, 473), already referred to in the section on the effects of sex steroids on epiphyseal closure. An important caveat in drawing conclusions from these two genetic cases, however, remains that in these individuals, loss of estrogen signaling began from conception and affected primarily the development and growth of the skeleton, rather than its maintenance during adulthood. Moreover, unlike the high bone turnover (reflected in high bone resorption and formation markers) caused by sex steroid deficiency after development and growth has been completed, the rate of remodeling and trabecular number in the man with the ERα mutation was normal in an iliac crest biopsy, and serum markers of bone resorption, but not formation, were high (474). These incongruences aside, periosteal circumference was normal in this man, consistent with the rest of the evidence for a predominant role of AR signaling in periosteal bone apposition in men.
The caveats with the ERα genetic mutations notwithstanding, the skeletal phenotype of humans with aromatase deficiency also supports a role of estrogens in skeletal homeostasis in men. Apart from an abnormal growth phenotype, these patients have low BMD and high turnover that can be reversed with estrogen replacement (67, 283, 428). Notably, in a man with concomitant mild hypogonadism from bilateral cryptorchidism, addition of T to estrogen replacement resulted in incremental gains in cortical expansion and thickness compared with estrogens alone (429), once again lending support to a model in which optimal periosteal bone expansion during growth requires both AR and ERα actions (523).
In contrast to human ERα mutations, AR mutations in patients with androgen insensitivity syndrome (AIS) have only a very mild effect on bone (122, 191), and treatment with estrogens restores cortical and trabecular vBMD and endosteal, but not periosteal circumference (489). Collectively, these genetic findings reinforce the notion that estrogens and ERα play an important role not only for female but also for male skeletal development and probably the maintenance of bone mass during adulthood, whereas AR has only selective but important effects on periosteal expansion during growth.
Finally, in (usually older) men with prostate cancer, near-total androgen deprivation therapy (ADT) is clearly associated with bone and muscle loss, micro-architectural bone decay, and increased fracture risk (182, 188, 456). However, AR antagonists alone (which increase serum T and also E2 concentrations) can control prostate tumors effectively without increasing bone turnover or BMD loss (477, 539). Whether this will remain true with the latest and more powerful AR antagonist enzalutamide is unknown. In male-to-female transsexuals, estrogen therapy maintains cortical and trabecular vBMD despite decreased muscle mass and strength and increased fat mass (511). Conversely, female-to-male transsexuals not only have higher muscle mass, muscle strength, and lower fat mass, but also display increased hip BMD (512) as well as greater cortical bone circumference and lower cortical vBMD at the radius and tibia (510). Thus the deleterious effects of androgen deficiency on bone are again mainly determined by the degree of concomitant estrogen deprivation.
In summary, a large body of epidemiological evidence suggests that bioavailable/free E2 and/or SHBG, but not T, concentrations in the serum are associated with bone loss in men. Bone maintenance appears to be compromised when bioavailable E2 falls below certain threshold(s), but the cut-off level is not clearly defined. Much stronger support for a role of estrogens in the maintenance of the male skeleton is provided by genetic evidence in men and women with loss of function mutations of the ERα or aromatase deficiency, as well as genome-wide association studies indicating a role of the ERα, but not the AR, in skeletal homeostasis. Hence, the hypothesis that estrogen deficiency plays a role in the development of osteoporosis in both sexes holds, but it does require refinement and needs to integrate the profound effects of skeletal aging and its sex monomorphic effects in the involution of the skeleton (258, 326). Be that as it may, observational studies cannot prove causality, and ultimately, other lines of evidence will be necessary. Recent findings from mouse genetic studies indicating that estrogens (derived from androgen aromatization) are the sex steroid responsible for the protection of cortical bone mass in both sexes, whereas nonaromatizable androgens are responsible for the protection of cancellous bone in males should help to clarify some of these issues (229, 504). Lastly, at present, the extent to which sex steroid deficiency in either sex contributes to the adverse effects of aging, per se, on the skeleton, remains unclear.
X. SEX STEROIDS IN THE TREATMENT OF OSTEOPOROSIS
A. Estrogens
Replacement therapy with estrogens, commonly known as hormone replacement therapy (HRT), was the mainstay treatment for postmenopausal osteoporosis (and chronic disease prevention in older women in general) for almost 50 years. However, during the last decade, the use of HRT has diminished dramatically for two reasons: 1) the risk of adverse effects, such as breast cancer, venous thromboembolism, stroke, and coronary heart disease revealed by the Women's Health Initiative (WHI) trial (435); and 2) the availability of more potent and effective antiresorptive drugs that work regardless of whether increased resorption is the result of estrogen deficiency or other pathogenetic mechanisms.
Nonetheless, more recent reanalysis and extended follow-up of the WHI data and other studies suggest that starting HRT early after menopause and maintaining it for a limited time may have beneficial effects without increasing the risk of major complications (442). The higher risk for breast cancer and cardiovascular diseases with HRT was identified in women receiving conjugated equine estrogen (CEE) plus progestin, but not CEE alone. Still, both CEE and CEE with progestin increased the risk of stroke, deep vein thrombosis, urinary incontinence, gallbladder disease, and breast tenderness. On the other hand, both CEE and CEE with progestin significantly decreased hip and clinical vertebral fractures, vasomotor symptoms and diabetes risk, while CEE alone tended to decrease invasive breast cancer risk (336). Notably, there was a significant interaction with age, with a tendency for cardiovascular disease protection, lower mortality, and a favorable global disease index in women 50–59 years of age. Nevertheless, these younger postmenopausal women were also at low fracture risk, especially because the WHI study population consisted mainly of women without prior fracture risk (336). Therefore, the strong evidence for prevention of hip and clinical vertebral fractures by HRT is all the more remarkable (338).
Postmenopausal women with higher bioavailable E2 concentrations have higher trabecular vBMD and cortical vBMD and area (262) and lower hip fracture risk (300). Nonetheless, the fracture benefits of HRT are independent of baseline E2 (or SHBG) (90), possibly because all women with E2 levels in the postmenopausal range may benefit. Interestingly, however, moderate to severe vasomotor symptoms are independent predictors of BMD and hip fracture risk (109). Potentially, these symptoms may better reflect biological estrogen status than the conventional radioimmunoassays and bioavailable E2 calculations.
Nowadays, HRT is used for the symptomatic management of menopausal symptoms like flushing, and no longer for general chronic disease prevention in asymptomatic elderly women. In women less than 60 years of age, however, the benefit-risk ratio of CEE appears favorable, especially compared with alternative antiresorptive therapies and their risk for long-term side effects. Women on HRT for symptomatic reasons generally do not require additional anti-osteoporotic drugs, unless fracture risk is very high (e.g., when fractures occur under HRT). For women seeking fracture prevention, more effective antiresorptive drugs are available, e.g., bisphosphonates. HRT may still be an alternative when other therapies are contraindicated or in women who are younger than 60 years of age and have increased fracture risk and/or menopausal symptoms that compromise their quality of life.
B. Androgens
Some evidence from animal models, as well as results from studies with hyperandrogenic women with polycystic ovarian syndrome (PCO) (85, 246, 449, 522), had suggested that androgen therapy may benefit the maintenance of female bone mass. However, observational studies in patients with PCO are confounded by altered body weight, insulin resistance, and estrogen deficiency (248). T replacement may also have small benefits on sexual function in postmenopausal women, but this remains controversial because of uncertainty with its effectiveness and safety (141). Nonetheless, several drug trials testing the benefit of adding T to estrogen replacement therapy found no significant increase in BMD (35, 124, 141, 550), and virilization may be problematic at least in some subjects (171).
T replacement in young or adult men with organic causes of hypogonadism increased BMD (21, 172, 480). Similarly, T administration to adult men with primary hypogonadism increased vertebral cortical and trabecular volumetric BMD, but cortical area was not affected (150, 306). Importantly, most of the men in that study had low-turnover osteopenia, so the risk of osteoporosis was probably due to compromised PBM acquisition. Cortical area was not affected by T administration, but because of the low-turnover osteopenia, the osteoporosis risk was probably due to compromised PBM acquisition. Nevertheless, other men with severely compromised androgen and estrogen levels may develop high turnover osteopenia, similar to what is seen in animal models (150).
In states of milder T deficiency in older men, so-called late-onset hypogonadism, the situation is less clear. Several randomized trials have demonstrated that androgens have anabolic musculoskeletal effects in normal middle-aged and elderly men. Albeit, the efficacy of androgens in this population was demonstrable in individuals with endogenous T levels below 200–250 ng/dl at baseline and was dependent on the duration and intensity of the treatment (172). Transdermal replacement may be less effective than depot injections (498), possibly due to higher androgen peak concentrations (466).
In a post hoc analysis, lumbar spine BMD increased with a T patch in older men, but this benefit was again limited to patients with pretreatment serum T levels significantly below 200–300 ng/dl (481). In contrast, several trials with short-term follow-up (∼6 months) and lower androgen doses showed no BMD differences compared with placebo (103, 143). Interestingly, adding a 5α-reductase inhibitor to T replacement prevents the adverse effects of T on the prostate without compromising the musculoskeletal benefits (22, 23, 50, 231, 343).
By far, the greatest concern with T replacement therapy is the potential of increased risk of cardiovascular events which were observed in a frail, older population (38, 39). The T Trial in Older Men is expected to shed new light on some of the previous equivocal musculoskeletal benefits as well as the cardiovascular safety of T in the near future (479).
The potential benefits of T or dehydroepiandrosterone (DHEA) therapy on bone and perhaps other tissues, could be due to conversion to estrogens via aromatization and stimulation of the ERα. To address this possibility, several trials have used either nonaromatizable androgens like DHT, or GnRH analogs combined with T replacement and aromatase inhibition. In a seminal study, both T and E2 had independent effects on bone turnover in older men (147). Although not significant by stringent two-way ANOVA, the effect on bone resorption was greater with combined T and E2 replacement than with E2 alone (Figure 11) (255). This conclusion is supported by another study in younger adult men, in which both androgens and estrogens independently suppressed resorption markers; bone formation markers initially declined but later increased, probably due to effects on resorption (299). Similarly, aromatase inhibition in men with borderline T levels at baseline resulted in higher T and lower E2 concentrations without affecting bone turnover markers over 12 wk, suggesting that high T levels may compensate for mildly decreased E2, perhaps when the latter is still above the putative skeletal estrogen threshold (298). In men with already low E2 levels, however, aromatase inhibition did not alter bone turnover markers but still decreased spinal BMD (78). Transdermal DHT, which suppresses endogenous T and E2, produced a 1.4% decrease in spinal but not femoral BMD (220). In men receiving long-term glucocorticoids, both T and nandrolone improved muscle mass and strength but only T increased lumbar BMD (111). Thus, despite an anabolic effect on muscle, nonaromatizable androgens may not be beneficial for bone in subjects with relatively normal endogenous estrogens. Finally, in the most recent trial with gonadotropin and aromatase inhibition (153), estrogen deficiency largely explained the increased bone turnover, spinal trabecular bone loss, and peripheral cortical bone loss in men with low T, while androgens contributed to increased bone turnover. Serum concentrations of E2 <10 pg/ml or T <200 ng/dl were required before bone loss became evident. This study, however, was also of limited duration and perhaps underpowered to detect effects of androgens on HR-pQCT changes.
FIGURE 11.

A: percent changes in urinary NTx excretion in men made acutely hypogonadal, treated with an aromatase inhibitor, and replaced with estrogen, testosterone, both, or neither. *P < 0.05, **P < 0.01, and ***P < 0.001 for change from baseline. The estrogen and testosterone effects were analyzed using a 2-factor ANOVA model: E effect, P = 0.0002; T effect, P = 0.085. B: data from A but now depicting postulated changes in cancellous bone resorption (testosterone effect) and in cortical bone resorption (estrogen effect) based on the mouse genetic studies of Ucer and colleagues. [From Khosla et al. (255), with permission from John Wiley & Sons, Inc.]
In conclusion, the results of trials with nonaromatizable androgens show that profound estrogen deficiency leads to male bone loss, and the adverse effects of estrogen deficiency cannot be overcome by stimulation of the AR. This conclusion is perfectly aligned with the observational evidence described above. However, heretofore an effect of varying androgen levels in the face of unaffected estrogen levels has not been examined (152). On the basis of the results of bone turnover markers in men, as well as evidence from mice that the effects of androgens on cancellous bone maintenance result from AR signaling, not via aromatization and ER signaling, it is very likely that androgens do, indeed, have independent effects on cancellous bone resorption (504). Albeit, the key physiological effect of androgen action on bone is on periosteal bone formation during puberty and possibly further into midlife.
C. Selective Estrogen Receptor Modulators
SERMs are a group of compounds synthesized and tested during the last 40 years for the purpose of maintaining the beneficial effects of estrogens on bone and other nonreproductive organs while diminishing or eliminating their adverse effects on reproductive organs like the uterus and the breast. SERMs bind to the ERα ligand binding pocket with high affinity but, due to the presence of a bulky side chain and steric hindrance, prevent approximation of helix 12 and availability of the AF-2 cofactor surface on the LBD (Figure 12) (509). In addition, they preclude more specific conformational changes required for ligand-induced effects via the AF-1 domain. As discussed earlier, mice with specific deletion of the ERα AF-1 display a normal response to E2 in cortical bone, but fail to exhibit the effects of SERMs (64, 66). It is theoretically possible that SERMs may selectively activate the AF-1 domain in ovariectomized ER AF-2 null mice, because ICI 182,780, a potent ERα antagonist, acts as an agonist on trabecular bone and the uterus and as an inverse agonist on the growth plate of these mice (363). Thus SERMs competitively inhibit estrogen binding to ERα, and in the estrogen-deficient state they trigger ERE-mediated signaling with decreased cofactor recruitment to AF-1 and AF-2. The net result is agonistic activity in some tissues, manifested as protection against bone resorption, increased tromboembolic risk, and decreased LDL cholesterol, but antagonistic activity in other tissues, manifested as increased vasomotor symptoms and protection against breast cancer. However, the bone-sparing effect of SERMs is seen only in postmenopausal women; in premenopausal women SERMs, e.g., tamoxifen, cause bone loss (525).
FIGURE 12.

Cartoon representations of the 3D crystal structure of ERα ligand-binding domain (LBD) homodimers (red-blue or blue-blue) in complex with estradiol (E2) (75) (A), 4-OH-tamoxifen (459) (B), raloxifene (75) (C), and lasofoxifene (509) (D). In A, occupancy of the ligand-binding pocket by E2 stabilizes helix 12 in a folded position (red arrow), revealing an interaction surface (AF-2) for coactivators (yellow star). In B–D, SERMs occupy the ligand-binding pocket but additionally contain a bulky side chain (underlined in red) which pushes helix 12 (light blue arrows) in front of AF-2, rendering it unavailable (red stars). Note that the crystal structure of ERα in complex with bazedoxifene is not yet available. Created using JSmol using structural data from the Protein Data Bank.
Randomized trials have produced favorable results for the use of raloxifene (145), bazedoxifene (461), and lasofoxifene (113) in the prevention of vertebral fractures. However, evidence of breast cancer chemoprophylaxis is lacking for bazedoxifene (or bazedoxifine with conjugated estrogen). Lasofoxifene also reduced nonvertebral fractures as well as coronary heart disease and stroke (113). Raloxifene, on the other hand, reduced nonvertebral fractures in a post hoc subgroup analysis in high-risk women with baseline severe vertebral fractures (126), but increased the risk of stroke with no effect on coronary heart disease (34). Similarly, a post hoc analysis has suggested that bazedoxifene might prevent nonvertebral fractures in high-risk women (460). Compared with raloxifene, tamoxifen is more effective against breast cancer but carries greater risks of endometrial cancer, uterine hyperplasia, thromboembolic events, and cataracts (538). Like all SERMs, tamoxifen, another ERα antagonist and degrader, induces bone loss in premenopausal breast cancer patients, but its fracture-prevention properties in postmenopausal women may be similar to raloxifene (537). Individuals with the highest risk of fracture are most likely to benefit from SERMs, but the lack of demonstrated efficacy in hip fractures remains a concern. Of note, the bisphosphonate alendronate has greater efficacy on BMD than raloxifene in a direct comparison trial, which was however underpowered to ascertain fracture outcomes (421). Based on these results, raloxifene and lasofoxifene may be clinically useful in osteoporotic women with low thromboembolic and hip fracture risk, and high breast cancer risk or contraindications to other drugs.
SERMs may also be beneficial in men, but only in those with very low E2 concentrations, i.e., lower than most elderly men. Indeed, a randomized trial in elderly men showed that men in whom bone turnover markers decreased with raloxifene had lower E2 levels (22 pg/ml) (133). Similarly, in a randomized trial of middle-aged men, a high dose of raloxifene reduced bone turnover markers only in those with quite low bioavailable E2 concentrations (<4.79 pM) (508). SERMs also raise gonadotropin levels and consequently the circulating concentrations of T and E2, but the skeletal effects may still be mediated via ERα (139, 508).
The efficacy of SERMs in the treatment of low bone mass in men can be better demonstrated in men with prostate cancer receiving androgen deprivation therapy, which also potently deprives estrogens. In this setting, raloxifene attenuates the loss of BMD (476) while toremifene (no longer available due to thromboembolic risk) also reduces fracture risk (478). Interestingly, estrogen therapy suppresses androgens and may be effective in prostate cancer treatment without the skeletal adverse effects of ADT (285).
Collectively, the results of these studies demonstrate that SERMs may have similarly beneficial skeletal effects in men as they do in postmenopausal women, but only if E2 bioactivity is severely compromised.
D. Selective Androgen Receptor Modulators
Potential adverse effects of androgen-based therapies, due to a lack of tissue selectivity, together with their poor oral bioavailability and pharmacokinetic profile, have prompted the development of selective AR modulators (SARMs). SARMs belong to one of the following four major structural classes: aryl propionamide analogs, hydantoin analogs, indoles, and quinoline analogs (357, 367). An ideal SARM should elicit anabolic effects on muscle and bone while sparing reproductive tissues such as the prostate. A considerable body of evidence has documented in animal models that several SARM compounds have the required in vivo tissue selectivity (357). In contrast to SERMs, however, the molecular mechanisms behind this tissue selectivity remain elusive. Plausible mechanisms include resistance to 5α-reduction or aromatization, differential recruitment of co-regulator proteins, as well as use of diverse intracellular signaling cascades by the same ligand in different tissues (367). Notably, whereas SERMs act as ER agonists in some tissues while showing ER antagonistic activity in other tissues, the SARMs studied to date only show agonistic activity. The degree of their stimulatory effect on the AR varies across tissues, with a more pronounced effect on muscle and bone compared with reproductive tissues (106). In other words, SARMs may be better characterized as selective AR marginators, i.e., widening the therapeutic margin, rather than true modulators. In a recent study the muscle-anabolic actions of enobosarm (GTx-024) did not require myocyte AR and may involve a paracrine effect on AR-expressing muscle-resident fibroblasts (138).
The dual anabolic effect of SARMs on muscle and bone makes them good candidates for the treatment of sarcopenia and osteoporosis in both men and women. Other potential applications include their use in muscle wasting syndromes, cancer cachexia, prostate cancer, and male contraception (369). Several preclinical studies provide proof of concept for the beneficial effects of SARMs on bone. The aryl propionamide SARM S-4 (andarine) was able to maintain bone mass and strength to the levels of intact controls and exhibited greater efficacy than DHT in both ovariectomized (251) and orchidectomized (163) rat models. Similarly, a dose-dependent increase in bone formation rate was observed upon administration of MK-0773 to ovariectomized rats (448). Biomechanical testing of bones from gonadectomized animals treated with the quinolines LGD-2226 and its derivative LGD-2941 showed strong enhancement of bone strength above sham levels (341, 352). S-101479, a quinoline reported to increase femoral BMD in ovariectomized rats, even has an additive effect with raloxifene treatment (159).
Thus far, few SARMs have advanced to the clinical trial stage. S-22 (ostarine or enobosarm) is the furthest ahead. Indeed, in phase II trials of healthy elderly men and postmenopausal women (119) as well as cancer patients with muscle wasting (131), S-22 increased lean body mass and improved physical function. However, neither of these studies showed increased bone mass with SARM administration. In another phase II trial evaluating the efficacy of MK-0773 in female subjects with sarcopenia, the drug improved BMC, but this was not statistically significant (397).
In conclusion, whereas several SARMs can increase bone mass in rodent models, evidence for beneficial effects in humans is lacking. Moreover, the observation that some SARMs lower E2 levels by suppressing FSH and LH (97, 119) requires a careful consideration of their use in osteoporosis patients, given the importance of aromatization for male skeletal maintenance. Adverse effects of AR agonists in general on the cardiovascular system have further dampened the enthusiasm about SARMs.
XI. SUMMARY AND CONCLUSIONS
Bone is a highly dynamic, structurally complex, and heterogeneous tissue, comprising specialized cell types and a mineralized matrix that forms the compact cortical compartment (80% of the entire skeleton) and the interconnected plates and strands of the cancellous compartment lying within the cortex. Bone is formed and resorbed by osteoblasts and osteoclasts, arising from mesenchymal and hematopoietic precursors, respectively. Osteocytes, former osteoblasts embedded in the mineralized matrix, are responsible for orchestrating the acquisition and removal of bone in response to mechanical demands and perhaps other signals. Signals arising from apoptotic and/or old dysfunctional osteocytes may be seminal culprits in the pathogenesis of metabolic bone diseases, including osteoporosis. During growth, bone is shaped by modeling. Afterwards, bone mass and functional integrity is maintained primarily by remodeling, periodical cycles of resorption and formation at the same site accomplished by teams of osteoclasts and osteoblasts assembled in the BMU.
Estrogens and androgens promote the acquisition of bone mass during puberty and are responsible for the sexual dimorphism of the skeleton. Low levels of estrogens and androgens at the early stages of puberty stimulate the pubertal growth spurt. The stimulatory effects of estrogens and androgens on the pubertal growth spurt are mediated indirectly, via effects on the secretion patterns of GH. In boys, pubertal growth spurt starts two years later than in girls, allowing more time for prepubertal appendicular growth. This accounts for the ultimate greater height (∼8%) in men. At the end of puberty, the higher estrogen concentrations, but not androgens, stimulate the closure of the epiphyseal growth plates and shut longitudinal growth in both girls and boys. The effects of estrogens on the closure of the epiphyses are mediated by direct actions on chondrocytes.
By the end of skeletal maturation, men achieve higher PBM and greater long bone width (i.e., cortical diameter). The latter is accomplished by greater accrual of bone at the outer surface, placing the cortex further away from its neutral axis, a biomechanically advantageous distribution. The higher PBM and greater long bone width in men, along with lower bone losses later in life, are the most likely reasons why men have stronger bones and lower incidence of osteoporotic fractures.
Both AR and ERα are required for the optimal expansion of cortical bone during puberty in males, acting largely but not exclusively via systemic GH/IGF-I signaling. Estrogens, but not androgens, have direct effects on hepatic IGF-I release. In females, estrogens acting through the ERα limit periosteal expansion and maintain the endosteal perimeter; both of these effects are independent of the somatotropic axis. The cellular target of the effects of AR on periosteal bone formation remains unknown.
In addition to their influence on skeletal growth, sex steroids help to maintain bone mass and strength during adult life by slowing the rate of bone remodeling and maintaining a balance between resorption and formation. Epidemiologic evidence in women along with mechanistic studies in rodents indicate that an abrupt decline of estrogens (at menopause or following pharmacological or surgical OVX) leads to a transient increase in the rate of bone remodeling and accelerates the age-dependent decline of cancellous bone mass that begins independently of sex steroid changes sometime around the third decade of life in humans and at 4–5 months of age in mice. Abrupt loss of estrogens contributes to the thinning of the cortices of long bones by increasing resorption of the endocortical surface. In contrast to the inevitability of menopause, a true andropause does not exist. Nonetheless, old age in men is associated with a gradual decrease in bioavailable E2 caused by a mild to moderate increase in SHBG, but not T. The decrease in bioavailable E2 in men with advancing age is associated with a decrease of bone mass. Therefore, in either sex, estrogen deficiency contributes to the profound and sex monomorphic effects of aging on skeletal involution and the development of osteoporosis in humans. Presently, the extent to which sex steroid deficiency in either sex contributes to the adverse effects of aging, per se, on the human skeleton, remains unclear. Nonetheless, in the mouse the effects of age and sex steroid deficiency are independent.
Strong support of the evidence that estrogens and ERα play an important role in male skeletal growth and probably the maintenance of bone mass during adulthood has been provided by two case reports of a man and a woman with loss of function ERα mutations. These individuals exhibited lack of clear pubertal growth spurt, continued growth into adulthood, and osteopenia. In contrast to human ERα mutations, AR mutations in patients with AIS cause very minor bone phenotype, and treatment of such patients with estrogens restores cortical and cancellous BMD as well as endosteal, but not periosteal, circumference. The inability of estrogens to restore periosteal circumference in AIS patients supports the notion that androgens and AR play an important role on periosteal expansion during growth.
Consistent with the role of estrogens on the maintenance of cortical bone mass in both women and men, recent mouse genetic studies suggest that estrogens (derived from androgen aromatization in males) are responsible for the protection of cortical bone mass in both sexes, whereas nonaromatizable androgens are responsible for the protection of cancellous bone in males. Evidence from the mouse model also suggests that increased H2O2 generation in cells of the osteoclast lineage is the culprit of the loss of cortical bone caused by acute sex steroid deficiency in both females and males, but not old age. In contrast, increased H2O2 generation in cells of the mesenchymal lineage is a contributing culprit of the loss of cortical bone with old age. Hence, the molecular culprits responsible for the loss of cortical bone loss in the two conditions may be distinct. If that were confirmed in humans, it would suggest that the acute effects of estrogen deficiency (as in menopause) are drastically modified and perhaps overridden by aging monomorphic, as opposed to sex steroid dimorphic, mechanisms intrinsic to bone.
The effects of estrogens and androgens on bone mass are mediated by the ERα and AR expressed in osteoblast and osteoclast progenitors and their descendants, as well as B lymphocytes and perhaps other extraskeletal targets. Some of the effects of estrogens and androgens on bone may result from direct binding to EREs or AREs, but the target genes of these actions remain unknown. The protective effect of estrogens on cortical bone mass maintenance may be the result of nonnuclear initiated signaling of the ERα. The ERα in mesenchymal/stromal cells residing in the periosteum may promote periosteal bone apposition in both females and males independently of ligand binding. Additionally, ERα may be a critical node on which hormonal and mechanical signals are integrated.
Estrogens slow the rate of bone remodeling by attenuating the birth rate of osteoclasts and osteoblasts. Additionally, estrogens shorten the lifespan of osteoclasts and prolong the lifespan of osteoblasts and osteocytes. Genetic evidence for the function of ER and AR in the mouse suggests that in females the osteoclast ERα mediates the protective effect of estrogens on the cancellous, but not the cortical bone compartment. The ERα in committed osteoblast progenitors promotes cortical bone accrual at the periosteum in both females and males, while the ERα in uncommitted mesenchymal progenitors mediates the protective effect of estrogens against endocortical, but not cancellous, bone resorption in females. The effects of androgens on the cancellous and cortical bone compartment in the male are also mediated via different cell types. Nonetheless, whereas the antiresorptive effects of estrogens on cancellous bone result from direct actions on osteoclasts, the antiresorptive effects of androgens on cancellous bone are exerted indirectly via osteoblasts and osteocytes. In cortical bone, estrogens protect against resorption in both females and males, at least in part, via ERα-mediated actions (upon aromatization of androgens to estrogens in males) on uncommitted mesenchymal progenitors.
Replacement therapy with estrogens or combinations of estrogens and progesterone was the treatment of choice for postmenopausal osteoporosis for almost 50 years. In the last 20 years, the use of estrogens for the prevention and treatment of osteoporosis for women over the age of 60 has decreased because of the appreciation of the side effects of estrogens on breast cancer and cardiovascular diseases, as well as the emergence of alternative and very potent antiresorptive agents, such as bisphosphonates and denosumab, a neutralizing antibody against RANKL. SERMs, like raloxifene and lasofoxifene, can overcome at least some of the adverse effects of estrogen replacement therapy and are a useful alternative in women with high breast cancer risk and/or contraindications to the other drugs. However, contrary to the situation for other antiresorptive therapies, their efficacy in hip fractures has not been demonstrated, and their effects on nonvertebral fractures are less convincing, at least for some of the SERMs.
T effectively maintains bone density in young and adult hypogonadal men with overt hypogonadism, but has little osteoanabolic activity in postmenopausal women. The benefits or risks of T replacement in older men with milder late-onset hypogonadism, however, are currently unclear. SARMs have a dual anabolic effect on muscle and bone in rodent models, which makes them appealing candidates for the treatment of sarcopenia and osteoporosis in both men and women. There is, however, no evidence of their efficacy in humans, and their appeal is decreased by evidence that AR agonists may have adverse effects on the cardiovascular system.
In the last 50 years, major research advances, genetic insights from humans and rodents, and newer imaging technologies have changed considerably the landscape of our understanding of the relationship between menopause and osteoporosis. Along with the appreciation of the side effects of estrogen-related therapies on breast cancer and concerns for increased risk of cardiovascular diseases for women over the age of 60, this progress has also drastically altered the treatment of the condition. Future work aiming to elucidate the target genes of sex steroid on bone and the overlap, or lack thereof, of the pathogenetic mechanisms of estrogen deficiency to those of aging represent, in the authors' opinion, two of the most important remaining frontiers in this area. Knowledge gained from this future quest should yield critical insights for optimizing the prevention and treatment of osteoporosis.
GRANTS
The authors' research is supported by National Institutes of Health Grants P01 AG13918 (to S. C. Manolagas), R01 AR56679 (to M. Almeida), and R01 AR049794 (to C. A. O'Brien); Biomedical Laboratory Research and Development Service of the Veteran's Administration Office of Research and Development Grants I01 BX001405 (to S. C. Manolagas) and 1I01BX000294 (to C. A. O'Brien); University of Arkansas for Medical Sciences Tobacco Funds and Translational Research Institute Grant 1UL1RR029884; Research Foundation Flanders (FWO) Grants G.0858.11 and G.0854.13N and KU Leuven Grant GOA/15/017 (to M. R. Laurent, F. Claessens, R. Bouillon, and V. Dubois). M. R. Laurent and V. Dubois are supported by fellowship grants from FWO.
DISCLOSURES
S. C. Manolagas is a founder, and serves on the scientific advisory board, of Radius Health, Inc. He has ownership of equity in this company and receives $10,000 per annum for his SAB service.
ACKNOWLEDGMENTS
We thank Katie H. Poe for help with the preparation of the manuscript and Robert L. Jilka and Robert S. Weinstein for their contribution of histologic and microCT images used in the figures. We regret that many important contributions to this subject could not be cited because of space limitations.
M. Almeida and M. R. Laurent contributed equally to this article.
Address for reprint requests and other correspondence: S. C. Manolagas, 4301 W. Markham St. #587, Little Rock, AR 72205-7199 (e-mail: manolagasstavros@uams.edu).
REFERENCES
- 1.National Institute of Health Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. JAMA 285: 785–795, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Osteoporosis in Men: The Effects of Gender on Skeletal Health. London: Elsevier, 2010. [Google Scholar]
- 3.Aarden EM, Burger EH, Nijweide PJ. Function of osteocytes in bone. J Cell Biochem 55: 287–299, 1994. [DOI] [PubMed] [Google Scholar]
- 4.Adams GB, Scadden DT. The hematopoietic stem cell in its place. Nat Immunol 7: 333–337, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Aguirre JI, Plotkin LI, Gortazar AR, Millan MM, O'Brien CA, Manolagas SC, Bellido T. A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J Biol Chem 282: 25501–25508, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Ahlborg HG, Johnell O, Turner CH, Rannevik G, Karlsson MK. Bone loss and bone size after menopause. N Engl J Med 349: 327–334, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Albers J, Keller J, Baranowsky A, Beil FT, Catala-Lehnen P, Schulze J, Amling M, Schinke T. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J Cell Biol 200: 537–549, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albright F, Bloomberg E, Smith PH. Postmenopausal osteoporosis. Trans Assoc Am Physicians 298–205, 1940. [Google Scholar]
- 9.Allen MR, Hock JM, Burr DB. Periosteum: biology, regulation, and response to osteoporosis therapies. Bone 35: 1003–1012, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Almeida M, Ucer SS, Iyer S, Kim HN, Han L, Rutlen C, Bartell SM, Warren A, Crawford JA, Jilka R, Manolagas S. Restraining mitochondrial H2O2 generation in cells of the mesenchymal lineage abrogates the adverse effects of aging on the murine skeleton (Abstract). J Bone Miner Res 30: S193, 2015. [Google Scholar]
- 11.Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased PPARγ expression and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem 284: 27438–27448, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem 280: 41342–41351, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem 282: 27298–27305, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O'Brien CA, Bellido T, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 282: 27285–27297, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, Onal M, Xiong J, Weinstein RS, Jilka RL, O'Brien CA, Manolagas SC. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest 123: 394–404, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almeida M, O'Brien CA. Basic biology of skeletal aging: role of stress response pathways. J Gerontol A Biol Sci Med Sci 68: 1197–1208, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altuwaijri S, Chuang KH, Lai KP, Lai JJ, Lin HY, Young FM, Bottaro A, Tsai MY, Zeng WP, Chang HC, Yeh S, Chang C. Susceptibility to autoimmunity and B cell resistance to apoptosis in mice lacking androgen receptor in B cells. Mol Endocrinol 23: 444–453, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ambrogini E, Almeida M, Martin-Millan M, Paik J, dePinho R, Han L, Goellner J, Weinstein R, Jilka R, O'Brien C, Manolagas S. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab 11: 136–146, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amin S, Zhang Y, Felson DT, Sawin CT, Hannan MT, Wilson PW, Kiel DP. Estradiol, testosterone, and the risk for hip fractures in elderly men from the Framingham Study. Am J Med 119: 426–433, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Amin S, Zhang Y, Sawin CT, Evans SR, Hannan MT, Kiel DP, Wilson PW, Felson DT. Association of hypogonadism and estradiol levels with bone mineral density in elderly men from the Framingham study. Ann Intern Med 133: 951–963, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Aminorroaya A, Kelleher S, Conway AJ, Ly LP, Handelsman DJ. Adequacy of androgen replacement influences bone density response to testosterone in androgen-deficient men. Eur J Endocrinol 152: 881–886, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Amory JK, Anawalt BD, Matsumoto AM, Page ST, Bremner WJ, Wang C, Swerdloff RS, Clark RV. The effect of 5alpha-reductase inhibition with dutasteride and finasteride on bone mineral density, serum lipoproteins, hemoglobin, prostate specific antigen and sexual function in healthy young men. J Urol 179: 2333–2338, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amory JK, Watts NB, Easley KA, Sutton PR, Anawalt BD, Matsumoto AM, Bremner WJ, Tenover JL. Exogenous testosterone or testosterone with finasteride increases bone mineral density in older men with low serum testosterone. J Clin Endocrinol Metab 89: 503–510, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Araujo AB, Travison TG, Leder BZ, McKinlay JB. Correlations between serum testosterone, estradiol, and sex hormone-binding globulin and bone mineral density in a diverse sample of men. J Clin Endocrinol Metab 93: 2135–2141, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Argoud T, Boutroy S, Claustrat B, Chapurlat R, Szulc P. Association between sex steroid levels and bone microarchitecture in men: the STRAMBO study. J Clin Endocrinol Metab 99: 1400–1410, 2014. [DOI] [PubMed] [Google Scholar]
- 26.Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE. Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J Biol Chem 282: 20715–20727, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Aubry-Rozier B, Stoll D, Krieg MA, Lamy O, Hans D. What was your fracture risk evaluated by FRAX(R) the day before your osteoporotic fracture? Clin Rheumatol 32: 219–223, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Bailey AJ, Knott L. Molecular changes in bone collagen in osteoporosis and osteoarthritis in the elderly. Exp Gerontol 34: 337–351, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Bala Y, Zebaze R, Ghasem-Zadeh A, Atkinson EJ, Iuliano S, Peterson JM, Amin S, Bjornerem A, Melton LJ III, Johansson H, Kanis JA, Khosla S, Seeman E. Cortical porosity identifies women with osteopenia at increased risk for forearm fractures. J Bone Miner Res 29: 1356–1362, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bala Y, Zebaze R, Seeman E. Role of cortical bone in bone fragility. Curr Opin Rheumatol 27: 406–413, 2015. [DOI] [PubMed] [Google Scholar]
- 31.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, aging. Cell 120: 483–495, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med 19: 179–192, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Barrett-Connor E, Laughlin GA, Li H, Nielson CM, Wang PY, Dam TT, Cauley JA, Ensrud KE, Stefanick ML, Lau E, Hoffman AR, Orwoll ES. The association of concurrent vitamin D and sex hormone deficiency with bone loss and fracture risk in older men: the osteoporotic fractures in men (MrOS) study. J Bone Miner Res 27: 2306–2313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, McNabb MA, Wenger NK. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 355: 125–137, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Barrett-Connor E, Young R, Notelovitz M, Sullivan J, Wiita B, Yang HM, Nolan J. A two-year, double-blind comparison of estrogen-androgen and conjugated estrogens in surgically menopausal women. Effects on bone mineral density, symptoms and lipid profiles. J Reprod Med 44: 1012–1020, 1999. [PubMed] [Google Scholar]
- 36.Bartell SM, Han L, Kim HN, Kim SH, Katzenellenbogen JA, Katzenellenbogen BS, Chambliss KL, Shaul PW, Roberson PK, Weinstein RS, Jilka RL, Almeida M, Manolagas SC. Non-nuclear-initiated actions of the estrogen receptor protect cortical bone mass. Mol Endocrinol 27: 649–656, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartell SM, Kim HN, Ambrogini E, Han L, Iyer S, Serra US, Rabinovitch P, Jilka RL, Weinstein RS, Zhao H, O'Brien CA, Manolagas SC, Almeida M. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun 5: 3773, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basaria S, Coviello AD, Travison TG, Storer TW, Farwell WR, Jette AM, Eder R, Tennstedt S, Ulloor J, Zhang A, Choong K, Lakshman KM, Mazer NA, Miciek R, Krasnoff J, Elmi A, Knapp PE, Brooks B, Appleman E, Aggarwal S, Bhasin G, Hede-Brierley L, Bhatia A, Collins L, LeBrasseur N, Fiore LD, Bhasin S. Adverse events associated with testosterone administration. N Engl J Med 363: 109–122, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basaria S, Davda MN, Travison TG, Ulloor J, Singh R, Bhasin S. Risk factors associated with cardiovascular events during testosterone administration in older men with mobility limitation. J Gerontol A Biol Sci Med Sci 68: 153–160, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bass S, Delmas PD, Pearce G, Hendrich E, Tabensky A, Seeman E. The differing tempo of growth in bone size, mass, and density in girls is region-specific. J Clin Invest 104: 795–804, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bass SL, Saxon L, Daly RM, Turner CH, Robling AG, Seeman E, Stuckey S. The effect of mechanical loading on the size and shape of bone in pre-, peri-, and postpubertal girls: a study in tennis players. J Bone Miner Res 17: 2274–2280, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Baxter-Jones AD, Faulkner RA, Forwood MR, Mirwald RL, Bailey DA. Bone mineral accrual from 8 to 30 years of age: an estimation of peak bone mass. J Bone Miner Res 26: 1729–1739, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Beato M, Klug J. Steroid hormone receptors: an update. Hum Reprod Update 6: 225–236, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Berger C, Goltzman D, Langsetmo L, Joseph L, Jackson S, Kreiger N, Tenenhouse A, Davison KS, Josse RG, Prior JC, Hanley DA. Peak bone mass from longitudinal data: implications for the prevalence, pathophysiology, and diagnosis of osteoporosis. J Bone Miner Res 25: 1948–1957, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertelloni S, Baroncelli GI, Sorrentino MC, Costa S, Battini R, Saggese G. Androgen-receptor blockade does not impair bone mineral density in adolescent females. Calcif Tissue Int 61: 1–5, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Berthold LD, Haras G, Mann M, Alzen G. Trabecular bone mineral density measured by quantitative CT of the lumbar spine in children and adolescents: reference values and peak bone mass. Rofo 178: 1235–1242, 2006. [DOI] [PubMed] [Google Scholar]
- 47.Bevan C, Parker M. The role of coactivators in steroid hormone action. Exp Cell Res 253: 349–356, 1999. [DOI] [PubMed] [Google Scholar]
- 48.Bhasin S, Pencina M, Jasuja GK, Travison TG, Coviello A, Orwoll E, Wang PY, Nielson C, Wu F, Tajar A, Labrie F, Vesper H, Zhang A, Ulloor J, Singh R, D'Agostino R, Vasan RS. Reference ranges for testosterone in men generated using liquid chromatography tandem mass spectrometry in a community-based sample of healthy nonobese young men in the Framingham Heart Study and applied to three geographically distinct cohorts. J Clin Endocrinol Metab 96: 2430–2439, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhasin S, Storer TW, Berman N, Callegari C, Clevenger B, Phillips J, Bunnell TJ, Tricker R, Shirazi A, Casaburi R. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med 335: 1–7, 1996. [DOI] [PubMed] [Google Scholar]
- 50.Bhasin S, Travison TG, Storer TW, Lakshman K, Kaushik M, Mazer NA, Ngyuen AH, Davda MN, Jara H, Aakil A, Anderson S, Knapp PE, Hanka S, Mohammed N, Daou P, Miciek R, Ulloor J, Zhang A, Brooks B, Orwoll K, Hede-Brierley L, Eder R, Elmi A, Bhasin G, Collins L, Singh R, Basaria S. Effect of testosterone supplementation with and without a dual 5alpha-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial. JAMA 307: 931–939, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhasin S, Woodhouse L, Casaburi R, Singh AB, Mac RP, Lee M, Yarasheski KE, Sinha-Hikim I, Dzekov C, Dzekov J, Magliano L, Storer TW. Older men are as responsive as young men to the anabolic effects of graded doses of testosterone on the skeletal muscle. J Clin Endocrinol Metab 90: 678–688, 2005. [DOI] [PubMed] [Google Scholar]
- 52.Bianco P. “Mesenchymal” stem cells. Annu Rev Cell Dev Biol 30: 677–704, 2014. [DOI] [PubMed] [Google Scholar]
- 53.Biro FM, McMahon RP, Striegel-Moore R, Crawford PB, Obarzanek E, Morrison JA, Barton BA, Falkner F. Impact of timing of pubertal maturation on growth in black and white female adolescents: the National Heart, Lung, and Blood Institute Growth and Health Study. J Pediatr 138: 636–643, 2001. [DOI] [PubMed] [Google Scholar]
- 54.Bjornerem A, Bui QM, Ghasem-Zadeh A, Hopper JL, Zebaze R, Seeman E. Fracture risk and height: an association partly accounted for by cortical porosity of relatively thinner cortices. J Bone Miner Res 28: 2017–2026, 2013. [DOI] [PubMed] [Google Scholar]
- 55.Blin-Wakkach C, Wakkach A, Rochet N, Carle GF. Characterization of a novel bipotent hematopoietic progenitor population in normal and osteopetrotic mice. J Bone Miner Res 19: 1137–1143, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Boland GM, Perkins G, Hall DJ, Tuan RS. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J Cell Biochem 93: 1210–1230, 2004. [DOI] [PubMed] [Google Scholar]
- 57.Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann NY Acad Sci 1116: 281–290, 2007. [DOI] [PubMed] [Google Scholar]
- 58.Bonewald LF. The amazing osteocyte. J Bone Miner Res 26: 229–238, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone 42: 606–615, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonjour JP, Chevalley T. Pubertal timing, bone acquisition, and risk of fracture throughout life. Endocr Rev 35: 820–847, 2014. [DOI] [PubMed] [Google Scholar]
- 61.Boonen S, Pye SR, O'Neill TW, Szulc P, Gielen E, Borghs H, Verschueren S, Claessens F, Adams JE, Ward KA, Bartfai G, Casanueva F, Finn JD, Forti G, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Labrie F, Lean ME, Pendleton N, Punab M, Silman AJ, Tajar A, Wu FC, Vanderschueren D. Influence of bone remodelling rate on quantitative ultrasound parameters at the calcaneus and DXA BMDa of the hip and spine in middle-aged and elderly European men: the European Male Ageing Study (EMAS). Eur J Endocrinol 165: 977–986, 2011. [DOI] [PubMed] [Google Scholar]
- 62.Boonen S, Rosen C, Bouillon R, Sommer A, McKay M, Rosen D, Adams S, Broos P, Lenaerts J, Raus J, Vanderschueren D, Geusens P. Musculoskeletal effects of the recombinant human IGF-I/IGF binding protein-3 complex in osteoporotic patients with proximal femoral fracture: a double-blind, placebo-controlled pilot study. J Clin Endocrinol Metab 87: 1593–1599, 2002. [DOI] [PubMed] [Google Scholar]
- 63.Boot AM, de Ridder MA, van der Sluis IM, van S I, Krenning EP, Keizer-Schrama SM. Peak bone mineral density, lean body mass and fractures. Bone 46: 336–341, 2010. [DOI] [PubMed] [Google Scholar]
- 64.Borjesson AE, Farman HH, Engdahl C, Koskela A, Sjogren K, Kindblom JM, Stubelius A, Islander U, Carlsten H, Antal MC, Krust A, Chambon P, Tuukkanen J, Lagerquist MK, Windahl SH, Ohlsson C. The role of activation functions 1 and 2 of estrogen receptor-alpha for the effects of estradiol and selective estrogen receptor modulators in male mice. J Bone Miner Res 28: 1117–1126, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borjesson AE, Lagerquist MK, Liu C, Shao R, Windahl SH, Karlsson C, Sjogren K, Moverare-Skrtic S, Antal MC, Krust A, Mohan S, Chambon P, Savendahl L, Ohlsson C. The role of estrogen receptor alpha in growth plate cartilage for longitudinal bone growth. J Bone Miner Res 25: 2690–2700, 2010. [DOI] [PubMed] [Google Scholar]
- 66.Borjesson AE, Windahl SH, Lagerquist MK, Engdahl C, Frenkel B, Moverare-Skrtic S, Sjogren K, Kindblom JM, Stubelius A, Islander U, Antal MC, Krust A, Chambon P, Ohlsson C. Roles of transactivating functions 1 and 2 of estrogen receptor-alpha in bone. Proc Natl Acad Sci USA 108: 6288–6293, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bouillon R, Bex M, Vanderschueren D, Boonen S. Estrogens are essential for male pubertal periosteal bone expansion. J Clin Endocrinol Metab 89: 6025–6029, 2004. [DOI] [PubMed] [Google Scholar]
- 68.Bouillon R, Koledova E, Bezlepkina O, Nijs J, Shavrikhova E, Nagaeva E, Chikulaeva O, Peterkova V, Dedov I, Bakulin A, Oganov V, Attanasio AF. Bone status and fracture prevalence in Russian adults with childhood-onset growth hormone deficiency. J Clin Endocrinol Metab 89: 4993–4998, 2004. [DOI] [PubMed] [Google Scholar]
- 69.Boyde A, Ali NN, Jones SJ. Resorption of dentine by isolated osteoclasts in vitro. Br Dent J 156: 216–220, 1984. [DOI] [PubMed] [Google Scholar]
- 70.Boyle WJ, Kung Y, Lacey DL, Sarosi I, Dunstan C, Timms E, Tan HL, Elliott G, Kelley MJ, Colombero A, Elliott R, Scully S, Capparelli C, Morony S, Penninger J. Osteoprotegerin ligand (OPGL) is required for murine osteoclastogenesis. Bone 23: S189, 1998. [Google Scholar]
- 71.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 423: 337–342, 2003. [DOI] [PubMed] [Google Scholar]
- 72.Bradney M, Karlsson MK, Duan Y, Stuckey S, Bass S, Seeman E. Heterogeneity in the growth of the axial and appendicular skeleton in boys: implications for the pathogenesis of bone fragility in men. J Bone Miner Res 15: 1871–1878, 2000. [DOI] [PubMed] [Google Scholar]
- 73.Brandi ML, Collin-Osdoby P. Vascular biology and the skeleton. J Bone Miner Res 21: 183–192, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Brown M, Ning J, Ferreira JA, Bogener JL, Lubahn DB. Estrogen receptor-alpha and -beta and aromatase knockout effects on lower limb muscle mass and contractile function in female mice. Am J Physiol Endocrinol Metab 296: E854–E861, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, Öhman L, Greene GL, Gustafsson JÅ, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389: 753–758, 1997. [DOI] [PubMed] [Google Scholar]
- 76.Burghardt AJ, Kazakia GJ, Ramachandran S, Link TM, Majumdar S. Age- and gender-related differences in the geometric properties and biomechanical significance of intracortical porosity in the distal radius and tibia. J Bone Miner Res 25: 983–993, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burkhardt R, Kettner G, Bohm W, Schmidmeier M, Schlag R, Frisch B, Mallmann B, Eisenmenger W, Gilg TH. Changes in trabecular bone, hematopoiesis and bone marrow vessels in aplastic anemia, primary osteoporosis, and old age: a comparative histomorphometric study. Bone 8: 157–164, 1987. [DOI] [PubMed] [Google Scholar]
- 78.Burnett-Bowie SA, McKay EA, Lee H, Leder BZ. Effects of aromatase inhibition on bone mineral density and bone turnover in older men with low testosterone levels. J Clin Endocrinol Metab 94: 4785–4792, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burr DB, Turner CH, Naick P, Forwood MR, Ambrosius W, Hasan MS, Pidaparti R. Does microdamage accumulation affect the mechanical properties of bone? J Biomech 31: 337–345, 1998. [DOI] [PubMed] [Google Scholar]
- 80.Callewaert F, Bakker A, Schrooten J, Van MB, Verhoeven G, Boonen S, Vanderschueren D. Androgen receptor disruption increases the osteogenic response to mechanical loading in male mice. J Bone Miner Res 25: 124–131, 2010. [DOI] [PubMed] [Google Scholar]
- 81.Callewaert F, Sinnesael M, Gielen E, Boonen S, Vanderschueren D. Skeletal sexual dimorphism: relative contribution of sex steroids, GH-IGF1, and mechanical loading. J Endocrinol 207: 127–134, 2010. [DOI] [PubMed] [Google Scholar]
- 82.Callewaert F, Venken K, Kopchick JJ, Torcasio A, van Lenthe GH, Boonen S, Vanderschueren D. Sexual dimorphism in cortical bone size and strength but not density is determined by independent and time-specific actions of sex steroids and IGF-1: evidence from pubertal mouse models. J Bone Miner Res 25: 617–626, 2010. [DOI] [PubMed] [Google Scholar]
- 83.Callewaert F, Venken K, Ophoff J, De Gendt K, Torcasio A, van Lenthe GH, Van Oosterwyck H, Boonen S, Bouillon R, Verhoeven G, Vanderschueren D. Differential regulation of bone and body composition in male mice with combined inactivation of androgen and estrogen receptor-alpha. FASEB J 23: 232–240, 2009. [DOI] [PubMed] [Google Scholar]
- 84.Callewaert L, Christiaens V, Haelens A, Verrijdt G, Verhoeven G, Claessens F. Implications of a polyglutamine tract in the function of the human androgen receptor. Biochem Biophys Res Commun 306: 46–52, 2003. [DOI] [PubMed] [Google Scholar]
- 85.Carmina E, Guastella E, Longo RA, Rini GB, Lobo RA. Correlates of increased lean muscle mass in women with polycystic ovary syndrome. Eur J Endocrinol 161: 583–589, 2009. [DOI] [PubMed] [Google Scholar]
- 86.Carson JA, Manolagas SC. Effects of sex steroids on bones and muscles: Similarities, parallels, and putative interactions in health and disease. Bone 80: 67–78, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Casanova G, Bossardi RR, Ziegelmann P, Spritzer PM. Effects of low-dose versus placebo or conventional-dose postmenopausal hormone therapy on variables related to cardiovascular risk: a systematic review and meta-analyses of randomized clinical trials. J Clin Endocrinol Metab 100: 1028–1037, 2015. [DOI] [PubMed] [Google Scholar]
- 88.Catherino WH, Wolf DM, Jordan VC. A naturally occurring estrogen receptor mutation results in increased estrogenicity of a tamoxifen analog. Mol Endocrinol 9: 1053–1063, 1995. [DOI] [PubMed] [Google Scholar]
- 89.Cauley JA, Ewing SK, Taylor BC, Fink HA, Ensrud KE, Bauer DC, Barrett-Connor E, Marshall L, Orwoll ES. Sex steroid hormones in older men: longitudinal associations with 4.5-year change in hip bone mineral density–the osteoporotic fractures in men study. J Clin Endocrinol Metab 95: 4314–4323, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cauley JA, LaCroix AZ, Robbins JA, Larson J, Wallace R, Wactawski-Wende J, Chen Z, Bauer DC, Cummings SR, Jackson R. Baseline serum estradiol and fracture reduction during treatment with hormone therapy: the Women's Health Initiative randomized trial. Osteoporos Int 21: 167–177, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cawthon PM, Schousboe JT, Harrison SL, Ensrud KE, Black D, Cauley JA, Cummings SR, LeBlanc ES, Laughlin GA, Nielson CM, Broughton A, Kado DM, Hoffman AR, Jamal SA, Barrett-Connor E, Orwoll ES. Sex hormones, sex hormone binding globulin, and vertebral fractures in older men. Bone 84: 271–278, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Center JR, Nguyen TV, Sambrook PN, Eisman JA. Hormonal and biochemical parameters in the determination of osteoporosis in elderly men. J Clin Endocrinol Metab 84: 3626–3635, 1999. [DOI] [PubMed] [Google Scholar]
- 93.Center JR, Nguyen TV, Sambrook PN, Eisman JA. Hormonal and biochemical parameters and osteoporotic fractures in elderly men. J Bone Miner Res 15: 1405–1411, 2000. [DOI] [PubMed] [Google Scholar]
- 94.Chambers TJ, Revell PA, Fuller K, Athanasou NA. Resorption of bone by isolated rabbit osteoclasts. J Cell Sci 66: 383–399, 1984. [DOI] [PubMed] [Google Scholar]
- 95.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest 120: 2319–2330, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527–605, 1979. [DOI] [PubMed] [Google Scholar]
- 97.Chen J, Hwang DJ, Bohl CE, Miller DD, Dalton JT. A selective androgen receptor modulator for hormonal male contraception. J Pharmacol Exp Ther 312: 546–553, 2005. [DOI] [PubMed] [Google Scholar]
- 98.Chevalley T, Bonjour JP, Ferrari S, Rizzoli R. Deleterious effect of late menarche on distal tibia microstructure in healthy 20-year-old and premenopausal middle-aged women. J Bone Miner Res 24: 144–152, 2009. [DOI] [PubMed] [Google Scholar]
- 99.Chevalley T, Bonjour JP, Ferrari S, Rizzoli R. Pubertal timing and body mass index gain from birth to maturity in relation with femoral neck BMD and distal tibia microstructure in healthy female subjects. Osteoporos Int 22: 2689–2698, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chevalley T, Bonjour JP, van RB, Rizzoli R, Ferrari S. Fractures in healthy females followed from childhood to early adulthood are associated with later menarcheal age and with impaired bone microstructure at peak bone mass. J Clin Endocrinol Metab 97: 4174–4181, 2012. [DOI] [PubMed] [Google Scholar]
- 101.Chiang C, Chiu M, Moore AJ, Anderson PH, Ghasem-Zadeh A, McManus JF, Ma C, Seeman E, Clemens TL, Morris HA, Zajac JD, Davey RA. Mineralization and bone resorption are regulated by the androgen receptor in male mice. J Bone Miner Res 24: 621–631, 2009. [DOI] [PubMed] [Google Scholar]
- 102.Christiaens V, Bevan CL, Callewaert L, Haelens A, Verrijdt G, Rombauts W, Claessens F. Characterization of the two coactivator-interacting surfaces of the androgen receptor and their relative role in transcriptional control. J Biol Chem 277: 49230–49237, 2002. [DOI] [PubMed] [Google Scholar]
- 103.Christmas C, O'Connor KG, Harman SM, Tobin JD, Munzer T, Bellantoni MF, Clair CS, Pabst KM, Sorkin JD, Blackman MR. Growth hormone and sex steroid effects on bone metabolism and bone mineral density in healthy aged women and men. J Gerontol A Biol Sci Med Sci 57: M12–M18, 2002. [DOI] [PubMed] [Google Scholar]
- 104.Cicognani A, Cacciari E, Tacconi M, Pascucci MG, Tonioli S, Pirazzoli P, Balsamo A. Effect of gonadectomy on growth hormone, IGF-I and sex steroids in children with complete and incomplete androgen insensitivity. Acta Endocrinol 121: 777–783, 1989. [DOI] [PubMed] [Google Scholar]
- 105.Claessens F, Denayer S, Van TN, Kerkhofs S, Helsen C, Haelens A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal 6: e008, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Clarke BL, Khosla S. New selective estrogen and androgen receptor modulators. Curr Opin Rheumatol 21: 374–379, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, Chernausek SD, Savage MO, Wit JM. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab 93: 4210–4217, 2008. [DOI] [PubMed] [Google Scholar]
- 108.Compston J. Obesity and fractures in postmenopausal women. Curr Opin Rheumatol 27: 414–419, 2015. [DOI] [PubMed] [Google Scholar]
- 109.Crandall CJ, Aragaki A, Cauley JA, Manson JE, LeBlanc E, Wallace R, Wactawski-Wende J, LaCroix A, O'Sullivan MJ, Vitolins M, Watts NB. Associations of menopausal vasomotor symptoms with fracture incidence. J Clin Endocrinol Metab 100: 524–534, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cranney A, Jamal SA, Tsang JF, Josse RG, Leslie WD. Low bone mineral density and fracture burden in postmenopausal women. CMAJ 177: 575–580, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Crawford BA, Liu PY, Kean MT, Bleasel JF, Handelsman DJ. Randomized placebo-controlled trial of androgen effects on muscle and bone in men requiring long-term systemic glucocorticoid treatment. J Clin Endocrinol Metab 88: 3167–3176, 2003. [DOI] [PubMed] [Google Scholar]
- 112.Crowne EC, Wallace WH, Moore C, Mitchell R, Robertson WH, Holly JM, Shalet SM. Effect of low dose oxandrolone and testosterone treatment on the pituitary-testicular and GH axes in boys with constitutional delay of growth and puberty. Clin Endocrinol 46: 209–216, 1997. [DOI] [PubMed] [Google Scholar]
- 113.Cummings SR, Ensrud K, Delmas PD, LaCroix AZ, Vukicevic S, Reid DM, Goldstein S, Sriram U, Lee A, Thompson J, Armstrong RA, Thompson DD, Powles T, Zanchetta J, Kendler D, Neven P, Eastell R. Lasofoxifene in postmenopausal women with osteoporosis. N Engl J Med 362: 686–696, 2010. [DOI] [PubMed] [Google Scholar]
- 114.Cummings SR, Karpf DB, Harris F, Genant HK, Ensrud K, LaCroix AZ, Black DM. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med 112: 281–289, 2002. [DOI] [PubMed] [Google Scholar]
- 115.Cummings SR, San MJ, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, Kutilek S, Adami S, Zanchetta J, Libanati C, Siddhanti S, Christiansen C. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361: 756–765, 2009. [DOI] [PubMed] [Google Scholar]
- 116.Cunningham GR, Stephens-Shields AJ, Rosen RC, Wang C, Ellenberg SS, Matsumoto AM, Bhasin S, Molitch ME, Farrar JT, Cella D, Barrett-Connor E, Cauley JA, Cifelli D, Crandall JP, Ensrud KE, Fluharty L, Gill TM, Lewis CE, Pahor M, Resnick SM, Storer TW, Swerdloff RS, Anton S, Basaria S, Diem S, Tabatabaie V, Hou X, Snyder PJ. Association of sex hormones with sexual function, vitality, and physical function of symptomatic older men with low testosterone levels at baseline in the testosterone trials. J Clin Endocrinol Metab 100: 1146–1155, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824, 2007. [DOI] [PubMed] [Google Scholar]
- 118.Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell… and more. Endocr Rev 34: 658–690, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dalton JT, Barnette KG, Bohl CE, Hancock ML, Rodriguez D, Dodson ST, Morton RA, Steiner MS. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J Cachexia Sarcopenia Muscle 2: 153–161, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dalzell N, Kaptoge S, Morris N, Berthier A, Koller B, Braak L, van RB, Reeve J. Bone micro-architecture and determinants of strength in the radius and tibia: age-related changes in a population-based study of normal adults measured with high-resolution pQCT. Osteoporos Int 20: 1683–1694, 2009. [DOI] [PubMed] [Google Scholar]
- 121.Damien E, Price JS, Lanyon LE. Mechanical strain stimulates osteoblast proliferation through the estrogen receptor in males as well as females. J Bone Miner Res 15: 2169–2177, 2000. [DOI] [PubMed] [Google Scholar]
- 122.Danilovic DL, Correa PH, Costa EM, Melo KF, Mendonca BB, Arnhold IJ. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos Int 18: 369–374, 2007. [DOI] [PubMed] [Google Scholar]
- 123.Darelid A, Ohlsson C, Nilsson M, Kindblom JM, Mellstrom D, Lorentzon M. Catch up in bone acquisition in young adult men with late normal puberty. J Bone Miner Res 27: 2198–2207, 2012. [DOI] [PubMed] [Google Scholar]
- 124.Davis SR, McCloud P, Strauss BJG, Burger H. Testosterone enhances estradiol's effects on postmenopausal bone density and sexuality. Maturitas 21: 227–236, 1995. [DOI] [PubMed] [Google Scholar]
- 125.Delmas PD, Bjarnason NH, Mitlak BH, Ravoux AC, Shah AS, Huster WJ, Draper M, Christiansen C. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med 337: 1641–1647, 1997. [DOI] [PubMed] [Google Scholar]
- 126.Delmas PD, Genant HK, Crans GG, Stock JL, Wong M, Siris E, Adachi JD. Severity of prevalent vertebral fractures and the risk of subsequent vertebral and nonvertebral fractures: results from the MORE trial. Bone 33: 522–532, 2003. [DOI] [PubMed] [Google Scholar]
- 127.Denger S, Bahr-Ivacevic T, Brand H, Reid G, Blake J, Seifert M, Lin CY, May K, Benes V, Liu ET, Gannon F. Transcriptome profiling of estrogen-regulated genes in human primary osteoblasts reveals an osteoblast-specific regulation of the insulin-like growth factor binding protein 4 gene. Mol Endocrinol 22: 361–379, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Destaing O, Saltel F, Geminard JC, Jurdic P, Bard F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol Biol Cell 14: 407–416, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dhindsa S, Furlanetto R, Vora M, Ghanim H, Chaudhuri A, Dandona P. Low estradiol concentrations in men with subnormal testosterone concentrations and type 2 diabetes. Diabetes Care 34: 1854–1859, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495: 231–235, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dobs AS, Boccia RV, Croot CC, Gabrail NY, Dalton JT, Hancock ML, Johnston MA, Steiner MS. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol 14: 335–345, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dobs AS, Nguyen T, Pace C, Roberts CP. Differential effects of oral estrogen versus oral estrogen-androgen replacement therapy on body composition in postmenopausal women. J Clin Endocrinol Metab 87: 1509–1516, 2002. [DOI] [PubMed] [Google Scholar]
- 133.Doran PM, Riggs BL, Atkinson EJ, Khosla S. Effects of raloxifene, a selective estrogen receptor modulator, on bone turnover markers and serum sex steroid and lipid levels in elderly men. J Bone Miner Res 16: 2118–2125, 2001. [DOI] [PubMed] [Google Scholar]
- 134.Downing JR, Rettenmier CW, Sherr CJ. Ligand-induced tyrosine kinase activity of the colony-stimulating factor 1 receptor in a murine macrophage cell line. Mol Cell Biol 8: 1795–1799, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Duan Y, Parfitt A, Seeman E. Vertebral bone mass, size, and volumetric density in women with spinal fractures. J Bone Miner Res 14: 1796–1802, 1999. [DOI] [PubMed] [Google Scholar]
- 136.Dubois V, Laurent M, Boonen S, Vanderschueren D, Claessens F. Androgens and skeletal muscle: cellular and molecular action mechanisms underlying the anabolic actions. Cell Mol Life Sci 69: 1651–1667, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dubois V, Laurent MR, Sinnesael M, Cielen N, Helsen C, Clinckemalie L, Spans L, Gayan-Ramirez G, Deldicque L, Hespel P, Carmeliet G, Vanderschueren D, Claessens F. A satellite cell-specific knockout of the androgen receptor reveals myostatin as a direct androgen target in skeletal muscle. FASEB J 28: 2979–2994, 2014. [DOI] [PubMed] [Google Scholar]
- 138.Dubois V, Simitsidellis I, Laurent MR, Jardi F, Saunders PT, Vanderschueren D, Claessens F. Enobosarm (GTx-024) modulates adult skeletal muscle mass independently of the androgen receptor in the satellite cell lineage. Endocrinology en20151479, 2015. [DOI] [PubMed] [Google Scholar]
- 139.Duschek EJ, Gooren LJ, Netelenbos C. Effects of raloxifene on gonadotrophins, sex hormones, bone turnover and lipids in healthy elderly men. Eur J Endocrinol 150: 539–546, 2004. [DOI] [PubMed] [Google Scholar]
- 140.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest 111: 1221–1230, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Elraiyah T, Sonbol MB, Wang Z, Khairalseed T, Asi N, Undavalli C, Nabhan M, Firwana B, Altayar O, Prokop L, Montori VM, Murad MH. Clinical review: the benefits and harms of systemic testosterone therapy in postmenopausal women with normal adrenal function: a systematic review and meta-analysis. J Clin Endocrinol Metab 99: 3543–3550, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, Jepsen KJ, Schaffler MB. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 46: 577–583, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Emmelot-Vonk MH, Verhaar HJ, Nakhai Pour HR, Aleman A, Lock TM, Bosch JL, Grobbee DE, van der Schouw YT. Effect of testosterone supplementation on functional mobility, cognition, and other parameters in older men: a randomized controlled trial. JAMA 299: 39–52, 2008. [DOI] [PubMed] [Google Scholar]
- 144.Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, Oei L, Albagha OM, Amin N, Kemp JP, Koller DL, Li G, Liu CT, Minster RL, Moayyeri A, Vandenput L, Willner D, Xiao SM, Yerges-Armstrong LM, Zheng HF, Alonso N, Eriksson J, Kammerer CM, Kaptoge SK, Leo PJ, Thorleifsson G, Wilson SG, Wilson JF, Aalto V, Alen M, Aragaki AK, Aspelund T, Center JR, Dailiana Z, Duggan DJ, Garcia M, Garcia-Giralt N, Giroux S, Hallmans G, Hocking LJ, Husted LB, Jameson KA, Khusainova R, Kim GS, Kooperberg C, Koromila T, Kruk M, Laaksonen M, LaCroix AZ, Lee SH, Leung PC, Lewis JR, Masi L, Mencej-Bedrac S, Nguyen TV, Nogues X, Patel MS, Prezelj J, Rose LM, Scollen S, Siggeirsdottir K, Smith AV, Svensson O, Trompet S, Trummer O, van Schoor NM, Woo J, Zhu K, Balcells S, Brandi ML, Buckley BM, Cheng S, Christiansen C, Cooper C, Dedoussis G, Ford I, Frost M, Goltzman D, Gonzalez-Macias J, Kahonen M, Karlsson M, Khusnutdinova E, Koh JM, Kollia P, Langdahl BL, Leslie WD, Lips P, Ljunggren O, Lorenc RS, Marc J, Mellstrom D, Obermayer-Pietsch B, Olmos JM, Pettersson-Kymmer U, Reid DM, Riancho JA, Ridker PM, Rousseau F, Slagboom PE, Tang NL, Urreizti R, Van HW, Viikari J, Zarrabeitia MT, Aulchenko YS, Castano-Betancourt M, Grundberg E, Herrera L, Ingvarsson T, Johannsdottir H, Kwan T, Li R, Luben R, Medina-Gomez C, Palsson ST, Reppe S, Rotter JI, Sigurdsson G, van Meurs JB, Verlaan D, Williams FM, Wood AR, Zhou Y, Gautvik KM, Pastinen T, Raychaudhuri S, Cauley JA, Chasman DI, Clark GR, Cummings SR, Danoy P, Dennison EM, Eastell R, Eisman JA, Gudnason V, Hofman A, Jackson RD, Jones G, Jukema JW, Khaw KT, Lehtimaki T, Liu Y, Lorentzon M, McCloskey E, Mitchell BD, Nandakumar K, Nicholson GC, Oostra BA, Peacock M, Pols HA, Prince RL, Raitakari O, Reid IR, Robbins J, Sambrook PN, Sham PC, Shuldiner AR, Tylavsky FA, van Duijn CM, Wareham NJ, Cupples LA, Econs MJ, Evans DM, Harris TB, Kung AW, Psaty BM, Reeve J, Spector TD, Streeten EA, Zillikens MC, Thorsteinsdottir U, Ohlsson C, Karasik D, Richards JB, Brown MA, Stefansson K, Uitterlinden AG, Ralston SH, Ioannidis JP, Kiel DP, Rivadeneira F. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet 44: 491–501, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ettinger B, Black DM, Mitlak BH, Knickerbocker RK, Nickelsen T, Genant HK, Christiansen C, Delmas PD, Zanchetta JR, Stakkestad J, Gluer CC, Krueger K, Cohen FJ, Eckert S, Ensrud KE, Avioli LV, Lips P, Cummings SR. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 282: 637–645, 1999. [DOI] [PubMed] [Google Scholar]
- 146.Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the big bang. Cell 157: 255–266, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Falahati-Nini A, Riggs BL, Atkinson EJ, O'Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest 106: 1553–1560, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Farr JN, Khosla S. Skeletal changes through the lifespan-from growth to senescence. Nat Rev Endocrinol 11: 513–521, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem 287: 4434–4440, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Finkelstein JS, Klibanski A, Neer RM, Doppelt SH, Rosenthal DI, Segre GV, Crowley WF Jr. Increases in bone density during treatment of men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 69: 776–783, 1989. [DOI] [PubMed] [Google Scholar]
- 151.Finkelstein JS, Lee H, Burnett-Bowie SA, Pallais JC, Yu EW, Borges LF, Jones BF, Barry CV, Wulczyn KE, Thomas BJ, Leder BZ. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med 369: 1011–1022, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Finkelstein JS, Lee H, Leder BZ, Burnett-Bowie SA, Goldstein DW, Hahn CW, Hirsch SC, Linker A, Perros N, Servais AB, Taylor AP, Webb ML, Youngner JM, Yu EW. Gonadal steroid-dependent effects on bone turnover and bone mineral density in men. J Clin Invest 126: 1114–1125, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Finkelstein JS, Yu EW, Burnett-Bowie SA. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med 369: 2457, 2013. [DOI] [PubMed] [Google Scholar]
- 154.Fritton JC, Emerton KB, Sun H, Kawashima Y, Mejia W, Wu Y, Rosen CJ, Panus D, Bouxsein M, Majeska RJ, Schaffler MB, Yakar S. Growth hormone protects against ovariectomy-induced bone loss in states of low circulating insulin-like growth factor (IGF-1). J Bone Miner Res 25: 235–246, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fritton JC, Myers ER, Wright TM, van der Meulen MC. Bone mass is preserved and cancellous architecture altered due to cyclic loading of the mouse tibia after orchidectomy. J Bone Miner Res 23: 663–671, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Frost HM. On the estrogen-bone relationship and postmenopausal bone loss: a new model. J Bone Miner Res 14: 1473–1477, 1999. [DOI] [PubMed] [Google Scholar]
- 157.Frost HM. Bone's mechanostat: a 2003 update. Anat Rec A Discov Mol Cell Evol Biol 275: 1081–1101, 2003. [DOI] [PubMed] [Google Scholar]
- 158.Fu J, Li S, Feng R, Ma H, Sabeh F, Roodman GD, Wang J, Robinson S, Guo XE, Lund T, Normolle D, Mapara MY, Weiss SJ, Lentzsch S. Multiple myeloma-derived MMP-13 mediates osteoclast fusogenesis and osteolytic disease. J Clin Invest 126: 1759–1772, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Furuya K, Yamamoto N, Ohyabu Y, Makino A, Morikyu T, Ishige H, Kuzutani K, Endo Y. The novel non-steroidal selective androgen receptor modulator S-101479 has additive effects with bisphosphonate, selective estrogen receptor modulator, and parathyroid hormone on the bones of osteoporotic female rats. Biol Pharm Bull 35: 1096–1104, 2012. [DOI] [PubMed] [Google Scholar]
- 160.Gabel L, Nettlefold L, Brasher PM, Moore SA, Ahamed Y, Macdonald HM, McKay HA. Reexamining the surfaces of bone in boys and girls during adolescent growth: a 12-year mixed longitudinal pQCT study. J Bone Miner Res 30: 2158–2167, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Galea GL, Meakin LB, Sugiyama T, Zebda N, Sunters A, Taipaleenmaki H, Stein GS, Van Wijnen AJ, Lanyon LE, Price JS. Estrogen receptor alpha mediates proliferation of osteoblastic cells stimulated by estrogen and mechanical strain, but their acute down-regulation of the Wnt antagonist Sost is mediated by estrogen receptor beta. J Biol Chem 288: 9035–9048, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Galien R, Garcia T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res 25: 2424–2429, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Selective androgen receptor modulator treatment improves muscle strength and body composition and prevents bone loss in orchidectomized rats. Endocrinology 146: 4887–4897, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Garcia AJ, Tom C, Guemes M, Polanco G, Mayorga ME, Wend K, Miranda-Carboni GA, Krum SA. ERalpha signaling regulates MMP3 expression to induce FasL cleavage and osteoclast apoptosis. J Bone Miner Res 28: 283–290, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Garnero P, Sornay-Rendu E, Delmas PD. Low serum IGF-1 and occurrence of osteoporotic fractures in postmenopausal women. Lancet 355: 898–899, 2000. [DOI] [PubMed] [Google Scholar]
- 166.Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest 85: 632–639, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gault EJ, Perry RJ, Cole TJ, Casey S, Paterson WF, Hindmarsh PC, Betts P, Dunger DB, Donaldson MD. Effect of oxandrolone and timing of pubertal induction on final height in Turner's syndrome: randomised, double blind, placebo controlled trial. BMJ 342: d1980, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gennari C, Agnusdei D, Nardi P, Civitelli R. Estrogen preserves a normal intestinal responsiveness to 1,25-dihydroxyvitamin D3 in oophorectomized women. J Clin Endocrinol Metab 71: 1288–1293, 1990. [DOI] [PubMed] [Google Scholar]
- 169.Gennari L, Merlotti D, Martini G, Gonnelli S, Franci B, Campagna S, Lucani B, Dal CN, Valenti R, Gennari C, Nuti R. Longitudinal association between sex hormone levels, bone loss, and bone turnover in elderly men. J Clin Endocrinol Metab 88: 5327–5333, 2003. [DOI] [PubMed] [Google Scholar]
- 170.Gennari L, Nuti R, Bilezikian JP. Aromatase activity and bone homeostasis in men. J Clin Endocrinol Metab 89: 5898–5907, 2004. [DOI] [PubMed] [Google Scholar]
- 171.Geusens P. Nandrolone decanoate: pharmacological properties and therapeutic use in osteoporosis. Clin Rheumatol 14 Suppl 3: 32–39, 1995. [DOI] [PubMed] [Google Scholar]
- 172.Giannoulis MG, Martin FC, Nair KS, Umpleby AM, Sonksen P. Hormone replacement therapy and physical function in healthy older men. Time to talk hormones? Endocr Rev 33: 314–377, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gillies GE, McArthur S. Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicines. Pharmacol Rev 62: 155–198, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8: 722–728, 2007. [DOI] [PubMed] [Google Scholar]
- 175.Glait-Santar C, Benayahu D. Regulation of SVEP1 gene expression by 17beta-estradiol and TNFalpha in pre-osteoblastic and mammary adenocarcinoma cells. J Steroid Biochem Mol Biol 130: 36–44, 2012. [DOI] [PubMed] [Google Scholar]
- 176.Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8: 751–764, 2005. [DOI] [PubMed] [Google Scholar]
- 177.Govoni KE, Wergedal JE, Chadwick RB, Srivastava AK, Mohan S. Prepubertal OVX increases IGF-I expression and bone accretion in C57BL/6J mice. Am J Physiol Endocrinol Metab 295: E1172–E1180, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Grassi F, Cristino S, Toneguzzi S, Piacentini A, Facchini A, Lisignoli G. CXCL12 chemokine up-regulates bone resorption and MMP-9 release by human osteoclasts: CXCL12 levels are increased in synovial and bone tissue of rheumatoid arthritis patients. J Cell Physiol 199: 244–251, 2004. [DOI] [PubMed] [Google Scholar]
- 179.Grassi F, Piacentini A, Cristino S, Toneguzzi S, Cavallo C, Facchini A, Lisignoli G. Human osteoclasts express different CXC chemokines depending on cell culture substrate: molecular and immunocytochemical evidence of high levels of CXCL10 and CXCL12. Histochem Cell Biol 120: 391–400, 2003. [DOI] [PubMed] [Google Scholar]
- 180.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495: 227–230, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Greendale GA, Edelstein S, Barrett-Connor E. Endogenous sex steroids and bone mineral density in older women and men: the Rancho Bernardo Study. J Bone Miner Res 12: 1833–1843, 1997. [DOI] [PubMed] [Google Scholar]
- 182.Greenspan SL, Wagner J, Nelson JB, Perera S, Britton C, Resnick NM. Vertebral fractures and trabecular microstructure in men with prostate cancer on androgen deprivation therapy. J Bone Miner Res 28: 325–332, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gunness M, Orwoll E. Early induction of alterations in cancellous and cortical bone histology after orchiectomy in mature rats. J Bone Miner Res 10: 1735–1744, 1995. [DOI] [PubMed] [Google Scholar]
- 184.Gustafsson A, Lindenfors P. Human size evolution: no evolutionary allometric relationship between male and female stature. J Hum Evol 47: 253–266, 2004. [DOI] [PubMed] [Google Scholar]
- 185.Ha H, Kwak HB, Lee SW, Jin HM, Kim HM, Kim HH, Lee ZH. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp Cell Res 301: 119–127, 2004. [DOI] [PubMed] [Google Scholar]
- 186.Haentjens P, Magaziner J, Colon-Emeric CS, Vanderschueren D, Milisen K, Velkeniers B, Boonen S. Meta-analysis: excess mortality after hip fracture among older women and men. Ann Intern Med 152: 380–390, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hagino H, Raab DM, Kimmel DB, Akhter MP, Recker RR. Effect of ovariectomy on bone response to in vivo external loading. J Bone Miner Res 8: 347–357, 1993. [DOI] [PubMed] [Google Scholar]
- 188.Hamilton EJ, Ghasem-Zadeh A, Gianatti E, Lim-Joon D, Bolton D, Zebaze R, Seeman E, Zajac JD, Grossmann M. Structural decay of bone microarchitecture in men with prostate cancer treated with androgen deprivation therapy. J Clin Endocrinol Metab 95: E456–E463, 2010. [DOI] [PubMed] [Google Scholar]
- 189.Hamilton JA. CSF1 signal transduction. J Leukoc Biol 62: 145–155, 1997. [DOI] [PubMed] [Google Scholar]
- 190.Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev 28: 726–741, 2007. [DOI] [PubMed] [Google Scholar]
- 191.Han TS, Goswami D, Trikudanathan S, Creighton SM, Conway GS. Comparison of bone mineral density and body proportions between women with complete androgen insensitivity syndrome and women with gonadal dysgenesis. Eur J Endocrinol 159: 179–185, 2008. [DOI] [PubMed] [Google Scholar]
- 192.Han ZH, Palnitkar S, Rao DS, Nelson D, Parfitt AM. Effects of ethnicity and age or menopause on the remodeling and turnover of iliac bone: implications for mechanisms of bone loss. J Bone Miner Res 12: 498–508, 1997. [DOI] [PubMed] [Google Scholar]
- 193.Hansen S, Shanbhogue V, Folkestad L, Nielsen MM, Brixen K. Bone microarchitecture and estimated strength in 499 adult Danish women and men: a cross-sectional, population-based high-resolution peripheral quantitative computed tomographic study on peak bone structure. Calcif Tissue Int 94: 269–281, 2014. [DOI] [PubMed] [Google Scholar]
- 194.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956. [DOI] [PubMed] [Google Scholar]
- 195.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 20: 491–502, 2006. [DOI] [PubMed] [Google Scholar]
- 196.Harris HA. Estrogen receptor-beta: recent lessons from in vivo studies. Mol Endocrinol 21: 1–13, 2007. [DOI] [PubMed] [Google Scholar]
- 197.Hassler N, Roschger A, Gamsjaeger S, Kramer I, Lueger S, van LA, Roschger P, Klaushofer K, Paschalis EP, Kneissel M, Papapoulos S. Sclerostin deficiency is linked to altered bone composition. J Bone Miner Res 29: 2144–2151, 2014. [DOI] [PubMed] [Google Scholar]
- 198.Hawse JR, Subramaniam M, Monroe DG, Hemmingsen AH, Ingle JN, Khosla S, Oursler MJ, Spelsberg TC. Estrogen receptor beta isoform-specific induction of transforming growth factor beta-inducible early gene-1 in human osteoblast cells: an essential role for the activation function 1 domain. Mol Endocrinol 22: 1579–1595, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.He Q, Karlberg J. BMI in childhood and its association with height gain, timing of puberty, and final height. Pediatr Res 49: 244–251, 2001. [DOI] [PubMed] [Google Scholar]
- 200.Heaney RP. Is the paradigm shifting? Bone 33: 457–465, 2003. [DOI] [PubMed] [Google Scholar]
- 201.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev 28: 778–808, 2007. [DOI] [PubMed] [Google Scholar]
- 202.Helsen C, Kerkhofs S, Clinckemalie L, Spans L, Laurent M, Boonen S, Vanderschueren D, Claessens F. Structural basis for nuclear hormone receptor DNA binding. Mol Cell Endocrinol 348: 411–417, 2012. [DOI] [PubMed] [Google Scholar]
- 203.Henning P, Ohlsson C, Engdahl C, Farman H, Windahl SH, Carlsten H, Lagerquist MK. The effect of estrogen on bone requires ERalpha in nonhematopoietic cells but is enhanced by ERalpha in hematopoietic cells. Am J Physiol Endocrinol Metab 307: E589–E595, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Henry YM, Eastell R. Ethnic and gender differences in bone mineral density and bone turnover in young adults: effect of bone size. Osteoporos Int 11: 512–517, 2000. [DOI] [PubMed] [Google Scholar]
- 205.Henry YM, Fatayerji D, Eastell R. Attainment of peak bone mass at the lumbar spine, femoral neck and radius in men and women: relative contributions of bone size and volumetric bone mineral density. Osteoporos Int 15: 263–273, 2004. [DOI] [PubMed] [Google Scholar]
- 206.Hernandez CJ, Beaupre GS, Carter DR. A theoretical analysis of the relative influences of peak BMD, age-related bone loss and menopause on the development of osteoporosis. Osteoporos Int 14: 843–847, 2003. [DOI] [PubMed] [Google Scholar]
- 207.Hernlund E, Svedbom A, Ivergard M, Compston J, Cooper C, Stenmark J, McCloskey EV, Jonsson B, Kanis JA. Osteoporosis in the European Union: medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch Osteoporos 8: 136, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hero M, Wickman S, Dunkel L. Treatment with the aromatase inhibitor letrozole during adolescence increases near-final height in boys with constitutional delay of puberty. Clin Endocrinol 64: 510–513, 2006. [DOI] [PubMed] [Google Scholar]
- 209.Hofbauer LC, Hicok KC, Chen D, Khosla S. Regulation of osteoprotegerin production by androgens and anti-androgens in human osteoblastic lineage cells. Eur J Endocrinol 147: 269–273, 2002. [DOI] [PubMed] [Google Scholar]
- 210.Hofbauer LC, Ten RM, Khosla S. The anti-androgen hydroxyflutamide and androgens inhibit interleukin-6 production by an androgen-responsive human osteoblastic cell line. J Bone Miner Res 14: 1330–1337, 1999. [DOI] [PubMed] [Google Scholar]
- 211.Horowitz MC, Fretz JA, Lorenzo JA. How B cells influence bone biology in health and disease. Bone 47: 472–479, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hsu B, Cumming RG, Seibel MJ, Naganathan V, Blyth FM, Bleicher K, Dave A, Le Couteur DG, Waite LM, Handelsman DJ. Reproductive hormones and longitudinal change in bone mineral density and incident fracture risk in older men: The Concord Health and Aging in Men Project. J Bone Miner Res 30: 1701–1708, 2015. [DOI] [PubMed] [Google Scholar]
- 213.Huang G, Basaria S, Travison TG, Ho MH, Davda M, Mazer NA, Miciek R, Knapp PE, Zhang A, Collins L, Ursino M, Appleman E, Dzekov C, Stroh H, Ouellette M, Rundell T, Baby M, Bhatia NN, Khorram O, Friedman T, Storer TW, Bhasin S. Testosterone dose-response relationships in hysterectomized women with or without oophorectomy: effects on sexual function, body composition, muscle performance and physical function in a randomized trial. Menopause 21: 612–623, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-β. Nat Med 2: 1132–1136, 1996. [DOI] [PubMed] [Google Scholar]
- 215.Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet 380: 1419–1428, 2012. [DOI] [PubMed] [Google Scholar]
- 216.Huhtaniemi IT, Pye SR, Limer KL, Thomson W, O'Neill TW, Platt H, Payne D, John SL, Jiang M, Boonen S, Borghs H, Vanderschueren D, Adams JE, Ward KA, Bartfai G, Casanueva F, Finn JD, Forti G, Giwercman A, Han TS, Kula K, Lean ME, Pendleton N, Punab M, Silman AJ, Wu FC. Increased estrogen rather than decreased androgen action is associated with longer androgen receptor CAG repeats. J Clin Endocrinol Metab 94: 277–284, 2009. [DOI] [PubMed] [Google Scholar]
- 217.Huhtaniemi IT, Tajar A, Lee DM, O'Neill TW, Finn JD, Bartfai G, Boonen S, Casanueva FF, Giwercman A, Han TS, Kula K, Labrie F, Lean ME, Pendleton N, Punab M, Silman AJ, Vanderschueren D, Forti G, Wu FC. Comparison of serum testosterone and estradiol measurements in 3174 European men using platform immunoassay and mass spectrometry; relevance for the diagnostics in aging men. Eur J Endocrinol 166: 983–991, 2012. [DOI] [PubMed] [Google Scholar]
- 218.Hui SL, Slemenda CW, Johnston CC Jr. Age and bone mass as predictors of fracture in a prospective study. J Clin Invest 81: 1804–1809, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hyldstrup L, Conway GS, Racz K, Keller A, Chanson P, Zacharin M, Lysgaard AL, Andreasen AH, Kappelgaard AM. Growth hormone effects on cortical bone dimensions in young adults with childhood-onset growth hormone deficiency. Osteoporos Int 23: 2219–2226, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Idan A, Griffiths KA, Harwood DT, Seibel MJ, Turner L, Conway AJ, Handelsman DJ. Long-term effects of dihydrotestosterone treatment on prostate growth in healthy, middle-aged men without prostate disease: a randomized, placebo-controlled trial. Ann Intern Med 153: 621–632, 2010. [DOI] [PubMed] [Google Scholar]
- 221.Ioannidis JP, Ng MY, Sham PC, Zintzaras E, Lewis CM, Deng HW, Econs MJ, Karasik D, Devoto M, Kammerer CM, Spector T, Andrew T, Cupples LA, Duncan EL, Foroud T, Kiel DP, Koller D, Langdahl B, Mitchell BD, Peacock M, Recker R, Shen H, Sol-Church K, Spotila LD, Uitterlinden AG, Wilson SG, Kung AW, Ralston SH. Meta-analysis of genome-wide scans provides evidence for sex- and site-specific regulation of bone mass. J Bone Miner Res 22: 173–183, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ireland A, Maden-Wilkinson T, McPhee J, Cooke K, Narici M, Degens H, Rittweger J. Upper limb muscle-bone asymmetries and bone adaptation in elite youth tennis players. Med Sci Sports Exerc 45: 1749–1758, 2013. [DOI] [PubMed] [Google Scholar]
- 223.Irvine RA, Ma H, Yu MC, Ross RK, Stallcup MR, Coetzee GA. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet 9: 267–274, 2000. [DOI] [PubMed] [Google Scholar]
- 224.Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, Ikeda K. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med 15: 259–266, 2009. [DOI] [PubMed] [Google Scholar]
- 225.Iuliano-Burns S, Hopper J, Seeman E. The age of puberty determines sexual dimorphism in bone structure: a male/female co-twin control study. J Clin Endocrinol Metab 94: 1638–1643, 2009. [DOI] [PubMed] [Google Scholar]
- 226.Iyer S, Kim HN, Ucer S, Han L, Warren A, Crawford JA, de Cabo R, Zhao H, Almeida M, Manolagas SC. From conditional ER alpha deletion mouse models to novel gene targets of the anti-resorptive effects of estrogens (Abstract). J Bone Miner Res S30, 2016. [Google Scholar]
- 227.Iyer S, Ambrogini E, Bartell SM, Han L, Roberson PK, Cabo R, Jilka RL, Weinstein RS, O'Brien CA, Manolagas SC, Almeida M. FoxOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest 123: 3404–3419, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Iyer S, Han L, Kim HN, Ucer SS, Bartell SM, Warren A, Crawford J, Skinner R, Dallas M, Johnson M, Weinstein R, Jilka R, O'Brien C, Almeida M, Manolagas S. ERalpha signaling in Osterix1 and Prx1 expressing cells, respectively, mediates the anabolic effect of mechanical loading in the murine periosteum and the protective effects of estrogens on endocortical resorption (Abstract). J Bone Miner Res 28: S5, 2013. [Google Scholar]
- 229.Iyer S, Ucer S, Kim HN, Han L, Warren A, Crawford J, Almeida M, Manolagas SC. Estrogens protect against endocortical bone resorption in both female and male mice; likely via an ERα-mediated suppression of SDF1/CXCL12 in uncommitted mesenchymal progenitors (Abstract). J Bone Miner Res 30: 148, 2015. [Google Scholar]
- 230.Jackowski SA, Erlandson MC, Mirwald RL, Faulkner RA, Bailey DA, Kontulainen SA, Cooper DM, Baxter-Jones AD. Effect of maturational timing on bone mineral content accrual from childhood to adulthood: evidence from 15 years of longitudinal data. Bone 48: 1178–1185, 2011. [DOI] [PubMed] [Google Scholar]
- 231.Jacobsen SJ, Cheetham TC, Haque R, Shi JM, Loo RK. Association between 5-alpha reductase inhibition and risk of hip fracture. JAMA 300: 1660–1664, 2008. [DOI] [PubMed] [Google Scholar]
- 232.Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res 21: 67–77, 2006. [DOI] [PubMed] [Google Scholar]
- 233.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45: 1–17, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jansson JO, Ekberg S, Isaksson O, Mode A, Gustafsson JA. Imprinting of growth hormone secretion, body growth, and hepatic steroid metabolism by neonatal testosterone. Endocrinology 117: 1881–1889, 1985. [DOI] [PubMed] [Google Scholar]
- 235.Jilka RL, Almeida M, Ambrogini E, Han L, Roberson PK, Weinstein RS, Manolagas SC. Decreased oxidative stress and greater bone anabolism in the aged, compared with the young, murine skeleton by parathyroid hormone. Aging Cell 9: 851–867, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jilka RL, O'Brien CA, Roberson PK, Bonewald LF, Weinstein RS, Manolagas SC. Dysapoptosis of osteoblasts and osteocytes increases cancellous bone formation but exaggerates cortical porosity with age. J Bone Miner Res 29: 103–117, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow: evidence for autonomy from factors released during bone resorption. J Clin Invest 101: 1942–1950, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Johansson H, Kanis JA, Oden A, McCloskey E, Chapurlat RD, Christiansen C, Cummings SR, Diez-Perez A, Eisman JA, Fujiwara S, Gluer CC, Goltzman D, Hans D, Khaw KT, Krieg MA, Kroger H, LaCroix AZ, Lau E, Leslie WD, Mellstrom D, Melton LJ III, O'Neill TW, Pasco JA, Prior JC, Reid DM, Rivadeneira F, van ST, Yoshimura N, Zillikens MC. A meta-analysis of the association of fracture risk and body mass index in women. J Bone Miner Res 29: 223–233, 2014. [DOI] [PubMed] [Google Scholar]
- 239.Jones HH, Priest JD, Hayes WC, Tichenor CC, Nagel DA. Humeral hypertrophy in response to exercise. J Bone Joint Surg Am 59: 204–208, 1977. [PubMed] [Google Scholar]
- 240.Kameda T, Mano H, Yuasa T, Mori Y, Miyazawa K, Shiokawa M, Nakamaru Y, Hiroi E, Hiura K, Kameda A, Yang NN, Hakeda Y, Kumegawa M. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J Exp Med 186: 489–495, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kamel-ElSayed SA, Tiede-Lewis LM, Lu Y, Veno PA, Dallas SL. Novel approaches for two and three dimensional multiplexed imaging of osteocytes. Bone 76: 129–140, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kanis JA, McCloskey EV. Risk factors in osteoporosis. Maturitas 30: 229–233, 1998. [DOI] [PubMed] [Google Scholar]
- 243.Kanis JA, Oden A, McCloskey EV, Johansson H, Wahl DA, Cooper C. A systematic review of hip fracture incidence and probability of fracture worldwide. Osteoporos Int 23: 2239–2256, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Karsenty G. The central regulation of bone remodeling. Trends Endocrinol Metab 11: 437–439, 2000. [DOI] [PubMed] [Google Scholar]
- 245.Kasperk C, Watt SM, Strong D, Mohan S, Jennings J, Wergedal J, Baylink D. Studies of the mechanism by which androgens enhance mitogenesis and differentiation in bone cells. J Clin Endocrinol Metab 71: 1322–1329, 1990. [DOI] [PubMed] [Google Scholar]
- 246.Kassanos D, Trakakis E, Baltas CS, Papakonstantinou O, Simeonidis G, Salamalekis G, Grammatikakis I, Basios G, Labos G, Skarantavos G, Balanika A. Augmentation of cortical bone mineral density in women with polycystic ovary syndrome: a peripheral quantitative computed tomography (pQCT) study. Hum Reprod 25: 2107–2114, 2010. [DOI] [PubMed] [Google Scholar]
- 247.Katavic V, Grcevic D, Lee SK, Kalinowski J, Jastrzebski S, Dougall W, Anderson D, Puddington L, Aguila HL, Lorenzo JA. The surface antigen CD45R identifies a population of estrogen-regulated murine marrow cells that contain osteoclast precursors. Bone 32: 581–590, 2003. [DOI] [PubMed] [Google Scholar]
- 248.Katulski K, Slawek S, Czyzyk A, Podfigurna-Stopa A, Paczkowska K, Ignaszak N, Podkowa N, Meczekalski B. Bone mineral density in women with polycystic ovary syndrome. J Endocrinol Invest 37: 1219–1224, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kawano H, Sato T, Yamada T, Matsumoto T, Sekine K, Watanabe T, Nakamura T, Fukuda T, Yoshimura K, Yoshizawa T, Aihara K, Yamamoto Y, Nakamichi Y, Metzger D, Chambon P, Nakamura K, Kawaguchi H, Kato S. Suppressive function of androgen receptor in bone resorption. Proc Natl Acad Sci USA 100: 9416–9421, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kawate H, Wu Y, Ohnaka K, Takayanagi R. Mutual transactivational repression of Runx2 and the androgen receptor by an impairment of their normal compartmentalization. J Steroid Biochem Mol Biol 105: 46–56, 2007. [DOI] [PubMed] [Google Scholar]
- 251.Kearbey JD, Gao W, Narayanan R, Fisher SJ, Wu D, Miller DD, Dalton JT. Selective androgen receptor modulator (SARM) treatment prevents bone loss and reduces body fat in ovariectomized rats. Pharm Res 24: 328–335, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Kelly A, Winer KK, Kalkwarf H, Oberfield SE, Lappe J, Gilsanz V, Zemel BS. Age-based reference ranges for annual height velocity in US children. J Clin Endocrinol Metab 99: 2104–2112, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 50: 1115–1122, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Khosla S. Update on estrogens and the skeleton. J Clin Endocrinol Metab 95: 3569–3577, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Khosla S. New insights into androgen and estrogen receptor regulation of the male skeleton. J Bone Miner Res 30: 1134–1137, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Khosla S, Melton LJ III, Atkinson EJ, O'Fallon WM. Relationship of serum sex steroid levels to longitudinal changes in bone density in young versus elderly men. J Clin Endocrinol Metab 86: 3555–3561, 2001. [DOI] [PubMed] [Google Scholar]
- 257.Khosla S, Melton LJ III, Atkinson EJ, O'Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid l evels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab 83: 2266–2274, 1998. [DOI] [PubMed] [Google Scholar]
- 258.Khosla S, Melton LJ III, Riggs BL. The unitary model for estrogen deficiency and the pathogenesis of osteoporosis: is a revision needed? J Bone Miner Res 26: 441–451, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Khosla S, Melton LJ III, Robb RA, Camp JJ, Atkinson EJ, Oberg AL, Rouleau PA, Riggs BL. Relationship of volumetric BMD and structural parameters at different skeletal sites to sex steroid levels in men. J Bone Miner Res 20: 730–740, 2005. [DOI] [PubMed] [Google Scholar]
- 260.Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab 23: 576–581, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Khosla S, Riggs BL, Atkinson EJ, Oberg AL, McDaniel LJ, Holets M, Peterson JM, Melton LJ III. Effects of sex and age on bone microstructure at the ultradistal radius: a population-based noninvasive in vivo assessment. J Bone Miner Res 21: 124–131, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Khosla S, Riggs BL, Robb RA, Camp JJ, Achenbach SJ, Oberg AL, Rouleau PA, Melton LJ III. Relationship of volumetric bone density and structural parameters at different skeletal sites to sex steroid levels in women. J Clin Endocrinol Metab 90: 5096–5103, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kim BT, Mosekilde L, Duan Y, Zhang XZ, Tornvig L, Thomsen JS, Seeman E. The structural and hormonal basis of sex differences in peak appendicular bone strength in rats. J Bone Miner Res 18: 150–155, 2003. [DOI] [PubMed] [Google Scholar]
- 264.Kim SH, Katzenellenbogen JA. Hormone-PAMAM dendrimer conjugates: polymer dynamics and tether structure affect ligand access to receptors. Angew Chem Int Ed Engl 45: 7243–7248, 2006. [DOI] [PubMed] [Google Scholar]
- 265.Kirmani S, Christen D, van Lenthe GH, Fischer PR, Bouxsein ML, McCready LK, Melton LJ III, Riggs BL, Amin S, Muller R, Khosla S. Bone structure at the distal radius during adolescent growth. J Bone Miner Res 24: 1033–1042, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kleerekoper M, Villanueva AR, Stanciu J, Rao DS, Parfitt AM. The role of three-dimensional trabecular microstructure in the pathogenesis of vertebral compression fractures. Calcif Tissue Int 37: 594–597, 1985. [DOI] [PubMed] [Google Scholar]
- 267.Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12: 657–664, 2006. [DOI] [PubMed] [Google Scholar]
- 268.Kondoh S, Inoue K, Igarashi K, Sugizaki H, Shirode-Fukuda Y, Inoue E, Yu T, Takeuchi JK, Kanno J, Bonewald LF, Imai Y. Estrogen receptor alpha in osteocytes regulates trabecular bone formation in female mice. Bone 60: 68–77, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kornreich L, Konen O, Schwarz M, Siegel Y, Horev G, Hershkovitz I, Laron Z. Abnormalities of the axial and proximal appendicular skeleton in adults with Laron syndrome (growth hormone insensitivity). Skeletal Radiol 37: 153–160, 2008. [DOI] [PubMed] [Google Scholar]
- 270.Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han K, DiGregorio G, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 104: 719–730, 2001. [PubMed] [Google Scholar]
- 271.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O'Brien C, Plotkin LI, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfit AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 298: 843–846, 2002. [DOI] [PubMed] [Google Scholar]
- 272.Kremer M, Judd J, Rifkin B, Auszmann J, Oursler MJ. Estrogen modulation of osteoclast lysosomal enzyme secretion. J Cell Biochem 57: 271–279, 1995. [DOI] [PubMed] [Google Scholar]
- 273.Kronenberg HM. Developmental regulation of the growth plate. Nature 423: 332–336, 2003. [DOI] [PubMed] [Google Scholar]
- 274.Krum SA. Direct transcriptional targets of sex steroid hormones in bone. J Cell Biochem 112: 401–408, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J 27: 535–545, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Krum SA, Miranda-Carboni GA, Lupien M, Eeckhoute J, Carroll JS, Brown M. Unique ERalpha cistromes control cell type-specific gene regulation. Mol Endocrinol 22: 2393–2406, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kuchuk NO, van Schoor NM, Pluijm SM, Smit JH, de RW, Lips P. The association of sex hormone levels with quantitative ultrasound, bone mineral density, bone turnover and osteoporotic fractures in older men and women. Clin Endocrinol 67: 295–303, 2007. [DOI] [PubMed] [Google Scholar]
- 278.Kumar MB, Tarpey RW, Perdew GH. Differential recruitment of coactivator RIP140 by Ah and estrogen receptors. Absence of a role for LXXLL motifs. J Biol Chem 274: 22155–22164, 1999. [DOI] [PubMed] [Google Scholar]
- 279.Lacasa D, Garcia E, Agli B, Giudicelli Y. Control of rat preadipocyte adipose conversion by ovarian status: regional specificity and possible involvement of the mitogen-activated protein kinase-dependent and c-fos signaling pathways. Endocrinology 138: 2729–2734, 1997. [DOI] [PubMed] [Google Scholar]
- 280.Lacey DL, Tan HL, Lu J, Kaufman S, Van G, Qiu W, Rattan A, Scully S, Fletcher F, Juan T, Kelley M, Burgess TL, Boyle WJ, Polverino AJ. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol 157: 435–448, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93: 165–176, 1998. [DOI] [PubMed] [Google Scholar]
- 282.Lambertini E, Tavanti E, Torreggiani E, Penolazzi L, Gambari R, Piva R. ERalpha and AP-1 interact in vivo with a specific sequence of the F promoter of the human ERalpha gene in osteoblasts. J Cell Physiol 216: 101–110, 2008. [DOI] [PubMed] [Google Scholar]
- 283.Lanfranco F, Zirilli L, Baldi M, Pignatti E, Corneli G, Ghigo E, Aimaretti G, Carani C, Rochira V. A novel mutation in the human aromatase gene: insights on the relationship among serum estradiol, longitudinal growth and bone mineral density in an adult man under estrogen replacement treatment. Bone 43: 628–635, 2008. [DOI] [PubMed] [Google Scholar]
- 284.Lang T, LeBlanc A, Evans H, Lu Y, Genant H, Yu A. Cortical and trabecular bone mineral loss from the spine and hip in long-duration spaceflight. J Bone Miner Res 19: 1006–1012, 2004. [DOI] [PubMed] [Google Scholar]
- 285.Langley RE, Cafferty FH, Alhasso AA, Rosen SD, Sundaram SK, Freeman SC, Pollock P, Jinks RC, Godsland IF, Kockelbergh R, Clarke NW, Kynaston HG, Parmar MK, Abel PD. Cardiovascular outcomes in patients with locally advanced and metastatic prostate cancer treated with luteinising-hormone-releasing-hormone agonists or transdermal oestrogen: the randomised, phase 2 MRC PATCH trial (PR09). Lancet Oncol 14: 306–316, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lanyon L, Skerry T. Postmenopausal osteoporosis as a failure of bone's adaptation to functional loading: a hypothesis. J Bone Miner Res 16: 1937–1947, 2001. [DOI] [PubMed] [Google Scholar]
- 287.Lapauw B, T'Sjoen G, Mahmoud A, Kaufman JM, Ruige JB. Short-term aromatase inhibition: effects on glucose metabolism and serum leptin levels in young and elderly men. Eur J Endocrinol 160: 397–402, 2009. [DOI] [PubMed] [Google Scholar]
- 288.Lapauw B, Taes Y, Goemaere S, Toye K, Zmierczak HG, Kaufman JM. Anthropometric and skeletal phenotype in men with idiopathic osteoporosis and their sons is consistent with deficient estrogen action during maturation. J Clin Endocrinol Metab 94: 4300–4308, 2009. [DOI] [PubMed] [Google Scholar]
- 289.Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958–2003. J Clin Endocrinol Metab 89: 1031–1044, 2004. [DOI] [PubMed] [Google Scholar]
- 290.Laron Z, Lilos P, Klinger B. Growth curves for Laron syndrome. Arch Dis Child 68: 768–770, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Laurent M, Gielen E, Claessens F, Boonen S, Vanderschueren D. Osteoporosis in older men: recent advances in pathophysiology and treatment. Best Pract Res Clin Endocrinol Metab 27: 527–539, 2013. [DOI] [PubMed] [Google Scholar]
- 292.Laurent MR, Dubois V, Claessens F, Verschueren SM, Vanderschueren D, Gielen E, Jardi F. Muscle-bone interactions: from experimental models to the clinic? A critical update. Mol Cell Endocrinol 432: 14–36, 2016. [DOI] [PubMed] [Google Scholar]
- 293.Laurent MR, Vanderschueren D. Reproductive endocrinology: functional effects of sex hormone-binding globulin variants. Nat Rev Endocrinol 10: 516–517, 2014. [DOI] [PubMed] [Google Scholar]
- 294.Lauretani F, Bandinelli S, Griswold ME, Maggio M, Semba R, Guralnik JM, Ferrucci L. Longitudinal changes in BMD and bone geometry in a population-based study. J Bone Miner Res 23: 400–408, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, Urry ZL, Chambers TJ. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest 112: 915–923, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.LeBlanc AD, Schneider VS, Evans HJ, Engelbretson DA, Krebs JM. Bone mineral loss and recovery after 17 weeks of bed rest. J Bone Miner Res 5: 843–850, 1990. [DOI] [PubMed] [Google Scholar]
- 297.LeBlanc ES, Nielson CM, Marshall LM, Lapidus JA, Barrett-Connor E, Ensrud KE, Hoffman AR, Laughlin G, Ohlsson C, Orwoll ES. The effects of serum testosterone, estradiol, and sex hormone binding globulin levels on fracture risk in older men. J Clin Endocrinol Metab 94: 3337–3346, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Leder BZ, Finkelstein JS. Effect of aromatase inhibition on bone metabolism in elderly hypogonadal men. Osteoporos Int 16: 1487–1494, 2005. [DOI] [PubMed] [Google Scholar]
- 299.Leder BZ, LeBlanc KM, Schoenfeld DA, Eastell R, Finkelstein JS. Differential effects of androgens and estrogens on bone turnover in normal men. J Clin Endocrinol Metab 88: 204–210, 2003. [DOI] [PubMed] [Google Scholar]
- 300.Lee JS, LaCroix AZ, Wu L, Cauley JA, Jackson RD, Kooperberg C, Leboff MS, Robbins J, Lewis CE, Bauer DC, Cummings SR. Associations of serum sex hormone-binding globulin and sex hormone concentrations with hip fracture risk in postmenopausal women. J Clin Endocrinol Metab 93: 1796–1803, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Lee K, Jessop H, Suswillo R, Zaman G, Lanyon L. Endocrinology: bone adaptation requires oestrogen receptor-alpha. Nature 424: 389, 2003. [DOI] [PubMed] [Google Scholar]
- 302.Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 106: 852–859, 2005. [DOI] [PubMed] [Google Scholar]
- 303.Lee SK, Kadono Y, Okada F, Jacquin C, Koczon-Jaremko B, Gronowicz G, Adams DJ, Aguila HL, Choi Y, Lorenzo JA. T lymphocyte-deficient mice lose trabecular bone mass with ovariectomy. J Bone Miner Res 21: 1704–1712, 2006. [DOI] [PubMed] [Google Scholar]
- 304.Lee SK, Kalinowski JF, Jacquin C, Adams DJ, Gronowicz G, Lorenzo JA. Interleukin-7 influences osteoclast function in vivo but is not a critical factor in ovariectomy-induced bone loss. J Bone Miner Res 21: 695–702, 2006. [DOI] [PubMed] [Google Scholar]
- 305.Legrand E, Hedde C, Gallois Y, Degasne I, Boux de CF, Mathieu E, Basle MF, Chappard D, Audran M. Osteoporosis in men: a potential role for the sex hormone binding globulin. Bone 29: 90–95, 2001. [DOI] [PubMed] [Google Scholar]
- 306.Leifke E, Korner HC, Link TM, Behre HM, Peters PE, Nieschlag E. Effects of testosterone replacement therapy on cortical and trabecular bone mineral density, vertebral body area and paraspinal muscle area in hypogonadal men. Eur J Endocrinol 138: 51–58, 1998. [DOI] [PubMed] [Google Scholar]
- 307.Li JY, Tawfeek H, Bedi B, Yang X, Adams J, Gao KY, Zayzafoon M, Weitzmann MN, Pacifici R. Ovariectomy disregulates osteoblast and osteoclast formation through the T-cell receptor CD40 ligand. Proc Natl Acad Sci USA 108: 768–773, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Li X, Masinde G, Gu W, Wergedal J, Mohan S, Baylink DJ. Genetic dissection of femur breaking strength in a large population (MRL/MpJ × SJL/J) of F2 Mice: single QTL effects, epistasis, and pleiotropy. Genomics 79: 734–740, 2002. [DOI] [PubMed] [Google Scholar]
- 309.Li X, Ominsky MS, Stolina M, Warmington KS, Geng Z, Niu QT, Asuncion FJ, Tan HL, Grisanti M, Dwyer D, Adamu S, Ke HZ, Simonet WS, Kostenuik PJ. Increased RANK ligand in bone marrow of orchiectomized rats and prevention of their bone loss by the RANK ligand inhibitor osteoprotegerin. Bone 45: 669–676, 2009. [DOI] [PubMed] [Google Scholar]
- 310.Li Y, Li A, Yang X, Weitzmann MN. Ovariectomy-induced bone loss occurs independently of B cells. J Cell Biochem 100: 1370–1375, 2007. [DOI] [PubMed] [Google Scholar]
- 311.Lippuner K, Johansson H, Kanis JA, Rizzoli R. Remaining lifetime and absolute 10-year probabilities of osteoporotic fracture in Swiss men and women. Osteoporos Int 20: 1131–1140, 2009. [DOI] [PubMed] [Google Scholar]
- 312.Liu CT, Estrada K, Yerges-Armstrong LM, Amin N, Evangelou E, Li G, Minster RL, Carless MA, Kammerer CM, Oei L, Zhou Y, Alonso N, Dailiana Z, Eriksson J, Garcia-Giralt N, Giroux S, Husted LB, Khusainova RI, Koromila T, Kung AW, Lewis JR, Masi L, Mencej-Bedrac S, Nogues X, Patel MS, Prezelj J, Richards JB, Sham PC, Spector T, Vandenput L, Xiao SM, Zheng HF, Zhu K, Balcells S, Brandi ML, Frost M, Goltzman D, Gonzalez-Macias J, Karlsson M, Khusnutdinova EK, Kollia P, Langdahl BL, Ljunggren O, Lorentzon M, Marc J, Mellstrom D, Ohlsson C, Olmos JM, Ralston SH, Riancho JA, Rousseau F, Urreizti R, Van HW, Zarrabeitia MT, Castano-Betancourt M, Demissie S, Grundberg E, Herrera L, Kwan T, Medina-Gomez C, Pastinen T, Sigurdsson G, Thorleifsson G, Vanmeurs JB, Blangero J, Hofman A, Liu Y, Mitchell BD, O'Connell JR, Oostra BA, Rotter JI, Stefansson K, Streeten EA, Styrkarsdottir U, Thorsteinsdottir U, Tylavsky FA, Uitterlinden A, Cauley JA, Harris TB, Ioannidis JP, Psaty BM, Robbins JA, Zillikens MC, Vanduijn CM, Prince RL, Karasik D, Rivadeneira F, Kiel DP, Cupples LA, Hsu YH. Assessment of gene-by-sex interaction effect on bone mineral density. J Bone Miner Res 27: 2051–2064, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Liu M, Goss PE, Ingle JN, Kubo M, Furukawa Y, Batzler A, Jenkins GD, Carlson EE, Nakamura Y, Schaid DJ, Chapman JA, Shepherd LE, Ellis MJ, Khosla S, Wang L, Weinshilboum RM. Aromatase inhibitor-associated bone fractures: a case-cohort GWAS and functional genomics. Mol Endocrinol 28: 1740–1751, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol 13: 27–38, 2012. [DOI] [PubMed] [Google Scholar]
- 315.Looker AC, Wahner HW, Dunn WL, Calvo MS, Harris TB, Heyse SP, Johnston CC Jr, Lindsay R. Updated data on proximal femur bone mineral levels of US adults. Osteoporos Int 8: 468–489, 1998. [DOI] [PubMed] [Google Scholar]
- 316.Lorentzon M, Mellstrom D, Ohlsson C. Age of attainment of peak bone mass is site specific in Swedish men–The GOOD study. J Bone Miner Res 20: 1223–1227, 2005. [DOI] [PubMed] [Google Scholar]
- 317.Lormeau C, Soudan B, d'Herbomez M, Pigny P, Duquesnoy B, Cortet B. Sex hormone-binding globulin, estradiol, and bone turnover markers in male osteoporosis. Bone 34: 933–939, 2004. [DOI] [PubMed] [Google Scholar]
- 318.Maatta JA, Buki KG, Gu G, Alanne MH, Vaaraniemi J, Liljenback H, Poutanen M, Harkonen P, Vaananen K. Inactivation of estrogen receptor alpha in bone-forming cells induces bone loss in female mice. FASEB J 27: 478–488, 2013. [DOI] [PubMed] [Google Scholar]
- 319.Maatta JA, Buki KG, Ivaska KK, Nieminen-Pihala V, Elo TD, Kahkonen T, Poutanen M, Harkonen P, Vaananen K. Inactivation of the androgen receptor in bone-forming cells leads to trabecular bone loss in adult female mice. Bonekey Rep 2: 440, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Macdonald HM, Nishiyama KK, Kang J, Hanley DA, Boyd SK. Age-related patterns of trabecular and cortical bone loss differ between sexes and skeletal sites: a population-based HR-pQCT study. J Bone Miner Res 26: 50–62, 2011. [DOI] [PubMed] [Google Scholar]
- 321.Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, Kikuchi Y, Takada I, Kato S, Kani S, Nishita M, Marumo K, Martin TJ, Minami Y, Takahashi N. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med 18: 405–412, 2012. [DOI] [PubMed] [Google Scholar]
- 322.Maheshwari HG, Bouillon R, Nijs J, Oganov VS, Bakulin AV, Baumann G. The Impact of congenital, severe, untreated growth hormone (GH) deficiency on bone size and density in young adults: insights from genetic GH-releasing hormone receptor deficiency. J Clin Endocrinol Metab 88: 2614–2618, 2003. [DOI] [PubMed] [Google Scholar]
- 323.Mano H, Yuasa T, Kameda T, Miyazawa K, Nakamaru Y, Shiokawa M, Mori Y, Yamada T, Miyata K, Shindo H, Azuma H, Hakeda Y, Kumegawa M. Mammalian mature osteoclasts as estrogen target cells. Biochem Biophys Res Commun 223: 637–642, 1996. [DOI] [PubMed] [Google Scholar]
- 324.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21: 115–137, 2000. [DOI] [PubMed] [Google Scholar]
- 325.Manolagas SC. Perspective: Choreography from the tomb: an emerging role of dying osteocytes in the purposeful, and perhaps not so purposeful, targeting of bone remodeling. BoneKey-Osteovision 3: 5–14, doi: 10.1138/20060193, 2006. [DOI] [Google Scholar]
- 326.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31: 266–300, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Manolagas SC, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol 21: 2605–2614, 2007. [DOI] [PubMed] [Google Scholar]
- 328.Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling: emerging insights into the pathophysiology of osteoporosis. N Engl J Med 332: 305–311, 1995. [DOI] [PubMed] [Google Scholar]
- 329.Manolagas SC, Jilka RL, Kousteni S, Bellido T, Weinstein RS, O'Brien CA, Plotkin L, Han L. Response to Windahl et al. J Clin Invest 116: 2834, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Manolagas SC, Kousteni S. Perspective: nonreproductive sites of action of reproductive hormones. Endocrinology 142: 2200–2204, 2001. [DOI] [PubMed] [Google Scholar]
- 331.Manolagas SC, Kousteni S, Chen JR, Schuller M, Plotkin L, Bellido T. Kinase-mediated transcription, activators of nongenotropic estrogen-like signaling (ANGELS), and osteoporosis: a different perspective on the HRT dilemma. Kidney Int Suppl 91: S41–S49, 2004. [DOI] [PubMed] [Google Scholar]
- 332.Manolagas SC, Kousteni S, Jilka RL. Sex steroids and bone. Recent Prog Horm Res 57: 385–409, 2002. [DOI] [PubMed] [Google Scholar]
- 333.Manolagas SC, O'Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol 9: 699–712, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab 21: 369–374, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Manolagas SC, Parfitt AM. For whom the bell tolls: Distress signals from long-lived osteocytes and the pathogenesis of metabolic bone diseases. Bone 54: 272–278, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Manson JE, Chlebowski RT, Stefanick ML, Aragaki AK, Rossouw JE, Prentice RL, Anderson G, Howard BV, Thomson CA, LaCroix AZ, Wactawski-Wende J, Jackson RD, Limacher M, Margolis KL, Wassertheil-Smoller S, Beresford SA, Cauley JA, Eaton CB, Gass M, Hsia J, Johnson KC, Kooperberg C, Kuller LH, Lewis CE, Liu S, Martin LW, Ockene JK, O'Sullivan MJ, Powell LH, Simon MS, Van HL, Vitolins MZ, Wallace RB. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA 310: 1353–1368, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Marcus R, Leary D, Schneider DL, Shane E, Favus M, Quigley CA. The contribution of testosterone to skeletal development and maintenance: lessons from the androgen insensitivity syndrome. J Clin Endocrinol Metab 85: 1032–1037, 2000. [DOI] [PubMed] [Google Scholar]
- 338.Marjoribanks J, Farquhar C, Roberts H, Lethaby A. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev 7: CD004143, 2012. [DOI] [PubMed] [Google Scholar]
- 339.Martin-Millan M, Almeida M, Ambrogini E, Han L, Zhao H, Weinstein RS, Jilka RL, O'Brien C, Manolagas SC. The estrogen receptor alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol 24: 323–334, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Martin-Millan M, Almeida M, Ambrogini E, Qui X, Warren A, Shelton R, Weinstein RS, Jilka RL, Bellido T, O'Brien CA, Manolagas SC. ERα deletion in cells of the monocyte/macrophage lineage increases osteoclastogenesis and abrogates the pro-apoptotic effect of E2 on osteoclasts (Abstract). J Bone Miner Res 23: S1, 2008. [Google Scholar]
- 341.Martinborough E, Shen Y, Oeveren A, Long YO, Lau TL, Marschke KB, Chang WY, Lopez FJ, Vajda EG, Rix PJ, Viveros OH, Negro-Vilar A, Zhi L. Substituted 6-(1-pyrrolidine)quinolin-2(1H)-ones as novel selective androgen receptor modulators. J Med Chem 50: 5049–5052, 2007. [DOI] [PubMed] [Google Scholar]
- 342.Masuzawa T, Miyaura C, Onoe Y, Kusano K, Ohta H, Nozawa S, Suda T. Estrogen deficiency stimulates B lymphopoiesis in mouse bone marrow. J Clin Invest 94: 1090–1097, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Matsumoto AM, Tenover L, McClung M, Mobley D, Geller J, Sullivan M, Grayhack J, Wessells H, Kadmon D, Flanagan M, Zhang GK, Schmidt J, Taylor AM, Lee M, Waldstreicher J. The long-term effect of specific type II 5alpha-reductase inhibition with finasteride on bone mineral density in men: results of a 4-year placebo controlled trial. J Urol 167: 2105–2108, 2002. [PubMed] [Google Scholar]
- 344.McInnes KJ, Smith LB, Hunger NI, Saunders PT, Andrew R, Walker BR. Deletion of the androgen receptor in adipose tissue in male mice elevates retinol binding protein 4 and reveals independent effects on visceral fat mass and on glucose homeostasis. Diabetes 61: 1072–1081, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Meakin LB, Price JS, Lanyon LE. The contribution of experimental in vivo models to understanding the mechanisms of adaptation to mechanical loading in bone. Front Endocrinol 5: 154, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Meier C, Nguyen TV, Handelsman DJ, Schindler C, Kushnir MM, Rockwood AL, Meikle AW, Center JR, Eisman JA, Seibel MJ. Endogenous sex hormones and incident fracture risk in older men: the Dubbo Osteoporosis Epidemiology Study. Arch Intern Med 168: 47–54, 2008. [DOI] [PubMed] [Google Scholar]
- 347.Mellstrom D, Vandenput L, Mallmin H, Holmberg AH, Lorentzon M, Oden A, Johansson H, Orwoll ES, Labrie F, Karlsson MK, Ljunggren O, Ohlsson C. Older men with low serum estradiol and high serum SHBG have an increased risk of fractures. J Bone Miner Res 23: 1552–1560, 2008. [DOI] [PubMed] [Google Scholar]
- 348.Melville KM, Kelly NH, Khan SA, Schimenti JC, Ross FP, Main RP, van der Meulen MC. Female mice lacking estrogen receptor-alpha in osteoblasts have compromised bone mass and strength. J Bone Miner Res 29: 370–379, 2014. [DOI] [PubMed] [Google Scholar]
- 349.Meunier PJ, Sellami S, Briancon D, Edouard C. Histologial heterogeneity of apparently idiopathic osteoporosis. In: Osteoporosis: Recent Advances in Pathogenesis and Treatment, edited by DeLuca HF. Baltimore, MD: University Park Press, 1981. [Google Scholar]
- 350.Miki Y, Suzuki T, Hatori M, Igarashi K, Aisaki KI, Kanno J, Nakamura Y, Uzuki M, Sawai T, Sasano H. Effects of aromatase inhibitors on human osteoblast and osteoblast-like cells: a possible androgenic bone protective effects induced by exemestane. Bone 40: 876–887, 2007. [DOI] [PubMed] [Google Scholar]
- 351.Millard WJ, Politch JA, Martin JB, Fox TO. Growth hormone-secretory patterns in androgen-resistant (testicular feminized) rats. Endocrinology 119: 2655–2660, 1986. [DOI] [PubMed] [Google Scholar]
- 352.Miner JN, Chang W, Chapman MS, Finn PD, Hong MH, Lopez FJ, Marschke KB, Rosen J, Schrader W, Turner R, van OA, Viveros H, Zhi L, Negro-Vilar A. An orally active selective androgen receptor modulator is efficacious on bone, muscle, and sex function with reduced impact on prostate. Endocrinology 148: 363–373, 2007. [DOI] [PubMed] [Google Scholar]
- 353.Miranda-Carboni GA, Guemes M, Bailey S, Anaya E, Corselli M, Peault B, Krum SA. GATA4 regulates estrogen receptor-alpha-mediated osteoblast transcription. Mol Endocrinol 25: 1126–1136, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, Suda T. Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci USA 94: 9360–9365, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Modder UI, Riggs BL, Spelsberg TC, Fraser DG, Atkinson EJ, Arnold R, Khosla S. Dose-response of estrogen on bone versus the uterus in ovariectomized mice. Eur J Endocrinol 151: 503–510, 2004. [DOI] [PubMed] [Google Scholar]
- 356.Modder UI, Sanyal A, Kearns AE, Sibonga JD, Nishihara E, Xu J, O'Malley BW, Ritman EL, Riggs BL, Spelsberg TC, Khosla S. Effects of loss of steroid receptor coactivator-1 on the skeletal response to estrogen in mice. Endocrinology 145: 913–921, 2004. [DOI] [PubMed] [Google Scholar]
- 357.Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, Miller DD. Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J Med Chem 52: 3597–3617, 2009. [DOI] [PubMed] [Google Scholar]
- 358.Monroe DG, Secreto FJ, Hawse JR, Subramaniam M, Khosla S, Spelsberg TC. Estrogen receptor isoform-specific regulation of the retinoblastoma-binding protein 1 (RBBP1) gene: roles of AF1 and enhancer elements. J Biol Chem 281: 28596–28604, 2006. [DOI] [PubMed] [Google Scholar]
- 359.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol 10: 384–391, 1998. [DOI] [PubMed] [Google Scholar]
- 360.Moriishi T, Maruyama Z, Fukuyama R, Ito M, Miyazaki T, Kitaura H, Ohnishi H, Furuichi T, Kawai Y, Masuyama R, Komori H, Takada K, Kawaguchi H, Komori T. Overexpression of Bcl2 in osteoblasts inhibits osteoblast differentiation and induces osteocyte apoptosis. PLoS One 6: e27487, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab 80: 3689–3698, 1995. [DOI] [PubMed] [Google Scholar]
- 362.Moverare S, Venken K, Eriksson AL, Andersson N, Skrtic S, Wergedal J, Mohan S, Salmon P, Bouillon R, Gustafsson JA, Vanderschueren D, Ohlsson C. Differential effects on bone of estrogen receptor alpha and androgen receptor activation in orchidectomized adult male mice. Proc Natl Acad Sci USA 100: 13573–13578, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Moverare-Skrtic S, Borjesson AE, Farman HH, Sjogren K, Windahl SH, Lagerquist MK, Andersson A, Stubelius A, Carlsten H, Gustafsson JA, Ohlsson C. The estrogen receptor antagonist ICI 182,780 can act both as an agonist and an inverse agonist when estrogen receptor alpha AF-2 is modified. Proc Natl Acad Sci USA 111: 1180–1185, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Moverare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Borjesson AE, Sjogren K, Windahl SH, Farman H, Kindlund B, Engdahl C, Koskela A, Zhang FP, Eriksson EE, Zaman F, Hammarstedt A, Isaksson H, Bally M, Kassem A, Lindholm C, Sandberg O, Aspenberg P, Savendahl L, Feng JQ, Tuckermann J, Tuukkanen J, Poutanen M, Baron R, Lerner UH, Gori F, Ohlsson C. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med 20: 1279–1288, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130: 811–823, 2007. [DOI] [PubMed] [Google Scholar]
- 366.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17: 1231–1234, 2011. [DOI] [PubMed] [Google Scholar]
- 367.Narayanan R, Mohler ML, Bohl CE, Miller DD, Dalton JT. Selective androgen receptor modulators in preclinical and clinical development. Nucl Recept Signal 6: e010, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344: 1434–1441, 2001. [DOI] [PubMed] [Google Scholar]
- 369.Negro-Vilar A. Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium. J Clin Endocrinol Metab 84: 3459–3462, 1999. [DOI] [PubMed] [Google Scholar]
- 370.Nelson AE, Meinhardt U, Hansen JL, Walker IH, Stone G, Howe CJ, Leung KC, Seibel MJ, Baxter RC, Handelsman DJ, Kazlauskas R, Ho KK. Pharmacodynamics of growth hormone abuse biomarkers and the influence of gender and testosterone: a randomized double-blind placebo-controlled study in young recreational athletes. J Clin Endocrinol Metab 93: 2213–2222, 2008. [DOI] [PubMed] [Google Scholar]
- 371.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112: 481–490, 2003. [DOI] [PubMed] [Google Scholar]
- 372.Nguyen ND, Ahlborg HG, Center JR, Eisman JA, Nguyen TV. Residual lifetime risk of fractures in women and men. J Bone Miner Res 22: 781–788, 2007. [DOI] [PubMed] [Google Scholar]
- 373.Nicks KM, Amin S, Atkinson EJ, Riggs BL, Melton LJ III, Khosla S. Relationship of age to bone microstructure independent of areal bone mineral density. J Bone Miner Res 27: 637–644, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Nicks KM, Fujita K, Fraser D, McGregor U, Drake MT, McGee-Lawrence ME, Westendorf JJ, Monroe DG, Khosla S. Deletion of estrogen receptor beta in osteoprogenitor cells increases trabecular but not cortical bone mass in female mice. J Bone Miner Res 31: 606–614, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Nishiyama KK, Macdonald HM, Moore SA, Fung T, Boyd SK, McKay HA. Cortical porosity is higher in boys compared with girls at the distal radius and distal tibia during pubertal growth: an HR-pQCT study. J Bone Miner Res 27: 273–282, 2012. [DOI] [PubMed] [Google Scholar]
- 376.Notini AJ, McManus JF, Moore A, Bouxsein M, Jimenez M, Chiu WS, Glatt V, Kream BE, Handelsman DJ, Morris HA, Zajac JD, Davey RA. Osteoblast deletion of exon 3 of the androgen receptor gene results in trabecular bone loss in adult male mice. J Bone Miner Res 22: 347–356, 2007. [DOI] [PubMed] [Google Scholar]
- 377.O'Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone 54: 258–263, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.O'Malley BW, Malovannaya A, Qin J. Minireview: nuclear receptor and coregulator proteomics–2012 and beyond. Mol Endocrinol 26: 1646–1650, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Ogden CL, Kuczmarski RJ, Flegal KM, Mei Z, Guo S, Wei R, Grummer-Strawn LM, Curtin LR, Roche AF, Johnson CL. Centers for Disease Control and Prevention 2000 growth charts for the United States: improvements to the 1977 National Center for Health Statistics version. Pediatrics 109: 45–60, 2002. [DOI] [PubMed] [Google Scholar]
- 380.Ohlsson C, Darelid A, Nilsson M, Melin J, Mellstrom D, Lorentzon M. Cortical consolidation due to increased mineralization and endosteal contraction in young adult men: a five-year longitudinal study. J Clin Endocrinol Metab 96: 2262–2269, 2011. [DOI] [PubMed] [Google Scholar]
- 381.Ohlsson C, Engdahl C, Borjesson AE, Windahl SH, Studer E, Westberg L, Eriksson E, Koskela A, Tuukkanen J, Krust A, Chambon P, Carlsten H, Lagerquist MK. Estrogen receptor-alpha expression in neuronal cells affects bone mass. Proc Natl Acad Sci USA 109: 983–988, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Ohlsson C, Mellstrom D, Carlzon D, Orwoll E, Ljunggren O, Karlsson MK, Vandenput L. Older men with low serum IGF-1 have an increased risk of incident fractures: the MrOS Sweden study. J Bone Miner Res 26: 865–872, 2011. [DOI] [PubMed] [Google Scholar]
- 383.Ohlsson C, Nilsson ME, Tivesten A, Ryberg H, Mellstrom D, Karlsson MK, Ljunggren O, Labrie F, Orwoll ES, Lee DM, Pye SR, O'Neill TW, Finn JD, Adams JE, Ward KA, Boonen S, Bartfai G, Casanueva FF, Forti G, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Lean ME, Pendleton N, Punab M, Vanderschueren D, Wu FC, Vandenput L. Comparisons of immunoassay and mass spectrometry measurements of serum estradiol levels and their influence on clinical association studies in men. J Clin Endocrinol Metab 98: E1097–E1102, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, Komatsu M, Selig M, Weinstein RS, Zhao H, Jilka RL, Almeida M, Manolagas SC, O'Brien CA. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem 288: 17432–17440, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Onal M, Xiong J, Chen X, Thostenson JD, Almeida M, Manolagas SC, O'Brien CA. Receptor activator of nuclear factor kappaB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem 287: 29851–29860, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270: 1354–1357, 1995. [DOI] [PubMed] [Google Scholar]
- 387.Onoe Y, Miyaura C, Ito M, Ohta H, Nozawa S, Suda T. Comparative effects of estrogen and raloxifene on B lymphopoiesis and bone loss induced by sex steroid deficiency in mice. J Bone Miner Res 15: 541–549, 2000. [DOI] [PubMed] [Google Scholar]
- 388.Ophoff J, Callewaert F, Venken K, De GK, Ohlsson C, Gayan-Ramirez G, Decramer M, Boonen S, Bouillon R, Verhoeven G, Vanderschueren D. Physical activity in the androgen receptor knockout mouse: evidence for reversal of androgen deficiency on cancellous bone. Biochem Biophys Res Commun 378: 139–144, 2009. [DOI] [PubMed] [Google Scholar]
- 389.Ophoff J, Van PK, Callewaert F, De GK, De BK, Vanden Bosch A, Verhoeven G, Hespel P, Vanderschueren D. Androgen signaling in myocytes contributes to the maintenance of muscle mass and fiber type regulation but not to muscle strength or fatigue. Endocrinology 150: 3558–3566, 2009. [DOI] [PubMed] [Google Scholar]
- 390.Orwoll ES. Toward an expanded understanding of the role of the periosteum in skeletal health. J Bone Miner Res 18: 949–954, 2003. [DOI] [PubMed] [Google Scholar]
- 391.Ottesen AM, Aksglaede L, Garn I, Tartaglia N, Tassone F, Gravholt CH, Bojesen A, Sorensen K, Jorgensen N, Rajpert-De ME, Gerdes T, Lind AM, Kjaergaard S, Juul A. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. Am J Med Genet A 152A: 1206–1212, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Otto C, Fuchs I, Altmann H, Klewer M, Schwarz G, Bohlmann R, Nguyen D, Zorn L, Vonk R, Prelle K, Osterman T, Malmstrom C, Fritzemeier KH. In vivo characterization of estrogen receptor modulators with reduced genomic versus nongenomic activity in vitro. J Steroid Biochem Mol Biol 111: 95–100, 2008. [DOI] [PubMed] [Google Scholar]
- 393.Oursler MJ, Osdoby P, Pyfferoen J, Riggs BL, Spelsberg TC. Avian osteoclasts as estrogen target cells. Proc Natl Acad Sci USA 88: 6613–6617, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Oz OK, Hirasawa G, Lawson J, Nanu L, Constantinescu A, Antich PP, Mason RP, Tsyganov E, Parkey RW, Zerwekh JE, Simpson ER. Bone phenotype of the aromatase deficient mouse. J Steroid Biochem Mol Biol 79: 49–59, 2001. [DOI] [PubMed] [Google Scholar]
- 395.Pacifici R. Role of T cells in ovariectomy induced bone loss–revisited. J Bone Miner Res 27: 231–239, 2012. [DOI] [PubMed] [Google Scholar]
- 396.Page ST, Marck BT, Tolliver JM, Matsumoto AM. Tissue selectivity of the anabolic steroid, 19-nor-4-androstenediol-3 beta,17 beta-diol in male Sprague Dawley rats: Selective stimulation of muscle mass and bone mineral density relative to prostate mass. Endocrinology 149: 1987–1993, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Papanicolaou DA, Ather SN, Zhu H, Zhou Y, Lutkiewicz J, Scott BB, Chandler J. A phase IIA randomized, placebo-controlled clinical trial to study the efficacy and safety of the selective androgen receptor modulator (SARM), MK-0773 in female participants with sarcopenia. J Nutr Health Aging 17: 533–543, 2013. [DOI] [PubMed] [Google Scholar]
- 398.Parfitt AM. Skeletal heterogeneity and the purposes of bone remodeling: implications for the understanding of osteoporosis. In: Osteoporosis, edited by Marcus R, Feldman D, Nelson D, Rosen C. San Diego, CA: Elsevier, 2007. [Google Scholar]
- 399.Parfitt AM. The coupling of bone formation to bone resorption: a critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab Bone Dis Relat Res 4: 1–6, 1982. [DOI] [PubMed] [Google Scholar]
- 400.Parfitt AM. Osteoporosis: 50 years of change, mostly in the right direction. In: Osteoporosis and Bone Biology: The State of the Art, edited by Compston RS. London: International Medical Press, 2000. [Google Scholar]
- 401.Parfitt AM. Misconceptions 2: turnover is always higher in cancellous than in cortical bone. Bone 30: 807–809, 2002. [DOI] [PubMed] [Google Scholar]
- 402.Parfitt AM. Misconceptions V: activation of osteoclasts is the first step in the bone remodeling cycle. Bone 39: 1170–1172, 2006. [DOI] [PubMed] [Google Scholar]
- 403.Parfitt AM, Mundy GR, Roodman GD, Hughes DE, Boyce BF. A new model for the regulation of bone resorption, with particular reference to the effects of bisphosphonates. J Bone Miner Res 11: 150–159, 1996. [DOI] [PubMed] [Google Scholar]
- 404.Parfitt AM, Villanueva AR, Foldes J, Rao DS. Relations between histologic indices of bone formation: implications for the pathogenesis of spinal osteoporosis. J Bone Miner Res 10: 466–473, 1995. [DOI] [PubMed] [Google Scholar]
- 405.Parfitt M, Qiu S, Palnitkar S, Rao DS. Abnormal bone remodeling in patients with spontaneous painful vertebral fracture. J Bone Miner Res 26: 475–485, 2011. [DOI] [PubMed] [Google Scholar]
- 406.Paternoster L, Lorentzon M, Lehtimaki T, Eriksson J, Kahonen M, Raitakari O, Laaksonen M, Sievanen H, Viikari J, Lyytikainen LP, Mellstrom D, Karlsson M, Ljunggren O, Grundberg E, Kemp JP, Sayers A, Nethander M, Evans DM, Vandenput L, Tobias JH, Ohlsson C. Genetic determinants of trabecular and cortical volumetric bone mineral densities and bone microstructure. PLoS Genet 9: e1003247, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Pead MJ, Skerry TM, Lanyon LE. Direct transformation from quiescence to bone formation in the adult periosteum following a single brief period of bone loading. J Bone Miner Res 3: 647–656, 1988. [DOI] [PubMed] [Google Scholar]
- 408.Pederson L, Kremer M, Judd J, Pascoe D, Spelsberg TC, Riggs BL, Oursler MJ. Androgens regulate bone resorption activity of isolated osteoclasts in vitro. Proc Natl Acad Sci USA 96: 505–510, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Perry RJ, Gault EJ, Paterson WF, Dunger DB, Donaldson MD. Effect of oxandrolone and timing of oral ethinylestradiol initiation on pubertal progression, height velocity and bone maturation in the UK Turner study. Horm Res Paediatr 81: 298–308, 2014. [DOI] [PubMed] [Google Scholar]
- 410.Pihlajamaa P, Sahu B, Janne OA. Determinants of receptor- and tissue-specific actions in androgen signaling. Endocr Rev 36: 357–384, 2015. [DOI] [PubMed] [Google Scholar]
- 411.Pugliese LS, Goncalves TO, Popi AF, Mariano M, Pesquero JB, Lopes JD. B-1 lymphocytes differentiate into functional osteoclast-like cells. Immunobiology 217: 336–344, 2012. [DOI] [PubMed] [Google Scholar]
- 412.Qiu S, Rao DS, Palnitkar S, Parfitt AM. Relationships between osteocyte density and bone formation rate in human cancellous bone. Bone 31: 709–711, 2002. [DOI] [PubMed] [Google Scholar]
- 413.Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol 8: 276–286, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Quaynor SD, Stradtman EW Jr, Kim HG, Shen Y, Chorich LP, Schreihofer DA, Layman LC. Delayed puberty and estrogen resistance in a woman with estrogen receptor alpha variant. N Engl J Med 369: 164–171, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Rai D, Frolova A, Frasor J, Carpenter AE, Katzenellenbogen BS. Distinctive actions of membrane-targeted versus nuclear localized estrogen receptors in breast cancer cells. Mol Endocrinol 19: 1606–1617, 2005. [DOI] [PubMed] [Google Scholar]
- 416.Rana K, Fam BC, Clarke MV, Pang TP, Zajac JD, MacLean HE. Increased adiposity in DNA binding-dependent androgen receptor knockout male mice associated with decreased voluntary activity and not insulin resistance. Am J Physiol Endocrinol Metab 301: E767–E778, 2011. [DOI] [PubMed] [Google Scholar]
- 417.Ranke MB. Treatment of children and adolescents with idiopathic short stature. Nat Rev Endocrinol 9: 325–334, 2013. [DOI] [PubMed] [Google Scholar]
- 418.Rariy CM, Ratcliffe SJ, Weinstein R, Bhasin S, Blackman MR, Cauley JA, Robbins J, Zmuda JM, Harris TB, Cappola AR. Higher serum free testosterone concentration in older women is associated with greater bone mineral density, lean body mass, and total fat mass: the cardiovascular health study. J Clin Endocrinol Metab 96: 989–996, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Raskin K, De GK, Duittoz A, Liere P, Verhoeven G, Tronche F, Mhaouty-Kodja S. Conditional inactivation of androgen receptor gene in the nervous system: effects on male behavioral and neuroendocrine responses. J Neurosci 29: 4461–4470, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Recker R, Lappe J, Davies K, Heaney R. Characterization of perimenopausal bone loss: a prospective study. J Bone Miner Res 15: 1965–1973, 2000. [DOI] [PubMed] [Google Scholar]
- 421.Recker RR, Kendler D, Recknor CP, Rooney TW, Lewiecki EM, Utian WH, Cauley JA, Lorraine J, Qu Y, Kulkarni PM, Gaich CL, Wong M, Plouffe L Jr, Stock JL. Comparative effects of raloxifene and alendronate on fracture outcomes in postmenopausal women with low bone mass. Bone 40: 843–851, 2007. [DOI] [PubMed] [Google Scholar]
- 422.Riggs BL, Khosla S, Melton LJ III. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res 13: 763–776, 1998. [DOI] [PubMed] [Google Scholar]
- 423.Riggs BL, Melton LJ III, Robb RA, Camp JJ, Atkinson EJ, Peterson JM, Rouleau PA, McCollough CH, Bouxsein ML, Khosla S. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. J Bone Miner Res 19: 1945–1954, 2004. [DOI] [PubMed] [Google Scholar]
- 424.Riggs BL, Melton LJ, Robb RA, Camp JJ, Atkinson EJ, McDaniel L, Amin S, Rouleau PA, Khosla S. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J Bone Miner Res 23: 205–214, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Riggs BL, Wahner HW, Dunn WL, Mazess RB, Offord KP, Melton LJ III. Differential changes in bone mineral density of the appendicular and axial skeleton with aging: relationship to spinal osteoporosis. J Clin Invest 67: 328–335, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Robling AG, Castillo AB, Turner CH. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng 8: 455–498, 2006. [DOI] [PubMed] [Google Scholar]
- 427.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283: 5866–5875, 2008. [DOI] [PubMed] [Google Scholar]
- 428.Rochira V, Carani C. Aromatase deficiency in men: a clinical perspective. Nat Rev Endocrinol 5: 559–568, 2009. [DOI] [PubMed] [Google Scholar]
- 429.Rochira V, Zirilli L, Madeo B, Aranda C, Caffagni G, Fabre B, Montangero VE, Roldan EJ, Maffei L, Carani C. Skeletal effects of long-term estrogen and testosterone replacement treatment in a man with congenital aromatase deficiency: evidences of a priming effect of estrogen for sex steroids action on bone. Bone 40: 1662–1668, 2007. [DOI] [PubMed] [Google Scholar]
- 430.Rochira V, Zirilli L, Maffei L, Premrou V, Aranda C, Baldi M, Ghigo E, Aimaretti G, Carani C, Lanfranco F. Tall stature without growth hormone: four male patients with aromatase deficiency. J Clin Endocrinol Metab 95: 1626–1633, 2010. [DOI] [PubMed] [Google Scholar]
- 431.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133: 3231–3244, 2006. [DOI] [PubMed] [Google Scholar]
- 432.Roelants M, Hauspie R, Hoppenbrouwers K. References for growth and pubertal development from birth to 21 years in Flanders, Belgium. Ann Hum Biol 36: 680–694, 2009. [DOI] [PubMed] [Google Scholar]
- 433.Ross FP, Teitelbaum SL. alphavbeta3 and macrophage colony-stimulating factor: partners in osteoclast biology. Immunol Rev 208: 88–105, 2005. [DOI] [PubMed] [Google Scholar]
- 434.Ross JL, Quigley CA, Cao D, Feuillan P, Kowal K, Chipman JJ, Cutler GB Jr. Growth hormone plus childhood low-dose estrogen in Turner's syndrome. N Engl J Med 364: 1230–1242, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA 288: 321–333, 2002. [DOI] [PubMed] [Google Scholar]
- 436.Rubin CT, Lanyon LE. Regulation of bone mass by mechanical strain magnitude. Calcif Tissue Int 37: 411–417, 1985. [DOI] [PubMed] [Google Scholar]
- 437.Rubin J, Rubin C, Jacobs CR. Molecular pathways mediating mechanical signaling in bone. Gene 367: 1–16, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Saintier D, Khanine V, Uzan B, Ea HK, De Vernejoul MC, Cohen-Solal ME. Estradiol inhibits adhesion and promotes apoptosis in murine osteoclasts in vitro. J Steroid Biochem Mol Biol 99: 165–173, 2006. [DOI] [PubMed] [Google Scholar]
- 439.Sakai D, Kii I, Nakagawa K, Matsumoto HN, Takahashi M, Yoshida S, Hosoya T, Takakuda K, Kudo A. Remodeling of actin cytoskeleton in mouse periosteal cells under mechanical loading induces periosteal cell proliferation during bone formation. PLoS One 6: e24847, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Salehpour S, Alipour P, Razzaghy-Azar M, Ardeshirpour L, Shamshiri A, Monfared MF, Gharib A. A double-blind, placebo-controlled comparison of letrozole to oxandrolone effects upon growth and puberty of children with constitutional delay of puberty and idiopathic short stature. Horm Res Paediatr 74: 428–435, 2010. [DOI] [PubMed] [Google Scholar]
- 441.Sanchez-Cardenas C, Fontanaud P, He Z, Lafont C, Meunier AC, Schaeffer M, Carmignac D, Molino F, Coutry N, Bonnefont X, Gouty-Colomer LA, Gavois E, Hodson DJ, Le TP, Robinson IC, Mollard P. Pituitary growth hormone network responses are sexually dimorphic and regulated by gonadal steroids in adulthood. Proc Natl Acad Sci USA 107: 21878–21883, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Sassarini J, Lumsden MA. Oestrogen replacement in postmenopausal women. Age Ageing 44: 551–558, 2015. [DOI] [PubMed] [Google Scholar]
- 443.Sato T, Shibata T, Ikeda K, Watanabe K. Generation of bone-resorbing osteoclasts from B220+ cells: its role in accelerated osteoclastogenesis due to estrogen deficiency. J Bone Miner Res 16: 2215–2221, 2001. [DOI] [PubMed] [Google Scholar]
- 444.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem 281: 23698–23711, 2006. [DOI] [PubMed] [Google Scholar]
- 445.Saxon LK, Galea G, Meakin L, Price J, Lanyon LE. Estrogen receptors alpha and beta have different gender-dependent effects on the adaptive responses to load bearing in cancellous and cortical bone. Endocrinology 153: 2254–2266, 2012. [DOI] [PubMed] [Google Scholar]
- 446.Saxon LK, Jackson BF, Sugiyama T, Lanyon LE, Price JS. Analysis of multiple bone responses to graded strains above functional levels, and to disuse, in mice in vivo show that the human Lrp5 G171V High Bone Mass mutation increases the osteogenic response to loading but that lack of Lrp5 activity reduces it. Bone 49: 184–193, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Saxon LK, Robling AG, Castillo AB, Mohan S, Turner CH. The skeletal responsiveness to mechanical loading is enhanced in mice with a null mutation in estrogen receptor-beta. Am J Physiol Endocrinol Metab 293: E484–E491, 2007. [DOI] [PubMed] [Google Scholar]
- 448.Schmidt A, Kimmel DB, Bai C, Scafonas A, Rutledge S, Vogel RL, McElwee-Witmer S, Chen F, Nantermet PV, Kasparcova V, Leu CT, Zhang HZ, Duggan ME, Gentile MA, Hodor P, Pennypacker B, Masarachia P, Opas EE, Adamski SA, Cusick TE, Wang J, Mitchell HJ, Kim Y, Prueksaritanont T, Perkins JJ, Meissner RS, Hartman GD, Freedman LP, Harada S, Ray WJ. Discovery of the selective androgen receptor modulator MK-0773 using a rational development strategy based on differential transcriptional requirements for androgenic anabolism versus reproductive physiology. J Biol Chem 285: 17054–17064, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Schmidt J, Dahlgren E, Brannstrom M, Landin-Wilhelmsen K. Body composition, bone mineral density and fractures in late postmenopausal women with polycystic ovary syndrome—a long-term follow-up study. Clin Endocrinol 77: 207–214, 2012. [DOI] [PubMed] [Google Scholar]
- 450.Seeman E. Modeling and remodeling. In: Principles of Bone Biology, edited by Bilezikian JP, Raisz LG, Martin TJ. New York: Academic, 2008. [Google Scholar]
- 451.Seeman E. Pathogenesis of bone fragility in women and men. Lancet 359: 1841–1850, 2002. [DOI] [PubMed] [Google Scholar]
- 452.Seeman E. Age- and menopause-related bone loss compromise cortical and trabecular microstructure. J Gerontol A Biol Sci Med Sci 68: 1218–1225, 2013. [DOI] [PubMed] [Google Scholar]
- 453.Seeman E, Hopper JL, Bach LA, Cooper ME, Parkinson E, McKay J, Jerums G. Reduced bone mass in daughters of women with osteoporosis. N Engl J Med 320: 554–558, 1989. [DOI] [PubMed] [Google Scholar]
- 454.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158–167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Sengupta A, Liu WK, Yeung YG, Yeung DCY, Frankelton ARJr, Stanley ER. Identification and subcellular localization of proteins that are rapidly phosphorylated in tyrosine in response to colony-stimulating factor 1. Proc Natl Acad Sci USA 85: 8062–8066, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Shahinian VB, Kuo YF, Freeman JL, Goodwin JS. Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med 352: 154–164, 2005. [DOI] [PubMed] [Google Scholar]
- 457.Shen J, Nielson CM, Marshall LM, Lee DC, Keaveny TM, Orwoll ES. The association between BMI and QCT-derived proximal hip structure and strength in older men: a cross-sectional study. J Bone Miner Res 30: 1301–1308, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Sherar LB, Mirwald RL, Baxter-Jones AD, Thomis M. Prediction of adult height using maturity-based cumulative height velocity curves. J Pediatr 147: 508–514, 2005. [DOI] [PubMed] [Google Scholar]
- 459.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95: 927–937, 1998. [DOI] [PubMed] [Google Scholar]
- 460.Silverman SL, Chines AA, Kendler DL, Kung AW, Teglbjaerg CS, Felsenberg D, Mairon N, Constantine GD, Adachi JD. Sustained efficacy and safety of bazedoxifene in preventing fractures in postmenopausal women with osteoporosis: results of a 5-year, randomized, placebo-controlled study. Osteoporos Int 23: 351–363, 2012. [DOI] [PubMed] [Google Scholar]
- 461.Silverman SL, Christiansen C, Genant HK, Vukicevic S, Zanchetta JR, de Villiers TJ, Constantine GD, Chines AA. Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo-, and active-controlled clinical trial. J Bone Miner Res 23: 1923–1934, 2008. [DOI] [PubMed] [Google Scholar]
- 462.Sims NA, Brennan K, Spaliviero J, Handelsman DJ, Seibel MJ. Perinatal testosterone surge is required for normal adult bone size but not for normal bone remodeling. Am J Physiol Endocrinol Metab 290: E456–E462, 2006. [DOI] [PubMed] [Google Scholar]
- 463.Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, Gaillard-Kelly M, Baron R. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone 30: 18–25, 2002. [DOI] [PubMed] [Google Scholar]
- 464.Sims NA, Martin TJ. Coupling signals between the osteoclast and osteoblast: how are messages transmitted between these temporary visitors to the bone surface? Front Endocrinol 6: 41, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Singer A, Exuzides A, Spangler L, O'Malley C, Colby C, Johnston K, Agodoa I, Baker J, Kagan R. Burden of illness for osteoporotic fractures compared with other serious diseases among postmenopausal women in the United States. Mayo Clin Proc 90: 53–62, 2015. [DOI] [PubMed] [Google Scholar]
- 466.Singh GK, Turner L, Desai R, Jimenez M, Handelsman DJ. Pharmacokinetic-pharmacodynamic study of subcutaneous injection of depot nandrolone decanoate using dried blood spots sampling coupled with ultrapressure liquid chromatography tandem mass spectrometry assays. J Clin Endocrinol Metab 99: 2592–2598, 2014. [DOI] [PubMed] [Google Scholar]
- 467.Singhal V, Goh BC, Bouxsein ML, Faugere MC, DiGirolamo DJ. Osteoblast-restricted disruption of the growth hormone receptor in mice results in sexually dimorphic skeletal phenotypes. Bone Res 1: 85–97, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Sinnesael M, Claessens F, Laurent M, Dubois V, Boonen S, Deboel L, Vanderschueren D. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: evidence from targeted AR disruption in mouse osteocytes. J Bone Miner Res 27: 2535–2543, 2012. [DOI] [PubMed] [Google Scholar]
- 469.Sinnesael M, Jardi F, Deboel L, Laurent MR, Dubois V, Zajac JD, Davey RA, Carmeliet G, Claessens F, Vanderschueren D. The androgen receptor has no direct antiresorptive actions in mouse osteoclasts. Mol Cell Endocrinol 411: 198–206, 2015. [DOI] [PubMed] [Google Scholar]
- 470.Sinnesael M, Laurent MR, Jardi F, Dubois V, Deboel L, Delisser P, Behets GJ, D'Haese PC, Carmeliet G, Claessens F, Vanderschueren D. Androgens inhibit the osteogenic response to mechanical loading in adult male mice. Endocrinology 156: 1343–1353, 2015. [DOI] [PubMed] [Google Scholar]
- 471.Sjogren K, Lagerquist M, Moverare-Skrtic S, Andersson N, Windahl SH, Swanson C, Mohan S, Poutanen M, Ohlsson C. Elevated aromatase expression in osteoblasts leads to increased bone mass without systemic adverse effects. J Bone Miner Res 24: 1263–1270, 2009. [DOI] [PubMed] [Google Scholar]
- 472.Slemenda CW, Longcope C, Zhou L, Hui SL, Peacock M, Johnston CC. Sex steroids and bone mass in older men. Positive associations with serum estrogens and negative associations with androgens. J Clin Invest 100: 1755–1759, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen- receptor gene in a man. N Engl J Med 331: 1056–1061, 1994. [DOI] [PubMed] [Google Scholar]
- 474.Smith EP, Specker B, Bachrach BE, Kimbro KS, Li XJ, Young MF, Fedarko NS, Abuzzahab MJ, Frank GR, Cohen RM, Lubahn DB, Korach KS. Impact on bone of an estrogen receptor-alpha gene loss of function mutation. J Clin Endocrinol Metab 93: 3088–3096, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Smith GI, Yoshino J, Reeds DN, Bradley D, Burrows RE, Heisey HD, Moseley AC, Mittendorfer B. Testosterone and progesterone, but not estradiol, stimulate muscle protein synthesis in postmenopausal women. J Clin Endocrinol Metab 99: 256–265, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Smith MR, Fallon MA, Lee H, Finkelstein JS. Raloxifene to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer: a randomized controlled trial. J Clin Endocrinol Metab 89: 3841–3846, 2004. [DOI] [PubMed] [Google Scholar]
- 477.Smith MR, Goode M, Zietman AL, McGovern FJ, Lee H, Finkelstein JS. Bicalutamide monotherapy versus leuprolide monotherapy for prostate cancer: effects on bone mineral density and body composition. J Clin Oncol 22: 2546–2553, 2004. [DOI] [PubMed] [Google Scholar]
- 478.Smith MR, Morton RA, Barnette KG, Sieber PR, Malkowicz SB, Rodriguez D, Hancock ML, Steiner MS. Toremifene to reduce fracture risk in men receiving androgen deprivation therapy for prostate cancer. J Urol 184: 1316–1321, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Snyder PJ, Ellenberg SS, Cunningham GR, Matsumoto AM, Bhasin S, Barrett-Connor E, Gill TM, Farrar JT, Cella D, Rosen RC, Resnick SM, Swerdloff RS, Cauley JA, Cifelli D, Fluharty L, Pahor M, Ensrud KE, Lewis CE, Molitch ME, Crandall JP, Wang C, Budoff MJ, Wenger NK, Mohler ER III, Bild DE, Cook NL, Keaveny TM, Kopperdahl DL, Lee D, Schwartz AV, Storer TW, Ershler WB, Roy CN, Raffel LJ, Romashkan S, Hadley E. The Testosterone Trials: seven coordinated trials of testosterone treatment in elderly men. Clin Trials 11: 362–375, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Snyder PJ, Peachey H, Berlin JA, Hannoush P, Haddad G, Dlewati A, Santanna J, Loh L, Lenrow DA, Holmes JH, Kapoor SC, Atkinson LE, Strom BL. Effects of testosterone replacement in hypogonadal men. J Clin Endocrinol Metab 85: 2670–2677, 2000. [DOI] [PubMed] [Google Scholar]
- 481.Snyder PJ, Peachey H, Hannoush P, Berlin JA, Loh L, Holmes JH, Dlewati A, Staley J, Santanna J, Kapoor SC, Attie MF, Haddad JG Jr, Strom BL. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab 84: 1966–1972, 1999. [DOI] [PubMed] [Google Scholar]
- 482.Specker BL, Wey HE, Binkley TL, Beare TM, Minett M, Weidauer L. Rural vs non-rural differences and longitudinal bone changes by DXA and pQCT in men aged 20–66 years: a population-based study. Bone 79: 79–87, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Sundh D, Mellstrom D, Nilsson M, Karlsson M, Ohlsson C, Lorentzon M. Increased cortical porosity in older men with fracture. J Bone Miner Res 30: 1692–1700, 2015. [DOI] [PubMed] [Google Scholar]
- 484.Sunters A, Armstrong VJ, Zaman G, Kypta RM, Kawano Y, Lanyon LE, Price JS. Mechano-transduction in osteoblastic cells involves strain-regulated estrogen receptor alpha-mediated control of insulin-like growth factor (IGF) I receptor sensitivity to ambient IGF, leading to phosphatidylinositol 3-kinase/AKT-dependent Wnt/LRP5 receptor-independent activation of beta-catenin signaling. J Biol Chem 285: 8743–8758, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Sunyer T, Lewis J, Collin-Osdoby P, Osdoby P. Estrogen's bone-protective effects may involve differential IL-1 receptor regulation in human osteoclast-like cells. J Clin Invest 103: 1409–1418, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Szulc P, Munoz F, Claustrat B, Garnero P, Marchand F, Duboeuf F, Delmas PD. Bioavailable estradiol may be an important determinant of osteoporosis in men: the MINOS study. J Clin Endocrinol Metab 86: 192–199, 2001. [DOI] [PubMed] [Google Scholar]
- 487.Szulc P, Seeman E, Duboeuf F, Sornay-Rendu E, Delmas PD. Bone fragility: failure of periosteal apposition to compensate for increased endocortical resorption in postmenopausal women. J Bone Miner Res 21: 1856–1863, 2006. [DOI] [PubMed] [Google Scholar]
- 488.Szulc P, Uusi-Rasi K, Claustrat B, Marchand F, Beck TJ, Delmas PD. Role of sex steroids in the regulation of bone morphology in men. The MINOS study. Osteoporos Int 15: 909–917, 2004. [DOI] [PubMed] [Google Scholar]
- 489.Taes Y, Lapauw B, Vandewalle S, Zmierczak H, Goemaere S, Vanderschueren D, Kaufman JM, T'Sjoen G. Estrogen-specific action on bone geometry and volumetric bone density: longitudinal observations in an adult with complete androgen insensitivity. Bone 45: 392–397, 2009. [DOI] [PubMed] [Google Scholar]
- 490.Tam CS, Heersche JNM, Murray TM, Parsons JA. Parathyroid hormone stimulates the bone apposition rate independently of its resorptive action: differential effects of intermittent and continuous administration. Endocrinology 110: 506–512, 1982. [DOI] [PubMed] [Google Scholar]
- 491.Tate MLK, Niederer P, Knothe U. In vivo tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading. Bone 22: 107–117, 1998. [DOI] [PubMed] [Google Scholar]
- 492.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5: 464–475, 2007. [DOI] [PubMed] [Google Scholar]
- 493.Taxel P, Kaneko H, Lee SK, Aguila HL, Raisz LG, Lorenzo JA. Estradiol rapidly inhibits osteoclastogenesis and RANKL expression in bone marrow cultures in postmenopausal women: a pilot study. Osteoporos Int 19: 193–199, 2008. [DOI] [PubMed] [Google Scholar]
- 494.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet 4: 638–649, 2003. [DOI] [PubMed] [Google Scholar]
- 495.Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res 13: 1243–1250, 1998. [DOI] [PubMed] [Google Scholar]
- 496.Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab 82: 3128–3135, 1997. [DOI] [PubMed] [Google Scholar]
- 497.Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN. IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci USA 100: 125–130, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Tracz MJ, Sideras K, Bolona ER, Haddad RM, Kennedy CC, Uraga MV, Caples SM, Erwin PJ, Montori VM. Testosterone use in men and its effects on bone health. A systematic review and meta-analy sis of randomized placebo-controlled trials. J Clin Endocrinol Metab 91: 2011–2016, 2006. [DOI] [PubMed] [Google Scholar]
- 499.Travison TG, Shackelton R, Araujo AB, Morley JE, Williams RE, Clark RV, McKinlay JB. Frailty, serum androgens, and the CAG repeat polymorphism: results from the Massachusetts Male Aging Study. J Clin Endocrinol Metab 95: 2746–2754, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, Bellido T. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50: 209–217, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Tuck SP, Scane AC, Fraser WD, Diver MJ, Eastell R, Francis RM. Sex steroids and bone turnover markers in men with symptomatic vertebral fractures. Bone 43: 999–1005, 2008. [DOI] [PubMed] [Google Scholar]
- 502.Turner CH, Owan I, Alvey T, Hulman J, Hock JM. Recruitment and proliferative responses of osteoblasts after mechanical loading in vivo determined using sustained-release bromodeoxyuridine. Bone 22: 463–469, 1998. [DOI] [PubMed] [Google Scholar]
- 503.Tyagi AM, Srivastava K, Sharan K, Yadav D, Maurya R, Singh D. Daidzein prevents the increase in CD4+CD28null T cells and B lymphopoesis in ovariectomized mice: a key mechanism for anti-osteoclastogenic effect. PLoS One 6: e21216, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Ucer S, Iyer S, Bartell SM, Martin-Millan M, Han L, Kim HN, Weinstein RS, Jilka RL, O'Brien CA, Almeida M, Manolagas SC. The effects of androgens on murine cortical bone do not require AR or ERalpha signaling in osteoblasts and osteoclasts. J Bone Miner Res 30: 1138–1149, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Ucer S, Iyer S, Han L, Bartell SM, Warren A, Crawford JA, Rutlen C, Jilka R, Almeida M, Manolagas S. H2O2 generated in the mitochondria of osteoclasts is required for the loss of cortical bone mass caused by estrogen or androgen deficiency, but not aging (Abstract). J Bone Miner Res 30: S26, 2015. [Google Scholar]
- 506.Ucer S, Iyer S, Kim HN, Han L, Thostenson J, Rutlen C, Allison K, Jilka RL, O'Brien CA, Manolagas SC. The effects of aging and sex steroid deficiency on the murine skeleton are independent and mechanistically distinct. J Bone Miner Res. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA 94: 7239–7244, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Uebelhart B, Herrmann F, Pavo I, Draper MW, Rizzoli R. Raloxifene treatment is associated with increased serum estradiol and decreased bone remodeling in healthy middle-aged men with low sex hormone levels. J Bone Miner Res 19: 1518–1524, 2004. [DOI] [PubMed] [Google Scholar]
- 509.Vajdos FF, Hoth LR, Geoghegan KF, Simons SP, LeMotte PK, Danley DE, Ammirati MJ, Pandit J. The 2.0 A crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Sci 16: 897–905, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Van Caenegem E, Wierckx K, Taes Y, Dedecker D, Van de Peer F, Toye K, Kaufman JM, T'Sjoen G. Bone mass, bone geometry, and body composition in female-to-male transsexual persons after long-term cross-sex hormonal therapy. J Clin Endocrinol Metab 97: 2503–2511, 2012. [DOI] [PubMed] [Google Scholar]
- 511.Van Caenegem E, Wierckx K, Taes Y, Schreiner T, Vandewalle S, Toye K, Kaufman JM, T'Sjoen G. Preservation of volumetric bone density and geometry in trans women during cross-sex hormonal therapy: a prospective observational study. Osteoporos Int 26: 35–47, 2015. [DOI] [PubMed] [Google Scholar]
- 512.Van Caenegem E, Wierckx K, Taes Y, Schreiner T, Vandewalle S, Toye K, Lapauw B, Kaufman JM, T'Sjoen G. Body composition, bone turnover, and bone mass in trans men during testosterone treatment: 1-year follow-up data from a prospective case-controlled study (ENIGI). Eur J Endocrinol 172: 163–171, 2015. [DOI] [PubMed] [Google Scholar]
- 513.Van den Beld AW, de Jong FH, Grobbee DE, Pols HA, Lamberts SW. Measures of bioavailable serum testosterone and estradiol and their relationships with muscle strength, bone density, and body composition in elderly men. J Clin Endocrinol Metab 85: 3276–3282, 2000. [DOI] [PubMed] [Google Scholar]
- 514.Van PI, Goemaere S, Kaufman JM. Bioavailable estradiol and an aromatase gene polymorphism are determinants of bone mineral density changes in men over 70 years of age. J Clin Endocrinol Metab 88: 3075–3081, 2003. [DOI] [PubMed] [Google Scholar]
- 515.Van PI, Goemaere S, Zmierczak H, Kaufman JM. Perturbed sex steroid status in men with idiopathic osteoporosis and their sons. J Clin Endocrinol Metab 89: 4949–4953, 2004. [DOI] [PubMed] [Google Scholar]
- 516.Van PI, Lumbroso S, Goemaere S, Sultan C, Kaufman JM. Lack of influence of the androgen receptor gene CAG-repeat polymorphism on sex steroid status and bone metabolism in elderly men. Clin Endocrinol 55: 659–666, 2001. [DOI] [PubMed] [Google Scholar]
- 517.Vandenput L, Boonen S, Van HE, Swinnen JV, Bouillon R, Vanderschueren D. Evidence from the aged orchidectomized male rat model that 17beta-estradiol is a more effective bone-sparing and anabolic agent than 5alpha-dihydrotestosterone. J Bone Miner Res 17: 2080–2086, 2002. [DOI] [PubMed] [Google Scholar]
- 518.Vandenput L, Lorentzon M, Sundh D, Nilsson ME, Karlsson MK, Mellstrom D, Ohlsson C. Serum estradiol levels are inversely associated with cortical porosity in older men. J Clin Endocrinol Metab 99: E1322–E1326, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Vandenput L, Mellstrom D, Kindmark A, Johansson H, Lorentzon M, Leung J, Redlund-Johnell I, Rosengren BE, Karlsson MK, Wang YX, Kwok T, Ohlsson C. High serum SHBG predicts incident vertebral fractures in elderly men. J Bone Miner Res 31: 683–689, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Vanderschueren D, Laurent MR, Claessens F, Gielen E, Lagerquist MK, Vandenput L, Borjesson AE, Ohlsson C. Sex steroid actions in male bone. Endocr Rev 35: 906–960, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Vanderschueren D, Pye SR, Venken K, Borghs H, Gaytant J, Huhtaniemi IT, Adams JE, Ward KA, Bartfai G, Casanueva FF, Finn JD, Forti G, Giwercman A, Han TS, Kula K, Labrie F, Lean ME, Pendleton N, Punab M, Silman AJ, Wu FC, O'Neill TW, Boonen S. Gonadal sex steroid status and bone health in middle-aged and elderly European men. Osteoporos Int 21: 1331–1339, 2010. [DOI] [PubMed] [Google Scholar]
- 522.Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C. Androgens and bone. Endocr Rev 25: 389–425, 2004. [DOI] [PubMed] [Google Scholar]
- 523.Vanderschueren D, Venken K, Ophoff J, Bouillon R, Boonen S. Clinical Review: Sex steroids and the periosteum–reconsidering the roles of androgens and estrogens in periosteal expansion. J Clin Endocrinol Metab 91: 378–382, 2006. [DOI] [PubMed] [Google Scholar]
- 524.Vedi S, Compston JE. The effects of long-term hormone replacement therapy on bone remodeling in postmenopausal women. Bone 19: 535–539, 1996. [DOI] [PubMed] [Google Scholar]
- 525.Vehmanen L, Elomaa I, Blomqvist C, Saarto T. Tamoxifen treatment after adjuvant chemotherapy has opposite effects on bone mineral density in premenopausal patients depending on menstrual status. J Clin Oncol 24: 675–680, 2006. [DOI] [PubMed] [Google Scholar]
- 526.Veldhuis JD, Metzger DL, Martha PM Jr, Mauras N, Kerrigan JR, Keenan B, Rogol AD, Pincus SM. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-insulin-like growth factor I axis in the human: evidence from pubertal pathophysiology and sex-steroid hormone replacement. J Clin Endocrinol Metab 82: 3414–3420, 1997. [DOI] [PubMed] [Google Scholar]
- 527.Veldhuis JD, Mielke KL, Cosma M, Soares-Welch C, Paulo R, Miles JM, Bowers CY. Aromatase and 5alpha-reductase inhibition during an exogenous testosterone clamp unveils selective sex steroid modulation of somatostatin and growth hormone secretagogue actions in healthy older men. J Clin Endocrinol Metab 94: 973–981, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Veldhuis JD, Roemmich JN, Rogol AD. Gender and sexual maturation-dependent contrasts in the neuroregulation of growth hormone secretion in prepubertal and late adolescent males and females–a general clinical research center-based study. J Clin Endocrinol Metab 85: 2385–2394, 2000. [DOI] [PubMed] [Google Scholar]
- 529.Venken K, De Gendt K, Boonen S, Ophoff J, Bouillon R, Swinnen JV, Verhoeven G, Vanderschueren D. Relative impact of androgen and estrogen receptor activation in the effects of androgens on trabecular and cortical bone in growing male mice: a study in the androgen receptor knockout mouse model. J Bone Miner Res 21: 576–585, 2006. [DOI] [PubMed] [Google Scholar]
- 530.Venken K, Moverare-Skrtic S, Kopchick JJ, Coschigano KT, Ohlsson C, Boonen S, Bouillon R, Vanderschueren D. Impact of androgens, growth hormone, and IGF-I on bone and muscle in male mice during puberty. J Bone Miner Res 22: 72–82, 2007. [DOI] [PubMed] [Google Scholar]
- 531.Venken K, Schuit F, Van LL, Tsukamoto K, Kopchick JJ, Coschigano K, Ohlsson C, Moverare S, Boonen S, Bouillon R, Vanderschueren D. Growth without growth hormone receptor: estradiol is a major growth hormone-independent regulator of hepatic IGF-I synthesis. J Bone Miner Res 20: 2138–2149, 2005. [DOI] [PubMed] [Google Scholar]
- 532.Verna C, Melsen B, Melsen F. Differences in static cortical bone remodeling parameters in human mandible and iliac crest. Bone 25: 577–583, 1999. [DOI] [PubMed] [Google Scholar]
- 533.Vico L, Vanacker JM. Sex hormones and their receptors in bone homeostasis: insights from genetically modified mouse models. Osteoporos Int 21: 365–372, 2010. [DOI] [PubMed] [Google Scholar]
- 534.Vilayphiou N, Boutroy S, Szulc P, van RB, Munoz F, Delmas PD, Chapurlat R. Finite element analysis performed on radius and tibia HR-pQCT images and fragility fractures at all sites in men. J Bone Miner Res 26: 965–973, 2011. [DOI] [PubMed] [Google Scholar]
- 535.Villanueva AR, Ilnicki L, Duncan H, Frost HM. Bone and cell dynamics in the osteoporoses: a review of measurements by tetracycline bone labeling. Clin Orthop Relat Res 49: 135–150, 1966. [PubMed] [Google Scholar]
- 536.Vlahopoulos S, Zimmer WE, Jenster G, Belaguli NS, Balk SP, Brinkmann AO, Lanz RB, Zoumpourlis VC, Schwartz RJ. Recruitment of the androgen receptor via serum response factor facilitates expression of a myogenic gene. J Biol Chem 280: 7786–7792, 2005. [DOI] [PubMed] [Google Scholar]
- 537.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER Jr, Wade JL, Robidoux A 3rd, Margolese RG, James J, Lippman SM, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 295: 2727–2741, 2006. [DOI] [PubMed] [Google Scholar]
- 538.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Wade JL III, Robidoux A, Margolese RG, James J, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N. Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: preventing breast cancer. Cancer Prev Res 3: 696–706, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Wadhwa VK, Weston R, Mistry R, Parr NJ. Long-term changes in bone mineral density and predicted fracture risk in patients receiving androgen-deprivation therapy for prostate cancer, with stratification of treatment based on presenting values. BJU Int 104: 800–805, 2009. [DOI] [PubMed] [Google Scholar]
- 540.Wainwright SA, Marshall LM, Ensrud KE, Cauley JA, Black DM, Hillier TA, Hochberg MC, Vogt MT, Orwoll ES. Hip fracture in women without osteoporosis. J Clin Endocrinol Metab 90: 2787–2793, 2005. [DOI] [PubMed] [Google Scholar]
- 541.Walsh JS, Henry YM, Fatayerji D, Eastell R. Lumbar spine peak bone mass and bone turnover in men and women: a longitudinal study. Osteoporos Int 20: 355–362, 2009. [DOI] [PubMed] [Google Scholar]
- 542.Walsh JS, Paggiosi MA, Eastell R. Cortical consolidation of the radius and tibia in young men and women. J Clin Endocrinol Metab 97: 3342–3348, 2012. [DOI] [PubMed] [Google Scholar]
- 543.Wang J, Stern PH. Sex-specific effects of estrogen and androgen on gene expression in human monocyte-derived osteoclasts. J Cell Biochem 112: 3714–3721, 2011. [DOI] [PubMed] [Google Scholar]
- 544.Wang Q, Seeman E. Skeletal growth and peak bone strength. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, edited by Rosen CJ. Oxford, UK: Wiley-Blackwell, 2013. [Google Scholar]
- 545.Wang Q, Wang XF, Iuliano-Burns S, Ghasem-Zadeh A, Zebaze R, Seeman E. Rapid growth produces transient cortical weakness: a risk factor for metaphyseal fractures during puberty. J Bone Miner Res 25: 1521–1526, 2010. [DOI] [PubMed] [Google Scholar]
- 546.Ward KA, Pye SR, Adams JE, Boonen S, Vanderschueren D, Borghs H, Gaytant J, Gielen E, Bartfai G, Casanueva FF, Finn JD, Forti G, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Labrie F, Lean ME, Pendleton N, Punab M, Silman AJ, Wu FC, O'Neill TW. Influence of age and sex steroids on bone density and geometry in middle-aged and elderly European men. Osteoporos Int 22: 1513–1523, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Warden SJ, Galley MR, Hurd AL, Richard JS, George LA, Guildenbecher EA, Barker RG, Fuchs RK. Cortical and trabecular bone benefits of mechanical loading are maintained long term in mice independent of ovariectomy. J Bone Miner Res 29: 1131–1140, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Warden SJ, Mantila Roosa SM, Kersh ME, Hurd AL, Fleisig GS, Pandy MG, Fuchs RK. Physical activity when young provides lifelong benefits to cortical bone size and strength in men. Proc Natl Acad Sci USA 111: 5337–5342, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Watts NB. Insights from the Global Longitudinal Study of Osteoporosis in Women (GLOW). Nat Rev Endocrinol 10: 412–422, 2014. [DOI] [PubMed] [Google Scholar]
- 550.Watts NB, Notelovitz M, Timmons MC, Addison WA, Wiita B, Downey LJ. Comparison of oral estrogens and estrogens plus androgen on bone mineral density, menopausal symptoms, and lipid-lipoprotein profiles in surgical menopause. Obstet Gynecol 85: 529–537, 1995. [DOI] [PubMed] [Google Scholar]
- 551.Wehkalampi K, Pakkila K, Laine T, Dunkel L. Adult height in girls with delayed pubertal growth. Horm Res Paediatr 76: 130–135, 2011. [DOI] [PubMed] [Google Scholar]
- 552.Wei W, Zeve D, Suh JM, Wang X, Du Y, Zerwekh JE, Dechow PC, Graff JM, Wan Y. Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin. Mol Cell Biol 31: 4706–4719, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Weinstein RS. The histological heterogeneity of osteopenia in the middle-aged and elderly patient. In: Nutrition in the Middle and Later Years, edited by Feldman ED. Boston, MA: PSG Publishing, 1983. [Google Scholar]
- 553a.Weinstein RS. Atlas of Osteoporosis (3rd ed), edited by Orwoll ES. Philadelphia, PA: Springer, 2009, p. 13–19. [Google Scholar]
- 554.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O'Brien C, Thostenson J, Roberson P, Boskey A, Clemens T, Manolagas SC. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell 9: 147–161, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Weinstein RS, Jia D, Powers CC, Stewart SA, Jilka RL, Parfitt AM, Manolagas SC. The skeletal effects of glucocorticoid excess override those of orchidectomy in mice. Endocrinology 145: 1980–1987, 2004. [DOI] [PubMed] [Google Scholar]
- 556.Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest 116: 1186–1194, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Wergedal JE, Kesavan C, Brommage R, Das S, Mohan S. Role of WNT16 in the regulation of periosteal bone formation in female mice. Endocrinology 156: 1023–1032, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Wergedal JE, Sheng MH, Ackert-Bicknell CL, Beamer WG, Baylink DJ. Genetic variation in femur extrinsic strength in 29 different inbred strains of mice is dependent on variations in femur cross-sectional geometry and bone density. Bone 36: 111–122, 2005. [DOI] [PubMed] [Google Scholar]
- 559.Wessler S, Otto C, Wilck N, Stangl V, Fritzemeier KH. Identification of estrogen receptor ligands leading to activation of non-genomic signaling pathways while exhibiting only weak transcriptional activity. J Steroid Biochem Mol Biol 98: 25–35, 2006. [DOI] [PubMed] [Google Scholar]
- 560.Whyte MP, Bergfeld MA, Murphy WA, Avioli LV, Teitelbaum SL. Postmenopausal osteoporosis. A heterogeneous disorder as assessed by histomorphometric analysis of Iliac crest bone from untreated patients. Am J Med 72: 193–202, 1982. [DOI] [PubMed] [Google Scholar]
- 561.Wickman S, Sipila I, Ankarberg-Lindgren C, Norjavaara E, Dunkel L. A specific aromatase inhibitor and potential increase in adult height in boys with delayed puberty: a randomised controlled trial. Lancet 357: 1743–1748, 2001. [DOI] [PubMed] [Google Scholar]
- 562.Wilhelmson AS, Stubelius A, Borjesson AE, Wu J, Stern A, Malin S, Martensson IL, Ohlsson C, Carlsten H, Tivesten A. Androgens regulate bone marrow B lymphopoiesis in male mice by targeting osteoblast-lineage cells. Endocrinology 156: 1228–1236, 2015. [DOI] [PubMed] [Google Scholar]
- 563.Wilson CA, Mrose SA, Thomas DW. Enhanced production of B lymphocytes after castration. Blood 85: 1535–1539, 1995. [PubMed] [Google Scholar]
- 564.Windahl SH, Borjesson AE, Farman HH, Engdahl C, Moverare-Skrtic S, Sjogren K, Lagerquist MK, Kindblom JM, Koskela A, Tuukkanen J, Divieti PP, Feng JQ, Dahlman-Wright K, Antonson P, Gustafsson JA, Ohlsson C. Estrogen receptor-alpha in osteocytes is important for trabecular bone formation in male mice. Proc Natl Acad Sci USA 110: 2294–2299, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Windahl SH, Galien R, Chiusaroli R, Clement-Lacroix P, Morvan F, Lepescheux L, Nique F, Horne WC, Resche-Rigon M, Baron R. Bone protection by estrens occurs through non-tissue-selective activation of the androgen receptor. J Clin Invest 116: 2500–2509, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Windahl SH, Saxon L, Borjesson AE, Lagerquist MK, Frenkel B, Henning P, Lerner UH, Galea GL, Meakin LB, Engdahl C, Sjogren K, Antal MC, Krust A, Chambon P, Lanyon LE, Price JS, Ohlsson C. Estrogen receptor-alpha is required for the osteogenic response to mechanical loading in a ligand-independent manner involving its activation function 1 but not 2. J Bone Miner Res 28: 291–301, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Wiren KM, Semirale AA, Zhang XW, Woo A, Tommasini SM, Price C, Schaffler MB, Jepsen KJ. Targeting of androgen receptor in bone reveals a lack of androgen anabolic action and inhibition of osteogenesis: a model for compartment-specific androgen action in the skeleton. Bone 43: 440–451, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Woo J, Kwok T, Leung JC, Ohlsson C, Vandenput L, Leung PC. Sex steroids and bone health in older Chinese men. Osteoporos Int 23: 1553–1562, 2012. [DOI] [PubMed] [Google Scholar]
- 569.Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, Amin N, Buchkovich ML, Croteau-Chonka DC, Day FR, Duan Y, Fall T, Fehrmann R, Ferreira T, Jackson AU, Karjalainen J, Lo KS, Locke AE, Magi R, Mihailov E, Porcu E, Randall JC, Scherag A, Vinkhuyzen AA, Westra HJ, Winkler TW, Workalemahu T, Zhao JH, Absher D, Albrecht E, Anderson D, Baron J, Beekman M, Demirkan A, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Fraser RM, Goel A, Gong J, Justice AE, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Lui JC, Mangino M, Mateo L I, Medina-Gomez C, Nalls MA, Nyholt DR, Palmer CD, Pasko D, Pechlivanis S, Prokopenko I, Ried JS, Ripke S, Shungin D, Stancakova A, Strawbridge RJ, Sung YJ, Tanaka T, Teumer A, Trompet S, van der Laan SW, van SJ, Van Vliet-Ostaptchouk JV, Wang Z, Yengo L, Zhang W, Afzal U, Arnlov J, Arscott GM, Bandinelli S, Barrett A, Bellis C, Bennett AJ, Berne C, Bluher M, Bolton JL, Bottcher Y, Boyd HA, Bruinenberg M, Buckley BM, Buyske S, Caspersen IH, Chines PS, Clarke R, Claudi-Boehm S, Cooper M, Daw EW, De Jong PA, Deelen J, Delgado G, Denny JC, Dhonukshe-Rutten R, Dimitriou M, Doney AS, Dorr M, Eklund N, Eury E, Folkersen L, Garcia ME, Geller F, Giedraitis V, Go AS, Grallert H, Grammer TB, Grassler J, Gronberg H, de Groot LC, Groves CJ, Haessler J, Hall P, Haller T, Hallmans G, Hannemann A, Hartman CA, Hassinen M, Hayward C, Heard-Costa NL, Helmer Q, Hemani G, Henders AK, Hillege HL, Hlatky MA, Hoffmann W, Hoffmann P, Holmen O, Houwing-Duistermaat JJ, Illig T, Isaacs A, James AL, Jeff J, Johansen B, Johansson A, Jolley J, Juliusdottir T, Junttila J, Kho AN, Kinnunen L, Klopp N, Kocher T, Kratzer W, Lichtner P, Lind L, Lindstrom J, Lobbens S, Lorentzon M, Lu Y, Lyssenko V, Magnusson PK, Mahajan A, Maillard M, McArdle WL, McKenzie CA, McLachlan S, McLaren PJ, Menni C, Merger S, Milani L, Moayyeri A, Monda KL, Morken MA, Muller G, Muller-Nurasyid M, Musk AW, Narisu N, Nauck M, Nolte IM, Nothen MM, Oozageer L, Pilz S, Rayner NW, Renstrom F, Robertson NR, Rose LM, Roussel R, Sanna S, Scharnagl H, Scholtens S, Schumacher FR, Schunkert H, Scott RA, Sehmi J, Seufferlein T, Shi J, Silventoinen K, Smit JH, Smith AV, Smolonska J, Stanton AV, Stirrups K, Stott DJ, Stringham HM, Sundstrom J, Swertz MA, Syvanen AC, Tayo BO, Thorleifsson G, Tyrer JP, van DS, van Schoor NM, van d V, van HD, van Oort FV, Vermeulen SH, Verweij N, Vonk JM, Waite LL, Waldenberger M, Wennauer R, Wilkens LR, Willenborg C, Wilsgaard T, Wojczynski MK, Wong A, Wright AF, Zhang Q, Arveiler D, Bakker SJ, Beilby J, Bergman RN, Bergmann S, Biffar R, Blangero J, Boomsma DI, Bornstein SR, Bovet P, Brambilla P, Brown MJ, Campbell H, Caulfield MJ, Chakravarti A, Collins R, Collins FS, Crawford DC, Cupples LA, Danesh J, de FU, den Ruijter HM, Erbel R, Erdmann J, Eriksson JG, Farrall M, Ferrannini E, Ferrieres J, Ford I, Forouhi NG. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46: 1173–1186, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Wright LM, Maloney W, Yu X, Kindle L, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 binding to its chemokine receptor CXCR4 on precursor cells promotes the chemotactic recruitment, development and survival of human osteoclasts. Bone 36: 840–853, 2005. [DOI] [PubMed] [Google Scholar]
- 571.Wronski TJ, Walsh CC, Ignaszewski LA. Histologic evidence for osteopenia and increased bone turnover in ovariectomized rats. Bone 7: 119–123, 1986. [DOI] [PubMed] [Google Scholar]
- 572.Wu FC, Tajar A, Beynon JM, Pye SR, Silman AJ, Finn JD, O'Neill TW, Bartfai G, Casanueva FF, Forti G, Giwercman A, Han TS, Kula K, Lean ME, Pendleton N, Punab M, Boonen S, Vanderschueren D, Labrie F, Huhtaniemi IT. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 363: 123–135, 2010. [DOI] [PubMed] [Google Scholar]
- 573.Wyce A, Bai Y, Nagpal S, Thompson CC. Research Resource: the androgen receptor modulates expression of genes with critical roles in muscle development and function. Mol Endocrinol 24: 1665–1674, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nature Med 17: 1235–1241, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Xu L, Wang Q, Wang Q, Lyytikainen A, Mikkola T, Volgyi E, Cheng S, Wiklund P, Munukka E, Nicholson P, Alen M, Cheng S. Concerted actions of insulin-like growth factor 1, testosterone, and estradiol on peripubertal bone growth: a 7-year longitudinal study. J Bone Miner Res 26: 2204–2211, 2011. [DOI] [PubMed] [Google Scholar]
- 576.Yamada T, Kawano H, Sekine K, Matsumoto T, Fukuda T, Azuma Y, Itaka K, Chung UI, Chambon P, Nakamura K, Kato S, Kawaguchi H. SRC-1 is necessary for skeletal responses to sex hormones in both males and females. J Bone Miner Res 19: 1452–1461, 2004. [DOI] [PubMed] [Google Scholar]
- 577.Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci USA 99: 13498–13503, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Yu EW, Bouxsein ML, Roy AE, Baldwin C, Cange A, Neer RM, Kaplan LM, Finkelstein JS. Bone loss after bariatric surgery: discordant results between DXA and QCT bone density. J Bone Miner Res 29: 542–550, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Miner Res 18: 1404–1418, 2003. [DOI] [PubMed] [Google Scholar]
- 580.Zebaze R, Seeman E. Cortical bone: a challenging geography. J Bone Miner Res 30: 24–29, 2015. [DOI] [PubMed] [Google Scholar]
- 581.Zebaze RM, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, Mackie EJ, Seeman E. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet 375: 1729–1736, 2010. [DOI] [PubMed] [Google Scholar]
- 582.Zhang J, Link DC. Targeting of mesenchymal stromal cells by Cre-recombinase transgenes commonly used to target osteoblast lineage cells. J Bone Miner Res. In press, doi: 10.1002/jbmr.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Zhang XZ, Kalu DN, Erbas B, Hopper JL, Seeman E. The effects of gonadectomy on bone size, mass, and volumetric density in growing rats are gender-, site-, and growth hormone-specific. J Bone Miner Res 14: 802–809, 1999. [DOI] [PubMed] [Google Scholar]