

Abstract
Early life stress increases risk for depression. Here we establish a “two-hit” stress model in mice wherein stress at a specific postnatal period increases susceptibility to adult social defeat stress and causes long-lasting transcriptional alterations that prime the ventral tegmental area (VTA)—a brain reward region—to be in a depression-like state. We identify a role for the developmental transcription factor orthodenticle homeobox 2 (Otx2) as an upstream mediator of these enduring effects. Transient juvenile—but not adult—knockdown of Otx2 in VTA mimics early life stress by increasing stress susceptibility, whereas its overexpression reverses the effects of early life stress. This work establishes a mechanism by which early life stress encodes lifelong susceptibility to stress via long-lasting transcriptional programming in VTA mediated by Otx2.
Studies in humans and animals suggest that early life stress increases risk for depression by heightening sensitivity to stressful events later in life (1–5), with early stress exerting prominent effects on the brain’s reward circuitry across species (6–8). We hypothesized that early life stress in a sensitive period of postnatal development might alter transcriptional regulation in the ventral tegmental area (VTA), a dopaminergic reward region implicated in mood and depression (9).
We adapted a maternal separation protocol of early life stress in C57BL/6J mice (10–12) and tested two different postnatal exposure windows (fig. S1A), such that mice were either standard-reared or subjected to stress in the early (postnatal day PND2 to 12) or late (PND10 to 20 or PND10 to 17) postnatal period. Early life stress in either time window did not affect offspring survival, although both early and late postnatal stress initially slowed weight gain measured at weaning, followed by increased weight gain by adulthood among late postnatal stress mice only (fig. S1B). We assessed the impact of early life stress on adult male stress susceptibility using an established chronic social defeat stress paradigm (13). Neither early nor late postnatal stress produced detectable behavioral effects before adult defeat stress (Fig. 1 and fig. S1). However, late, but not early, postnatal stress (see fig. S1) rendered adult mice more susceptible to depression-like behaviors after social defeat, including social avoidance [reduced exploration ratio of social versus nonsocial targets (Fig. 1, A and B)], anhedonia [preference for sucrose over water in a two-bottle choice test (Fig. 1D)], and immobility upon forced swimming (Fig. 1E). Late postnatal stress before social defeat also increased the proportion of “susceptible” mice according to a validated measure of social interaction (Fig. 1C) (13) and a composite “depression” score (fig. S1F). Increased risk for depressionlike behavior was independent of differences in basal corticosterone levels (fig. S1G) and anxietylike and locomotor behavior in an open field (Fig. 1F and fig. S1H).
Fig. 1. Early life stress enhances susceptibility to depression-like behavior.
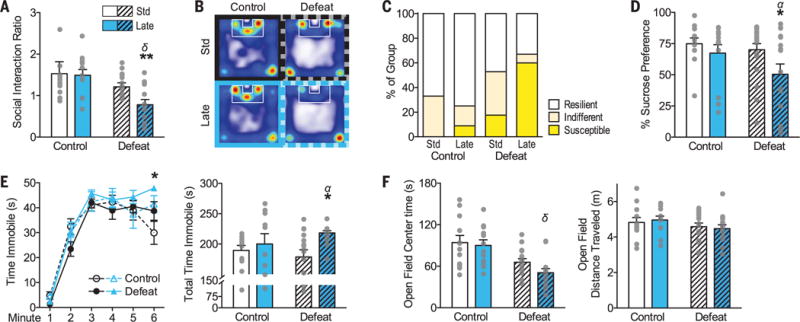
(A) Social interaction ratio and (B) exploration heat maps with social target present within the outlined zone. (C) Proportions of susceptible, indifferent, and resilient mice in each group. (D) Percentage preference for 1% sucrose solution in a two-bottle choice test. (E) Time immobile in a forced swim test (left) at each min and (right) total. (F) Open field center (left) exploration time and (right) total distance traveled. See table S2 for group sizes. Bars show means ± SEM overlaid with individual data points. *P < 0.05, **P < 0.01 indicate post hoc differences between socially defeated groups; α and δ indicate main effects (P < 0.05) of late postnatal stress (Late) or adult social defeat stress (Defeat), respectively.
We used RNA-sequencing to capture transcriptome-wide alterations in adult male VTA after late postnatal stress or after adult defeat stress. Late postnatal stress altered expression of 225 genes (P < 0.05, |fold-change| >1.3), and adult defeat stress altered expression of 428 genes, compared with standard-reared, unstressed controls. Differentially expressed genes (DEGs) were enriched for several relevant gene ontology terms (fig. S2A). Although only 69 genes (table S1) were altered at criteria by both late postnatal and adult defeat stress, a union heat map of both DEG lists revealed that genes are altered in the same direction by both stressors (Fig. 2A). We then used rank-rank hypergeometric overlap analysis to identify patterns and strength of genome-wide overlap in a threshold-free manner (Fig. 2B), which confirmed co-regulation of genes by late postnatal and defeat stresses. These analyses suggest that late postnatal stress transcriptionally primes VTA to be in a “depression-like” state. The transcriptional alterations induced by late postnatal stress are long-lasting and occur in the absence of detectable depression-like behavior and instead reflect latent sensitivity to adult stress.
Fig. 2. RNA-seq reveals long-lasting transcriptional alterations in VTA that predict OTX2 as an upstream regulator.
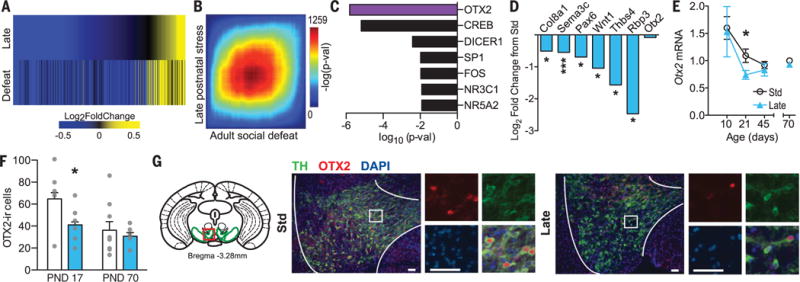
(A) Union heat map of late postnatal stress and social defeat stress DEGs in VTA compared with standard-reared (Std)–Control (|fold-change| >1.3, P < 0.05). (B) Threshold-free comparison of DEGs by rank-rank hypergeometric overlap. Pixels represent the overlap between the transcriptome of each comparison (versus Std-Control), with the significance of overlap [−log10(P value) of a hypergeometric test] color coded. Genes along each axis are sorted from most significantly upregulated to most down-regulated from the lower left corner. (C) Top predicted upstream regulators of DEGs by late postnatal stress. (D) Log2 fold-change of OTX2-predicting genes and Otx2 by late postnatal stress. (E) Relative Otx2 mRNA levels across postnatal development. (F) OTX2-immunoreactive cells in VTA at PND17 and PND70 among Std (white) and late postnatal stress (blue) mice. (G) Mouse VTA (diagram, left) imaged at PND17. Scale bars, 50 μm. Data are means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 between Std and Late Control groups.
Ingenuity Pathway Analysis (Qiagen) identified orthodenticle homeobox 2 (OTX2) as the highest-ranked upstream regulator of this prodepressive transcriptional signature (Fig. 2C). OTX2, a transcription factor, plays a role in neurodevelopment, specifically in VTA (14–16). Otx2 mRNA (17) and OTX2 protein are enriched in VTA, where >70% of OTX2-immunoreactive (+) cells are dopaminergic (tyrosine hydroxylase, TH+) (18). All six genes that led to the prediction of OTX2 as an upstream regulator were down-regulated by late postnatal stress (Fig. 2D, fig. S2B, and table S1). Five of the six were also down-regulated (P < 0.05 level) by adult defeat stress in standard-reared mice, whereas other predicted upstream regulators shared proportionally fewer genes (fig. S2, C and D). We also found that the proportion of genes with an OTX2 binding motif within 1 kb of their transcription start site is significantly enriched among DEGs after late postnatal stress and adult defeat stress (fig. S2F). Together, these analyses implicate OTX2 as a key mediator of the transcriptional signature underlying late postnatal stress–induced lifelong depression vulnerability. As a control, we examined expression of OTX2-predicting genes in nucleus accumbens—a main projection target of VTA—given the paracrine mechanisms of OTX2 action in visual cortex plasticity (16) but found no effect (fig. S2E).
Otx2 mRNA levels were not altered in adult VTA by prior postnatal stress. We therefore sought to understand how OTX2 acts as an upstream transcriptional regulator after late postnatal stress without being differentially expressed in adulthood. A time course of Otx2 mRNA levels in VTA showed that Otx2 is down-regulated relative to that in standard-reared mice at PND21 after late postnatal stress but not by early postnatal stress and not at later time points (Fig. 2E and fig. S2G). This was confirmed at the protein level by a decreased number of OTX2+ cells in VTA at PND17 but not at PND70 (Fig. 2, F and G), without any difference in the number of TH+ neurons (fig. S2H).
To test the prediction that transient down-regulation of Otx2 in VTA mediates the effect of late postnatal stress on transcriptional and behavioral end points in adulthood, we developed herpes simplex viruses (HSVs) to transiently overexpress or suppress (knockdown) Otx2 (fig. S2I). The short expression window (on within 12 hours, off after 7 days) of HSVs enabled us to have temporal control over Otx2 expression. To rescue the effects of late postnatal stress, mice were standard-reared or stressed from PND10 to 17, injected intra-VTA bilaterally with HSV-Otx2 or HSV-Gfp, and subjected to control or adult defeat stress conditions. Transient juvenile rescue (PND15 to 17 HSV delivery) of Otx2 in postnatally stressed mice rescued the enhanced vulnerability to adult defeat stress based upon individual and composite measures of behavioral abnormalities (Fig. 3A, fig. S3A, and table S3). Transient juvenile Otx2 overexpression in VTA also rescued the down-regulation of several OTX2-predicting target genes in this region, without having lasting effects on Otx2 expression itself (Fig. 3A). These findings support the hypothesis that transient suppression of Otx2 in VTA programs long-lasting changes in gene expression and stress susceptibility. Otx2 over-expression in adult (PND60 to 70) postnatally stressed mice caused a partial rescue of behavioral and gene expression abnormalities (Fig. 3B and fig. S3B).
Fig. 3. Otx2 expression in VTA bidirectionally controls depression-like behavior.
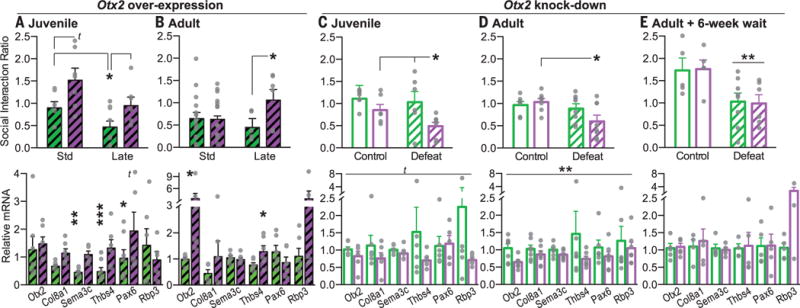
Depression-like behavior indicated by social interaction ratio (top), and VTA mRNA levels of Otx2 and OTX2-predicting genes (bottom), after each manipulation. HSV-Gfp (solid green) or HSV-Otx2 (solid purple) were overexpressed at (A) juvenile PND15 to 17 or (B) adult PND60 to 70 time points. HSV-miR-LacZ (open green) or HSV-miR-Otx2 (open purple) were injected for knockdown at (C) juvenile or (D and E) adult time points. Social defeat and behavioral testing was begun in adulthood 6 weeks after (A and C) juvenile or (E) adult manipulation or (B and D) <1 week after adult manipulation. Hashing indicates postdefeat quantification. Bars are means ± SEM, t < 0.1, *P < 0.05, **P < 0.01, ***P < 0.001; horizontal bars indicate main effect of condition [two-way analysis of variance (ANOVA)] (see table S3).
To test whether transient juvenile suppression of Otx2 recapitulates the effects of late postnatal stress on susceptibility to depression-like behavior, we generated HSVs encoding microRNAs targeting Otx2 or LacZ as a control. As predicted, transient juvenile Otx2 knockdown in VTA of standard-reared mice sensitized mice to subthreshold adult social defeat on the basis of individual and composite behavioral measures (Fig. 3C and fig. S3C). Adult Otx2 knockdown produced a subset of these abnormalities (Fig. 3D and fig. S3D). Adult knockdown of Otx2 significantly reduced mRNA levels of Otx2 and its target genes during the viral expression period (Fig. 3D). There was a trend for juvenile Otx2 knockdown to also reduce target gene expression in adult VTA.
To address whether the long-lasting effects of transient Otx2 suppression are specific to the juvenile period, we knocked down Otx2 in adulthood and waited 6 weeks before social defeat and behavior testing, akin to the maturation period after juvenile Otx2 manipulation. In contrast to juvenile knockdown, transient adult knockdown had no lasting effect on stress susceptibility or OTX2 target gene levels (Fig. 3E and fig. S3E). Together, these data establish that transient Otx2 suppression in the juvenile VTA is necessary and sufficient to enhance stress susceptibility in adulthood and that there is a sensitive window in early postnatal development for this lasting molecular and behavioral programming.
To determine whether late postnatal stress disrupts OTX2 regulation of target genes, we performed chromatin immunoprecipitation (ChIP) for OTX2 in PND21 or PND70 VTA. Given the presence of numerous OTX2 binding motifs within and upstream of the selected target genes, we identified putative regulatory regions by the presence of monomethylated histone 3 Lys4 (H3K4me1), which is associated with promoter and enhancer regions (19). Because of the tissue-specificity of H3K4me1 (20), we determined VTA enrichment by ChIP sequencing (ChIP-seq) from VTA of standard-reared and late postnatal stress adult mice (Fig. 4 and fig. S4B). OTX2 bound target genes within H3K4me1+ regulatory regions at PND21 in standard-reared but not postnatally stressed mice (Fig. 4 and fig. S4B). In contrast, no differences in OTX2 binding were seen at PND70 or in H3K4me1– regions at PND21. These findings are consistent with reduced OTX2 protein levels in VTA at PND21 but not adulthood. Further work is needed to understand how transient reductions in OTX2 binding within specific regulatory regions (21) of its target genes cause lasting effects on gene expression in VTA.
Fig. 4. Late postnatal stress transiently alters OTX2 binding at target genes.
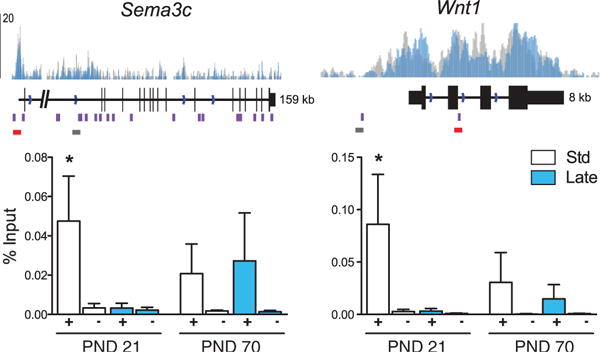
ChIP for OTX2 (+) and IgG (−) in VTA at PND21 and PND70 for representative OTX2 target genes, Sema3c and Wnt1, after standard rearing (white/gray) or late postnatal stress (blue). Binding at Col8a1, Pax6, Thbs4, and Rbp3 shows a similar pattern (see fig. S4). (Top) Regulatory regions illustrated by H3K4me1 ChIP-seq enrichment, above gene features. OTX2 binding motif sites (purple), and primer pairs for qChIP amplification, are indicated (red bar, H3K4me1+ region corresponding to data below; gray bar, H3K4me1– region) (see fig. S4). (Bottom) Bars (means ± SEM) represent percentage input (n = 4 to 6), *P < 0.05.
In sum, we demonstrate a postnatal sensitive window for stress, mediated by Otx2 in mouse VTA, to increase lifetime risk for depression-like behavior, which becomes manifest only after additional stress in adulthood. Understanding how Otx2 programs lasting stress susceptibility will provide insight into ways of reducing the deleterious effects of early life adversity.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Mental Health, NIH (P50MH096890) and Hope for Depression Research Foundation. RNA-seq and ChIP-seq data are available at Gene Expression Omnibus, accession number GSE89692. Supplementary materials contain additional data.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/356/6343/1185/suppl/DC1
Materials and Methods
References (22–43)
REFERENCES AND NOTES
- 1.Jonson-Reid M, Kohl PL, Drake B. Pediatrics. 2012;129:839–845. doi: 10.1542/peds.2011-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammen C, Henry R, Daley SE. J Consult Clin Psychol. 2000;68:782–787. [PubMed] [Google Scholar]
- 3.Kendler KS, Kuhn JW, Prescott CA. Psychol Med. 2004;34:1475–1482. doi: 10.1017/s003329170400265x. [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZY, et al. Behav Brain Res. 2016;306:154–159. doi: 10.1016/j.bbr.2016.03.040. [DOI] [PubMed] [Google Scholar]
- 5.Williams LM, Debattista C, Duchemin A-M, Schatzberg AF, Nemeroff CB. Transl Psychiatry. 2016;6:e799. doi: 10.1038/tp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabib S, Puglisi-Allegra S, D’Amato FR. Brain Res. 1993;604:232–239. doi: 10.1016/0006-8993(93)90374-v. [DOI] [PubMed] [Google Scholar]
- 7.Brake WG, Zhang TY, Diorio J, Meaney MJ, Gratton A. Eur J Neurosci. 2004;19:1863–1874. doi: 10.1111/j.1460-9568.2004.03286.x. [DOI] [PubMed] [Google Scholar]
- 8.Hanson JL, et al. Soc Cogn Affect Neurosci. 2016;11:405–412. doi: 10.1093/scan/nsv124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo SJ, Nestler EJ. Nat Rev Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotsky PM, Meaney MJ. Brain Res Mol Brain Res. 1993;18:195–200. doi: 10.1016/0169-328x(93)90189-v. [DOI] [PubMed] [Google Scholar]
- 11.Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. Endocrinology. 2008;149:4892–4900. doi: 10.1210/en.2008-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rincón-Cortés M, Sullivan RM. Front Endocrinol (Lausanne) 2014;5:33. doi: 10.3389/fendo.2014.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnan V, et al. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 14.Puelles E, et al. Development. 2004;131:2037–2048. doi: 10.1242/dev.01107. [DOI] [PubMed] [Google Scholar]
- 15.Vernay B, et al. J Neurosci. 2005;25:4856–4867. doi: 10.1523/JNEUROSCI.5158-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugiyama S, et al. Cell. 2008;134:508–520. doi: 10.1016/j.cell.2008.05.054. [DOI] [PubMed] [Google Scholar]
- 17.Lein ES, et al. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 18.Di Salvio M, Di Giovannantonio LG, Omodei D, Acampora D, Simeone A. Int J Dev Biol. 2010;54:939–945. doi: 10.1387/ijdb.092974ms. [DOI] [PubMed] [Google Scholar]
- 19.Kim TK, et al. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kundaje A, et al. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SH, et al. Cell Reports. 2014;7:1968–1981. doi: 10.1016/j.celrep.2014.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.