

Abstract
Acid-base homeostasis is critical to maintenance of normal health. Renal ammonia excretion is the quantitatively predominant component of renal net acid excretion, both under basal conditions and in response to acid-base disturbances. Although titratable acid excretion also contributes to renal net acid excretion, the quantitative contribution of titratable acid excretion is less than that of ammonia under basal conditions and is only a minor component of the adaptive response to acid-base disturbances. In contrast to other urinary solutes, ammonia is produced in the kidney and then is selectively transported either into the urine or the renal vein. The proportion of ammonia that the kidney produces that is excreted in the urine varies dramatically in response to physiological stimuli, and only urinary ammonia excretion contributes to acid-base homeostasis. As a result, selective and regulated renal ammonia transport by renal epithelial cells is central to acid-base homeostasis. Both molecular forms of ammonia, NH3 and NH4+, are transported by specific proteins, and regulation of these transport processes determines the eventual fate of the ammonia produced. In this review, we discuss these issues, and then discuss in detail the specific proteins involved in renal epithelial cell ammonia transport.
I. REVIEW OF ROLE OF AMMONIA IN ACID-BASE BALANCE
A. Importance of Acid-Base Homeostasis
Maintaining acid-base homeostasis is critical for normal health. Acid-base disorders lead to such clinical problems as growth retardation in neonates and children, nausea and vomiting, electrolyte disturbances, increased susceptibility to cardiac arrhythmias, decreased cardiovascular catecholamine sensitivity, bone disorders including osteoporosis and osteomalacia, recurrent nephrolithiasis, skeletal muscle atrophy, paresthesia, and coma in adults (191). Recent studies suggest important correlations between abnormal acid-base homeostasis and mortality, with both an elevated and a lowered serum bicarbonate predicting increased mortality in patients both with and without chronic kidney disease (CKD) (155, 213, 235).
The kidneys have two major functions in acid-base homeostasis: 1) reabsorbing filtered bicarbonate and 2) generating new bicarbonate. In the typical adult, the kidneys filter ∼4,200 mmol/day of bicarbonate. Renal epithelial cells reabsorb almost all of this in the process termed “bicarbonate reabsorption.” Kidneys also produce new bicarbonate in a process termed “bicarbonate generation.” Kidneys can also excrete alkali, in the form of bicarbonate and organic anions, such as citrate and 2-oxoglutarate. This is necessary for recovery from both respiratory (91) and metabolic alkalosis (82, 83, 244, 293) to prevent metabolic alkalosis in humans consuming vegetarian diets, which presents an alkali load (264, 326), and in rabbits (135, 151, 294). Excretion of the organic anion citrate is also important for prevention of renal stone formation (2, 113, 297). Multiple excellent reviews have been published regarding the mechanisms and regulation of bicarbonate reabsorption and of organic anion transport (28, 54, 111, 225, 329), and the interested reader is referred to these for additional details.
Complete reabsorption of filtered bicarbonate, while critical, is not sufficient to maintain acid-base homeostasis. Metabolism of the normal amino-acid content of the typical Western diet results in significant rates of endogenous acid production, averaging 0.8-1.0 mmol·kg−1·day−1. This continuous acid load is buffered rapidly by intracellular and extracellular buffers, of which the CO2-HCO3− buffer system is the most relevant. Protons (H+) are buffered by bicarbonate (HCO3−), forming carbonic acid (H2CO3), which then rapidly dissociates to CO2 and water. CO2 is eliminated through normal respiration. While highly effective at buffering endogenous acid production, equimolar bicarbonate is used in the process, thereby depleting total body bicarbonate levels. A critical function of the kidneys is to generate “new” bicarbonate to replenish that bicarbonate utilized for buffering acid loads.
Renal new bicarbonate generation involves both ammonia1 metabolism and titratable acid excretion. Under basal conditions, ammonia metabolism, which includes net ammoniagenesis and renal epithelial cell ammonia transport leading to urinary ammonia excretion, is the quantitatively greater component of new bicarbonate generation. In response to exogenous acid loads, changes in ammonia excretion substantially exceed changes in titratable acid excretion (310, 312, 313, 325). In the electrolyte disorder of hypokalemia there is increased ammonia production and excretion, and this appears to be an important factor in the genesis of the resulting metabolic alkalosis (15, 184, 263, 285). Organic anion excretion also contributes to acid-base homeostasis, but appears to be a substantially smaller component (114, 311). Titratable acid excretion and organic anion excretion will not be discussed further in this review.
There are many conditions of altered ammonia metabolism. Because the focus of this review is on ammonia transporters and their role in acid-base homeostasis, we will review the contribution of ammonia metabolism and excretion to maintenance or disturbance of acid-base homeostasis in two common clinical conditions. These two conditions are CKD and renal tubular acidosis (RTA). Following a discussion of these conditions, we will present an overview of renal ammonia transport, and then we will provide in-depth review of the specific proteins involved in renal ammonia transport.
B. Ammonia Metabolism in CKD
Ammonia metabolism and acid-base homeostasis in individuals with CKD is of critical importance. CKD currently affects 10–13% of adults in Western countries (50), lifetime risks of developing CKD appear to exceed 50% (105), and chronic kidney disease is an important risk factor for the development of metabolic acidosis (194). Indeed, as shown in Figure 1, as many as 40% of individuals with severe CKD develop metabolic acidosis. A positive feedback cycle may develop, whereby metabolic acidosis leads to worsening of CKD, and worsening CKD causes worsening of metabolic acidosis. In particular, randomized controlled clinical trials indicate that correction of the metabolic acidosis, either with oral sodium bicarbonate (60) or with alkali administration through increased dietary ingestion of foods that are alkali precursors (101, 104), slows the worsening of CKD and decreases the likelihood of development of end-stage renal disease. Mechanisms through which metabolic acidosis leads to worsening of CKD are incompletely understood, but may involve angiotensin II, endothelin, and aldosterone (44, 101–103).
FIGURE 1.
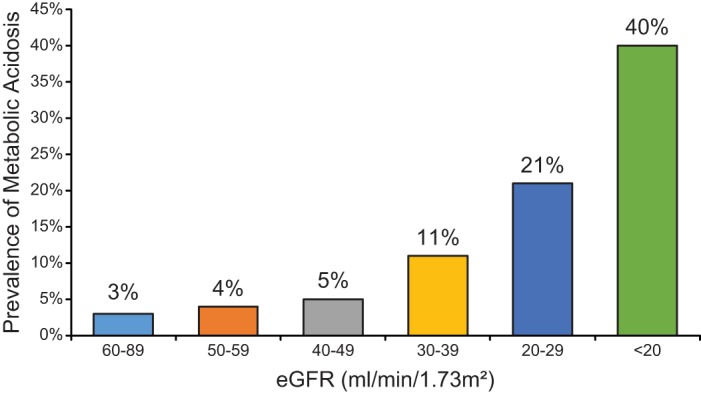
Correlation of metabolic acidosis with severity of chronic kidney disease. Progressive decreases in renal function correlate with an increased risk of metabolic acidosis. [Data from Moranne et al. (194).]
Adaptive responses in ammonia metabolism appear to facilitate maintenance of urinary ammonia and net acid excretion despite the decreased number of functional nephron units. These responses likely include residual renal hypertrophy (125, 158), increased proximal tubule NHE3 and thick ascending limb NKCC2 expression (158), increased apical polarization of H+-ATPase (18), and increased apical and basolateral Rhcg polarization (148). Several studies show that ammonia excretion, when adjusted for glomerular filtration rate (GFR) as a measure of functional nephron number, actually increases with CKD (20, 58, 59, 74, 148, 181, 253).
However, with worsening of CKD the ability to compensate for decreased nephron number is gradually lost, and impairments in urinary ammonia excretion likely contribute to the increased risk of metabolic acidosis with progressive CKD (237, 292). The decreased ammonia excretion seen in a rat model of advanced CKD is associated with impaired expression of multiple proteins involved in renal ammonia metabolism, including the glutamine transporter SN1, the ammoniagenic enzymes PDG and PEPCK, and the ammonia transporter Rhcg (37).
Whether ammonia itself directly contributes to the progression of CKD is an important but as yet unresolved question. A classic study, one of the early studies to show that metabolic acidosis contributes to CKD progression, suggested that ammonia itself, possibly by activating complement, accelerated the progression of renal injury (212). A proposed mechanism was that ammonia reacted with C3 to form a convertase that activates the alternative complement pathway (212).
However, several lines of evidence suggest that interstitial ammonia may not be a primary pathogenic factor causing progressive CKD. First, a subsequent study from the same research group that suggested that interstitial ammonia was pathogenic showed that hyperosmolality induced by sodium chloride and urea, as present in the renal medulla, blocked cellular injury mediated by the alternative complement pathway (47). The authors concluded that hyperosmolality protected the renal medulla from immune injury triggered by ammonia's modification of C3 and activation of the alternative pathway (47). Second, the recent evidence that interstitial sulfatides reversibly bind interstitial ammonia (281) identifies a mechanism that would minimize unbound interstitial ammonia concentrations and thereby decrease ammonia-mediated modification of C3 and alternative pathway activation. Finally, after controlling for the degree of metabolic acidosis, worsening renal function actually correlates with low total urinary ammonia content and low urinary ammonia concentration, not high urinary ammonia concentration (292). Thus maintenance of adequate renal ammonia metabolism may be protective for patients with CKD.
C. Ammonia Metabolism in RTA
Ammonia metabolism appears to be abnormal in all three major forms of RTA. However, the pathophysiological mechanisms involved in abnormal ammonia metabolism, transport, and excretion are likely different in type I (distal), type II (proximal), and type IV (hyperkalemic) RTA.
1. Type I (distal) RTA
Type I (distal) RTA is a condition characterized by non-anion gap metabolic acidosis, which is often very severe, hypokalemia, and failure of the normal ability to acidify urine pH despite severe metabolic acidosis (140, 242, 260, 276). In addition, urinary ammonia excretion is impaired (108, 238, 325). The primary defect in genetic forms of type I RTA involves proteins involved in urine acidification, either H+-ATPase or AE1 (63, 136, 138, 139, 261, 262, 272). To date, mutations in proteins involved in collecting duct ammonia transport have not been reported in patients with RTA. Paralleling this genetic information, the correlation between urinary ammonia excretion and urine pH is similar to if not greater in patients with type I RTA than in normal individuals (238). Thus the impaired ammonia excretion likely results from the higher urine pH, which increases the proportion of urinary NH3 relative to NH4+, which would decrease the rate of collecting duct facilitated NH3 secretion.
2. Incomplete RTA
Incomplete distal RTA is identified by normal basal acid-base homeostasis and normal urinary pH, but accompanied by an impaired ability to acidify urine in response to acute metabolic acidosis, typically induced by oral ammonium chloride loading (325). Most of these patients come to medical attention because of either nephrocalcinosis or recurrent nephrolithiasis. Interestingly, these patients have normal basal urinary ammonia excretion and normal, if not accentuated, ammonia excretion in response to acute metabolic acidosis (220, 325). They also exhibit hypocitraturia (66, 220), which likely contributes to the nephrolithiasis and/or nephrocalcinosis that brought them to medical attention. At present, the molecular mechanisms underlying incomplete distal RTA are not fully understood.
Incomplete RTA, associated with impaired urine acidification in response to acid-loading, should be differentiated from conditions associated with accentuated ability to decrease urine pH. The latter is observed in mouse models in response to deletion of either of the Rh glycoproteins, Rhbg or Rhcg, where there is an exaggerated decrease in urine pH associated with decreased ammonia excretion in response to acid-loading (23, 24, 160, 165, 167). The greater decrease in urine pH likely reflects intact collecting duct H+ secretion via H+-ATPase and H+-K+-ATPase in the setting of impaired Rhbg/Rhcg-mediated NH3 secretion.
Because the urinary findings and the underlying causes differ between Rhbg or Rhcg deletion in mice and those observed in classic “incomplete RTA” in humans, we propose using terminology that reflects these differences. We suggest that the “classic” form of “incomplete RTA” (an impaired ability to decrease urine pH after an exogenous acid load) should be termed “incomplete RTA, type A” and that that an accentuated decrease in urinary pH in combination with impaired ammonia excretion, as seen with genetic deletion of either Rhbg or Rhcg, should be termed “incomplete RTA, type B.” This addition to our terminology would recognize, and emphasize, the different pathophysiologic mechanisms involved.
3. Type II (proximal) RTA
Isolated proximal RTA is characterized typically by chronic metabolic acidosis, hypokalemia, elevated fractional excretion of bicarbonate when serum bicarbonate is normal, and intact ability to acidify the urine in the absence of exogenous alkali administration (131, 143, 211, 242, 275, 276). Less well recognized is that isolated proximal RTA may also be associated with abnormal ammonia metabolism. Small clinical studies suggest that patients with isolated proximal RTA that are not receiving oral alkali therapy have a urinary ammonia excretion rate unchanged from that observed in unaffected control individuals with a normal serum bicarbonate (32, 169). Because the normal response to metabolic acidosis is a dramatic increase in ammonia excretion, this absence of increased urinary ammonia excretion in untreated type II RTA indicates abnormal ammonia metabolism. In addition, following an oral acid load, the increase in urinary ammonia excretion in individuals with isolated proximal RTA is significantly less than in unaffected control individuals (32). Thus isolated proximal RTA may also be associated with an impaired urinary ammonia response to both the basal level of acidosis and to acute acid loads.
We have recently suggested that the defect in ammonia metabolism in isolated proximal RTA may reflect a critical role of the basolateral sodium bicarbonate cotransporter NBCe1. Genetic defects in NBCe1 are the only identified genetic causes of isolated proximal RTA (62, 65, 130, 132, 290, 331). As observed in individuals with isolated proximal RTA, mice with genetic deletion of NBCe1 develop spontaneous metabolic acidosis (86, 123) despite an intact ability to lower urinary pH (123). However, mice with NBCe1 deletion have a substantial impairment of urinary ammonia excretion, with urinary ammonia suppressed ∼70% compared with wild-type nonacidotic mice (123). The impaired urinary ammonia excretion in these mice is due at least in part to abnormal expression of multiple enzymes involved in proximal tubule ammonia generation, including phosphate-dependent glutaminase, glutamate dehydrogenase, phosphoenolpyruvate carboxykinase, and glutamine synthetase (163, 219). There is no apparent abnormality in expression of the thick ascending limb ammonia transporter, NKCC2, or the collecting duct ammonia transporters, Rhbg or Rhcg (163). These findings indicate that NBCe1 expression is necessary for normal proximal tubule ammonia metabolism and provide a mechanistic explanation of the abnormal ammonia metabolism in individuals with isolated proximal RTA.
4. Type IV RTA
Type IV RTA, perhaps the most common form of RTA, is characterized by hyperkalemia and non-anion gap metabolic acidosis in the absence of abnormal acid-base loads (141, 276). The ability to acidify the urine is generally intact (154). Renal function, as assessed by the GFR, is often suppressed, but not sufficiently to explain the degree of metabolic acidosis observed. Urinary ammonia excretion is typically suppressed in type IV RTA (57, 154). Likely causes of suppressed urinary ammonia excretion include hypoaldosteronism, concomitant interstitial fibrosis limiting ammonia transfer between the thick ascending limb and the collecting duct, and possible impairment of proximal tubule ammonia generation from the hyperkalemia itself (141, 236, 250, 257).
II. INTEGRATED OVERVIEW OF RENAL AMMONIA METABOLISM
Renal ammonia metabolism differs significantly from the renal handling of other solutes. For most urinary constituents, urinary excretion derives from arterial delivery. For these, glomerular filtration and regulated tubular transport determine urinary excretion, and the renal arterial delivery exceeds the renal venous return. This is not the case for ammonia. Essentially all urinary ammonia is generated in the kidney from amino acid metabolism; quantitative estimates indicate that only 2–3% of urinary ammonia derives from glomerular filtration (114, 310, 312). Also in contrast to most other urinary solutes, even though the kidneys excrete ammonia, significant amounts of ammonia produced in the kidney enter the renal vein and add to systemic ammonia loads (61, 81, 222, 289). The importance of this differential distribution, urine versus the renal vein, will be discussed below.
A. Mechanisms of Renal Ammonia Generation
In contrast to most urinary solutes, the majority of urinary ammonia is generated in the kidney, and does not derive from arterial delivery. Renal epithelial cells produce ammonia from the metabolism of amino acids, with glutamine serving as the primary metabolic substrate (187, 221, 229). Several other amino acids can also be used, but they are quantitatively less important in basal ammonia generation and do not appear to contribute to adaptive changes, such as occur in response to metabolic acidosis (221, 229). Because the proximal tubule is the site of the majority of ammonia generation and is the main site of adaptive changes (96, 215, 216), the majority of evidence regarding the molecular mechanisms of ammonia generation derives from studies that primarily address the proximal tubule. Figure 2 shows a summary of mechanisms involved that will be discussed below. However, space limitations preclude a complete discussion of all transport and enzymatic mechanisms involved in ammonia generation. Below we summarize the mechanisms thought currently to have primary roles in this process. The interested reader is directed to several excellent reviews on this specific topic for additional information (26, 54, 88, 286, 288).
FIGURE 2.

Overview of renal glutamine metabolism leading to ammonia generation. Filtered glutamine is essentially 100% reabsorbed by uptake by the apical amino acid transporter, B°AT1. It can then either enter mitochondria via a variety of glutamine transporters, or alternatively, glutamine can be transported across the basolateral membrane via the heterotrimeric amino acid exchangers, LAT2-4F2hc or Y+LAT1-4F2hc, resulting in net glutamine reabsorption. These basolateral glutamine transporters are likely functionally coupled to aromatic amino acid recycling involving TAT1. The basolateral glutamine transporter, SN1, also contributes to glutamine uptake. Under basal conditions, this transporter contributes to glutamine uptake primarily in the straight proximal tubule segments in the outer stripe and deep inner cortex, but in response to several conditions that increase glutamine uptake, SN1 expression increases and is evident in the proximal convoluted tubules and proximal straight tubules throughout the cortex as well. Mitochondrial glutamine is then metabolized through PDG and GDH, resulting in release of two NH4+ molecules and generation of 2-oxoglutarate. 2-Oxoglutarate is then metabolized through a series of enzymatic reactions forming malate. Malate is then transported out of mitochondria via a malate-phosphate exchange activity. Cytoplasmic malate is metabolized via the combined actions of malate dehydrogenase (MDH) and phosphoenolpyruvate carboxykinase (PEPCK), forming phosphoenolpyruvate (PEP). PEP can be metabolized by pyruvate kinase (PK) to form pyruvate, which can enter mitochondria and the tricarboxylic acid cycle, where it is metabolized to carbon dioxide and bicarbonate. Alternatively, PEP can be metabolized to fructose-1,6-bisphosphate (F-1,6-P), which is converted to fructose-6-phosphate (F-6-P) by fructose-1,6-bisphosphatase (F-1,6,P2ase). Fructose-6-phosphate is converted to glucose-6-phosphate (G-6-P), which is then converted to glucose by glucose-6-phosphatase (G-6-Pase). Glucose then exits across the basolateral plasma membrane by either Glut2 or Glut1. Dashed lines indicate enzymatic processes involving several steps, the details of which are not shown.
1. Glutamine transport
Glutamine uptake appears to involve both apical and basolateral transport mechanisms in the proximal tubule. Glutamine is small enough in molecular size that it is essentially completely filtered by the glomerulus. Luminal uptake appears to occur primarily via the broad specificity, Na+-dependent amino acid cotransporter BoAT1 (SLC6A19) (243). The proximal tubule reabsorbs essentially 100% of filtered glutamine (265), and adaptive changes in apical amino acid transporter expression do not appear to occur in conditions of altered glutamine uptake, such as metabolic acidosis. Filtered glutamine transported across the apical plasma membrane and not utilized for ammoniagenesis can be transported across the basolateral plasma membrane via LAT2-4F2hc (SLC7A8-SLC3A2) and Y+LAT1-4F2hc (SLC7A7-SLC3A2), two heterodimeric neutral amino acid exchangers (19, 228, 245); this enables net glutamine reabsorption. Because both LAT2-4F2hc and Y+LAT1-4F2hc are obligatory amino acid exchangers, the amino acid transported into the cell may exit via basolateral TAT1 (Slc16a10), a facilitated aromatic amino acid transporter (19, 233, 234).
During metabolic acidosis, proximal tubule glutamine uptake can rise to as much as 35% of arterial glutamine (226). Because this is greater than filtered glutamine, activation of a basolateral glutamine uptake mechanism is necessary. This appears to involve the Na+-coupled glutamine transporter SN1 (SLC38A3) (38, 142, 274). This protein is expressed primarily in the proximal straight tubule in the outer stripe of the outer medulla under basal conditions (38, 274). In response to metabolic acidosis, hypokalemia, and high dietary protein intake, three conditions which are associated with stimulation of renal ammoniagenesis, SN1 expression increases and becomes prominent in the proximal convoluted tubule segments and increased in the proximal straight segment in the outer stripe (38, 142, 195, 274). During metabolic acidosis, there is a simultaneous decreased expression of mRNA for the basolateral glutamine efflux transporter Y+LAT1 (195). This coordinate regulation of basolateral glutamine uptake and efflux transporters facilitates the increased net cellular glutamine uptake necessary for its role as a substrate for ammoniagenesis.
2. Enzymes involved in ammonia generation
Proximal tubule glutamine metabolism involves a number of enzymatic steps, and a number of alternative pathways. This review, because its focus is on ammonia transport mechanisms, will confine discussion of ammonia metabolism to a summary of the primary metabolic pathway. Glutamine uptake into mitochondria is the initial step, and appears to involve at least two functionally distinct glutamine antiporters (1, 270). During metabolic acidosis, increased mitochondrial glutamine uptake may occur via a glutamine uniporter (10). The kidney-type glutaminase (KGA) isoform of phosphate-dependent glutaminase (PDG) is present in the inner mitochondrial membrane (52, 53, 56, 231) and catalyzes the reaction: Glutamine Glutamate + NH4+. Glutamate is converted to 2-oxoglutarate (also known as α-ketoglutarate) via the mitochondrial enzyme glutamate dehydrogenase (GDH): Glutamate 2-Oxoglutarate + NH4+. 2-Oxoglutarate is then metabolized through 2-oxoglutarate dehydrogenase complex and succinyl-CoA synthetase to form succinate. Succinate is metabolized via succinate dehydrogenase and fumarase to form malate. Malate is transported out of the mitochondria into the cytoplasm; this may occur via a malate-phosphate exchange activity (43, 252). Malate is then converted to oxaloacetic acid (OAA) by the enzyme malate dehydrogenase (230). OAA is a substrate for phosphoenolpyruvate carboxykinase (PEPCK) (254). Phosphoenolpyruvate that is generated can then either be used for gluconeogenesis via a glucose-6-phosphatase-dependent pathway (27, 189, 251, 254) or it can be converted to pyruvate via pyruvate kinase and enter the tricarboxylic acid pathway (251). Both pathways result in generation of two HCO3− molecules. The net reaction for complete renal glutamine metabolism is either 2 Glutamine (C5H10O3N2) + 6 H2O + 3 O2 → Glucose (C6H12O6) + 4 HCO3− + 4 NH4+ or 2 Glutamine (C5H10O3N2) + 9 O2 → 6 CO2 + 4 HCO3− + 4 NH4+.
Conditions that increase ammonia generation, such as metabolic acidosis and hypokalemia, cause coordinated stimulation of multiple components of this pathway, including SN1, PDG, GDH, and PEPCK (3, 38, 51, 52, 55, 142, 178, 219, 227, 274, 333).
3. Ammonia recycling
The proximal tubule also can counter ammoniagenesis, at least at the level immediately following PDG, through the action of glutamine synthetase. The enzyme glutamine synthetase catalyzes the reaction: Glutamate + NH4+ → Glutamine + H+ (296), thereby effectively reversing the PDG reaction. Glutamine synthetase is a cytoplasmic enzyme, and its expression is normally regulated in the opposite direction as the enzymes involved in ammonia generation in a variety of conditions (49, 123, 160, 162–164, 296). Deletion of glutamine synthetase specifically from the proximal tubule increases renal ammonia excretion, indicating glutamine synthetase-mediated ammonia recycling is active under basal conditions and contributes to basal ammonia metabolism homeostasis (163). Its deletion also impairs the response to experimental metabolic acidosis, indicating that decreasing glutamine synthetase expression is a key component of the integrated response necessary to increase net ammonia generation (163).
4. Renal gluconeogenesis in association with ammoniagenesis
As mentioned previously, glucose is generated during the process of ammoniagenesis. Although the liver is generally considered the most important source of glucose under most physiological circumstances, the kidney can generate substantial amounts of blood glucose under certain circumstances. Examples include starvation, metabolic acidosis, and diabetic ketoacidosis (145, 223). Under basal conditions, renal gluconeogenesis may account for ∼20% of total glucose release in normal postabsorptive humans (88). During prolonged starvation, renal gluconeogenesis may provide as much as 45% of total blood glucose (223). Renal gluconeogenesis increases in many conditions, such as metabolic acidosis and hypokalemia, in parallel with changes in ammonia generation and excretion (46, 46, 100, 137). This increase in renal glucose generation likely contributes to the impaired glucose tolerance seen in these two conditions (182, 183, 246). The rate-limiting enzymes are likely to include PEPCK, fructose-1,6-bisphosphatase, and glucose-6-phosphatase (254). However, the finding that pharmacological inhibitors of renal gluconeogenesis do not alter ammoniagenesis (46) suggests renal gluconeogenesis can be dissociated from ammoniagenesis. A complete discussion of renal gluconeogenesis is beyond the scope of this manuscript, and the interested reader is referred to several more comprehensive publications on this topic (192, 254, 282, 323).
5. Bicarbonate transport in ammoniagenesis
The bicarbonate produced during ammoniagenesis is preferentially transported across the basolateral plasma membrane in the proximal tubule. This bicarbonate is then delivered to the systemic circulation via the peritubular capillaries and renal veins. As such, this is “new” bicarbonate generated by the kidney and enables renal venous bicarbonate to exceed renal arterial delivery.
B. Role of Renal Ammonia Transport
The ammonia produced in the kidney then undergoes transport by renal epithelial cells, and this transport determines the proportion of ammonia that is excreted into the urine or delivered to the systemic circulation via the renal veins. Studies in humans indicate that under basal conditions ∼50% of renal ammonia is excreted in the urine and 50% enters the renal veins and results in systemic ammonia loads (61, 85, 221). However, in response to metabolic acidosis, the proportion that is excreted increases significantly, to 70–80% (61, 221) (Figure 3).
FIGURE 3.
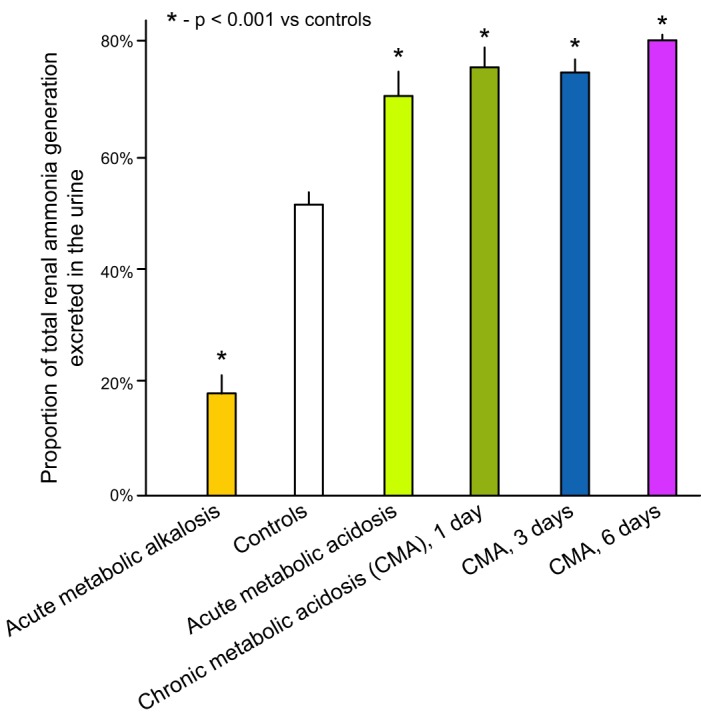
Regulation of proportional excretion of renal ammonia generated by acid-base conditions. Acute metabolic acidosis and alkalosis change the proportion of ammonia generated in the kidney that is excreted in the urine, where it contributes to net new bicarbonate generation, as opposed to systemic addition by transport into the renal veins, where it does not. During the chronic phase of metabolic acidosis, there is no further increase in the proportion of ammonia generated that is excreted in the urine; changes in net ammonia excretion are the result of increased net ammoniagenesis in chronic metabolic acidosis. [Data from Deferrari et al. (61).]
1. Adverse effects of renal ammonia delivered to the systemic circulation
Ammonia generated in the kidneys and added to the systemic circulation via the renal veins has two important adverse effects. First, ammonia is a neurotoxin, and excessive systemic ammonia levels can lead to altered mental status, lethargy, cerebral edema, and even death (283, 287). Under normal conditions, hepatocytes prevent this through nearly 100% first-pass ammonia clearance and metabolism. However, in the presence of liver disease, this process is impaired, and systemic renal ammonia addition can lead to hyperammonemic encephalopathy. Second, hepatic ammonia metabolism negates the beneficial effect of renal ammonia generation from an acid-base perspective. There are two predominant pathways of ammonia metabolism in the liver, a ureagenic pathway and a glutamine-producing pathway (124). The ureagenic pathway utilizes equimolar bicarbonate as a co-substrate with ammonia to produce urea. The glutamine-producing pathway synthesizes glutamine from equimolar glutamate and NH4+ via the enzyme glutamine synthetase and in the process releases a H+. This H+ reacts with bicarbonate forming carbonic acid. Carbonic acid undergoes spontaneous dehydration, resulting in CO2 generation, and the CO2 is eliminated by respiration. Notably, both pathways of hepatic NH4+ metabolism utilize equimolar bicarbonate. Since renal ammonia generation produces equimolar bicarbonate (54, 288), renal ammonia that enters the systemic circulation and undergoes hepatic ammonia metabolism results in no net bicarbonate gain. Accordingly, only the ammonia excreted in the urine is associated with actual new bicarbonate generation.
2. Beneficial effect of renal ammonia transport
Two distinct mechanisms regulate urinary ammonia excretion: net ammonia generation and renal epithelial cell ammonia transport. Changes in net renal ammonia generation occur from coordinate regulation of extrarenal glutamine release, renal tubular cell glutamine uptake, cellular ammonia generation, and cellular ammonia recycling. The rate of renal ammoniagenesis can increase severalfold over baseline, but maximal increases in ammoniagenesis require days to develop, at least in the proximal tubule S1 segment of acid-loaded rats (64). In contrast, changes in renal epithelial cell transport can alter the proportion of ammonia generated in the kidney that is excreted in the urine within minutes (61). However, the capacity of renal tubular cell ammonia transport to alter ammonia excretion, independent of changes in net ammonia generation, is limited. Under basal conditions, ∼50% of ammonia generated in the kidney is excreted in the urine (61, 85, 221), and in response to acute acidosis this can increase to 70–80% of ammonia generated in the kidney (61, 221). Based on these observations, the relative increase in ammonia excretion that can occur from changes in renal epithelial cell ammonia transport alone appears to be limited to a 40–60% increase.
3. Overview of renal ammonia transport
Ammonia produced in the kidney undergoes transport in essentially all renal epithelial segments. Figure 4 summarizes an integrated view of this movement. In the kidney, ammonia is generated predominantly in the proximal tubule and is secreted preferentially into the luminal fluid. At the end of the proximal tubule amenable to micropuncture, which is the late proximal convoluted tubule, the ammonia content is similar to total urinary ammonia excretion (35, 36, 97, 248, 266). There is ammonia secretion in the proximal straight tubule and thin descending limb of the loop of Henle, but ammonia reabsorption in the thin and thick ascending limb of the loop of Henle, and the net effect of this ammonia recycling is to induce an interstitial ammonia gradient that increases from cortex to outer medulla to inner medulla (8, 224, 241, 247). Overall, there is net ammonia reabsorption in the loop of Henle. Quantitative assessment, using in vivo micropuncture, shows that luminal ammonia delivery at the end of the thick ascending limb, as assessed by ammonia content in the early distal tubule, is substantially less than ammonia delivery at the end of the proximal convoluted tubule, and accounts for only 20–40% of urinary ammonia (248, 266). Ammonia is then secreted, predominantly in the collecting duct, to reach final urinary ammonia excretion rates.
FIGURE 4.

Overall model of renal epithelial ammonia transport under basal conditions. Numbers in blue show ammonia amounts relative to total urinary ammonia (100%). Under basal conditions, relatively little urinary ammonia derives from glomerular filtration. The proximal tubule generates ammonia. This occurs primarily via metabolism of glutamine, with generation of 2 NH4+ and 2 HCO3− from each glutamine completely metabolized. NH4+ is secreted preferentially into the luminal fluid through a mechanism that appears to involve NHE3-mediated Na+/NH4+ exchange. Between the end of the proximal tubule that is accessible to micropuncture and the bend of the loop of Henle, there is ammonia secretion; this likely involves components of secretion in the proximal tubule straight tubule and in the thin descending limb of the loop of Henle. There is sufficient ammonia secretion such that ammonia delivery to the bend of the loop of Henle averages 160% of total urinary ammonia. In the ascending limb of the loop of Henle, there is ammonia reabsorption; this primarily occurs in the thick ascending limb of the loop of Henle via NKCC2-mediated NH4+ uptake. At the early distal tubule, luminal ammonia accounts for only 20–40% of urinary ammonia excretion. The collecting duct, and possibly the DCT and the CNT, secrete ammonia, and account for the remaining 60–80% of urinary ammonia. Collecting duct ammonia secretion involves parallel H+ and NH3 transport, with no measureable transepithelial NH4+ permeability. Interstitial sulfatides (represented by the molecule encircled in green area) reversibly bind ammonia, limiting ionized interstitial ammonia concentrations.
Early models of renal ammonia metabolism suggested that ammonia transport could be explained through the simple paradigm of NH4+ trapping and NH3 diffusion equilibrium. In this model, NH4+ could not be transported across plasma membranes, and NH3, because it was a small and uncharged molecule, diffused essentially instantaneously across lipid bilayers and was in diffusion equilibrium throughout the kidney. Both aspects of this previous paradigm are now known to be incorrect, and that specific membrane proteins have critical roles in the movement of both NH4+ and NH3 across renal epithelial cell plasma membranes.
III. BIOPHYSICAL CHARACTERISTICS OF THE AMMONIA MOLECULAR SPECIES, NH3 AND NH4+
Understanding the molecular mechanisms of ammonia transport requires recognizing that ammonia exists in two different molecular forms, NH3 and NH4+, and that these two molecular forms have different transport characteristics. NH3 and NH4+ are in nearly instantaneous equilibrium with each other via the buffer reaction, NH3 + H+ → NH4+. This buffer reaction has a pKa of ∼9.15 at biologically relevant temperature and osmolality. Accordingly, in biological systems, the vast majority of ammonia is present as NH4+, and only a very small amount is present as NH3. Figure 5 summarizes the effects of pH on the relative amounts of NH3 and NH4+ in a biological fluid. Because this is a proton-dependent buffer reaction, the actual amounts of NH3 and NH4+ in any biological fluids vary as a function of pH. Because the biologically relevant pH is typically significantly below the pKa of the buffer reaction, the amount present as NH4+ does not vary substantially. However, the amount present as NH3 varies almost exponentially as a function of pH. Thus mechanisms that mediate transmembrane movement of NH3 are influenced significantly by changes in extracellular and intracellular pH.
FIGURE 5.
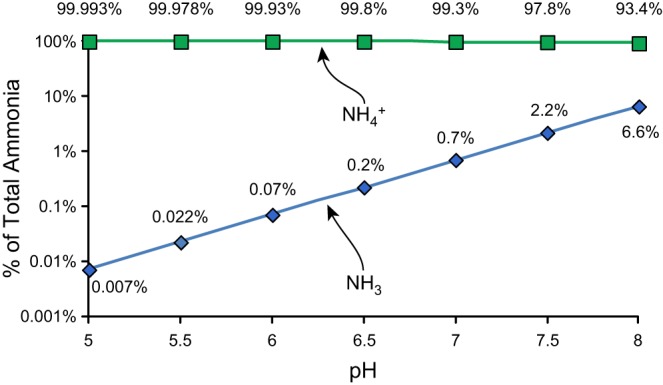
Effect of biological fluid pH on proportion of ammonia as NH3 and NH4+. Ammonia consists of two molecular species, NH3 and NH4+, which are in equilibrium with each other in the reaction NH3 + H+ ↔ NH4+. The pKa' of this reaction is ∼9.15 at biologically relevant ionic concentrations. As a result, at biologically relevant pH levels, the majority of total ammonia is in the molecular form NH4+. The proportion of total ammonia present as NH3 is very dependent on actual pH, whereas the proportion present as NH4+ does not change substantially.
NH4+ is a small, positively charged molecule, and hence was initially thought to be unable to cross cell membranes. However, detailed analysis has shown that NH4+ has essentially identical biophysical characteristics in aqueous solutions as potassium ion (K+) (9, 73, 197, 307). Table 1 summarizes these characteristics. Identical biophysical characteristics suggest that NH4+ might be able to substitute for potassium ion in many proteins; evidence now indicates that NH4+ can substitute for potassium in essentially all potassium transporters (21, 45, 67, 92, 126, 149, 170, 307). NH4+ is also transported in place of H+ by the sodium-hydrogen exchanger isoforms NHE3 and NHE4 (31, 150). The molecular mechanism through which NH4+ is transported at the proton binding site of specific isoforms is not understood at present. One possible, but untested, mechanism is that certain isoforms transport hydronium (H3O+), not H+, and that NH4+ substitutes for hydronium, which is closer in biophysical characteristics to NH4+. Nevertheless, multiple ion transporters are known to transport NH4+ across lipid membranes; this transport is important in renal epithelial ammonia metabolism.
Table 1.
Biophysical characteristics of common cations
| Cation | Atomic Weight | Ionic Radius, Å | Hydrodynamic Radius, Å | Mobility in H2O, 10−4 cm2·s−1·V−1 | TiH2O |
|---|---|---|---|---|---|
| Li+ | 6 | 0.060 | 1.73 | 4.01 | 0.33 |
| Na+ | 23 | 0.095 | 1.67 | 5.19 | 0.39 |
| NH4+ | 18 | 0.133 | 1.14 | 7.60 | 0.49 |
| K+ | 39 | 0.143 | 1.14 | 7.62 | 0.49 |
NH3 is a small, uncharged molecule, and this led initially to the conclusion that it had a high permeability across lipid membranes and rapid diffusion across lipid bilayers. However, although NH3 is uncharged, it is actually a polar molecule. The second orbital shell around the central nitrogen has eight electrons; there are three sets of paired electrons shared with the three hydrogen atoms (three electrons deriving from the nitrogen atom and three electrons from the three hydrogen atoms) and two unshared electrons that derive from the nitrogen. The negatively charged electrons and the positively charged hydrogen nuclei are asymmetrically distributed around the central nitrogen (Figure 6). Consequently, NH3, while an uncharged molecule, is actually a polar molecule. Quantitatively, NH3 has a molecular dipole moment, a standard measure of molecular polarity, measured at 1.47–1.56 (109, 190, 284), a value close to that of water, 1.85 (16, 48). Thus, in many respects, NH3 is similar to H2O and urea, two other small, uncharged, yet polar molecules that undergo specific and regulated transmembrane movement in renal epithelial cells.
FIGURE 6.

Molecular model of NH3, H2O, and urea showing electrostatic charge distribution. NH3, H2O, and urea are all uncharged molecules, but because of asymmetric distributions of charged elements have significant molecular polarity. Shown are space-filling models of each, with electrostatic charge distribution shown. Models were generated using Avogadro, v1.0.3.
Several studies have directly examined NH3 permeability across lipid membranes. First, direct measurements show that the cortex, outer medulla, and inner medulla of the kidney have significantly different NH3 concentrations (98). This can only occur if there are significant barriers, i.e., limited permeability, to NH3 movement. Second, direct measurements of NH3 transport have shown differences in NH3 permeability between the renal proximal tubule and the collecting duct (115). Finally, studies in the medullary thick ascending limb have shown that apical NH3 permeability is very low, and that apical ammonia permeability is dominated by NH4+ permeability (146, 240). Consistent with this is the finding that the apical membranes of gastric and colonic epithelial cells have little to no NH3 permeability (271, 298). Thus multiple lines of evidence indicate that NH3 permeability across lipid bilayers is finite. As a consequence, integral membrane proteins that transport NH3 facilitate and can regulate transmembrane NH3 movement. As will be discussed in more detail below, two integral membrane protein families, Rhesus glycoproteins and aquaporins, are capable of transporting NH3 and thereby contributing to regulated ammonia transport.
IV. PROXIMAL TUBULE AMMONIA TRANSPORT
A. Overview of Proximal Tubule Ammonia Transport
Ammonia is produced by almost all renal epithelial cells, but the proximal tubule is the primary site of ammoniagenesis. Studies examining dissected renal segments [including the glomeruli; proximal tubule segments S1, S2, and S3; descending thin limb of the loop of Henle; medullary and cortical thick ascending limb of the loop of Henle; distal convoluted tubule; cortical collecting duct (CCD); outer medullary collecting duct (OMCD) and inner medullary collecting duct (IMCD)] show that all have the capability to produce ammonia using glutamine as their primary metabolic substrate (96). Although essentially all renal epithelial cells can produce ammonia, metabolic acidosis increases only proximal tubule ammoniagenesis (96, 203, 324). Ammonia produced in proximal tubule segments is then secreted preferentially into the tubule lumen (200, 201) This combination of proximal tubule ammonia generation and preferential luminal secretion results in ammonia delivery to the end of the proximal tubule amenable to micropuncture that is essentially equivalent to urinary ammonia excretion (114). Figure 7 shows an integrated model of ammonia transporters in the proximal tubule. We discuss below the evidence of the involvement of each of the major transport mechanisms involved in proximal tubule ammonia secretion.
FIGURE 7.

Ammonia transport mechanisms in the proximal tubule. Glutamine serves as the primary metabolic substrate for ammoniagenesis. Proximal tubule glutamine uptake (influx) involves transport across the apical plasma membrane, primarily via the broad specificity, Na+-dependent amino acid cotransporter BOAT-1 (SLC6A19), and across the basolateral plasma membrane by the Na+-coupled neutral amino acid transporter SN1 (SLC38A3). Complete metabolism of each glutamine results in mitochondrial generation of two NH4+ and two HCO3−. At least a component of mitochondrial ammonia appears to be transported from mitochondria to the cytosol via AQP8. Although shown as NH4+ transport, whether this occurs as NH4+ transport or as AQP8-mediated NH3 transport with H+ transport through an alternative mechanism has not been examined. Bicarbonate is transported across the basolateral plasma membrane via the electrogenic sodium bicarbonate cotransporter, isoform 1A, NBCe-1A (SLC4A4). Ammonium secretion across the apical plasma membrane occurs primarily via NHE3-mediated Na+/NH4+exchange, with a lesser contribution by parallel H+ and NH3 transport. The mechanism enabling NH3 transport, whether diffusive transport across the apical plasma membrane or via a specific transporter, has not been identified.
An additional mechanism regulating proximal tubule ammonia metabolism and transport likely involves the enzyme glutamine synthetase. This is a cytosolic protein predominantly expressed in the proximal tubule which mediates the synthesis of glutamine from NH4+ and glutamate, i.e., “recycling” ammonia (49). During metabolic acidosis there is decreased glutamine synthetase expression and activity (49, 162, 163, 296), and proximal tubule-specific glutamine synthetase deletion impairs both basal and acidosis-stimulated renal ammonia metabolism and excretion (163). This likely leads to increased cytosolic ammonia, which facilitates ammonia secretion and blunts ammonia reabsorption.
B. NHE3
1. Evidence of involvement
At present, substantial evidence indicates that the apical Na+/H+ exchanger, NHE3, mediates a substantial component of proximal tubule ammonia secretion. In this model, NH4+ substitutes for cytosolic H+, resulting in functional Na+/NH4+ exchange activity. Data supporting this model include evidence that proximal tubule brush-border membrane vesicles exhibit NH4+/Na+ exchange activity (150), that combining a low luminal Na+ concentration with the Na+/H+ exchange inhibitor amiloride decreases proximal tubule ammonia secretion (199) and that the Na+/H+ exchange inhibitor EIPA, when used in combination with the K+-channel inhibitor BaCl2, inhibits proximal tubule ammonia transport (269).
However, there is also evidence inconsistent with NHE3 being the primary mechanism of proximal tubule luminal ammonia secretion. First, in vivo microperfusion studies of the rat proximal tubule suggested that NH3 transport in parallel with H+ secretion is a primary mechanism of proximal tubule ammonia secretion (267). Another study found that inhibiting NHE3 in rats with chronic metabolic acidosis did not alter proximal tubule ammonia secretion despite inhibiting bicarbonate reabsorption (268). Finally, a recent study examined mice with proximal tubule-specific NHE3 deletion (171). Urinary ammonia excretion was similar in wild-type mice and proximal tubule-specific NHE3 deletion mice, both under basal conditions and following induction of metabolic acidosis. This was despite an ∼30% decrease in proximal tubule bicarbonate reabsorption (equivalent to apical proton secretion). Unfortunately, direct measurements of proximal tubule ammonia transport were not performed. One interpretation, which incorporates all available data, is that NHE3 does have an important role in proximal tubule ammonia secretion, acting via a Na+/NH4+ exchange mode, but in addition there exists both a barium-sensitive NH4+ transport mechanism and concurrent parallel H+ and NH3 secretion.
2. Regulation in models of acid-base disturbances
Changes in NHE3 expression generally parallel proximal tubule ammonia transport. In response to chronic metabolic acidosis, changes in extracellular potassium, and Ang II stimulation, changes in NHE3 expression and activity parallel changes in ammonia secretion (4, 69, 205). In both the S2 and S3 segments, chronic metabolic acidosis increases AT1 receptor-mediated stimulation of NHE3 (202, 203, 204). Other studies show that increased endothelin-1 expression with subsequent activation of the endothelin-B receptor mediates an important role in increasing NHE3 expression and renal ammonia excretion in metabolic acidosis (159).
B. Ba2+-Sensitive Transport Mechanisms
The primary evidence that K+ channels contribute to proximal tubule ammonia transport comes from in vitro microperfusion studies showing that barium, a nonspecific K+ channel inhibitor, can inhibit proximal tubule ammonia transport (269). Interestingly, luminal barium by itself does not block ammonia secretion but does inhibit reabsorption significantly (269). The barium-sensitive pathway likely involves K+ channels that are able to transport NH4+ because of the previously described molecular mimicry between NH4+ and K+ (307). As such, the preferential role of apical K channels in NH4+ reabsorption is consistent with intracellular electronegativity facilitating NH4+ uptake rather than secretion. Multiple K+ channels are present in the apical plasma membrane of the proximal tubule, including KCNA10, KCNQ1/KCNE1, and TWIK-1; which of these contribute to ammonia transport is not currently known.
C. Parallel NH3 and H+ Secretion
Parallel H+ and NH3 secretion may also contribute to proximal tubule ammonia secretion. Apical proton secretion may involve either NHE3, acting in a Na+ for H+ exchange mode, or apical H+-ATPase, while NH3 secretion may occur through lipid phase diffusion across the apical plasma membrane. Direct measurements of proximal tubule ammonia permeability are consistent with this mechanism. A role for proton secretion via apical H+-ATPase and parallel NH3 secretion may well explain the observation that inhibiting both apical NHE3, with EIPA, and apical K+ channels, with BaCl2, decreased proximal tubule ammonia secretion by only 50% (269). It could also explain the observation that proximal tubule-specific NHE3 deletion did not apparently alter ammonia excretion (171). However, to our knowledge, H+-ATPase's specific role in proximal tubule ammonia secretion has not been experimentally tested using either specific inhibitors or genetically modified animals. Furthermore, whether other transport mechanisms, such as AQP1, enable NH3 transport has not been examined specifically.
D. Basolateral Plasma Membrane Transport
Mathematical modeling studies suggest that peritubular NH4+ uptake may occur via either basolateral Na+-K+-ATPase or by basolateral K+ channels (314). This ammonia is then available for apical secretion. The exact contribution of this pathway to proximal tubule ammonia secretion has not been examined experimentally.
E. Mitochondrial Ammonia Transport
The majority of ammonia generation occurs inside of the inner membrane of mitochondria in the proximal tubule. Thus there is a need for ammonia transport from mitochondria to cytoplasm. Current evidence supports a role for aquaporin-8 (AQP8) in this process. AQP8 is a member of the aquaporin family. In addition to AQP8's ability to transport H2O, it also transports ammonia and ammonia analogs when expressed in heterologous systems (127, 173, 249). In cultured proximal tubule cells, AQP8 is present in the inner mitochondrial membrane, and AQP8 knock-down decreased ammonia secretion by these cells (193). Finally, in vivo metabolic acidosis increases AQP8 expression (193). All of these findings support an important role of AQP8 in proximal tubule mitochondrial ammonia transport. Evidence against this role, however, is that AQP8 genetic deletion is reported to not alter urinary ammonia excretion either under basal conditions or following either acute or chronic acid loading (327).
V. THIN DESCENDING LIMB
There is a small component of ammonia transport in the thin descending limb (75). Interstitial ammonia concentrations are elevated in both the outer medulla and the inner medulla, primarily resulting from ammonia reabsorption by the thick ascending limb (see below). Both in vitro microperfused tubule studies (75) and mathematical modeling studies support the existence of ammonia secretion that likely has a role in maintaining the corticomedullary interstitial ammonia gradient (214, 316). Ammonia secretion appears to involve predominantly an NH3 permeability, with a smaller component of NH4+ permeability (75). The molecular mechanisms of thin descending limb NH3 and NH4+ transport have not been well characterized to date.
VI. THICK ASCENDING LIMB OF THE LOOP OF HENLE
The thick ascending limb of the loop of Henle (TAL) is an important site for reabsorbing luminal ammonia. This appears critical for generating the medullary interstitial ammonia concentration gradient, whereby ammonia concentration in the inner medulla is greater than in the outer medulla, which is greater than in the cortex. Ammonia reabsorption also decreases delivery to distal cortical segments. Quantitatively, TAL ammonia absorption results in ammonia delivery to the distal tubule accounting for only ∼20–40% of final urinary ammonia content (248, 266). Figure 8 shows a current model of thick ascending limb ammonia transport.
FIGURE 8.

Ammonia transporters in the thick ascending limb. The primary mechanism of apical ammonium absorption occurs through substitution of NH4+ for K+ and transport by the loop diuretic-sensitive, apical plasma membrane transporter NKCC2. Cytoplasmic NH4+ is transported across the basolateral membrane either via Na+/NH4+ exchange mediated by NHE4 or via a bicarbonate shuttling mechanism involving NH3 transport and NBCn1.
A. NKCC2
The primary mechanism of ammonia reabsorption in the TAL appears to involve NKCC2. NKCC2 is a kidney-specific Na+-K+-2Cl− cotransporter expressed in the apical plasma membrane of the TAL. K+ and NH4+ appear to compete for the same binding site; coupled transport with sodium and chloride ions results in a secondarily active NH4+ reabsorption. Because luminal NH4+ competes with K+ for binding to the K+-transport site, alterations in luminal K+ in hypokalemia and hyperkalemia alter net NH4+ reabsorption. Specifically, increased luminal K+ inhibits ammonia reabsorption and decreased luminal K+ increases reabsorption (92, 93). Pharmacological inhibitor studies show NKCC2 is the primary mechanism of TAL ammonia reabsorption. Specifically, inhibiting NKCC2 in vitro with luminal furosemide completely inhibited medullary TAL ammonia reabsorption and in cortical TAL segments converted net reabsorption to net secretion (99). In some studies, metabolic acidosis increases NKCC2 expression, which contributes to the increased TAL ammonia reabsorption observed (12, 94); these changes appear related to an increase in systemic glucocorticoid levels (13). However, in our experience, NKCC2 protein expression does not routinely change in the mouse kidney in response to metabolic acidosis (unpublished observations). Other mechanisms that regulate NKCC2 besides steady-state protein expression include membrane trafficking, phosphorylation, and protein-protein interactions (7); the extent to which these contribute to the regulation of TAL ammonia transport in response to metabolic acidosis, or other conditions of altered TAL ammonia transport, remains incompletely understood at present.
NKCC2-mediated NH4+ transport may also contribute to NaCl reabsorption in this site. Because NH4+ effectively substitutes for K+ in NKCC2-mediated transport, NH4+ transport directly contributes to NaCl reabsorption. Moreover, mathematical modeling suggests that luminal K+ concentrations in the TAL may be insufficient to account completely for the observed rates of NaCl reabsorption when considering luminal concentrations of Na+ versus K+ and observed rates of K+ recycling via apical K+ channels (317). This ability of luminal NH4+ to support NKCC2-mediated Na+ reabsorption may explain the clinical observation that Bartters syndrome due to defects in ROMK (Type 2), which would not alter NaCl reabsorption coupled to NH4+ reabsorption, produces less severe phenotypes than that due to defects in NKCC2, which would block NaCl reabsorption coupled to either K+ or NH4+ transport (317).
The effects of inhibiting NKCC2 on whole kidney ammonia handling are somewhat complicated. Acutely, NKCC2 inhibition increases urinary ammonia excretion (128, 129, 321), whereas chronic inhibition does not (321). These results are suggestive that acute NKCC2 blockade decreases ammonia reabsorption in the TAL, increasing ammonia delivery to more distal segments and thereby increasing urinary ammonia excretion. During chronic NKCC2 inhibition, interstitial ammonia levels decrease (224, 299). It is possible that decreased interstitial ammonia leads to decreased collecting duct ammonia secretion, which counterbalances the increased ammonia delivery resulting from decreased reabsorption in the TAL, and thereby prevents chronic increases in urinary ammonia excretion.
B. NHE3
Apical NHE3 is also present in the TAL (5). However, NHE3 secretes ammonium, whereas the thick ascending limb is a site of net ammonia reabsorption. Thus NHE3 does not appear to have an important role in TAL ammonia transport.
C. Ba2+-Sensitive K+ Channels
In the TAL, electrogenic luminal NH4+ uptake can occur via apical K+ channels when NKCC2 is inhibited (11). However, because inhibiting NKCC2 almost completely inhibits TAL ammonia reabsorption, apical K+ channels are unlikely to mediate a quantitatively important role in TAL ammonia transport (95, 99).
D. K+/NH4+ Exchange
Physiological studies have demonstrated that the TAL contains an apical electroneutral K+-NH4+ (H+) activity that can be inhibited by barium and verapamil (6). As yet, the specific protein that mediates this activity is unidentified, nor has the contribution of this activity to transepithelial ammonia transport been determined. However, indirect evidence from studies that have demonstrated that inhibition of NKCC2 essentially abolishes transepithelial TAL ammonia transport (99, 304) suggests that apical K+-NH4+ exchange activity is unlikely to contribute significantly to ammonia reabsorption by the TAL.
E. NHE4
Luminal ammonia reabsorbed by the TAL must then be transported across the basolateral plasma membrane to enable net ammonia reabsorption. Studies of the TAL basolateral membrane vesicles show that a basolateral Na+ for NH4+ exchange activity is present that may have an important role in basolateral NH4+ exit (25). Na+/H+ exchanger isoform 4 (NHE4) is expressed in the TAL basolateral plasma membrane (42) and appears to mediate this activity (31). Both NHE4-specific inhibitors and NHE4 gene deletion significantly decrease medullary thick ascending limb ammonia reabsorption (31). Mice with NHE4 deletion have spontaneous metabolic acidosis with inappropriate urinary net acid excretion, and in response to exogenous acid loads, NHE4 deletion blocks the ability to increase urinary ammonia excretion and it reduces medullary ammonia content (31). Finally, metabolic acidosis increases NHE4 expression, and it enhances basolateral Na+/H+ exchange activity in isolated perfused rat medullary TAL segments (31). Thus basolateral NHE4 appears to be necessary for basal TAL ammonia reabsorption and increased NHE4 expression in response to metabolic acidosis likely contributes to the increased TAL ammonia transport observed. However, NHE4 deletion does not completely block ammonia reabsorption by the thick ascending limb (31). This indicates that other mechanisms of basolateral ammonia transport are present.
This role of NHE4 in TAL basolateral ammonia transport appears to be specific to NHE4. Another member of the Na+/H+ exchanger family, NHE1, is also expressed in the basolateral plasma membrane, yet inhibiting NHE1 does not alter TAL ammonia transport (31). This lack of a role for NHE1 could indicate either that it does not have a Na+ for NH4+ exchange activity or that it is not active under these conditions, possibly due to differences in the cytosolic pH sensitivity between NHE1 and NHE4 (42).
F. NBCn1
A second mechanism of TAL basolateral ammonia transport appears to involve the electroneutral sodium-bicarbonate cotransporter, NBCn1. This transport mechanism has been termed the “bicarbonate shuttling” pathway. In this pathway, NH4+ transported from the luminal fluid into the cytosol dissociates to NH3 and H+, causing intracellular acidification. NH3 exits the cell across the basolateral plasma membrane. This presumably occurs via lipid phase diffusion, but this mechanism has not been carefully examined. The cytosolic H+ generated is buffered by bicarbonate which enters across the basolateral plasma membrane via the electroneutral sodium-coupled bicarbonate cotransporter, NBCn1. Evidence supporting this model includes the findings that: 1) luminal ammonia reabsorption results in rapid intracellular acidification of such a magnitude that rapid NH3 exit is necessary to allow continued apical NH4+ reabsorption with subsequent H+ release (317); 2) two conditions that increase TAL ammonia reabsorption, metabolic acidosis and hypokalemia, increase NBCn1 expression and activity (134, 157, 218); and 3) NBCn1 expression increases ammonia transport rates (measured using the radiolabeled ammonia analog [14C]methylammonia) (168).
VII. DISTAL CONVOLUTED TUBULE
The specific role of the distal convoluted tubule (DCT) in ammonia transport is incompletely understood. Because of technical issues, isolated perfused tubule studies examining DCT ammonia transport have not been performed. A limited number of micropuncture studies have examined ammonia transport between the “early” and “late” micropuncturable distal tubule, a region which includes both the distal convoluted tubule and portions of the connecting segment. There appear to be low rates of ammonia secretion, and the rate of ammonia secretion is not increased by in vivo chronic metabolic acidosis (266). Mathematical modeling studies also suggest the DCT secretes ammonia (315).
VIII. CONNECTING SEGMENT
Similar to the DCT, the specific role of the connecting segment (CNT) in ammonia transport is incompletely understood. Technical difficulties preclude the study of CNT segments using in vitro microperfusion. Studies examining in vivo free flow micropuncture were discussed in the previous paragraph, and in mathematical modeling resulted in similar conclusions regarding CNT and DCT ammonia transport.
However, the ammonia transporters expressed in the CNT are very different than in the DCT. The CNT has a substantial number of type A and non-A, non-B intercalated cells, which likely have an important role in urinary acidification and ammonia excretion. Moreover, the expression of the ammonia transporter family members, Rhbg and Rhcg, is substantially higher in the CNT than in the DCT (68, 258, 295). Rhbg and Rhcg are expressed abundantly in both the type A and non-A, non-B intercalated cells as well as in the majority cell type in the CNT, CNT cells. To the extent that Rhbg and Rhcg have important roles in transepithelial ammonia transport, these observations suggest the CNT may have a substantially greater role in ammonia transport than the DCT. Whether conditions that alter ammonia excretion cause adaptive changes in CNT ammonia secretion has not been reported.
IX. COLLECTING DUCT
Collecting duct ammonia secretion is a complex process. Ammonia secretion throughout the collecting duct appears to occur almost entirely by parallel NH3 and H+ transport (76, 115, 152, 153, 279, 280, 328). Both of the apical proton pumps, H+-ATPase and H+-K+-ATPase, are likely to mediate the H+ secretion. Carbonic anhydrase is necessary for ammonia secretion, probably by supplying cytosolic H+ for secretion (300). Figure 9 shows a current model summarizing the mechanisms of collecting duct ammonia secretion.
FIGURE 9.

Model of collecting duct ammonia secretion. Ammonia uptake across the basolateral plasma membrane primarily involves transporter-mediated uptake by either Rhbg or Rhcg, with a component of diffusive NH3 absorption. Cytosolic NH3 is transported across the apical plasma membrane by a combination of Rhcg and diffusive transport. In the IMCD, but not the CCD, basolateral Na+-K+-ATPase also contributes to basolateral plasma membrane NH4+ uptake. Cytosolic H+ is generated by a carbonic anhydrase II-mediated mechanism and is secreted across the apical plasma membrane via H+-ATPase and H+-K+-ATPase. Luminal H+ titrates luminal NH3, forming NH4+ and maintaining a low luminal NH3 concentration necessary for NH3 secretion. Rhbg has been shown in heterologous expression systems to transport both NH3 and NH4+. The relative contributions of the transport of each to net basolateral uptake is not known. The model is not meant to indicate coupled transport of these two molecular forms of ammonia; only that both transport mechanisms may be present.
Transmembrane NH3 movement theoretically can involve either diffusive or transporter-mediated movement. Studies using cultured collecting duct cells grown on polarized support membranes in combination with [14C]methylammonia as an ammonia analog have addressed this issue. These studies showed the presence of both transporter-mediated and diffusive components across both the apical plasma membrane and the basolateral plasma membrane (120, 121). The inhibitable component was Na+- and K+-independent and exhibited characteristics consistent with electroneutral NH3 transport (120, 121). Thus collecting duct cells appear to have both diffusive and transporter-mediated NH3 movement.
A. Na+-K+-ATPase
Na+-K+-ATPase was the first integral membrane protein to be identified as specifically involved in collecting duct ammonia secretion. It is present in the basolateral plasma membrane of essentially all renal epithelial cells. NH4+ binds to and is transported at the K+-binding site, enabling active Na+-NH4+ exchange (156, 302). In the IMCD, Na+-K+-ATPase-mediated basolateral NH4+ uptake is critical for ammonia and H+ secretion (299, 301, 302). Moreover, decreased interstitial K+ levels during hypokalemia facilitate increased basolateral NH4+ uptake by Na+-K+-ATPase, contribute to increased NH4+ secretion rates (301), and thereby likely contribute to the increased urinary ammonia excretion during this condition. Na+-K+-ATPase's role in ammonia secretion outside of the IMCD is unclear. In the CCD, pharmacological inhibition of Na+-K+-ATPase with ouabain did not alter ammonia secretion (152). The role of Na+-K+-ATPase in NH4+ transport in the OMCD has not been specifically examined.
B. Rhesus Glycoproteins
A fundamental advance in our understanding of mammalian ammonia transport has been the identification of Rhesus glycoproteins as ammonia-specific transporters. Evolutionarily, Rhesus glycoproteins are related to ammonia transporters found in yeast (Mep proteins), bacteria (AMT proteins), and plants (AMT proteins), and they are present throughout the animal kingdom. Three families of Rhesus glycoproteins have been identified. The first identified was the erythrocyte-specific protein, Rhesus-associated glycoprotein, also known as Rhag (186). Following identification of Rhag as an ammonia transporter, identification and cloning of Rhesus B glycoprotein (Rhbg) and Rhesus C glycoprotein (Rhcg) quickly followed (174, 175).
1. Rhag
Rh A glycoprotein (Rhag) is a component of the Rh complex, which consists of the nonglycosylated Rh proteins RhD and RhCE in humans and Rh30 in nonhuman mammals, in association with Rhag. Although the erythrocyte Rhesus complex was initially thought to comprise one RhD subunit, one RhCE subunit, and two Rhag subunits, current evidence, based on x-ray crystallography of related proteins, suggests that the erythrocyte Rhesus complex comprises one RHD, one RhCE, and one Rhag subunit (106). Substantial evidence shows that Rhag mediates electroneutral, NH3 transport (186, 239, 319, 320); however, it may also transport NH4+ (39).
The role of Rhag in renal ammonia metabolism and transport appears to be limited. In particular, studies in human and mouse kidney (305, 312) found no evidence of nonerythroid Rhag expression. Studies in the liver, another important site of ammonia metabolism and transport, similarly did not detect Rhag expression (308). Thus current evidence indicates that Rhag is unlikely to contribute to renal ammonia metabolism.
2. Rhbg
Rhbg was the second Rhesus glycoprotein to be identified as an ammonia transporter. Initial studies identified Rhbg expression in kidneys, liver, and skin (175). In the kidney, Rhbg was initially thought to be expressed in the proximal tubule, based on in situ hybridization results (175). However, development of effective antibodies led to the recognition that Rhbg protein is found exclusively in distal epithelial cell populations. In rat and mouse kidney, Rhbg immunoreactivity is exclusively basolateral but variable in intensity in specific epithelial cells from the DCT through the IMCD. Rhbg expression in DCT cells is relatively weak, but in the heterogeneous epithelial populations of the connecting segment (CNT) through the IMCD, Rhbg is strongly expressed in specific cell types (118, 232, 295). Type A intercalated cells in the CNT through the IMCD, non-A, non-B intercalated cells in the DCT and CNT, CNT cells, and principal cells in the initial collecting duct (iCT) and CCD have intense Rhbg immunoreactivity. Type B intercalated cells, medullary principal cells, and IMCD cells do not express detectable Rhbg protein.
Whether Rhbg protein is expressed in the human kidney has been the subject of some controversy. Although multiple studies agree that Rhbg mRNA is expressed in the human kidney (33, 118, 175), one report concluded that Rhbg protein was not expressed (33). In contrast, another study, using different antibodies, identified Rhbg protein expression in human kidney tissue (118). Immunohistochemistry demonstrated distinct basolateral Rhbg expression in the CNT cells, non-A, non-B intercalated cells, and in collecting duct type A intercalated cells, but not in type B intercalated cells or principal cells (118).
The second study also identified that two different amino acid sequences for human Rhbg have been suggested. This discrepancy appears to result from a discrepancy in the mRNA coding sequence for Rhbg. Specifically, in the terminal portion of the coding region, beginning at nucleotide 1265, there is a sequence of multiple consecutive cytosines. One reported sequence for human Rhbg contained seven consecutive cytosines and another reported sequence contained eight. The difference in the number of these consecutive cytosines results in a frameshift change in the amino acid sequence downstream of this region, causing significantly different carboxyl tails in the two different predicted proteins. Sequencing both kidney and liver mRNA from human isolates identified eight consecutive cytosines, which is consistent with the DNA sequence from the published human genome database (118). Importantly, a vector commonly used for heterologous expression of human Rhbg was found to have only seven consecutive cytosines (118). The report that Rhbg protein was not expressed in human tissue used antibodies validated against the presumptive Rhbg expressed using this vector (33). Thus it is possible that the inability to detect Rhbg protein in human tissue reported in Reference 33 was related to use of antibodies that recognize a peptide sequence not present in human Rhbg.
Rhbg's role in renal ammonia excretion has been examined in a variety of studies of ammonia metabolism under basal conditions and in response to experimental stimuli. These studies have examined both the effects of Rhbg deletion and changes in Rhbg expression.
Under basal conditions, Rhbg deletion, whether global genomic deletion or intercalated cell-specific deletion, did not alter ammonia excretion (23, 41). However, mice with intercalated cell-specific Rhbg deletion have decreased glutamine synthetase expression (23). Glutamine synthetase catalyzes reaction of NH4+ with glutamate and regenerates glutamine, effectively the reverse of renal ammoniagenic processes. Recent studies show that decreased glutamine synthetase expression increases ammonia excretion (163). Thus the decreased glutamine synthetase expression with Rhbg deletion likely increases net ammoniagenesis and is an adaptive response to the effects of Rhbg deletion to enable maintenance of normal rates of ammonia excretion (23). Thus Rhbg appears to have a role in basal ammonia metabolism, but adaptive changes in other aspects of renal ammonia metabolism can compensate for its absence in genetic deletion models.
Rhbg appears to have important roles in the increased ammonia excretion that occurs in response to both metabolic acidosis and hypokalemia. Both conditions increase Rhbg protein expression, and genetic deletion of Rhbg from intercalated cells impairs the increase in ammonia excretion normally seen in both conditions (22, 23). It is important to note that one study, using a different method of acid-loading that resulted in lesser stimulation of ammonia excretion, found no effect of Rhbg deletion on ammonia excretion (41). It may be that other mechanisms promoting ammonia excretion can compensate for the lack of Rhbg if only modest increases in ammonia excretion are needed, but that greater degrees of adaptation require Rhbg expression. It is also possible that the different methods of acid-loading or other genetic differences in the mouse strains used caused the different conclusions regarding the role of Rhbg in ammonia excretion.
Several studies have addressed molecular mechanisms regulating Rhbg expression. One study reported that Tyr-429 mediates phosphorylation-regulated transport activity and membrane insertion and that a PDZ-binding motif is present at 455-DTQA-458 (273). Another study reported an ankyrin G-interaction domain at the 419-FLD-421 sequence (177). Most recently, evidence has been presented for a multi-protein complex of Rhbg, ankyrin G, and AE1 that contributes to plasma membrane stability (87).
However, each of these studies used the Rhbg expressed using a vector with seven cytosines (see above discussion), and examined interactions involving the carboxy tail encoded by nucleotides distal to the controversial section of multiple sequential cytosines. In the native protein expressed from an mRNA with eight consecutive cytosines at nucleotide 1265, these amino acid residues are not present. Thus these molecular regulatory mechanisms may not be relevant to Rhbg expressed from mRNA with eight consecutive cytosines beginning at nucleotide 1265, which appears to be the mRNA sequence present in human kidney (118).
3. Rhcg
Rhcg is the third member of the mammalian Rhesus glycoprotein family. Initial studies identified Rhcg expression in the kidney, brain, and testes (174). Subsequent studies have demonstrated a much more widespread pattern of expression, including epithelial cells throughout the entire gastrointestinal tract, epithelial cells in the testis and epididymis, bronchial epithelial cells, and skeletal muscle (117, 122, 161). In the human, mouse, and rat kidney, Rhcg is expressed in the kidney exclusively in distal epithelial cells (68, 116, 258, 295). Rhcg expression is prominent in the CNT, iCT, CCD, OMCD, and IMCD; weakly expressed in late DCT cells; and exhibits both apical and basolateral expression in the majority of these cells (116, 147, 258, 259). Rhcg expression differs among renal epithelial cell types. In general, type A intercalated cells express higher levels of Rhcg than do principal cells, and intercalated cells have a larger proportion of Rhcg in apical cytoplasmic vesicles under basal conditions than do principal cells (259). Rhcg is not detectable by immunohistochemistry in type B intercalated cells (147, 295). In the CNT, non-A, non-B intercalated cells have strong apical Rhcg, but no detectable basolateral Rhcg, whereas CNT cells have both apical and basolateral Rhcg. IMCD cells do not express detectable Rhcg (147, 295).
Rhcg has an important role in renal ammonia excretion in all conditions of altered ammonia excretion that have been studied. Gene deletion studies show that both global and collecting duct-specific Rhcg deletion impair basal ammonia excretion (24, 160). Both metabolic acidosis and hypokalemia increase Rhcg expression, and Rhcg expression is necessary for the normal increase in ammonia excretion in these conditions (24, 160, 166, 258, 259). Urinary ammonia excretion decreases during low protein feeding, and this is associated with decreased Rhcg expression (162). Feeding high dietary protein rich in acid-producing sulfur amino acids increases urinary ammonia excretion and Rhcg mRNA expression in wild-type mice, but in global Rhcg deletion mice the ammonia excretion response is delayed, and only normalizes concurrent with adaptive increases in ammoniagenic proteins (29). With reduced renal mass there is increased single-nephron ammonia excretion and increased apical and basolateral polarization of Rhcg (148). Cyclosporine A decreases Rhcg expression, which likely contributes to the renal tubular acidosis and subsequent metabolic acidosis often observed when this drug is administered (172). Aldosterone increases renal ammonia excretion, and mineralocorticoids increase Rhcg expression both in vivo and in cell culture (133).
Although both Rhbg and Rhcg contribute to increased ammonia excretion in multiple conditions, their relative importance appears to be condition-specific. For example, in studies in our laboratory, Rhcg deletion had a quantitatively greater effect on the ammonia excretion response to metabolic acidosis than did Rhbg, whereas the converse was true for hypokalemia (22, 23, 160, 166).
Rhcg expression appears to be regulated through a variety of mechanisms. There are changes in total protein expression in many conditions, as detailed in the previous paragraphs. In response to chronic metabolic acidosis, these changes appear to be posttranscriptional, as there is no change in steady-state mRNA expression (313). However, with high dietary protein loads, Rhcg mRNA expression increases (29), indicating that at least in some conditions there is a component of transcriptional regulation.
Changes in the subcellular localization of Rhcg are a second regulatory mechanism. Rhcg is found in both the apical and basolateral plasma membranes and in subapical cytoplasmic vesicles (147, 259). Ultrastructural studies have shown that changes in Rhcg's subcellular distribution occur in response to metabolic acidosis, with increased apical plasma membrane expression, increased basolateral plasma membrane expression, and a decrease in subapical vesicular Rhcg expression (259). Light microscopic studies suggest that similar patterns in Rhcg subcellular distribution occur with hypokalemia and reduced renal mass (22, 119, 148, 259).
These changes in Rhcg protein expression and in its subcellular distribution are renal epithelial cell and plasma membrane specific. In intercalated cells, redistribution of cytoplasmic vesicle-associated Rhcg to the apical plasma membrane, rather than changes in total cellular expression, appears to be the major mechanism increasing apical plasma membrane Rhcg expression (259). In contrast, in principal cells, changes in Rhcg's subcellular distribution are only a minor component of the increase in apical plasma membrane Rhcg expression (259). Also, metabolic acidosis increases basolateral plasma membrane Rhcg in principal cells, whereas in intercalated cells basolateral plasma membrane Rhcg expression is unchanged (259).
4. Molecular mechanism of ammonia transport by Rhesus glycoproteins
Several studies have examined which molecular species of ammonia, NH3 and NH4+, Rh glycoproteins transport. All results are consistent with Rh glycoproteins mediating facilitated, ATP-independent movement of ammonia across plasma membranes using the electrochemical gradient; there is no evidence for coupling of transport with other ions, with the possible exception of H+. A wide variety of expression systems and approaches to assess transport have been used, and in some cases the findings in different studies are inconsistent. Tables 2 and 3 summarize these studies, their approaches, and their conclusions.
Table 2.
NH3 and NH4+ transport characteristics of Rhbg
| Electroneutral |
Electrogenic |
|||||
|---|---|---|---|---|---|---|
| Expression System | NH3 transport | CH3NH2 transport | NH4+ transport | CH3NH3+ transport | Reference Nos. | Year |
| Murine Rhbg expression in Xenopus oocyte | Absent | Present | 209 | 2004 | ||
| Human RhBG in Xenopus oocyte | Present | Absent | 179 | 2004 | ||
| Human RhBG in HEK293 and MDCK cells | Present | Absent | 332 | 2005 | ||
| Murine Rhbg | Present | Absent | 185 | 2006 | ||
| Murine Rhbg in Xenopus oocyte | Present | Present | Present | 208 | 2010 | |
| Murine Rhbg in Xenopus oocyte | Absent | Present | Present | Present | 207 | 2010 |
| Human RhBG in Xenopus oocyte | Present | 89 | 2013 | |||
| Murine Rhbg in Xenopus oocyte | Present | Present | 39 | 2015 | ||
Table 3.
NH3 and NH4+ transport characteristics of Rhcg
| Electroneutral |
Electrogenic |
|||||
|---|---|---|---|---|---|---|
| Expression System | NH3 transport | CH3NH2 transport | NH4+ transport | CH3NH3+ transport | Reference Nos. | Year |
| Human RhCG expressed in Xenopus oocyte | Present | Present | 17 | 2004 | ||
| Human RhCG in HEK293 and MDCK cells | Present | Absent | 332 | 2005 | ||
| Murine Rhcg in Xenopus oocyte | Present | Absent | 185 | 2006 | ||
| Human RhCG in Xenopus oocyte and in S. cerevisae | Present | Absent | 188 | 2006 | ||
| Rhcg gene deletion, perfused collecting duct segment, apical transport | Present | Absent | 24 | 2008 | ||
| Purified RhCG-HA reconstituted in liposomes | Present | Absent | 196 | 2010 | ||
| Human RhCG reconstituted in liposomes | Present | Absent | 106 | 2010 | ||
| Rhcg gene deletion, perfused collecting duct segment, basolateral transport | Present | Absent | 30 | 2012 | ||
| Human RhCG in Xenopus oocyte | Present | 89 | 2013 | |||
| Murine Rhcg in Xenopus oocyte | Absent | Present | 39 | 2015 | ||
Several recent studies suggest that Rhbg can transport both NH3 and NH4+. Importantly, these studies show that results using the radiolabeled ammonia analog [14C]methylammonia may not accurately reflect the relative components of NH3 and NH4+ transport. The apparent ability of the Rhesus glycoprotein Rhbg to transport both NH3 and NH4+ means future studies examining ammonia transport by Rhesus glycoproteins should include methods that discriminate between the two moieties. However, from the perspective of basolateral ammonia uptake, the electrochemical gradient across the basolateral plasma membranes in the collecting duct is such that both electroneutral NH3 transport and electrogenic NH4+ transport modes result in ammonia transport from the interstitium into the cell cytoplasm.
Similar studies have addressed the molecular form of ammonia that Rhcg transports. In contrast to the results for Rhbg, essentially all have found that Rhcg transports ammonia in the form of NH3. Again, Tables 2 and 3 summarize these studies. Rhcg's transport of NH3 rather than NH4+ is critical for Rhcg to enable ammonia secretion across the apical plasma membrane of collecting duct cells. Specifically, high luminal NH4+ concentrations in the collecting duct and intracellular electronegativity of collecting duct cells result in an electrochemical gradient for NH4+ reabsorption, not secretion. The low luminal pH and the high pKa' of the NH3 + H+ ↔ NH4+ buffer reaction keeps collecting duct luminal NH3 concentration low, which facilitates continued NH3 secretion across collecting duct apical plasma membranes. Thus the specificity of Rhcg to transport NH3, but not NH4+, is essential for it to contribute to collecting duct ammonia secretion.
A wide variety of experimental approaches have been used to determine the molecular mechanisms of ammonia transport by Rh glycoproteins. These include amino acid substitution analysis, X-ray crystallography, in silico computational analysis, and molecular dynamic simulations. Mammalian Rhesus glycoproteins are integral membrane proteins containing 12 transmembrane spanning segments, with cytoplasmic carboxy and amino termini. X-ray crystallography studies of human Rhcg, nonmammalian Rhesus glycoproteins, and bacterial AMT orthologs consistently show that Rhesus glycoproteins are present in a homotrimeric form (106, 144, 180, 330). Each monomer has both a cytoplasmic and external vestibule as well as an ∼20 Å long, narrow, hydrophobic central channel. There appears to be stabilization of NH3 through this hydrophobic channel that occurs as a result of NH3 interaction with two in-line histidines. These in-line histidines appear to enable the NH3 selectivity of Rhesus glycoproteins (110). Between the extracellular vestibule and the central channel a phenylalanine ring is present and interrupts communication between these important structural sites. Specific amino acid residues present in the extracellular and the cytoplasmic vestibules are likely to facilitate electrogenic interaction with NH4+. In addition, in these vestibules, changes in the NH3-NH4+ buffer reaction pKa' appear to develop that facilitates NH4+ dissociation to NH3 and H+ (144). This causes development of locally high NH3 concentrations, which favor NH3 transport through the central channel.
The crystal structure of human Rhcg revealed an unexpected finding, the presence of a shunt pathway or pocket (106) extending from the cytosolic vestibule to the lateral surface of the Rhcg molecule and contacting the lipid bilayer. Because the amino acids lining this shunt are conserved among human Rh glycoproteins and a similar shunt is also observed in the bacterial Rh glycoprotein ortholog NeRh, it likely has a specific function. Although the functional significance of this shunt pathway has not been experimentally determined, it may represent an alternative path for NH3 transport (106).
The molecular mechanism(s) that might mediate electrogenic NH4+ transport by Rhesus glycoproteins are not fully understood at present. As noted previously, Rhbg appears to transport both NH3 and NH4+. Whether NH4+ transport occurs as NH4+ transport or as cotransport of NH3 with H+ has been the subject of several investigations. Molecular dynamics simulation studies show that NH4+ movement through the central channel is highly energetically unfavorable (14, 217, 303) and support a model of NH3-H+ cotransport. One possible H+ transport mechanism could involve a water chain through the central pore (14). Another possibility involves H+ transport involving the twin-inline histidines (303). Finally, H+ transport involving alternative pathways cannot be excluded at this time. Importantly, these interpretations result from molecular dynamic simulation studies examining the bacterial ortholog AmtB and not mammalian Rh glycoproteins. The high degree of tertiary structural similarities among ammonia transporter family member suggests that findings from AmtB can be extrapolated to mammalian Rh glycoproteins, but this needs to be experimentally verified.
5. CO2 transport by Rh glycoproteins
Rhesus glycoproteins may also transport CO2, in addition to ammonia. Quantitative studies using human erythrocytes deficient in RhAG show decreased CO2 transport (71, 72) and studies utilizing heterologous expression in Xenopus oocytes show all Rh glycoproteins can transport CO2 (89, 198). In the green algae, Chlamydomonas rheinhardtii, Rhesus glycoprotein Rh1 expression is regulated by CO2, and Rh1 deletion impairs the normal response to changes in CO2, supporting a role of Rh1 as an essential CO2 transporter in this species (277, 278). However, whether Rh glycoproteins have a significant role in CO2 transport in the kidney is not clear. Intercalated cells use cytoplasmic CO2 to generate, through a carbonic anhydrase II-catalyzed process, the intracellular H+ used for urinary acidification, and impaired CO2 transport would be predicted to impair urine acidification. However, in several studies using either isolated or combined Rhbg and Rhcg deletion, there was no impairment of urine acidification (23, 24, 41, 160, 165, 167). However, it certainly is possible that either altered intrarenal CO2 concentrations, which could promote transmembrane CO2 diffusion in the absence of Rhbg and Rhcg, or adaptations in other CO2 transport mechanisms could compensate for the absence of Rh glycoprotein-mediated CO2 transport in these gene deletion studies.
C. HCN2
HCN2, a hyperpolarization-activated cyclic nucleotide-gated channel, subtype 2, may be another mechanism of basolateral ammonia transport by collecting duct cells (40). In Xenopus oocytes, heterologous HCN2 expression increased NH4+ transport, in addition to K+ transport, and both were transported significantly better than sodium (40). In microperfused rat outer medullary collecting duct segments, the initial rate of acidification upon exposure to a basolateral ammonium chloride pulse, an indirect measure of NH4+ entry, was greater in intercalated cells than in principal cells. A HCN2-specific inhibitor, ZD7288, blunted these intracellular pH changes in intercalated cells from control rats, but not in acid-loaded rats. Thus HCN2 may be an intercalated cell basolateral NH4+ transport pathway that contributes to basal, but not acidosis-stimulated, ammonia secretion.
D. H+-K+-ATPase
H+-K+-ATPase proteins are P-type ATPases and may contribute to collecting duct ammonia transport. Potassium deficiency increases expression of the HKα1 and HKα2 isoforms of H+-K+-ATPase (107). This has been postulated to mediate increased NH4+ secretion via binding of NH4+ to and transport at the H+ binding site (206). Other studies, however, have suggested that NH4+ binds to and is transported at the K+ binding site (84).
In addition to being a substrate for H+-K+-ATPase transport, ammonia also appears to regulate H+-K+-ATPase-mediated H+ secretion. In studies examining in vitro microperfused CCD segments, ammonia causes concentration-dependent stimulation of net H+-K+-ATPase-mediated, but not H+-ATPase-mediated, H+ secretion (80). In the OMCD, ammonia may stimulate net H+ secretion by as much as ∼50% (77). Ammonia's stimulation of CCD H+-K+-ATPase is independent of changes in intracellular pH and involves changes in intracellular calcium, microtubules, and SNARE protein-mediated vesicle fusion (78–80). This stimulation of H+-K+-ATPase activity, by increasing unidirectional K+ reabsorption, likely contributes to ammonia's ability to regulate CCD net K+ secretion (112).
E. Aquaporins
The aquaporins are integral membrane water channels present in many tissues and cell types. In the renal tubules, numerous aquaporin isoforms are expressed with specific segmental, cellular, and subcellular distribution and isoform-specific regulation. The knowledge that the electrostatic properties and size of H2O and NH3 are similar led to explorations of whether aquaporins may contribute to NH3 transport. These studies have demonstrated that specific aquaporins, as detailed below, do transport ammonia (198). However, whether all aquaporins transport ammonia is unclear, as some studies have come to differing conclusions regarding the ammonia transport capacity of specific aquaporin isoforms. For example, one carefully performed study suggested AQP3 and AQP8 do not transport ammonia (90), whereas another study showed that AQP3 and AQP8 could (127).
The earliest studies showing that aquaporins can transport ammonia demonstrated that AQP1 expression increases NH3 transport in Xenopus oocytes (198, 210). However, NH3 transport by AQP1 has not been detected by others (127). AQP1 is present in the descending thin limb of the loop of Henle and in both the apical and basolateral plasma membranes of the proximal tubule. Thus AQP1 may contribute to the ammonia permeability observed in the descending thin limb (75). Whether AQP1 contributes to proximal tubule ammonia transport has not been determined experimentally, but it is certainly ideally located to mediate a component of the Ba2+- and NHE3-independent apical NH3 secretion.
AQP3's role in collecting duct ammonia transport is unclear. AQP3 is present in the principal cells of the medullary collecting duct as well as in inner medullary collecting duct cells and is located in the basolateral plasma membrane. Some studies (127), but not others (90), indicate AQP3 transports NH3. Thus AQP3 may contribute to principal cell basolateral NH3 transport, but this hypothesis has not yet been tested.
AQP8 is expressed in intracellular sites in the proximal tubule, CCD, and OMCD, but not in the plasma membrane (70). In cultured proximal tubule cells, AQP8 is present in the inner mitochondrial membrane, and its expression is necessary for normal ammonia secretion (193). In vivo studies show that metabolic acidosis increases AQP8 expression (193). All of these findings support an important role of AQP8 in proximal tubule mitochondrial ammonia transport. Evidence against this role, however, is that AQP8 genetic deletion is reported to not alter urinary ammonia excretion either under basal conditions or following either acute or chronic acid loading (327). Since NH4+ produced from glutamine metabolism is generated in mitochondria, ammonia transport from mitochondria to cytoplasm is a critical step in proximal tubule ammonia generation. AQP8 may play an important role in this process.
F. Carbonic Anhydrase
Carbonic anhydrase (CA), in addition to its role in bicarbonate reabsorption, also contributes to ammonia secretion. Studies examining isolated perfused OMCD segments have shown that CA inhibition blocks ammonia secretion (300). This is likely to occur because cytosolic H+ production via CA II is necessary to enable parallel H+ and NH3 across the apical plasma membrane.
The membrane-bound isoform CA IV likely contributes to the regulation of both bicarbonate reabsorption and ammonia secretion. Collecting duct CA IV, although functioning to increase bicarbonate reabsorption, likely decreases collecting duct ammonia secretion by preventing a luminal disequilibrium pH. Apical CA IV expression has been demonstrated in the rabbit CCD type A intercalated cell, in the rabbit OMCD and IMCD (256), and in the human CCD and OMCD (176), but has not been found in the rat collecting duct (34). This pattern is consistent with the evidence of luminal disequilibrium pH in the rabbit CCD and OMCD outer stripe segments (279, 280). Whether collecting duct CA IV expression is regulated by physiological stimuli has not been rigorously examined. Studies examining tissue homogenates have demonstrated that metabolic acidosis increases CA IV mRNA expression (291, 322). This observation is consistent with the observation of basolateral CA IV expression in the proximal tubule, where it may contribute to bicarbonate reabsorption (255).
G. Diffusive NH3 Movement
The contribution of diffusive NH3 movement across collecting duct apical and basolateral plasma membranes remains unclear. As discussed previously, studies of methylammonia transport in cultured collecting duct epithelia suggested that diffusive components contribute to both apical and basolateral transmembrane NH3 movement (120, 121). Studies of intercalated cells in isolated perfused collecting ducts, identified by luminal 2',7'-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF) accumulation (306), from mice with genetic deletion of Rhcg also address this question. Rhcg deletion decreased intercalated cell apical NH3 permeability, confirming Rhcg's role in ammonia secretion, but did not abolish it (24). Thus there is Rhcg-independent apical plasma membrane NH3 transport. At present, Rhcg is the only apical plasma membrane NH3-transporting protein known in intercalated cells, suggesting that diffusive NH3 transport mediates this Rhcg-independent NH3 permeability.
The relative contributions of Rhcg-mediated and diffusive NH3 transport to apical ammonia secretion are somewhat difficult to determine. The studies examining the effect of Rhcg deletion on apical plasma membrane NH3 permeability used collecting duct segments from mice exposed to chronic acid loads (24) and showed that Rhcg deletion decreased apical intercalated cell NH3 permeability by ∼60% (24). However, because these studies used collecting duct segments from mice with metabolic acidosis, which is known to increase apical plasma membrane Rhcg expression by approximately fourfold (259), the likely contribution under basal conditions of Rhcg to NH3 transport is likely to be <60%. Of course, these considerations of Rhcg-independent apical plasma membrane NH3 permeability only reflect non-carrier-mediated diffusive NH3 permeability to the extent that other, currently unrecognized, NH3 transporters are not present.
X. RENAL INTERSTITIUM
A. Sulfatides
Recent studies show that sulfatides have an important role in renal ammonia metabolism. Sulfatides are highly charged anion glycosphingolipids that can reversibly bind NH4+. They are expressed throughout the kidney, but are expressed at highest levels in the outer and inner medulla (281), and appear to have an important role in maintaining the high inner medullary ammonium content and the increase in urinary acid elimination that develop during metabolic acidosis. Disruption of renal sulfatide synthesis, by a genetic approach along the entire renal tubule, leads to lower urinary pH accompanied by lower ammonia excretion (281). After acid loading, mice deficient in renal sulfatide synthesis have impaired ammonia excretion, decreased ammonia accumulation in the papilla, and chronic hyperchloremic metabolic acidosis (281). Thus sulfatides, likely through their ability to reversibly bind interstitial NH4+, have an important role in renal ammonia handling, urinary acidification, and acid-base homeostasis.
XI. SUMMARY
For years, an ammonia transport model involving passive, non-carrier-mediated NH3 diffusion in combination with NH4+ trapping due to an absence of NH4+ transport mechanisms was the established model of renal ammonia transport. We have summarized data that show that this model requires substantial changes. Passive, non-protein-mediated NH3 movement across plasma membranes is likely to be only a minor component of ammonia transport. A wide variety of integral membrane proteins, with specific abilities to transport either NH3 or NH4+, and in rare cases, both, are present in renal epithelial cells. These proteins exhibit cell- and membrane-specific expression and are regulated in response to essentially all stimuli that alter renal ammonia excretion. Thus protein-mediated NH3 and NH4+ transport appears to be the primary mechanism underlying renal ammonia excretion.
GRANTS
Generation and publication of this review was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK045788 and R01-DK107798 and Department of Veterans Affairs Grant 1I01BX000818.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: David Weiner, PO Box 100224, Div. of Nephrology, Hypertension and Renal Transplantation, Univ. of Florida College of Medicine, Gainesville, FL 32610 (e-mail: david.weiner@medicine.ufl.edu).
Footnotes
1Ammonia exists in two molecular forms, NH3 and NH4+, which are in equilibrium with each other. We use the term ammonia to refer to the combination of these two molecular forms. When referring to a specific molecular form, we state specifically either “NH3” or “NH4+.”
REFERENCES
- 1.Adam W, Simpson DP. Glutamine transport in rat kidney mitochondria in metabolic acidosis. J Clin Invest 54: 165, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal KP, Narula S, Kakkar M, Tandon C. Nephrolithiasis: molecular mechanism of renal stone formation and the critical role played by modulators. Biomed Res Int 2013: 292953, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alleyne GA, Scullard GH. Renal metabolic response to acid base changes. I. Enzymatic control of ammoniagenesis in the rat. J Clin Invest 48: 364–370, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ambuhl PM, Amemiya M, Danczkay M, Lotscher M, Kaissling B, Moe OW, Preisig PA, Alpern RJ. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 271: F917–F925, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Amemiya M, Loffing J, Lotscher M, Kaissling B, Alpern RJ, Moe OW. Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney Int 48: 1206–1215, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Amlal H, Paillard M, Bichara M. NH4+ transport pathways in cells of medullary thick ascending limb of rat kidney. NH4+ conductance and K+/NH4+(H+) antiport. J Biol Chem 269: 21962–21971, 1994. [PubMed] [Google Scholar]
- 7.Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol 301: F1143–F1159, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atherton JC, Hai MA, Thomas S. The time course of changes in renal tissue composition during mannitol diuresis in the rat. J Physiol 197: 411–428, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atkins PW. Molecules in motion: ion transport and molecular diffusion. In: Physical Chemistry, edited by Atkins PW. Oxford, UK: Oxford Univ. Press, 1978, p. 819–848. [Google Scholar]
- 10.Atlante A, Passarella S, Minervini GM, Quagliariello E. Glutamine transport in normal and acidotic rat kidney mitochondria. Arch Biochem Biophys 315: 369–381, 1994. [DOI] [PubMed] [Google Scholar]
- 11.Attmane-Elakeb A, Amlal H, Bichara M. Ammonium carriers in medullary thick ascending limb. Am J Physiol Renal Physiol 280: F1–F9, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Attmane-Elakeb A, Mount DB, Sibella V, Vernimmen C, Hebert SC, Bichara M. Stimulation by in vivo and in vitro metabolic acidosis of expression of rBSC-1, the Na+-K+ (NH4+)-2Cl− cotransporter of the rat medullary thick ascending limb. J Biol Chem 273: 33681–33691, 1998. [DOI] [PubMed] [Google Scholar]
- 13.Attmane-Elakeb A, Sibella V, Vernimmen C, Belenfant X, Hebert SC, Bichara M. Regulation by glucocorticoids of expression and activity of rBSC1, the Na+-K+(NH4+)-2Cl− cotransporter of medullary thick ascending limb. J Biol Chem 275: 33548–33553, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Baday S, Wang S, Lamoureux G, Bernèche S. Different hydration patterns in the pores of AmtB and RhCG could determine their transport mechanisms. Biochemistry 52: 7091–7098, 2013. [DOI] [PubMed] [Google Scholar]
- 15.Baertl JM, Sancetta SM, Gabuzda GJ. Relation of acute potassium depletion to renal ammonium metabolism in patients with cirrhosis. J Clin Invest 42: 696–706, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bako I, Mayer I. On dipole moments and hydrogen bond identification in water clusters. J Phys Chem A 120: 4408–4417, 2016. [DOI] [PubMed] [Google Scholar]
- 17.Bakouh N, Benjelloun F, Hulin P, Brouillard F, Edelman A, Cherif-Zahar B, Planelles G. NH3 is involved in the NH4+ transport induced by the functional expression of the human Rh C glycoprotein. J Biol Chem 279: 15975–15983, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Bastani B, Gluck SL. Adaptational changes in renal vacuolar H+-ATPase in the rat remnant kidney. J Am Soc Nephrol 8: 868–879, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Bauch C, Forster N, Loffing-Cueni D, Summa V, Verrey F. Functional cooperation of epithelial heteromeric amino acid transporters expressed in madin-darby canine kidney cells. J Biol Chem 278: 1316–1322, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Benyajati S, Goldstein L. Relation of ammonia excretion adaptation to glutaminase activity in acidotic, subtotal nephrectomized rats. Kidney Int 14: 50–57, 1978. [DOI] [PubMed] [Google Scholar]
- 21.Bergeron MJ, Gagnon E, Wallendorff B, Lapointe JY, Isenring P. Ammonium transport and pH regulation by K+-Cl− cotransporters. Am J Physiol Renal Physiol 285: F68–F78, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Bishop JM, Lee HW, Handlogten ME, Han KH, Verlander JW, Weiner ID. Intercalated cell-specific Rh B glycoprotein deletion diminishes renal ammonia excretion response to hypokalemia. Am J Physiol Renal Physiol 304: F422–F431, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bishop JM, Verlander JW, Lee HW, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the Rhesus glycoprotein, Rh B Glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol 299: F1065–F1077, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biver S, Belge H, Bourgeois S, Van Vooren P, Nowik M, Scohy S, Houillier P, Szpirer J, Szpirer C, Wagner CA, Devuyst O, Marini AM. A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature 456: 339–343, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Blanchard A, Eladari D, Leviel F, Tsimaratos M, Paillard M, Podevin RA. NH4+ as a substrate for apical and basolateral Na+-H+ exchangers of thick ascending limbs of rat kidney: evidence from isolated membranes. J Physiol 506: 689–698, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bode BP. Recent molecular advances in mammalian glutamine transport. J Nutr 131: 2475S–2485S, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Bontig SL, Toodle AD, DeBruin H, Mayren BR. Quantitative histochemistry of the nephron. VIII. Various phosphatases in the healthy human kidney. Arch Biochem Biophys 91: 130–137, 1960. [Google Scholar]
- 28.Boron WF. Acid-base transport by the renal proximal tubule. J Am Soc Nephrol 17: 2368–2382, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Bounoure L, Ruffoni D, Muller R, Kuhn GA, Bourgeois S, Devuyst O, Wagner CA. The role of the renal ammonia transporter Rhcg in metabolic responses to dietary protein. J Am Soc Nephrol 25: 2040–2052, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourgeois S, Bounoure L, Christensen EI, Ramakrishnan SK, Houillier P, Devuyst O, Wagner CA. Haploinsufficiency of the ammonia transporter Rhcg predisposes to chronic acidosis. Rhcg is critical for apical and basolateral ammonia transport in the mouse collecting duct. J Biol Chem 288: 5518–5529, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bourgeois S, Meer LV, Wootla B, Bloch-Faure M, Chambrey R, Shull GE, Gawenis LR, Houillier P. NHE4 is critical for the renal handling of ammonia in rodents. J Clin Invest 120: 1895–1904, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brenes LG, Sanchez MI. Impaired urinary ammonium excretion in patients with isolated proximal renal tubular acidosis. J Am Soc Nephrol 4: 1073–1078, 1993. [DOI] [PubMed] [Google Scholar]
- 33.Brown ACN, Hallouane D, Mawby WJ, Karet FE, Saleem MA, Howie AJ, Toye AM. RhCG is the major putative ammonia transporter expressed in human kidney and RhBG is not expressed at detectable levels. Am J Physiol Renal Physiol 296: F1279–F1290, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown D, Zhu XL, Sly WS. Localization of membrane-associated carbonic anhydrase type IV in kidney epithelial cells. Proc Natl Acad Sci USA 87: 7457–7461, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buerkert J, Martin D, Trigg D. Ammonium handling by superficial and juxtamedullary nephrons in the rat. Evidence for an ammonia shunt between the loop of Henle and the collecting duct. J Clin Invest 70: 1–12, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buerkert J, Martin D, Trigg D. Segmental analysis of the renal tubule in buffer production and net acid formation. Am J Physiol Renal Fluid Electrolyte Physiol 244: F442–F454, 1983. [DOI] [PubMed] [Google Scholar]
- 37.Burki R, Mohebbi N, Bettoni C, Wang X, Serra AL, Wagner CA. Impaired expression of key molecules of ammoniagenesis underlies renal acidosis in a rat model of chronic kidney disease. Nephrol Dial Transplant 30: 770–781, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Busque SM, Wagner CA. Potassium restriction, high protein intake, and metabolic acidosis increase expression of the glutamine transporter SNAT3 (Slc38a3) in mouse kidney. Am J Physiol Renal Physiol 297: F440–F450, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Caner T, Abdulnour-Nakhoul S, Brown K, Islam MT, Hamm LL, Nakhoul NL. Mechanisms of ammonia and ammonium transport by rhesus associated glycoproteins. Am J Physiol Cell Physiol 309: C747–C758, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carrisoza-Gaytan R, Rangel C, Salvador C, Saldana-Meyer R, Escalona C, Satlin LM, Liu W, Zavilowitz B, Trujillo J, Bobadilla NA, Escobar LI. The hyperpolarization-activated cyclic nucleotide-gated HCN2 channel transports ammonium in the distal nephron. Kidney Int 80: 832–840, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chambrey R, Goossens D, Bourgeois S, Picard N, Bloch-Faure M, Leviel F, Geoffroy V, Cambillau M, Colin Y, Paillard M, Houillier P, Cartron JP, Eladari D. Genetic ablation of Rhbg in mouse does not impair renal ammonium excretion. Am J Physiol Renal Physiol 289: F1281–F1290, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Chambrey R, John PL, Eladari D, Quentin F, Warnock DG, Abrahamson DR, Podevin RA, Paillard M. Localization and functional characterization of Na+/H+ exchanger isoform NHE4 in rat thick ascending limbs. Am J Physiol Renal Physiol 281: F707–F717, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Cheema-Dhadli S, Halperin ML. Ammoniagenesis in kidney cortex mitochondria of the rat: role of the mitochondrial dicarboxylate anion transporter. Can J Biochem 56: 23–28, 1978. [DOI] [PubMed] [Google Scholar]
- 44.Chen W, Abramowitz MK. Metabolic acidosis and the progression of chronic kidney disease. BMC Nephrol 15: 55, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choe H, Sackin H, Palmer LG. Permeation properties of inward-rectifier potassium channels and their molecular determinants. J Gen Physiol 115: 391–404, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Churchill PC, Malvin RL. Relation of renal gluconeogenesis to ammonia production in the rat. Am J Physiol 218: 353–357, 1970. [DOI] [PubMed] [Google Scholar]
- 47.Clark EC, Nath KA, Hostetter TH, Hostetter MK. Hyperosmolality impairs ammonia-mediated inflammation: Implications for the renal medulla. Am J Physiol Regul Integr Comp Physiol 263: R148–R155, 1992. [DOI] [PubMed] [Google Scholar]
- 48.Clough SA, Beers Y, Klein GP, Rothman LS. Dipole moment of water from Stark measurements of H2O, HDO, and D2O. J Chem Phys 59: 2254–2259, 1973. [Google Scholar]
- 49.Conjard A, Komaty O, Delage H, Boghossian M, Martin M, Ferrier B, Baverel G. Inhibition of glutamine synthetase in the mouse kidney: a novel mechanism of adaptation to metabolic acidosis. J Biol Chem 278: 38159–38166, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Coresh J, Selvin E, Stevens LA. Prevalence of chronic kidney disease in the united states. JAMA 298: 2038–2047, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Curthoys NP, Godfrey SS. Properties of rat kidney glutaminase enzymes and their role in renal ammoniagenesis. Curr Probl Clin Biochem 6: 346–356, 1976. [PubMed] [Google Scholar]
- 52.Curthoys NP, Kuhlenschmidt T, Godfrey SS, Weiss RF. Phosphate-dependent glutaminase from rat kidney. Cause of increased activity in response to acidosis and identity with glutaminase from other tissues. Arch Biochem Biophys 172: 162–167, 1976. [DOI] [PubMed] [Google Scholar]
- 53.Curthoys NP, Lowry OH. The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J Biol Chem 248: 162–168, 1973. [PubMed] [Google Scholar]
- 54.Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol 9: 1627–1638, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Curthoys NP, Taylor L, Hoffert JD, Knepper MA. Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. Am J Physiol Renal Physiol 292: F140–F147, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Curthoys NP, Weiss RF. Regulation of renal ammoniagenesis: subcellular localization of rat kidney glutaminase isoenzymes. J Biol Chem 249: 3261–3266, 1974. [PubMed] [Google Scholar]
- 57.D'Asaro S, Lo PR, Caimi G. Physiopathology and clinical aspects of type IV renal tubular acidosis. Minerva Med 78: 1151–1161, 1987. [PubMed] [Google Scholar]
- 58.Dass PD, Kurtz I. Renal ammonia and bicarbonate production in chronic renal failure. Miner Electrolyte Metab 16: 308–314, 1990. [PubMed] [Google Scholar]
- 59.Dass PD, Martin D. Adaptive ammoniagenesis in chronic renal failure. Ren Physiol Biochem 13: 259–263, 1990. [DOI] [PubMed] [Google Scholar]
- 60.De Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20: 2075–2084, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deferrari G, Robaudo C, Garibotto G, Saffioti S, Sala MR, Tizianello A. Determinants of the partition of renal ammonia production between urine and venous blood in man with metabolic acid-base disturbances. Contrib Nephrol 92: 109–113, 1991. [DOI] [PubMed] [Google Scholar]
- 62.Demirci FY, Chang MH, Mah TS, Romero MF, Gorin MB. Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol Vis 12: 324–330, 2006. [PubMed] [Google Scholar]
- 63.Devonald MA, Smith AN, Poon JP, Ihrke G, Karet FE. Non-polarized targeting of AE1 causes autosomal dominant distal renal tubular acidosis. Nat Genet 33: 125–127, 2003. [DOI] [PubMed] [Google Scholar]
- 64.DiGiovanni SR, Madsen KM, Luther AD, Knepper MA. Dissociation of ammoniagenic enzyme adaptation in rat S1 proximal tubules and ammonium excretion response. Am J Physiol Renal Fluid Electrolyte Physiol 267: F407–F414, 1994. [DOI] [PubMed] [Google Scholar]
- 65.Dinour D, Chang MH, Ji Satoh Smith BL, Angle N, Knecht A, Serban I, Holtzman EJ, Romero MF. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem 279: 52238–52246, 2004. [DOI] [PubMed] [Google Scholar]
- 66.Donnelly S, Kamel KS, Vasuvattakul S, Narins RG, Halperin ML. Might distal renal tubular acidosis be a proximal tubular cell disorder? Am J Kidney Dis 19: 272–281, 1992. [DOI] [PubMed] [Google Scholar]
- 67.Eisenman G, Latorre R, Miller C. Multi-ion conduction and selectivity in the high-conductance Ca2+-activated K+ channel from skeletal muscle. Biophys J 50: 1025–1034, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eladari D, Cheval L, Quentin F, Bertrand O, Mouro I, Cherif-Zahar B, Cartron JP, Paillard M, Doucet A, Chambrey R. Expression of RhCG, a new putative NH3/NH4+ transporter, along the rat nephron. J Am Soc Nephrol 13: 1999–2008, 2002. [DOI] [PubMed] [Google Scholar]
- 69.Elkjar ML, Kwon TH, Wang W, Nielsen J, Knepper MA, Frokiar J, Nielsen S. Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am J Physiol Renal Physiol 283: F1376–F1388, 2002. [DOI] [PubMed] [Google Scholar]
- 70.Elkjar ML, Nejsum LN, Gresz V, Kwon TH, Jensen UB, Frokiar J, Nielsen S. Immunolocalization of aquaporin-8 in rat kidney, gastrointestinal tract, testis, and airways. Am J Physiol Renal Physiol 281: F1047–F1057, 2001. [DOI] [PubMed] [Google Scholar]
- 71.Endeward V, Cartron JP, Ripoche P, Gros G. Red cell membrane CO2 permeability in normal human blood and in blood deficient in various blood groups, and effect of DIDS. Transfusion Clin Biol 13: 123–127, 2006. [DOI] [PubMed] [Google Scholar]
- 72.Endeward V, Cartron JP, Ripoche P, Gros G. RhAG protein of the Rhesus complex is a CO2 channel in the human red cell membrane. FASEB J 22: 64–73, 2007. [DOI] [PubMed] [Google Scholar]
- 73.Falk KG. Transference numbers of electrolytes in aqueous solutions. In: International Critical Tables of Numerical Data, Physics, Chemistry and Technology, edited by Washburn EW. New York: McGraw-Hill, 1929, p. 309–311. [Google Scholar]
- 74.Finkelstein FO, Hayslett JP. Role of medullary structures in the functional adaptation of renal insufficiency. Kidney Int 6: 419–425, 1974. [DOI] [PubMed] [Google Scholar]
- 75.Flessner MF, Mejia R, Knepper MA. Ammonium and bicarbonate transport in isolated perfused rodent long-loop thin descending limbs. Am J Physiol Renal Fluid Electrolyte Physiol 264: F388–F396, 1993. [DOI] [PubMed] [Google Scholar]
- 76.Flessner MF, Wall SM, Knepper MA. Permeabilities of rat collecting duct segments to NH3 and NH4+. Am J Physiol Renal Fluid Electrolyte Physiol 260: F264–F272, 1991. [DOI] [PubMed] [Google Scholar]
- 77.Flessner MF, Wall SM, Knepper MA. Ammonium and bicarbonate transport in rat outer medullary collecting ducts. Am J Physiol Renal Fluid Electrolyte Physiol 262: F1–F7, 1992. [DOI] [PubMed] [Google Scholar]
- 78.Frank AE, Weiner ID. Effects of ammonia on acid-base transport by the B-type intercalated cell. J Am Soc Nephrol 12: 1607–1614, 2001. [DOI] [PubMed] [Google Scholar]
- 79.Frank AE, Wingo CS, Andrews PM, Ageloff S, Knepper MA, Weiner ID. Mechanisms through which ammonia regulates cortical collecting duct net proton secretion. Am J Physiol Renal Physiol 282: F1120–F1128, 2002. [DOI] [PubMed] [Google Scholar]
- 80.Frank AE, Wingo CS, Weiner ID. Effects of ammonia on bicarbonate transport in the cortical collecting duct. Am J Physiol Renal Physiol 278: F219–F226, 2000. [DOI] [PubMed] [Google Scholar]
- 81.Gabuzda GJ, Hall II. Relation of potassium depletion to renal ammonium metabolism and hepatic coma. Medicine 45: 481–489, 1966. [DOI] [PubMed] [Google Scholar]
- 82.Galla JH, Bonduris DN, Luke RG. Correction of acute chloride-depletion alkalosis in the rat without volume expansion. Am J Physiol Renal Fluid Electrolyte Physiol 244: F217–F221, 1983. [DOI] [PubMed] [Google Scholar]
- 83.Galla JH, Rome L, Luke RG. Bicarbonate transport in collecting duct segments during chloride-depletion alkalosis. Kidney Int 48: 52–55, 1995. [DOI] [PubMed] [Google Scholar]
- 84.Garg LC, Narang N. Ouabain-insensitive K-adenosine triphosphatase in distal nephron segments of the rabbit. J Clin Invest 81: 1204–1208, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garibotto G, Sofia A, Robaudo C, Saffioti S, Sala MR, Verzola D, Vettore M, Russo R, Procopio V, Deferrari G, Tessari P. Kidney protein dynamics and ammoniagenesis in humans with chronic metabolic acidosis. J Am Soc Nephrol 15: 1606–1615, 2004. [DOI] [PubMed] [Google Scholar]
- 86.Gawenis LR, Bradford EM, Prasad V, Lorenz JN, Simpson JE, Clarke LL, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE. Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 cotransporter. J Biol Chem 282: 9042–9052, 2007. [DOI] [PubMed] [Google Scholar]
- 87.Genetet S, Ripoche P, Le Van Kim C, Colin Y, Lopez C. Evidence of a structural and functional ammonium transporter RhBG-+anion exchanger 1-+Ankyrin-G complex in kidney epithelial cells. J Biol Chem 290: 6925–6936, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis. Diabetes Care 24: 382–391, 2001. [DOI] [PubMed] [Google Scholar]
- 89.Geyer RR, Parker MD, Toye AM, Boron WF, Musa-Aziz R. Relative CO2/NH3 permeabilities of human RhAG, RhBG and RhCG. J Membr Biol 246: 915–926, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Geyer RR, Musa-Aziz R, Qin X, Boron WF. Relative CO2/NH3 selectivities of mammalian aquaporins 0–9. Am J Physiol Cell Physiol 304: C985–C994, 2013. [DOI] [PubMed] [Google Scholar]
- 91.Gonzalez NC, Albrecht T, Sullivan LP, Clancy RL. Compensation of respiratory alkalosis induced after acclimation to stimulated altitude. J Appl Physiol (1985) 69: 1380–1386, 1990. [DOI] [PubMed] [Google Scholar]
- 92.Good DW. Effects of potassium on ammonia transport by medullary thick ascending limb of the rat. J Clin Invest 80: 1358–1365, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Good DW. Active absorption of NH4+ by rat medullary thick ascending limb: inhibition by potassium. Am J Physiol Renal Fluid Electrolyte Physiol 255: F78–F87, 1988. [DOI] [PubMed] [Google Scholar]
- 94.Good DW. Adaptation of HCO3− and NH4+ transport in rat MTAL: effects of chronic metabolic acidosis and Na+ intake. Am J Physiol Renal Fluid Electrolyte Physiol 258: F1345–F1353, 1990. [DOI] [PubMed] [Google Scholar]
- 95.Good DW. Ammonium transport by the thick ascending limb of henles loop. Annu Rev Physiol 56: 623–647, 1994. [DOI] [PubMed] [Google Scholar]
- 96.Good DW, Burg MB. Ammonia production by individual segments of the rat nephron. J Clin Invest 73: 602–610, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Good DW, DuBose TD. Ammonia transport by early and late proximal convoluted tubule of the rat. J Clin Invest 79: 684–691, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Good DW, DuBose TD. Concentrations of NH3 in cortex and medulla of rat kidney. Contrib Nephrol 63: 16–20, 1988. [DOI] [PubMed] [Google Scholar]
- 99.Good DW, Knepper MA, Burg MB. Ammonia and bicarbonate transport by thick ascending limb of rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 247: F35–F44, 1984. [DOI] [PubMed] [Google Scholar]
- 100.Goodman AD, Fuisz RE, Cahill GF Jr. Renal gluconeogenesis in acidosis, alkalosis, and potassium deficiency: its possible role in regulation of renal ammonia production. J Clin Invest 45: 612–619, 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goraya N, Simoni J, Jo CH, Wesson DE. Treatment of metabolic acidosis in patients with stage 3 chronic kidney disease with fruits and vegetables or oral bicarbonate reduces urine angiotensinogen and preserves glomerular filtration rate. Kidney Int 86: 1031–1038, 2014. [DOI] [PubMed] [Google Scholar]
- 102.Goraya N, Wesson DE. Dietary management of chronic kidney disease: protein restriction and beyond. Curr Opin Nephrol Hypertens 21: 635–640, 2012. [DOI] [PubMed] [Google Scholar]
- 103.Goraya N, Wesson DE. Does correction of metabolic acidosis slow chronic kidney disease progression? Curr Opin Nephrol Hypertens 22: 193–197, 2013. [DOI] [PubMed] [Google Scholar]
- 104.Goraya N, Simoni J, Jo CH, Wesson DE. A comparison of treating metabolic acidosis in CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin J Am Soc Nephrol 8: 371–381, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grams ME, Chow EK, Segev DL, Coresh J. Lifetime incidence of CKD stages 3–5 in the United States. Am J Kidney Dis 62: 245–252, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gruswitz F, Chaudhary S, Ho JD, Schlessinger A, Pezeshki B, Ho CM, Sali A, Westhoff CM, Stroud RM. Function of human Rh based on structure of RhCG at 2.1 Å. Proc Natl Acad Sci USA 107: 9638–9643, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal-ATPases: physiology, regulation, structure. Am J Physiol Renal Physiol 298: F12–F21, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Györy ÁZ, Edwards KD. Renal tubular acidosis. Am J Med 45: 43–62, 1968. [DOI] [PubMed] [Google Scholar]
- 109.Halkier A, Taylor PR. A theoretical investigation of the equilibrium electric dipole moment of ammonia. Chem Phys Lett 285: 133–137, 1998. [Google Scholar]
- 110.Hall JA, Yan D. The molecular basis of K+ exclusion by the Escherichia coli ammonium channel AmtB. J Biol Chem 288: 14080–14086, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hamm LL. Renal handling of citrate. Kidney Int 38: 728–735, 1990. [DOI] [PubMed] [Google Scholar]
- 112.Hamm LL, Gillespie C, Klahr S. NH4Cl inhibition of transport in the rabbit cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 248: F631–F637, 1985. [DOI] [PubMed] [Google Scholar]
- 113.Hamm LL, Hering-Smith KS. Pathophysiology of hypocitraturic nephrolithiasis. Endocrinol Metab Clin North Am 31: 885–893, 2002. [DOI] [PubMed] [Google Scholar]
- 114.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol Renal Fluid Electrolyte Physiol 253: F595–F605, 1987. [DOI] [PubMed] [Google Scholar]
- 115.Hamm LL, Trigg D, Martin D, Gillespie C, Buerkert J. Transport of ammonia in the rabbit cortical collecting tubule. J Clin Invest 75: 478–485, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han KH, Croker BP, Clapp WL, Werner D, Sahni M, Kim J, Kim HY, Handlogten ME, Weiner ID. Expression of the ammonia transporter, Rh C Glycoprotein, in normal and neoplastic human kidney. J Am Soc Nephrol 17: 2670–2679, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Han KH, Mekala K, Babida V, Kim HY, Handlogten ME, Verlander JW, Weiner ID. Expression of the gas transporting proteins, Rh B Glycoprotein and Rh C Glycoprotein, in the murine lung. Am J Physiol Lung Cell Mol Physiol 297: L153–L163, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Han KH, Lee HW, Handlogten ME, Whitehill FM, Croker BP, Clapp W, Verlander JW, Weiner ID. Expression of the ammonia transporter family member, Rh B Glycoprotein, in the human kidney. Am J Physiol Renal Physiol 304: F972–F981, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han KH, Lee HW, Handlogten ME, Bishop JM, Levi M, Kim J, Verlander JW, Weiner ID. Effect of hypokalemia on renal expression of the ammonia transporter family members, Rh B Glycoprotein and Rh C Glycoprotein, in the rat kidney. Am J Physiol Renal Physiol 301: F823–F832, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Handlogten ME, Hong SP, Westhoff CM, Weiner ID. Basolateral ammonium transport by the mouse inner medullary collecting duct cell (mIMCD-3). Am J Physiol Renal Physiol 287: F628–F638, 2004. [DOI] [PubMed] [Google Scholar]
- 121.Handlogten ME, Hong SP, Westhoff CM, Weiner ID. Apical ammonia transport by the mouse inner medullary collecting duct cell (mIMCD-3). Am J Physiol Renal Physiol 289: F347–F358, 2005. [DOI] [PubMed] [Google Scholar]
- 122.Handlogten ME, Hong SP, Zhang L, Vander AW, Steinbaum ML, Campbell-Thompson M, Weiner ID. Expression of the ammonia transporter proteins, Rh B Glycoprotein and Rh C Glycoprotein, in the intestinal tract. Am J Physiol Gastrointest Liver Physiol 288: G1036–G1047, 2005. [DOI] [PubMed] [Google Scholar]
- 123.Handlogten ME, Osis G, Lee HW, Romero MF, Verlander JW, Weiner ID. NBCe1 expression is required for normal renal ammonia metabolism. Am J Physiol Renal Physiol 309: F658–F666, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Haussinger D, Lamers WH, Moorman AF. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme 46: 72–93, 1992. [DOI] [PubMed] [Google Scholar]
- 125.Hayslett JP. Functional adaptation to reduction in renal mass. Physiol Rev 59: 137–164, 1979. [DOI] [PubMed] [Google Scholar]
- 126.Heginbotham L, MacKinnon R. Conduction properties of the cloned Shaker K+ channel. Biophys J 65: 2089–2096, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holm LM, Jahn TP, Moller AL, Schjoerring JK, Ferri D, Klaerke DA, Zeuthen T. NH3 and NH4+ permeability in aquaporin-expressing Xenopus oocytes. Pflügers Arch 450: 415–428, 2005. [DOI] [PubMed] [Google Scholar]
- 128.Hropot M, Fowler N, Karlmark B, Giebisch G. Tubular action of diuretics: distal efects on electrolyte transport and acidification. Kidney Int 28: 477–489, 1985. [DOI] [PubMed] [Google Scholar]
- 129.Hutcheon DE, Vincent ME, Sandhu RS. Renal electrolyte excretion pattern in response to bumetanide in healthy volunteers. J Clin Pharmacol 21: 604–609, 1981. [DOI] [PubMed] [Google Scholar]
- 130.Igarashi T, Inatomi J, Sekine T, Cha SH, Kanai Y, Kunimi M, Tsukamoto K, Satoh H, Shimadzu M, Tozawa F, Mori T, Shiobara M, Seki G, Endou H. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet 23: 264–266, 1999. [DOI] [PubMed] [Google Scholar]
- 131.Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol 13: 2171–2177, 2002. [DOI] [PubMed] [Google Scholar]
- 132.Inatomi J, Horita S, Braverman N, Sekine T, Yamada H, Suzuki Y, Kawahara K, Moriyama N, Kudo A, Kawakami H, Shimadzu M, Endou H, Fujita T, Seki G, Igarashi T. Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflügers Arch 448: 438–444, 2004. [DOI] [PubMed] [Google Scholar]
- 133.Izumi Y, Hori K, Nakayama Y, Kimura M, Hasuike Y, Nanami M, Kohda Y, Otaki Y, Kuragano T, Obinata M, Kawahara K, Tanoue A, Tomita K, Nakanishi T, Nonoguchi H. Aldosterone requires vasopressin V1a receptors on intercalated cells to mediate acid-base homeostasis. J Am Soc Nephrol 22: 673–680, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jakobsen JK, Odgaard E, Wang W, Elkjaer ML, St Nielsen Aalkjaer C, Leipziger J. Functional up-regulation of basolateral Na+-dependent HCO3− transporter NBCn1 in medullary thick ascending limb of K+-depleted rats. Pflügers Arch 448: 571–578, 2004. [DOI] [PubMed] [Google Scholar]
- 135.Janicki RH, Goldstein L. Glutamine synthetase and renal ammonia metabolism. Am J Physiol 216: 1107–1110, 1969. [DOI] [PubMed] [Google Scholar]
- 136.Jarolim P, Shayakul C, Prabakaran D, Jiang L, Stuart-Tilley A, Rubin HL, Simova S, Zavadil J, Herrin JT, Brouillette J, Somers MJ, Seemanova E, Brugnara C, Guay-Woodford LM, Alper SL. Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band 3) Cl−/HCO3− exchanger. J Biol Chem 273: 6380–6388, 1998. [DOI] [PubMed] [Google Scholar]
- 137.Kamm DE, Strope GL. Glutamine and glutamate metabolism in renal cortex from potassium-depleted rats. Am J Physiol 224: 1241–1248, 1973. [DOI] [PubMed] [Google Scholar]
- 138.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999. [DOI] [PubMed] [Google Scholar]
- 139.Karet FE, Gainza FJ, Gyory AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, di Pietro A, Walker WG, Lifton RP. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA 95: 6337–6342, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Karet FE. Inherited distal renal tubular acidosis. J Am Soc Nephrol 13: 2178–2184, 2002. [DOI] [PubMed] [Google Scholar]
- 141.Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol 20: 251–254, 2009. [DOI] [PubMed] [Google Scholar]
- 142.Karinch AM, Lin CM, Wolfgang CL, Pan M, Souba WW. Regulation of expression of the SN1 transporter during renal adaptation to chronic metabolic acidosis in rats. Am J Physiol Renal Physiol 283: F1011–F1019, 2002. [DOI] [PubMed] [Google Scholar]
- 143.Katzir Z, Dinour D, Reznik-Wolf H, Nissenkorn A, Holtzman E. Familial pure proximal renal tubular acidosis—clinical and genetic study. Nephrol Dial Transplant 23: 1211–1215, 2008. [DOI] [PubMed] [Google Scholar]
- 144.Khademi S, O'Connell J III, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 A. Science 305: 1587–1594, 2004. [DOI] [PubMed] [Google Scholar]
- 145.Kida K, Nakajo S, Kamiya F, Toyama Y, Nishio T, Nakagawa H. Renal net glucose release in vivo and its contribution to blood glucose in rats. J Clin Invest 62: 721–726, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kikeri D, Sun A, Zeidel ML, Hebert SC. Cell membranes impermeable to NH3. Nature 339: 478–480, 1989. [DOI] [PubMed] [Google Scholar]
- 147.Kim HY, Verlander JW, Bishop JM, Cain BD, Han KH, Igarashi P, Lee HW, Handlogten ME, Weiner ID. Basolateral expression of the ammonia transporter family member, Rh C Glycoprotein, in the mouse kidney. Am J Physiol Renal Physiol 296: F545–F555, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol 293: F1238–F1247, 2007. [DOI] [PubMed] [Google Scholar]
- 149.Kinne R, Kinne-Saffran E, Schutz H, Schölermann B. Ammonium transport in medullary thick ascending limb of rabbit kidney: involvement of the Na+,K+,Cl−-cotransporter. J Membr Biol 94: 279–284, 1986. [DOI] [PubMed] [Google Scholar]
- 150.Kinsella JL, Aronson PS. Interaction of NH4+ and Li+ with the renal microvillus membrane Na+-H+ exchanger. Am J Physiol Cell Physiol 241: C220–C226, 1981. [DOI] [PubMed] [Google Scholar]
- 151.Kiwull-Schöne H, Kalhoff H, Manz F, Kiwull P. Food mineral compositionand acid-base balance in rabbits. Eur J Nutr 44: 499–508, 2005. [DOI] [PubMed] [Google Scholar]
- 152.Knepper MA, Good DW, Burg MB. Mechanism of ammonia secretion by cortical collecting ducts of rabbits. Am J Physiol Renal Fluid Electrolyte Physiol 247: F729–F738, 1984. [DOI] [PubMed] [Google Scholar]
- 153.Knepper MA, Good DW, Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am J Physiol Renal Fluid Electrolyte Physiol 249: F870–F877, 1985. [DOI] [PubMed] [Google Scholar]
- 154.Knochel JP. The syndrome of hyporeninemic hypoaldosteronism. Annu Rev Med 30: 145–153, 1979. [DOI] [PubMed] [Google Scholar]
- 155.Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrol Dial Transplant 24: 1232–1237, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kurtz I, Balaban RS. Ammonium as a substrate for Na+-K+-ATPase in rabbit proximal tubules. Am J Physiol Renal Fluid Electrolyte Physiol 250: F497–F502, 1986. [DOI] [PubMed] [Google Scholar]
- 157.Kwon TH, Fulton C, Wang W, Kurtz I, Frokiaer J, Aalkjaer C, Nielsen S. Chronic metabolic acidosis upregulates rat kidney Na-HCO cotransporters NBCn1 and NBC3 but not NBC1. Am J Physiol Renal Physiol 282: F341–F351, 2002. [DOI] [PubMed] [Google Scholar]
- 158.Kwon TH, Frokiaer J, Fernandez-Llama P, Maunsbach AB, Knepper MA, Nielsen S. Altered expression of Na transporters NHE-3, NaPi-II, Na-K-ATPase, BSC-1, and TSC in CRF rat kidneys. Am J Physiol Renal Physiol 277: F257–F270, 1999. [DOI] [PubMed] [Google Scholar]
- 159.Laghmani K, Preisig PA, Moe OW, Yanagisawa M, Alpern RJ. Endothelin-1/endothelin-B receptor-mediated increases in NHE3 activity in chronic metabolic acidosis. J Clin Invest 107: 1563–1569, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C Glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee HW, Verlander JW, Handlogten ME, Han KH, Cooke PS, Weiner ID. Expression of the rhesus glycoprotein, ammonia transporter family members, Rhcg and Rhbg, in male reproductive organs. J Reprod Fertil 146: 283–296, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee HW, Osis G, Handlogten ME, Guo H, Verlander JW, Weiner ID. Effect of dietary protein restriction on renal ammonia metabolism. Am J Physiol Renal Physiol 308: F1463–F1473, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lee HW, Osis G, Handlogten ME, Lamers WH, Chaudhry FA, Verlander JW, Weiner ID. Proximal tubule-specific glutamine synthetase deletion alters basal and acidosis-stimulated ammonia metabolism. Am J Physiol Renal Physiol 310: F1229–F1242, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee HW, Verlander JW, Bishop JM, Handlogten ME, Weiner ID. Effect of collecting duct-specific deletion of both Rh B glycoprotein (Rhbg) and Rh C glycoprotein (Rhcg) on renal response to metabolic acidosis (Abstract). J Am Soc Nephrol 23: 31A, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee HW, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID. Effect of intercalated cell-specific Rh C Glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 299: F369–F379, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lee HW, Verlander JW, Bishop JM, Handlogten ME, Han KH, Weiner ID. Renal ammonia excretion in response to hypokalemia: effects of collecting duct-specific Rh C Glycoprotein deletion. Am J Physiol Renal Physiol 304: F410–F421, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lee HW, Verlander JW, Handlogten ME, Han KH, Weiner ID. Effect of collecting duct-specific deletion of both Rh B Glycoprotein (Rhbg) and Rh C Glycoprotein (Rhcg) on renal response to metabolic acidosis. Am J Physiol Renal Physiol 306: F389–F400, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee S, Lee HJ, Yang HS, Thornell IM, Bevensee MO, Choi I. Na/bicarbonate Ccotransporter NBCn1 in the kidney medullary thick ascending limb cell line is upregulated under acidic conditions and enhances ammonium transport. Exp Physiol 95: 926–937, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int 58: 1267–1277, 2000. [DOI] [PubMed] [Google Scholar]
- 170.LeMasurier M, Heginbotham L, Miller C. KcsA: it's a potassium channel. J Gen Physiol 118: 303–314, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li HC, Du Z, Barone S, Rubera I, McDonough AA, Tauc M, Zahedi K, Wang T, Soleimani M. Proximal tubule specific knockout of the Na+/H+ exchanger NHE3: effects on bicarbonate absorption and ammonium excretion. J Mol Med 91: 951–963, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lim SW, Ahn KO, Kim WY, Han DH, Li C, Ghee JY, Han KH, Kim HY, Handlogten ME, Kim J, Yang CW, Weiner ID. Expression of ammonia transporters, Rhbg and Rhcg, in chronic cyclosporine nephropathy in rats. Nephron Exp Nephrol 110: e49–e58, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu K, Nagase H, Huang CG, Calamita G, Agre P. Purification and functional characterization of aquaporin-8. Biol Cell 98: 153–161, 2006. [DOI] [PubMed] [Google Scholar]
- 174.Liu Z, Chen Y, Mo R, Cc Hui Cheng JF, Mohandas N, Huang CH. Characterization of human RhCG and mouse Rhcg as novel nonerythroid Rh glycoprotein homologues predominantly expressed in kidney and testis. J Biol Chem 275: 25641–25651, 2000. [DOI] [PubMed] [Google Scholar]
- 175.Liu Z, Peng J, Mo R, Hui C, Huang CH. Rh type B glycoprotein is a new member of the Rh superfamily and a putative ammonia transporter in mammals. J Biol Chem 276: 1424–1433, 2001. [DOI] [PubMed] [Google Scholar]
- 176.Lonnerholm G, Wistrand PJ. Membrane-bound carbonic anhydrase CA IV in the human kidney. Acta Physiol Scand 141: 231–234, 2008. [DOI] [PubMed] [Google Scholar]
- 177.Lopez C, Metral S, Eladari D, Drevensek S, Gane P, Chambrey R, Bennett V, Cartron JP, Van Kim C, Colin Y. The ammonium transporter RhBG: requirement of a tyrosine-based signal and ankyrin-G for basolateral targeting and membrane anchorage in polarized kidney epithelial cells. J Biol Chem 280: 8221–8228, 2004. [DOI] [PubMed] [Google Scholar]
- 178.Lowry M, Ross BD. Activation of oxoglutarate dehydrogenase in the kidney in response to acute acidosis. Biochem J 190: 771–780, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ludewig U. Electroneutral ammonium transport by basolateral Rhesus B glycoprotein. J Physiol 559: 751–759, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lupo D, Li XD, Durand A, Tomizaki T, Cherif-Zahar B, Matassi G, Merrick M, Winkler FK. The-A. The A resolution structure of Nitrosomonas europaea Rh50 and mechanistic implications for NH3 transport by Rhesus family proteins. Proc Natl Acad Sci USA 104: 19303–19308, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.MacClean AJ, Hayslett JP. Adaptive change in ammonia excretion in renal insufficiency. Kidney Int 17: 595–606, 1980. [DOI] [PubMed] [Google Scholar]
- 182.Mackler B, Lichtenstein H, Guest GM. Effects of ammonium chloride acidosis on the action of insulin in dogs. Am J Physiol 166: 191–198, 1951. [DOI] [PubMed] [Google Scholar]
- 183.Mackler B, Lichtenstein H, Guest GM. Effects of ammonium chloride acidosis on glucose tolerance in dogs. Am J Physiol 168: 125–130, 1952. [DOI] [PubMed] [Google Scholar]
- 184.Maher T, Schambelan M, Kurtz I, Hulter HN, Jones JW, Sebastian A. Amelioration of metabolic acidosis by dietary potassium restriction in hyperkalemic patients with chronic renal insufficiency. J Lab Clin Med 103: 432–445, 1984. [PubMed] [Google Scholar]
- 185.Mak DO, Dang B, Weiner ID, Foskett JK, Westhoff CM. Characterization of transport by the kidney Rh glycoproteins, RhBG and RhCG. Am J Physiol Renal Physiol 290: F297–F305, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Marini AM, Matassi G, Raynal V, Andre B, Cartron JP, Cherif-Zahar B. The human Rhesus-associated RhAG protein and a kidney homologue promote ammonium transport in yeast. Nat Genet 26: 341–344, 2000. [DOI] [PubMed] [Google Scholar]
- 187.Matsutaka H, Aikawa T, Yamamoto H, Ishikawa E. Gluconeogenesis and amino acid metabolism. III. Uptake of glutamine and output of alanine and ammonia by non-hepatic splanchnic organs of fasted rats and their metabolic significance. J Biochem 74: 1019–1029, 1973. [PubMed] [Google Scholar]
- 188.Mayer M, Schaaf G, Mouro I, Lopez C, Colin Y, Neumann P, Cartron JP, Ludewig U. Different transport mechanisms in plant and human AMT/Rh-type ammonium transporters. J Gen Physiol 127: 133–144, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McCann WP. Intrarenal distribution of glucose-6-phosphatase: methodological and physiological considerations. Proc Soc Exp Biol Med 124: 185–187, 1971. [DOI] [PubMed] [Google Scholar]
- 190.McClellan AL. Tables of Experimental Dipole Moments. San Francisco, CA: Freeman, 1989. [Google Scholar]
- 191.Mitch WE. Metabolic and clinical consequences of metabolic acidosis. J Nephrol 19: S70–S75, 2006. [PubMed] [Google Scholar]
- 192.Mitrakou A. Kidney: its impact on glucose homeostasis and hormonal regulation. Diabetes Res Clin Pract 93: S66–S72, 2011. [DOI] [PubMed] [Google Scholar]
- 193.Molinas SM, Trumper L, Marinelli RA. Mitochondrial aquaporin-8 in renal proximal tubule cells: evidence for a role in the response to metabolic acidosis. Am J Physiol Renal Physiol 303: F458–F466, 2012. [DOI] [PubMed] [Google Scholar]
- 194.Moranne O, Froissart M, Rossert J, Gauci C, Boffa JJ, Haymann JP, M'rad MB, Jacquot C, Houillier P, Stengel B, Fouqueray B, NephroTest Study Group. Timing of onset of CKD-related metabolic complications. J Am Soc Nephrol 20: 164–171, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA. Regulation of renal amino acid transporters during metabolic acidosis. Am J Physiol Renal Physiol 292: F555–F566, 2007. [DOI] [PubMed] [Google Scholar]
- 196.Mouro-Chanteloup I, Cochet S, Chami M, Genetet S, Zidi-Yahiaoui N, Engel A, Colin Y, Bertrand O, Ripoche P. Functional reconstitution into liposomes of purified human rhcg ammonia channel. PLoS One 5: e8921, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mudry B, Guy RH, Gado-Charro MB. Transport numbers in transdermal iontophoresis. Biophys J 90: 2822–2830, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Musa-Aziz R, Chen LM, Pelletier MF, Boron WF. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc Natl Acad Sci USA 106: 5406–5411, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nagami GT. Luminal secretion of ammonia in the mouse proximal tubule perfused in vitro. J Clin Invest 81: 159–164, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nagami GT. Net luminal secretion of ammonia by the proximal tubule. Contrib Nephrol 63: 1–5, 1988. [DOI] [PubMed] [Google Scholar]
- 201.Nagami GT. Ammonia production and secretion by the proximal tubule. Am J Kidney Dis 14: 258–261, 1989. [DOI] [PubMed] [Google Scholar]
- 202.Nagami GT. Enhanced ammonia secretion by proximal tubules from mice receiving NH4Cl: role of angiotensin II. Am J Physiol Renal Physiol 282: F472–F477, 2002. [DOI] [PubMed] [Google Scholar]
- 203.Nagami GT. Ammonia production and secretion by S3 proximal tubule segments from acidotic mice: role of ANG II. Am J Physiol Renal Physiol 287: F707–F712, 2004. [DOI] [PubMed] [Google Scholar]
- 204.Nagami GT. Role of angiotensin II in the enhancement of ammonia production and secretion by the proximal tubule in metabolic acidosis. Am J Physiol Renal Physiol 294: F874–F880, 2008. [DOI] [PubMed] [Google Scholar]
- 205.Nagami GT, Sonu CM, Kurokawa K. Ammonia production by isolated mouse proximal tubules perfused in vitro: effect of metabolic acidosis. J Clin Invest 78: 124–129, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Nakamura S, Amlal H, Galla JH, Soleimani M. NH4+ secretion in inner medullary collecting duct in potassium deprivation: role of colonic H+-K+-ATPase. Kidney Int 56: 2160–2167, 1999. [DOI] [PubMed] [Google Scholar]
- 207.Nakhoul NL, Abdulnour-Nakhoul SM, Boulpaep EL, Rabon E, Schmidt E, Hamm LL. Substrate specificity of Rhbg: ammonium and methyl ammonium transport. Am J Physiol Cell Physiol 299: C695–C705, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Nakhoul NL, Abdulnour-Nakhoul SM, Schmidt E, Doetjes R, Rabon E, Hamm LL. pH sensitivity of ammonium transport by Rhbg. Am J Physiol Cell Physiol 299: C1386–C1397, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nakhoul NL, DeJong H, Abdulnour-Nakhoul SM, Boulpaep EL, Hering-Smith K, Hamm LL. Characteristics of renal Rhbg as an NH4+ transporter. Am J Physiol Renal Physiol 288: F170–F181, 2005. [DOI] [PubMed] [Google Scholar]
- 210.Nakhoul NL, Hering-Smith KS, Abdulnour-Nakhoul SM, Hamm LL. Transport of NH3/NH in oocytes expressing aquaporin-1. Am J Physiol Renal Physiol 281: F255–F263, 2001. [DOI] [PubMed] [Google Scholar]
- 211.Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann JR. Renal tubular acidosis in infants and children: clinical course, response to treatment, and prognosis. J Pediatr 80: 738–748, 1972. [DOI] [PubMed] [Google Scholar]
- 212.Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest 76: 667–675, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Navaneethan SD, Schold JD, Arrigain S, Jolly SE, Wehbe E, Raina R, Simon JF, Srinivas TR, Jain A, Schreiber MJ Jr, Nally JV Jr. Serum bicarbonate and mortality in stage 3 and stage 4 chronic kidney disease. Clin J Am Soc Nephrol 6: 2395–2402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Noiret L, Baigent S, Jalan R, Thomas SR. Mathematical model of ammonia handling in the rat renal medulla. PLoS One 10: e0134477, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nonoguchi H, Takehara Y, Endou H. Intra- and inter-nephron heterogeneity of ammoniagenesis in rats: effects of chronic metabolic acidosis and potassium depletion. Pflügers Arch 407: 245–251, 1986. [DOI] [PubMed] [Google Scholar]
- 216.Nonoguchi H, Uchida S, Shiigai T, Endou H. Effect of chronic metabolic acidosis on ammonia production from l-glutamine in microdissected rat nephron segments. Pflügers Arch 403: 229–235, 1985. [DOI] [PubMed] [Google Scholar]
- 217.Nygaard TP, Alfonso-Prieto M, Peters GH, Jensen MO, R ovira C. Substrate recognition in the Escherichia coli ammonia channel AmtB: A QM/MM Investigation. J Phys Chem 114: 11859–11865, 2010. [DOI] [PubMed] [Google Scholar]
- 218.Odgaard E, Jakobsen JK, Frische S, Praetorius J, Nielsen S, Aalkjaer C, Leipziger J. Basolateral Na+-dependent HCO3− transporter NBCn1-mediated HCO3− influx in rat medullary thick ascending limb. J Physiol 555: 205–218, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Osis G, Handlogten ME, Lee HW, Hering-Smith KS, Huang W, Romero MF, Verlander JW, Weiner ID. Effect of NBCe1 deletion on renal citrate and 2-oxoglutarate handling. Physiol Rep 4: e12778, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Osther PJ, Bollerslev J, Hansen AB, Engel K, Kildeberg P. Pathophysiology of incomplete renal tubular acidosis in recurrent renal stone formers: evidence of disturbed calcium, bone and citrate metabolism. Urol Res 21: 169–173, 1993. [DOI] [PubMed] [Google Scholar]
- 221.Owen EE, Robinson RR. Amino acid extraction and ammonia metabolism by the human kidney during the prolonged administration of ammonium chloride. J Clin Invest 42: 263–276, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Owen EE, Tyor MP, Flanagan JF, Berry JN. The kidney as a source of blood ammonia in patients with liver disease: the effect of acetazolamide. J Clin Invest 39: 288–294, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF. Liver and kidney metabolism during prolonged starvation. J Clin Invest 48: 574–583, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Packer RK, Desai SS, Hornbuckle K, Knepper MA. Role of countercurrent multiplication in renal ammonium handling: regulation of medullary ammonium accumulation. J Am Soc Nephrol 2: 77–83, 1991. [DOI] [PubMed] [Google Scholar]
- 225.Pajor AM. Citrate transport by the kidney and intestine. Semin Nephrol 19: 195–200, 1999. [PubMed] [Google Scholar]
- 226.Parry DM, Brosnan JT. Glutamine metabolism in the kidney during induction of, and recovery from, metabolic acidosis in the rat. Biochem J 174: 387–396, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Parry DM, Brosnan JT. Renal phosphoenolpyruvate carboxykinase during perturbations of acid-base homeostasis in rats. Immunochemical studies. Can J Biochem 58: 1298–1301, 1980. [DOI] [PubMed] [Google Scholar]
- 228.Pfeiffer R, Rossier G, Spindler B, Meier C, Kuhn L, Verrey F. Amino acid transport of y+L-type by heterodimers of 4F2hc/CD98 and members of the glycoprotein-associated amino acid transporter family. EMBO J 18: 49–57, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pitts RF. Metabolism of amino acids by the perfused rat kidney. Am J Physiol 220: 862–867, 1971. [DOI] [PubMed] [Google Scholar]
- 230.Pollak VE, Mattenheimer H, DeBruin H, Weinman KJ. Experimental metabolic acidosis: the enzymatic basis of ammonia production by the dog kidney. J Clin Invest 44: 169–181, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Porter LD, Ibrahim H, Taylor L, Curthoys NP. Complexity and species variation of the kidney-type glutaminase gene. Physiol Gen 9: 157–166, 2002. [DOI] [PubMed] [Google Scholar]
- 232.Quentin F, Eladari D, Cheval L, Lopez C, Goossens D, Colin Y, Cartron JP, Paillard M, Chambrey R. RhBG and RhCG, the putative ammonia transporters, are expressed in the same cells in the distal nephron. J Am Soc Nephrol 14: 545–554, 2003. [DOI] [PubMed] [Google Scholar]
- 233.Ramadan T, Camargo SMR, Herzog B, Bordin M, Pos KM, Verrey F. Recycling of aromatic amino acids via TAT1 allows efflux of neutral amino acids via LAT2-4F2hc exchanger. Pflügers Arch 454: 507–516, 2007. [DOI] [PubMed] [Google Scholar]
- 234.Ramadan T, Camargo SMR, Summa V, Hunziker P, Chesnov S, Pos KM, Verrey F. Basolateral aromatic amino acid transporter TAT1 (Slc16a10) functions as an efflux pathway. J Cell Physiol 206: 771–779, 2006. [DOI] [PubMed] [Google Scholar]
- 235.Raphael KL, Murphy RA, Shlipak MG, Satterfield S, Huston HK, Sebastian A, Sellmeyer DE, Patel KV, Newman AB, Sarnak MJ, Ix JH, Fried LF. Bicarbonate concentration, acid-base status, and mortality in the health, aging, and body composition study. Clin J Am Soc Nephrol 11: 308–316, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Rastogi S, Bayliss JM, Nascimento L, Arruda JA. Hyperkalemic renal tubular acidosis: effect of furosemide in humans and in rats. Kidney Int 28: 801–807, 1985. [DOI] [PubMed] [Google Scholar]
- 237.Relman AS. Renal acidosis and renal excretion of acid in health and disease. Adv Internal Med 12: 295–347, 1963. [PubMed] [Google Scholar]
- 238.Reynolds TB. Observations on the pathogenesis of renal tubular acidosis. Am J Med 25: 503–515, 1958. [DOI] [PubMed] [Google Scholar]
- 239.Ripoche P, Bertrand O, Gane P, Birkenmeier C, Colin Y, Cartron JP. Human Rhesus-associated glycoprotein mediates facilitated transport of NH3 into red blood cells. Proc Natl Acad Sci USA 101: 17222–17227, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Rivers R, Blanchard A, Eladari D, Leviel F, Paillard M, Podevin RA, Zeidel ML. Water and solute permeabilities of medullary thick ascending limb apical and basolateral membranes. Am J Physiol Renal Physiol 274: F453–F462, 1998. [DOI] [PubMed] [Google Scholar]
- 241.Robinson RR, Owen EE. Intrarenal distribution of ammonia during diuresis and antidiuresis. Am J Physiol 208: 1129–1134, 1965. [DOI] [PubMed] [Google Scholar]
- 242.Rocher LL, Tannen RL. The clinical spectrum of renal tubular acidosis. Annu Rev Med 37: 319–331, 1986. [DOI] [PubMed] [Google Scholar]
- 243.Romeo E, Dave MH, Bacic D, Ristic Z, Camargo SMR, Loffing J, Wagner CA, Verrey F. Luminal kidney and intestine SLC6 amino acid transporters of B0AT-cluster and their tissue distribution in Mus musculus. Am J Physiol Renal Physiol 290: F376–F383, 2006. [DOI] [PubMed] [Google Scholar]
- 244.Rosen RA, Julian BA, Dubovsky EV, Galla JH, Luke RG. On the mechanism by which chloride corrects metabolic alkalosis in man. Am J Med 84: 449–458, 1988. [DOI] [PubMed] [Google Scholar]
- 245.Rossier G, Meier C, Bauch C, Summa V, Sordat B, Verrey F, Kuhn LC. LAT2, a new basolateral 4F2hc/CD98-associated amino acid transporter of kidney and intestine. J Biol Chem 274: 34948–34954, 1999. [DOI] [PubMed] [Google Scholar]
- 246.Sagild U, Andersen V, Andreasen PB. Glucose tolerance and insulin responsiveness in experimental potassium depletion. Acta Med Scand 169: 243–251, 1961. [DOI] [PubMed] [Google Scholar]
- 247.Saikiat C. Composition of the renal cortex and medulla of rats during water diuresis and antidiuresis. Q J Exp Physiol Cogn Med Sci 50: 146–157, 1965. [DOI] [PubMed] [Google Scholar]
- 248.Sajo IM, Goldstein MB, Sonnenberg H, Stinebaugh BJ, Wilson DR, Halperin ML. Sites of ammonia addition to tubular fluid in rats with chronic metabolic acidosis. Kidney Int 20: 353–358, 1982. [DOI] [PubMed] [Google Scholar]
- 249.Saparov SM, Liu K, Agre P, Pohl P. Fast and selective ammonia transport by aquaporin-8. J Biol Chem 282: 5296–5301, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Schlueter W, Keilani T, Hizon M, Kaplan B, Batlle DC. On the mechanism of impaired distal acidification in hyperkalemic renal tubular acidosis: evaluation with amiloride and bumetanide. J Am Soc Nephrol 3: 953–964, 1992. [DOI] [PubMed] [Google Scholar]
- 251.Schmidt U, Guder WG. Sites of enzyme activity along the nephron. Kidney Int 9: 233–242, 1976. [DOI] [PubMed] [Google Scholar]
- 252.Schoolwerth AC, LaNoue KF. Transport of metabolic substrates in renal mitochondria. Annu Rev Physiol 47: 143–171, 1985. [DOI] [PubMed] [Google Scholar]
- 253.Schoolwerth AC, Sandler RS, Hoffman PM, Klahr S. Effects of nephron reduction and dietary protein content on renal ammoniagenesis in the rat. Kidney Int 7: 397–404, 1975. [DOI] [PubMed] [Google Scholar]
- 254.Schoolwerth AC, Smith BC, Culpepper RM. Renal gluconeogenesis. Miner Electrolyte Metab 14: 347–361, 1988. [PubMed] [Google Scholar]
- 255.Schwartz GJ. Physiology and molecular biology of renal carbonic anhydrase. J Nephrol 15 Suppl 5: S61–S74, 2002. [PubMed] [Google Scholar]
- 256.Schwartz GJ, Kittelberger AM, Barnhart DA, Vijayakumar S. Carbonic anhydrase IV is expressed in H+-secreting cells of rabbit kidney. Am J Physiol Renal Physiol 278: F894–F904, 2000. [DOI] [PubMed] [Google Scholar]
- 257.Sebastian A, Schambelan M, Sutton JM. Amelioration of hyperchloremic acidosis with furosemide therapy in patients with chronic renal insufficiency and type 4 renal tubular acidosis. Am J Nephrol 4: 287–300, 1984. [DOI] [PubMed] [Google Scholar]
- 258.Seshadri RM, Klein JD, Kozlowski S, Sands JM, Kim YH, Handlogten ME, Verlander JW, Weiner ID. Renal expression of the ammonia transporters, Rhbg and Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F397–F408, 2006. [DOI] [PubMed] [Google Scholar]
- 259.Seshadri RM, Klein JD, Smith T, Sands JM, Handlogten ME, Verlander JW, Weiner ID. Changes in the subcellular distribution of the ammonia transporter Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F1443–F1452, 2006. [DOI] [PubMed] [Google Scholar]
- 260.Shayakul C, Alper SL. Inherited renal tubular acidosis. Curr Opin Nephrol Hypertens 9: 541–546, 2000. [DOI] [PubMed] [Google Scholar]
- 261.Shayakul C, Alper SL. Defects in processing and trafficking of the AE1 Cl−/HCO3− exchanger associated with inherited distal renal tubular acidosis. Clin Exp Nephrol 8: 1–11, 2004. [DOI] [PubMed] [Google Scholar]
- 262.Shayakul C, Jarolim P, Zachlederova M, Prabakaran D, Cortez-Campeao D, Kalabova D, Stuart-Tilley AK, Ideguchi H, Haller C, Alper SL. Characterization of a highly polymorphic marker adjacent to the SLC4A1 gene and of kidney immunostaining in a family with distal renal tubular acidosis. Nephrol Dial Transplant 19: 371–379, 2004. [DOI] [PubMed] [Google Scholar]
- 263.Shear L, Gabuzda GJ. Potassium deficiency and endogenous ammonium overload from kidney. Am J Clin Nutr 23: 614–618, 1970. [DOI] [PubMed] [Google Scholar]
- 264.Siener R, Hesse A. The effect of different diets on urine composition and the risk of calcium oxalate crystallisation in healthy subjects. Eur Urol 42: 289–296, 2002. [DOI] [PubMed] [Google Scholar]
- 265.Silbernagl S. Tubular reabsorption of l-glutamine studied by free-flow micropuncture and microperfusion of rat kidney. Int J Biochem 12: 9–16, 1980. [DOI] [PubMed] [Google Scholar]
- 266.Simon E, Martin D, Buerkert J. Contribution of individual superficial nephron segments to ammonium handling in chronic metabolic acidosis in the rat. Evidence for ammonia disequilibrium in the renal cortex. J Clin Invest 76: 855–864, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Simon EE, Hamm LL. Ammonia entry along rat proximal tubule in vivo: effects of luminal pH and flow rate. Am J Physiol Renal Fluid Electrolyte Physiol 253: F760–F766, 1987. [DOI] [PubMed] [Google Scholar]
- 268.Simon EE, Merli C, Herndon J, Cragoe EJ Jr, Hamm LL. Determinants of ammonia entry along the rat proximal tubule during chronic metabolic acidosis. Am J Physiol Renal Fluid Electrolyte Physiol 256: F1104–F1110, 1989. [DOI] [PubMed] [Google Scholar]
- 269.Simon EE, Merli C, Herndon J, Cragoe EJ Jr, Hamm LL. Effects of barium and 5-(N-ethyl-N-isopropyl)-amiloride on proximal tubule ammonia transport. Am J Physiol Renal Fluid Electrolyte Physiol 262: F36–F39, 1992. [DOI] [PubMed] [Google Scholar]
- 270.Simpson DP, Adam W. Glutamine transport and metabolism by mitochondria from dog renal cortex. General properties and response to acidosis and alkalosis. J Biol Chem 250: 8148–8158, 1975. [PubMed] [Google Scholar]
- 271.Singh SK, Binder HJ, Geibel JP, Boron WF. An apical permeability barrier to NH3/NH4+ in isolated, perfused colonic crypts. Proc Natl Acad Sci USA 92: 11573–11577, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26: 71–75, 2000. [DOI] [PubMed] [Google Scholar]
- 273.Sohet F, Colin Y, Genetet S, Ripoche P, Metral S, Le Van Kim C, Lopez C. Phosphorylation and ankyrin-G binding of the C-terminal domain regulate targeting and function of the ammonium transporter RhBG. J Biol Chem 283: 26557–26567, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Solbu TT, Boulland JL, Zahid W, Lyamouri Bredahl MK, miry-Moghaddam M, Storm-Mathisen J, Roberg BA, Chaudhry FA. Induction and targeting of the glutamine transporter SN1 to the basolateral membranes of cortical kidney tubule cells during chronic metabolic acidosis suggest a role in pH regulation. J Am Soc Nephrol 16: 869–877, 2005. [DOI] [PubMed] [Google Scholar]
- 275.Soriano JR, Boichis H, Stark H, Edelmann CM. Proximal renal tubular acidosis. A defect in bicarbonate reabsorption with normal urinary acidification. Pediatr Res 1: 81–98, 1967. [DOI] [PubMed] [Google Scholar]
- 276.Soriano JR. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 13: 2160–2170, 2002. [DOI] [PubMed] [Google Scholar]
- 277.Soupene E, Inwood W, Kustu S. Lack of the Rhesus protein Rh1 impairs growth of the green alga Chlamydomonas reinhardtii at high CO2. Proc Natl Acad Sci USA 101: 7787–7792, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Soupene E, King N, Feild E, Liu P, Niyogi KK, Huang CH, Kustu S. Rhesus expression in a green alga is regulated by CO2. Proc Natl Acad Sci USA 99: 7769–7773, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Star RA, Burg MB, Knepper MA. Luminal disequilibrium pH and ammonia transport in outer medullary collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 252: F1148–F1157, 1987. [DOI] [PubMed] [Google Scholar]
- 280.Star RA, Kurtz I, Mejia R, Burg MB, Knepper MA. Disequilibrium pH and ammonia transport in isolated perfused cortical collecting ducts. Am J Physiol Renal Fluid Electrolyte Physiol 253: F1232–F1242, 1987. [DOI] [PubMed] [Google Scholar]
- 281.Stettner P, Bourgeois S, Marsching C, Traykova-Brauch M, Porubsky S, Nordstrom V, Hopf C, Koesters R, Sandhoff R, Wiegandt H, Wagner CA, Gröne HJ, Jennemann R. Sulfatides are required for renal adaptation to chronic metabolic acidosis. Proc Natl Acad Sci USA 110: 9998–10003, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Stumvoll M, Meyer C, Mitrakou A, Nadkarni V, Gerich EJ. Renal glucose production and utilization: new aspects in humans. Diabetologia 40: 749–757, 1997. [DOI] [PubMed] [Google Scholar]
- 283.Sturgeon JP, Shawcross DL. Recent insights into the pathogenesis of hepatic encephalopathy and treatments. Expert Rev Gastroenterol Hepatol 8: 83–100, 2014. [DOI] [PubMed] [Google Scholar]
- 284.Tannen RL. The relationship between urine pH and acid excretion–the influence of urine flow rate. J Lab Clin Med 74: 757–769, 1969. [PubMed] [Google Scholar]
- 285.Tannen RL. Relationship of renal ammonia production and potassium homeostasis. Kidney Int 11: 453–465, 1977. [DOI] [PubMed] [Google Scholar]
- 286.Tannen RL, Sahai A. Biochemical pathways and modulators of renal ammoniagenesis. Miner Electrolyte Metab 16: 249–258, 1990. [PubMed] [Google Scholar]
- 287.Tapper EB, Jiang ZG, Patwardhan VR. Refining the ammonia hypothesis: a physiology-driven approach to the treatment of hepatic encephalopathy. Mayo Clin Proc 90: 646–658, 2015. [DOI] [PubMed] [Google Scholar]
- 288.Taylor L, Curthoys NP. Glutamine metabolism: role in acid-base balance. Biochem Mol Biol Ed 32: 291–304, 2004. [DOI] [PubMed] [Google Scholar]
- 289.Tizianello A, Deferrari G, Garibotto G, Robaudo C, Acquarone N, Ghiggeri GM. Renal ammoniagenesis in an early stage of metabolic acidosis in man. J Clin Invest 69: 240–250, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Toye AM, Parker MD, Daly CM, Lu J, Virkki LV, Pelletier MF, Boron WF. The human NBCe1-A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. Am J Physiol Cell Physiol 291: C788–C801, 2006. [DOI] [PubMed] [Google Scholar]
- 291.Tsuruoka S, Kittelberger AM, Schwartz GJ. Carbonic anhydrase II and IV mRNA in rabbit nephron segments: stimulation during metabolic acidosis. Am J Physiol Renal Physiol 274: F259–F267, 1998. [DOI] [PubMed] [Google Scholar]
- 292.Vallet M, Metzger M, Haymann JP, Flamant M, Gauci C, Thervet E, Boffa JJ, Vrtovsnik F, Froissart M, Stengel B, Houillier P. Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int 88: 137–145, 2015. [DOI] [PubMed] [Google Scholar]
- 293.Verlander JW, Madsen KM, Galla JH, Luke RG, Tisher CC. Response of intercalated cells to chloride depletion metabolic alkalosis. Am J Physiol Renal Fluid Electrolyte Physiol 262: F309–F319, 1992. [DOI] [PubMed] [Google Scholar]
- 294.Verlander JW, Madsen KM, Tisher CC. Axial distribution of band 3-positive intercalated cells in the collecting duct of control and ammonium chloride-loaded rabbits. Kidney Int 57: S137–S147, 1996. [PubMed] [Google Scholar]
- 295.Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID. Localization of the ammonium transporter proteins, Rh B Glycoprotein and Rh C Glycoprotein, in the mouse kidney. Am J Physiol Renal Physiol 284: F323–F337, 2003. [DOI] [PubMed] [Google Scholar]
- 296.Verlander JW, Chu D, Lee HW, Handlogten ME, Weiner ID. Expression of glutamine synthetase in the mouse kidney: localization in multiple epithelial cell types and differential regulation by hypokalemia. Am J Physiol Renal Physiol 305: F701–F713, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wagner CA, Mohebbi N. Urinary pH and stone formation. J Nephrol 23 Suppl 16: S165–S169, 2010. [PubMed] [Google Scholar]
- 298.Waisbren SJ, Geibel JP, Modlin IM, Boron WF. Unusual permeability properties of gastric gland cells. Nature 368: 332–335, 1994. [DOI] [PubMed] [Google Scholar]
- 299.Wall SM, Davis BS, Hassell KA, Meh ta P, Park SJ. In rat tIMCD, NH4+ uptake by the Na+,K+-ATPase is critical to net acid secretion during chronic hypokalemia. Am J Physiol Renal Physiol 277: F866–F874, 1999. [DOI] [PubMed] [Google Scholar]
- 300.Wall SM, Fischer MP. Contribution of the Na+-K+-2Cl− cotransporter (NKCC1) to transepithelial transport of H+, NH4+, K+, and Na+ in rat outer medullary collecting duct. J Am Soc Nephrol 13: 827–835, 2002. [DOI] [PubMed] [Google Scholar]
- 301.Wall SM, Fischer MP, Kim GH, Nguyen BM, Hassell KA. In rat inner medullary collecting duct, NH4+ uptake by the Na,K-ATPase is increased during hypokalemia. Am J Physiol Renal Physiol 282: F91–F102, 2002. [DOI] [PubMed] [Google Scholar]
- 302.Wall SM, Koger LM. NH4+ transport mediated by Na+-K+-ATPase in rat inner medullary collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 267: F660–F670, 1994. [DOI] [PubMed] [Google Scholar]
- 303.Wang S, Orabi EA, Baday S, Berneche S, Lamoureux G. Ammonium transporters achieve charge transfer by fragmenting their substrate. J Am Chem Soc 134: 10419–10427, 2012. [DOI] [PubMed] [Google Scholar]
- 304.Watts BA, Good DW. Effects of ammonium on intracellular pH in rat medullary thick ascending limb: mechanisms of apical membrane NH4+ transport. J Gen Physiol 103: 917–936, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Weiner ID. The Rh gene family and renal ammonium transport. Curr Opin Nephrol Hypertens 13: 533–540, 2004. [DOI] [PubMed] [Google Scholar]
- 306.Weiner ID, Hamm LL. Use of fluorescent dye BCECF to measure intracellular pH in cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 256: F957–F964, 1989. [DOI] [PubMed] [Google Scholar]
- 307.Weiner ID, Hamm LL. Molecular mechanisms of renal ammonia transport. Annu Rev Physiol 69: 317–340, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Weiner ID, Miller RT, Verlander JW. Localization of the ammonium transporters, Rh B Glycoprotein and Rh C Glycoprotein in the mouse liver. Gastroenterology 124: 1432–1440, 2003. [DOI] [PubMed] [Google Scholar]
- 310.Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol 10: 1444–1458, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Weiner ID, Verlander JW. Renal acidification mechanisms. In: Brenner and Rector's The Kidney, edited by Tall MW, Chertow GM, Marsden PA, Skorecki K, Yu AS, Brenner BM. New York: Saunders, 2015, p. 234–257. [Google Scholar]
- 312.Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Comprehens Physiol 3: 201–220, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Weiner ID, Verlander JW. Ammonia transport in the kidney by Rhesus glycoproteins. Am J Physiol Renal Physiol 306: F1107–F1120, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Weinstein AM. Ammonia transport in a mathematical model of rat proximal tubule. Am J Physiol Renal Fluid Electrolyte Physiol 267: F237–F248, 1994. [DOI] [PubMed] [Google Scholar]
- 315.Weinstein AM. A mathematical model of the rat nephron: glucose transport. Am J Physiol Renal Physiol 308: F1098–F1118, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Weinstein AM. A mathematical model of rat proximal tubule and loop of Henle. Am J Physiol Renal Physiol 308: F1076–F1097, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Weinstein AM, Krahn TA. A mathematical model of rat ascending Henle limb. II. Epithelial function. Am J Physiol Renal Physiol 298: F525–F542, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Westhoff CM, Ferreri-Jacobia M, Mak DO, Foskett JK. Identification of the erythrocyte Rh-blood group glycoprotein as a mammalian ammonium transporter. J Biol Chem 277: 12499–12502, 2002. [DOI] [PubMed] [Google Scholar]
- 320.Westhoff CM, Siegel DL, Burd CG, Foskett JK. Mechanism of genetic complementation of ammonium transport in yeast by human erythrocyte Rh-associated glycoprotein (RhAG). J Biol Chem 279: 17443–17448, 2004. [DOI] [PubMed] [Google Scholar]
- 321.Wilcox CS, Loon NR, Kanthawatana S, Pham MA, Cannizzaro R. Generation of alkalosis with loop diuretics: roles of contraction and acid excretion. J Nephrol 2: 81–87, 1990. [Google Scholar]
- 322.Winkler CA, Kittelberger AM, Schwartz GJ. Expression of carbonic anhydrase IV mRNA in rabbit kidney: stimulation by metabolic acidosis. Am J Physiol Renal Physiol 272: F551–F560, 1997. [DOI] [PubMed] [Google Scholar]
- 323.Wirthensohn G, Guder WG. Renal substrate metabolism. Physiol Rev 66: 469–497, 1986. [DOI] [PubMed] [Google Scholar]
- 324.Wright PA, Knepper MA. Phosphate-dependent glutaminase activity in rat renal cortical and medullary tubule segments. Am J Physiol Renal Fluid Electrolyte Physiol 259: F961–F970, 1990. [DOI] [PubMed] [Google Scholar]
- 325.Wrong O, Davies HE. The excretion of acid in renal disease. Q J Med 28: 259–312, 1959. [PubMed] [Google Scholar]
- 326.Xu J, Yang S, Cai S, Dong J, Li X, Chen Z. Identification of biochemical changes in lactovegetarian urine using 1H NMR spectroscopy and pattern recognition. Anal Bioanal Chem 396: 1451–1463, 2010. [DOI] [PubMed] [Google Scholar]
- 327.Yang B, Zhao D, Solenov E, Verkman AS. Evidence from knockout mice against physiologically significant aquaporin 8-facilitated ammonia transport. Am J Physiol Cell Physiol 291: C417–C423, 2006. [DOI] [PubMed] [Google Scholar]
- 328.Yip KP, Kurtz I. NH3 permeability of principal cells and intercalated cells measured by confocal fluorescence imaging. Am J Physiol Renal Fluid Electrolyte Physiol 269: F545–F550, 1995. [DOI] [PubMed] [Google Scholar]
- 329.Zacchia M, Preisig P. Low urinary citrate: an overview. J Nephrol 23 Suppl 16: S49–S56, 2010. [PubMed] [Google Scholar]
- 330.Zheng L, Kostrewa D, Berneche S, Winkler FK, Li XD. The mechanism of ammonia transport based on the crystal structure of AmtB of Escherichia coli. Proc Natl Acad Sci USA 101: 17090–17095, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Zhu Q, Shao XM, Kao L, Azimov R, Weinstein AM, Newman D, Liu W, Kurtz I. Missense mutation T485S alters NBCe1-A electrogenicity causing proximal renal tubular acidosis. Am J Physiol Cell Physiol 305: C392–C405, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Zidi-Yahiaoui N, Mouro-Chanteloup I, D'Ambrosio AM, Lopez C, Gane P, Kim CVAN, Cartron JP, Colin Y, Ripoche P. Human Rhesus B and Rhesus C glycoproteins: properties of facilitated ammonium transport in recombinant kidney cells. Biochem J 391: 33–40, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Zipp T, Tannen RL. Influence of phosphoenolpyruvate carboxykinase inhibition on the response of NH3 production to acute acidosis. J Lab Clin Med 102: 198–204, 1983. [PubMed] [Google Scholar]