

Physical forces elicit biochemical signalling in a diverse array of cells, tissues and organisms1–3, helping to govern fundamental biological processes. Several hypotheses have been advanced that link physical forces to intracellular signalling pathways, but in many cases the molecular mechanisms of mechanotransduction remain elusive1–9. Here we find that compressive stress shrinks the lateral intercellular space surrounding epithelial cells, and triggers cellular signalling via autocrine binding of epidermal growth factor family ligands to the epidermal growth factor receptor. Mathematical analysis predicts that constant rate shedding of autocrine ligands into a collapsing lateral intercellular space leads to increased local ligand concentrations that are sufficient to account for the observed receptor signalling; direct experimental comparison of signalling stimulated by compressive stress versus exogenous soluble ligand supports this prediction. These findings establish a mechanism by which mechanotransduction arises from an autocrine ligand–receptor circuit operating in a dynamically regulated extracellular volume, not requiring induction of force-dependent biochemical processes within the cell or cell membrane.
Epithelial and endothelial cells line the surfaces of the body and frequently encounter physical stresses as pressures equilibrate across these cellular barriers10–13. We studied the morphological and biochemical responses of normal human bronchial epithelial (NHBE) cells to compressive stress: in vivo they experience such stress as a consequence of airway constriction14, and in vitro compressive stress triggers an integrated pro-fibrotic response reminiscent of that seen in chronic asthma15,16.
We used two-photon microscopy to visualize the morphology of living NHBE cells both before and during the application of physical stress. Cell-permeant cytoplasmic dyes (Celltracker and calcein AM) were used to visualize the cell bodies, and a cell-impermeant fluorophore-conjugated dextran was used to visualize the extracellular space directly. The cells, which were grown at the air–liquid interface to produce a pseudo-stratified, mucociliary epithelium, were joined by tight junctions at their apical surface, and separated along their basolateral surface by a lateral intercellular space (LIS) comprising 15 ± 3% (mean ± s.d., n = 5) of the total tissue volume. This morphology, including the LIS, is typical of stratified epithelium17 and is representative of the airways in vivo18,19.
To expose the cells to a compressive stress similar to that resulting from airway constriction we applied an apical to basal transcellular pressure gradient across the epithelium14,16. This compressive stress decreased the thickness of NHBE cell layers an average of 10.6 ± 1.9% (mean ± s.d., n = 7, Supplementary Table 1). However, total cell volume was not significantly altered by the compressive stress (decrease of 1.6 ± 2.3%, n = 7, Supplementary Table 1), suggesting that changes in cell layer height were a result of fluid leaving the LIS. This was confirmed qualitatively by visualizing the extracellular space (Fig. 1). Quantitative determination of LIS volume in tissue specimens before and after exposure to compressive stress demonstrated an 87.3 ± 10.0% reduction in LIS volume (mean ± s.d., n = 5, Supplementary Table 2). Our findings indicate that although epithelial cell volumes are maintained relatively constant under compressive stress, the LIS is highly compliant and susceptible to large percentage volume changes.
Figure 1.
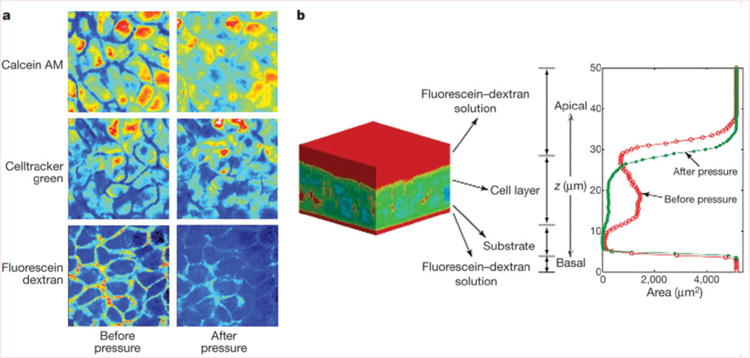
Compliance of the lateral intercellular space. a, Pseudo-colour x–y slices from identical mid-cell layers obtained by two-photon microscopy before and during application of 20 cm H2O transcellular pressure. b, A typical pseudo-colour three-dimensional reconstruction of bronchial epithelial cells negatively stained with cell-impermeant fluorescein–dextran. Red corresponds to dextran, whereas cooler colours (blue–green) correspond to the absence of dextran. The intensity of dextran staining is plotted along the z axis. The red contour indicates the profile of dextran staining under control conditions, whereas the green profile was obtained in the same tissue 30 min after the onset of a continuous 20 cm H2O transcellular pressure. Note the decrease in cell layer height and loss of LIS volume.
Collapse of the LIS under compressive stress brings the lateral surfaces of adjacent cells closer to one another. The basolateral surfaces of NHBE cells exhibit strong staining for the ErbB1 epidermal growth factor receptor (EGFR, Fig. 2a), in agreement with observations made in intact human airways and primary airway epithelial cells20,21. As previously shown22, application of compressive stress for 30 min elicited a robust increase in phosphorylation of the mitogen-activated protein kinase ERK (Fig. 2b). This response was preceded by an increase in phosphorylation of the EGFR at 5–20 min (Fig. 2a). Blockade of the tyrosine kinase domain of the EGFR with basolaterally applied tyrphostin AG 1478 strongly attenuated the mechanical stress-induced phosphorylation of ERK, whereas incubation with tyrphostin AG 1296, which inhibits the tyrosine kinase domain of the platelet-derived growth factor receptor, had no effect on the mechanically induced signal (Fig. 2b). Basolateral application of a monoclonal function-blocking EGFR antibody before compressive stress attenuated the compressive stress-induced ERK phosphorylation in a concentration-dependent manner (Fig. 2b), confirming a role for the extracellular ligand-binding domain of the EGFR. To identify the EGF family member responsible, we applied compressive stress in the presence of increasing basolateral amounts of neutralizing antibodies to EGF, transforming growth factor-α (TGF-α) and heparin-binding epidermal growth factor (HB-EGF). Whereas the EGF and TGF-α antibodies were ineffective at the highest concentration used, the antibody to HB-EGF attenuated the compressive stress-induced ERK phosphorylation in a dose-dependent manner (Fig. 2b). Incubation with GM 6001, a broad-spectrum inhibitor of matrix metalloprotease activity, was used to inhibit ectodomain shedding of transmembrane EGF family precursors from the basolateral cell surface23,24. Its presence attenuated both baseline and transcellular pressure-induced increases in ERK phosphorylation, demonstrating that constitutive sheddase activity for EGF family ligands is present in the LIS, and is required for transduction of the applied mechanical stress. These results demonstrate that compressive stress elicits ERK phosphorylation through a LIS-restricted autocrine pathway involving metalloprotease-dependent shedding of HB-EGF, leading to subsequent binding and activation of the EGFR.
Figure 2.
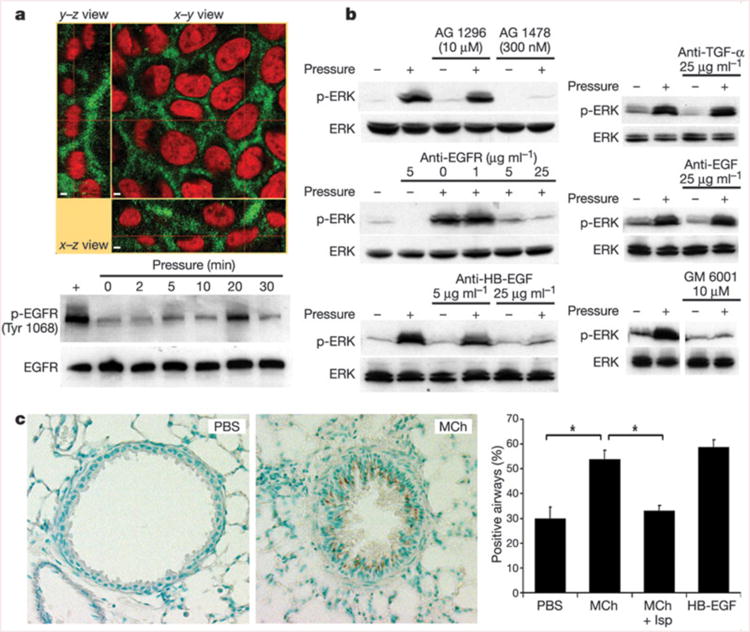
The EGFR mediates transduction of compressive stress. a, NHBE cells exhibit basolaterally polarized distribution of EGFR (green) lining the LIS; cell nuclei are counterstained with propidium iodide (red). Scale bars indicate 2 μm. The EGFR is rapidly phosphorylated (Tyr 1068) in response to transcellular compressive stress (pressure). b, NHBE cells exhibit ERK phosphorylation after 30 min exposure to transcellular pressure (20–30 cm H2O); this response is attenuated in the presence of the EGFR inhibitor AG 1478 but not the platelet-derived growth factor receptor inhibitor AG 1296, and is also attenuated by a function-blocking antibody that prevents ligand binding to the EGFR, a neutralizing antibody against HB-EGF, and the matrix metalloprotease inhibitor GM 6001, but not neutralizing antibodies against EGF and TGF-α. All blots are representative of at least two independent experiments. c, Representative images show the increased immunostaining (brown) for the phosphorylated EGFR (Tyr 1068) in mouse lungs perfused for 5 min with MCh (1 mM) relative to those perfused with PBS. The increased staining (percentage positive airways; asterisk = P < 0.0005, one-factorial analysis of variance and Bonferroni/Dunn test, n = 4–5, mean s.e.m.) was reversed by pre-treatment with isoproterenol (Mch + Isp), which abrogated the airway constriction. Perfusion with 10 ng ml−1 HB-EGF is included as a positive control.
To confirm the relevance of this signalling response in the native setting of the airway wall, we fixed isolated mouse lungs after administration of various bronchially active substances, and examined the distribution of epithelial staining for the active (phosphorylated) form of the EGFR. A 5 min tracheal perfusion with methacholine (MCh; 1 mM), which was sufficient to maximally constrict the airways, significantly increased the number of airways positive for the phosphorylated EGFR (Fig. 2c). This response to MCh was specific to the mechanical stress evoked by muscle constriction, as pre-treatment with isoproterenol (Isp), which greatly attenuated constriction, abrogated the EGFR response to MCh.
The localization of the EGFR autocrine loop at the perimeter of the collapsing LIS suggests a proximal role for this receptor system in the transduction of mechanical stress in epithelial cells. We considered the possibility that transient shear stresses generated as fluid is squeezed out of the collapsing LIS might trigger EGFR activation; however, when we applied transcellular pressure gradually as a ramp function over 30 min to minimize intercellular shear stress, signal transduction was stimulated as effectively as by a step increase in transcellular pressure (Supplementary Fig. 1). We also considered the possibility that the compressive stress response might result from EGFR transactivation secondary to G-protein-coupled receptor activation6,11,24 or ATP release8,25,26. Pre-treatment with either pertussis toxin or the extracellular ATP scavenger apyrase was incapable of attenuating the ERK phosphorylation response to compression (Supplementary Fig. 1).
We next considered whether it is feasible that the geometric alterations in the LIS are sufficient to stimulate EGFR signalling by modulating ligand availability and receptor occupancy27–29. EGF family ligands that are shed into LIS undergo molecular diffusion near the cell surface until either they are bound by cell-surface receptors, or they escape into the basal medium27. To describe the steady-state concentration profile of ligand molecules in the LIS, and the dependence of this concentration profile on LIS geometry, we solved the mass balance equation for ligand shedding and diffusion, leading to the expression c = cm + (h2 – X2)r/(Dw). The concentration c in the LIS exceeds the concentration cm in the medium, or in the underlying tissue in vivo, owing to the autocrine ligand release rate r. The epithelium allows outflux of the released ligand to the medium through the basal LIS opening but not across the tight junctions at the apical surface of the cells, and so the concentration depends on the distance X measured from the apex of the LIS (Fig. 3a). Assuming a uniform distribution of ligand shedding, constant in time, the concentration of ligands is highest adjacent to the tight junctions and decreases quadratically, reaching cm at the basal LIS opening, where X = h, the LIS height. The difference in ligand concentration from that in the basal medium is proportional to the shedding rate r and inversely proportional to the diffusion constant of the ligand, D. Notably, we find that the ligand concentration is inversely proportional to the LIS width w. Thus, our analysis predicts that the effective ligand concentration to which cells are exposed is strongly dependent on the geometry of the LIS, which is itself a function of the mechanical environment of the tissue. On the basis of this analysis we directly compared compressive stress to stimulation with soluble HB-EGF. In our imaging studies we measured an 87 ± 10% decrease in LIS volume, which should yield an approximately eightfold increase in HB-EGF concentration. Compressive stress produced a change in ERK phosphorylation equivalent to a roughly tenfold increase in HB-EGF concentration (Fig. 3b), consistent with our prediction.
Figure 3.
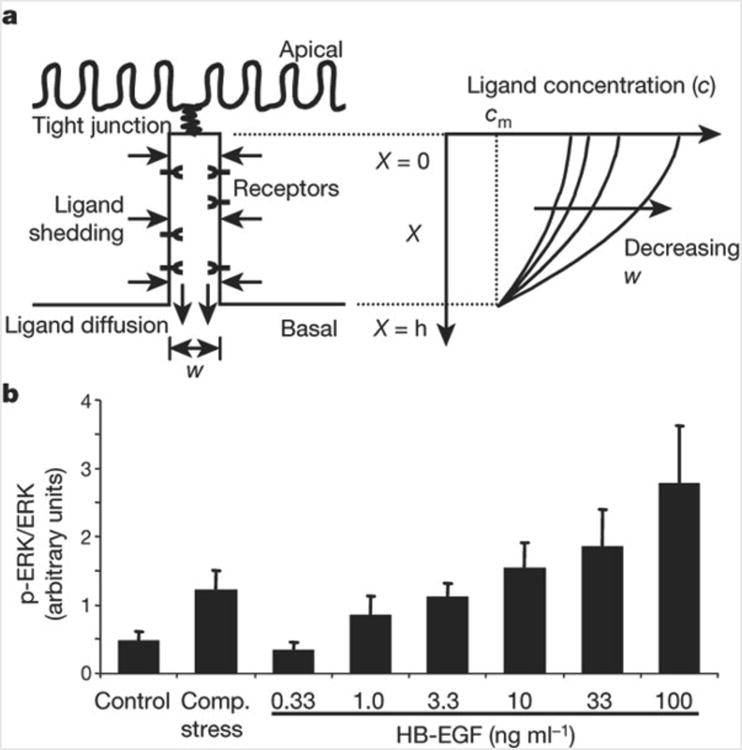
LIS width regulates autocrine signalling. a, Schematic of the epithelial LIS showing the boundary conditions regulating EGF family ligand diffusion, and example profiles of ligand concentration within the LIS. b, ERK phosphorylation elicited by compressive stress compared to 3.3-fold increasing concentrations of HB-EGF (mean ± s.e.m., n = 5). Concentrations of HB-EGF <0.33 ng ml−1 had no effect on ERK phosphorylation. We thus assume that the baseline local concentration of EGF family ligands in the LIS is between 0.33 and 1.0 ng ml−1. From equation (7) we predict that the result of compressive stress should be most comparable to the effect of adding exogenous HB-EGF to increase this concentration approximately tenfold, to between 3.3 and 10 ng ml−1.
Our findings support the hypothesis that cells can sense mechanical stress through autocrine loops localized to compliant extracellular spaces; this mechanism seems to be capable of acting both independently of other known mechanosensitive responses and in concert with other mechanisms to broaden and amplify the response. Spatially limited autocrine loops occur in a wide variety of cell types, especially in epithelia where they are frequently localized in the LIS. Our results emphasize the high relative compliance of the LIS, and indicate that cells respond to local changes in ligand availability as a result of geometric alterations in their intercellular spaces. A unique aspect of this mechanochemical transduction mechanism is that it requires no direct conformational or metabolic alteration in any protein by mechanical stress. We speculate that such a transduction system could be tuned to perceive a wide range of deformations and stresses, and may be important in the sensation of and responses to physical forces in a number of biological systems.
Methods
Cell culture
NHBE cells (Clonetics-BioWhittaker) were cultured as previously described on uncoated microporous polyester inserts (Transwell-Clear, 0.4-μm pore size, Costar)22. Experiments were carried out in minimal medium with no serum or growth factors except insulin (5.7 μg ml−1) and transferrin (5 μg ml−1). Function-blocking antibodies (all from R&D Systems except anti-EGFR clone LA1; Upstate Biotechnology), GM 6001, AG 1478, AG 1296 (all from Calbiochem), and soluble HB-EGF (R&D Systems) were applied to the basal surface of cultures only.
Two-photon imaging
NHBE cells were stained apically and basally with the fluorescent probes calcein AM (10 μM), Celltracker green (10 μM), or 70 kDa fluorescein–dextran (100 μM, all from Molecular Probes) diluted in culture medium. After incubation for 30 min each tissue culture insert was mounted on a custom-built two-photon microscope, and a silicon plug was used to create a pressure chamber over the apical surface of the cells. After a complete sequence of images was obtained under control conditions, an apical-to-basal transcellular pressure difference of 20 cm H2O was applied. Imaging was then repeated within the same region of cells. Images were acquired at intervals of 0.5 μm in the z direction, with each image acquired at 256 × 256 pixels, 0.28 μm pixel dimension. Image stacks were analysed using Spyglass Slicer (Spyglass).
Western blotting
Cell lysates were separated in 10% precast polyacrylamide gels (BioRad)22. Proteins were transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore), blocked with 5% milk, and incubated overnight (4 °C) in primary antibodies against ERK, phospho-ERK, EGFR, or phospho-EGFR (Tyr 1068; all from Cell Signaling Technology). Bands were visualized with horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000) and enhanced chemiluminescence.
Immunofluorescence
Cells were fixed with 3% paraformaldehyde and stained with a mouse monoclonal antibody to the extracellular domain of the human EGFR (clone LA1, Upstate Biotechnology) and a secondary goat anti-mouse IgG antibody conjugated to Alexa Fluor 488 (Molecular Probes). Cell nuclei were counterstained with propidium iodide. Three-dimensional stacks of confocal images were obtained using a Sarastro 2000 confocal laser-scanning microscope.
Isolated lung perfusion
Lungs of male BALB/C mice (age 8 weeks) were tracheally perfused following the method of ref. 30 with PBS at a flow rate of 20 ml h−1. Perfusate exited the lung via small holes (5 per lobe) poked in the pleura (27 gauge needle). The airway opening pressure (Pao) at the trachea was continuously measured as an index of airway constriction. Perfusion with MCh induced a transient, concentration-dependent increase in Pao, with a maximal response at 1 mM (ΔPao = 13 cm H2O at 5 min). Perfusion with 10−4 M Isp 1 min before and continuous with MCh perfusion attenuated MCh-induced bronchial constriction by 78%. Perfusion with Isp alone had little effect on Pao. Perfused lungs from each group (n = 4–5) were fixed by perfusion at 5min with 4% paraformaldehyde.
Immunohistochemistry
Paraffin-embedded lung sections were incubated with a rabbit polyclonal antibody against phopho-EGF receptor (Tyr 1068, Cell Signalling Technology); immunopositive areas were visualized with an avidin–biotin method using biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories), the chromagen diaminobenzidine and methyl green counterstaining. Irrelevant IgG antibodies substituted for the primary antiserum were the negative controls. All cross-sectioned airways in each section were counted, and airways with any staining in the epithelium were counted as phospho-EGFR positive.
Mass balance analysis of the LIS
We approximate the geometry of the LIS as a two-dimensional gap of width w and height h between two surfaces (Fig. 3a), and assume that the rate of ligand shedding r, width w, and diffusion coefficient D are independent of position X along the apical–basal axis. Transport by convection is ignored. At steady state the rate of ligand entering the LIS via shedding from the cells bounding the LIS will be exactly balanced by the ligand leaving the LIS by diffusion through the basal opening of the LIS into the bulk medium. If we examine the mass transfer in an infinitesimal slice of LIS of width w and thickness dX at position X, the flux into the slice can be written as:
| (1) |
where r is the density of ligand shedding per unit length, dX is the slice thickness, and the factor 2 comes from the two surfaces bounding the LIS in our idealized geometry. This influx is balanced by the net outflux due to diffusion, which is proportional to the spatial derivative of the ligand concentration gradient:
| (2) |
where c is the concentration averaged over y. Equating influx and outflux, assuming constant r, w and D, and integrating from 0 to X, yields:
| (3) |
Our boundary conditions are zero flux through the impermeable tight junction bounding the apical end of the LIS (X = 0), and ligand concentration equilibrates with the bulk medium (cm) at the basal opening of the LIS (X = h):
| (4) |
and
| (5) |
Integrating equation (3) and using the boundary condition at X = 0 (equation (4)), the following relation is obtained:
| (6) |
Solving this equation for c(X) and satisfying the second boundary condition (equation (5)) yields:
| (7) |
Supplementary Material
Acknowledgments
We thank J. Shively and J. Foley for technical assistance, G. Horsley for early work on EGFR signalling, and J.Lai for performing conventional confocal imaging. This work was supported by funding from the NHLBI (J.M.D. and R.D.K.), NIGMS (D.A.L.) and a Parker B. Francis Fellowship in Pulmonary Research (D.J.T.).
Footnotes
Supplementary Information accompanies the paper on www.nature.com/nature.
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Blount P. Molecular mechanisms of mechanosensation: big lessons from small cells. Neuron. 2003;37:731–734. doi: 10.1016/s0896-6273(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 2.Torday JS, Rehan VK. Mechanotransduction determines the structure and function of lung and bone: a theoretical model for the pathophysiology of chronic disease. Cell Biochem Biophys. 2003;37:235–246. doi: 10.1385/cbb:37:3:235. [DOI] [PubMed] [Google Scholar]
- 3.Farge E. Mechanical induction of Twist in the Drosophila foregut/stomodeal primordium. Curr Biol. 2003;13:1365–1377. doi: 10.1016/s0960-9822(03)00576-1. [DOI] [PubMed] [Google Scholar]
- 4.Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci STKE. 2002;2002:E6. doi: 10.1126/stke.2002.119.pe6. [DOI] [PubMed] [Google Scholar]
- 5.Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev. 2001;81:685–740. doi: 10.1152/physrev.2001.81.2.685. [DOI] [PubMed] [Google Scholar]
- 6.Gudi SR, Clark CB, Frangos JA. Fluid flow rapidly activates G proteins in human endothelial cells. Involvement of G proteins in mechanochemical signal transduction. Circ Res. 1996;79:834–839. doi: 10.1161/01.res.79.4.834. [DOI] [PubMed] [Google Scholar]
- 7.Shyy JY, Chien S. Role of integrins in endothelial mechanosensing of shear stress. Circ Res. 2002;91:769–775. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 8.Dull RO, Tarbell JM, Davies PF. Mechanisms of flow-mediated signal transduction in endothelial cells: kinetics of ATP surface concentrations. J Vasc Res. 1992;29:410–419. doi: 10.1159/000158959. [DOI] [PubMed] [Google Scholar]
- 9.Ali MH, Schumacker PT. Endothelial responses to mechanical stress: where is the mechanosensor? Crit Care Med. 2002;30:S198–S206. doi: 10.1097/00003246-200205001-00005. [DOI] [PubMed] [Google Scholar]
- 10.Han O, Li GD, Sumpio BE, Basson MD. Strain induces Caco-2 intestinal epithelial proliferation and differentiation via PKC and tyrosine kinase signals. Am J Physiol. 1998;275:G534–G541. doi: 10.1152/ajpgi.1998.275.3.G534. [DOI] [PubMed] [Google Scholar]
- 11.Correa-Meyer E, Pesce L, Guerrero C, Sznajder JI. Cyclic stretch activates ERK1/2 via G proteins and EGFR in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L883–L891. doi: 10.1152/ajplung.00203.2001. [DOI] [PubMed] [Google Scholar]
- 12.Ressler B, Lee RT, Randell SH, Drazen JM, Kamm RD. Molecular responses of rat tracheal epithelial cells to transmembrane pressure. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1264–L1272. doi: 10.1152/ajplung.2000.278.6.L1264. [DOI] [PubMed] [Google Scholar]
- 13.Lehoux S, Tedgui A. Signal transduction of mechanical stresses in the vascular wall. Hypertension. 1998;32:338–345. doi: 10.1161/01.hyp.32.2.338. [DOI] [PubMed] [Google Scholar]
- 14.Wiggs BR, Hrousis CA, Drazen JM, Kamm RD. On the mechanism of mucosal folding in normal and asthmatic airways. J Appl Physiol. 1997;83:1814–1821. doi: 10.1152/jappl.1997.83.6.1814. [DOI] [PubMed] [Google Scholar]
- 15.Tschumperlin DJ, Shively JD, Kikuchi T, Drazen JM. Mechanical stress triggers selective release of fibrotic mediators from bronchial epithelium. Am J Respir Cell Mol Biol. 2003;28:142–149. doi: 10.1165/rcmb.2002-0121OC. [DOI] [PubMed] [Google Scholar]
- 16.Swartz MA, Tschumperlin DJ, Kamm RD, Drazen JM. Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proc Natl Acad Sci USA. 2001;98:6180–6185. doi: 10.1073/pnas.111133298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spring KR, Hope A. Size and shape of the lateral intercellular spaces in a living epithelium. Science. 1978;200:54–58. doi: 10.1126/science.635571. [DOI] [PubMed] [Google Scholar]
- 18.Ohashi Y, Motojima S, Fukuda T, Makino S. Airway hyperresponsiveness, increased intracellular spaces of bronchial epithelium, and increased infiltration of eosinophils and lymphocytes in bronchial mucosa in asthma. Am Rev Respir Dis. 1992;145:1469–1476. doi: 10.1164/ajrccm/145.6.1469. [DOI] [PubMed] [Google Scholar]
- 19.Kondo M, Finkbeiner WE, Widdicombe JH. Changes in permeability of dog tracheal epithelium in response to hydrostatic pressure. Am J Physiol. 1992;262:L176–L182. doi: 10.1152/ajplung.1992.262.2.L176. [DOI] [PubMed] [Google Scholar]
- 20.Polosa R, et al. Expression of c-erbB receptors and ligands in human bronchial mucosa. Am J Respir Cell Mol Biol. 1999;20:914–923. doi: 10.1165/ajrcmb.20.5.3308. [DOI] [PubMed] [Google Scholar]
- 21.Vermeer PD, et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422:322–326. doi: 10.1038/nature01440. [DOI] [PubMed] [Google Scholar]
- 22.Tschumperlin DJ, et al. Bronchial epithelial compression regulates MAP kinase signaling and HB-EGF-like growth factor expression. Am J Physiol Lung Cell Mol Physiol. 2002;282:L904–L911. doi: 10.1152/ajplung.00270.2001. [DOI] [PubMed] [Google Scholar]
- 23.Dong J, et al. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci USA. 1999;96:6235–6240. doi: 10.1073/pnas.96.11.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prenzel N, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 25.Homolya L, Steinberg TH, Boucher RC. Cell to cell communication in response to mechanical stress via bilateral release of ATP and UTP in polarized epithelia. J Cell Biol. 2000;150:1349–1360. doi: 10.1083/jcb.150.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto K, et al. Endogenously released ATP mediates shear stress-induced Ca2+ influx into pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2003;285:H793–H803. doi: 10.1152/ajpheart.01155.2002. [DOI] [PubMed] [Google Scholar]
- 27.Shvartsman SY, Wiley HS, Deen WM, Lauffenburger DA. Spatial range of autocrine signaling: modeling and computational analysis. Biophys J. 2001;81:1854–1867. doi: 10.1016/S0006-3495(01)75837-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lauffenburger DA, Oehrtman GT, Walker L, Wiley HS. Real-time quantitative measurement of autocrine ligand binding indicates that autocrine loops are spatially localized. Proc Natl Acad Sci USA. 1998;95:15368–15373. doi: 10.1073/pnas.95.26.15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shvartsman SY, et al. Autocrine loops with positive feedback enable context-dependent cell signaling. Am J Physiol Cell Physiol. 2002;282:C545–C559. doi: 10.1152/ajpcell.00260.2001. [DOI] [PubMed] [Google Scholar]
- 30.Martins MA, Shore SA, Gerard NP, Gerard C, Drazen JM. Peptidase modulation of the pulmonary effects of tachykinins in tracheal superfused guinea pig lungs. J Clin Invest. 1990;85:170–176. doi: 10.1172/JCI114408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.