

This article describes a mouse model of the human disease complete congenital stationary night blindness in which the mutation reduces but does not eliminate GRM6 expression and bipolar cell function, a phenotype distinct from that seen in other Grm6 mouse models.
Keywords: bipolar cell, electroretinogram, ganglion cell, retina, retinal disease
Abstract
GRM6 encodes the metabotropic glutamate receptor 6 (mGluR6) used by retinal depolarizing bipolar cells (DBCs). Mutations in GRM6 lead to DBC dysfunction and underlie the human condition autosomal recessive complete congenital stationary night blindness. Mouse mutants for Grm6 are important models for this condition. Here we report a new Grm6 mutant, identified in an electroretinogram (ERG) screen of mice maintained at The Jackson Laboratory. The Grm6nob8 mouse has a reduced-amplitude b-wave component of the ERG, which reflects light-evoked DBC activity. Sequencing identified a missense mutation that converts a highly conserved methionine within the ligand binding domain to leucine (p.Met66Leu). Consistent with prior studies of Grm6 mutant mice, the laminar size and structure in the Grm6nob8 retina were comparable to control. The Grm6nob8 phenotype is distinguished from other Grm6 mutants that carry a null allele by a reduced but not absent ERG b-wave, decreased but present expression of mGluR6 at DBC dendritic tips, and mislocalization of mGluR6 to DBC somas. Consistent with a reduced but not absent b-wave, there were a subset of retinal ganglion cells whose responses to light onset have times to peak within the range of those in control retinas. These data indicate that the p.Met66Leu mutant mGluR6 is trafficked less than control. However, the mGluR6 that is localized to the DBC dendritic tips is able to initiate DBC signal transduction. The Grm6nob8 mouse extends the Grm6 allelic series and will be useful for elucidating the role of mGluR6 in DBC signal transduction and in human disease.
NEW & NOTEWORTHY This article describes a mouse model of the human disease complete congenital stationary night blindness in which the mutation reduces but does not eliminate GRM6 expression and bipolar cell function, a distinct phenotype from that seen in other Grm6 mouse models.
the metabotropic glutamate receptor 6 (mGluR6) initiates a signal transduction cascade used exclusively by depolarizing bipolar cells (DBCs) to convey the light-induced decline in photoreceptor glutamate release to retinal ganglion cells (RGCs; Shiells et al. 1981; Slaughter and Miller 1981). DBC activity can be monitored noninvasively, using the electroretinogram (ERG) b-wave (Hood and Birch 1996; Kofuji et al. 2000; Robson and Frishman 1995, 1996; Sharma et al. 2005). Accordingly, b-wave abnormalities have identified mice and humans with mutations in the gene encoding mGluR6 (Dryja et al. 2005; Maddox et al. 2008; Masu et al. 1995; Pinto et al. 2007; Qian et al. 2015; Zeitz et al. 2005). In all cases, mutations in GRM6 in humans or its homologous gene in mice (Grm6) result in an elimination of the b-wave component of the ERG, consistent with a loss of DBC function. These models have been helpful in understanding the components of the DBC signaling complex (Gregg et al. 2014), the impact of mGluR6 dysfunction on the response properties of RGCs (Maddox et al. 2008; Rentería et al. 2006), and the interactions that occur between the primary and secondary rod pathways that result in destructive interference at 15 Hz (Zeitz et al. 2005). A comparable “no b-wave” (nob) ERG phenotype is present in models that encode other components of the DBC signal transduction cascade including Trpm1, Gpr179, Nyx, and Lrit3 (reviewed by Pardue and Peachey 2014) and in human patients with the complete form of congenital stationary night blindness (cCSNB) due to mutations in TRPM1, GPR179, NYX, or LRIT3 (reviewed by Zeitz et al. 2015).
Here we report a novel animal model in the form of a Grm6 mouse mutant in which the ERG b-wave is reduced but is not abolished. The identified missense mutation, p.Met66Leu, is at a highly conserved residue of the mGluR6 glutamate binding domain. The mutation (hereafter Grm6nob8) is associated with retention of mGluR6 in the DBC soma and reduction of mGluR6 expression on the DBC dendritic tips. This expression is sufficient to support function in the form of a small ERG b-wave at both scotopic and photopic levels. Consistent with reduced b-wave amplitudes, there are fewer visually responsive Grm6nob8 RGCs, albeit more than in Grm6−/− mice. In addition, the proportion of ON RGCs with time to peak firing timings in the control range is greater in the Grm6nob8 mutant than in Grm6−/− mice.
METHODS
Mice.
CBA/CaJ (stock no. 000654) and C57BL/6J (stock no. 000664; control) mice were obtained from the Jackson Laboratory production facility. We also recovered a CBA/CaJ line that was cryopreserved from the breeding colony maintained in 2002 (now designated CBA/Ca2J; stock no. 027000). AXB6/PgnJ (Cacna1fnob2; Chang et al. 2006), 129S6.129S(Cg)-Grm6tm1Nak (Masu et al. 1995; hereafter Grm6−/−), and B6.B10(D2)-Grm6nob3/BOC mice (Maddox et al. 2008; hereafter Grm6nob3) were obtained from local breeding colonies. Mice were housed in conventional microisolator cages and had free access to food and water. Mice were examined by ERG from 6 wk to 9 mo of age and analyzed anatomically at 6–8 wk of age. RGC electrophysiology was examined at 12–14 wk of age. All procedures involving live animals were approved by the Institutional Animal Care and Use Committees of the Jackson Laboratory, the University of Louisville, and the Cleveland Clinic. Animals of either sex were used in experiments, and no differences were observed.
Histology.
Mice were killed by carbon dioxide inhalation, and enucleated eyes were fixed overnight in cold methanol-acetic acid solution [3:1 (vol/vol)]. The paraffin-embedded eyes were cut into 6-μm sections, stained with hematoxylin and eosin, and examined by light microscopy. Images shown were taken between 0.5 and 1.0 mm along the vertical meridian from the optic nerve head.
Immunohistochemistry.
Dissected retinas were immersion fixed for 15 min in 4% (wt/vol) paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 (PB), then washed in PB, cryoprotected through a graded sucrose series (5%, 10%, 15%, and 20% in PB), and frozen in OCT (Sakura Finetek, Torrance, CA)-20% sucrose (2:1) (Barthel and Raymond 1990). Sixteen-micrometer sections were cut on a cryostat, mounted onto Superfrost glass slides, air-dried, and stored at −80°C.
Sections were brought to room temperature, washed in PB saline (PBS) for 5 min and PBS containing 0.5% (vol/vol) Triton X-100 (PBX) for 5 min, and then incubated in blocking solution [PBX, containing 5% (vol/vol) normal goat serum] for 1 h. Primary antibodies were diluted in blocking solution and incubated on retinal sections at room temperature overnight. The antibody against mouse mGluR6 (amino acids 853–871; KKTSTMAAPPKSENSEDAK) was generated in sheep (Cao et al. 2011). Other primary antibodies and dilutions used were anti-calbindin (1:1,000; Swant, Marly, Switzerland); anti-PKCα (1:1,000; Sigma-Aldrich, St. Louis, MO); anti-PSD95 (1:1,000; Chemicon, Temecula, CA); anti-Ribeye (1:1,000; BD Biosciences, San Jose, CA), anti-pikachurin (1:1,000; Wako Chemicals, Richmond, VA), anti-GPR179 (1:4,000; Peachey et al. 2012a), anti-TRPM1 (1:2,000; Cao et al. 2012), anti-RGS11 (1:2,000; Cao et al. 2012), and anti-RGS7 (1:2,000; Cao et al. 2008). After incubation with the primary antibody, sections were washed three times in PBS for 5 min each and subsequently incubated with fluorescently labeled secondary antibodies (1:1,000 in blocking solution) at room temperature for 1 h. Secondary antibodies were Alexa 488 goat anti-rabbit and Alexa 555 goat anti-mouse (Invitrogen, Carlsbad, CA). Slides were then washed three times in PBS and coverslipped with Immunomount (ThermoShandon, Pittsburgh, PA). An antibody against LRIT3 was generated by immunizing guinea pigs with a peptide (GELWTRRHRDDGERLLLC) conjugated to KLH by Biosynthesis (Lewisville, TX) and purified by affinity chromatography. Sections were imaged on an Olympus (Center Valley, PA) FV1000 confocal microscope with a ×60 oil objective (1.45 NA). Images shown are maximum projections of confocal stacks, adjusted for contrast and brightness with Fluoview software.
Western blot.
Mouse retinas were isolated and homogenized in lysis buffer [1% Nonidet P-40, 2 mM EDTA, and 20 mM HEPES, pH 7.4, supplemented with protease inhibitor cocktail (P8340, Sigma-Aldrich)] and lysed further by rotating at 4°C for 45 min. Homogenates were cleared by centrifugation at 17,000 g for 20 min at 4°C. Protein samples were separated by SDS-PAGE and transferred to PVDF membranes, and immunoblotting was performed as described previously (Hasan et al. 2016). Deglycosylation was achieved by incubating homogenates with PNGase F (New England Biolabs, Ipswich, MA; 30 U/μg protein) for 48 h at 37°C. Protein bands were visualized by scanning the membranes in an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE) using 700-nm and 800-nm channels. Band densities were quantified with Image Studio Version 5.0 software (LI-COR).
Electroretinography.
ERGs were recorded at The Jackson Laboratory (JAX), the University of Louisville (UL), and the Cleveland Clinic (CC) with published procedures (Chang et al. 2007; Noel et al. 2012; Yu et al. 2015, respectively). At each location, dark-adapted mice were anesthetized with ketamine (80 mg/kg) and xylazine (16 mg/kg) diluted in saline and delivered intraperitoneally. The pupil of the test eye was dilated with mydriatic eye drops (JAX: 1% atropine; UL and CC: 1% tropicamide; 2.5% phenylephrine HCl), and the corneal surface was anesthetized with a 1% proparacaine HCl eye drop. Mice were maintained on a temperature-regulated heating pad throughout the recording session.
At each site, ERGs were first recorded to stimuli presented in darkness and in order of increasing flash strength. The interstimulus interval was increased with increasing flash strength, and the number of responses that were averaged ranged from 20 to the low-luminance stimuli to 2 at the highest flash levels. Light-adapted cone ERGs were obtained to flash stimuli delivered at 2 Hz and superimposed upon a steady rod-desensitizing adapting field and after a minimum 5-min adaptation period. At JAX, responses were amplified and filtered (0.03–10,000 Hz) and digitized with an I/O board (model PCI-1200; National Instruments, Austin, TX) in a personal computer. At UL and CC, responses were amplified and filtered (0.03–2,000 Hz) and digitized with an LKC (Gaithersburg, MD) signal averaging system (UL: UTAS Big Shot; CC: UTAS E-3000). At each site, white light stimuli were delivered in an LKC ganzeld and ranged in luminance from −3.6 to 2.1 log cd s/m2.
The amplitude of the a-wave was measured from the prestimulus baseline to 8 ms after the stimulus flash, to minimize contributions from postreceptoral neurons (Mao et al. 2016; Robson et al. 2003). For low-luminance stimuli, the amplitude of the b-wave was measured from the baseline to the positive peak. For high-luminance stimuli, the b-wave was measured from the negative trough to the b-wave peak that followed the high-frequency oscillatory potentials. The amplitude of the cone ERG b-wave was measured from the initial negative trough to the b-wave peak.
Dark-adapted multielectrode array retinal ganglion cell recordings.
The recording procedures for the multielectrode array (MEA) have been previously published (Fransen et al. 2015). MEAs contained 60 electrodes (60MEA200/30Ti; Multi Channel Systems, Reutlingen, Germany), each with a diameter of 30 µm and separated from each other by 200 µm. Mice were dark-adapted overnight, deeply anesthetized [ketamine-xylazine (see Electroretinography)], and cervically dislocated. Both eyes were removed and placed in an oxygenated Ringer solution. Under dim red illumination, their anterior segments were removed and retinas were dissected. The vitreous was removed by incubation (10 min) in Ringer solution containing collagenase (12 U/µl) and hyaluronidase (50 U/µl) (Worthington Biochemicals, Lakewood, NJ). Dorsal or ventral retinal pieces were placed in the MEA recording chamber so that the RGCs directly contacted the electrodes. The retina was covered with a transparent cell culture membrane (Thermo Fisher Scientific, Waltham, MA) and held in place with a platinum ring. The recording chamber was continuously perfused with oxygenated Ringer solution at 36°C. Spontaneous activity was recorded for 45 min in darkness before visually evoked responses were recorded.
Visual stimulation for retinal ganglion cell responses.
Stimuli were focused onto the photoreceptor from a miniprojector (Hewlett-Packard, Palo Alto, CA; Notebook Consumer Projection Companion model no. AX325AA) mounted on a microscope. Stimuli were generated with custom code written in MATLAB with the cogent graphics toolbox (MathWorks, Natick, MA). Ten trials of full-field light stimuli (2-s duration; 20-s interstimulus interval) were presented to the dark-adapted retina at three levels and in increasing order (0.01, 0.03, 1.58 cd/m2).
Analyses of retinal ganglion cell responses.
Signals were band-pass filtered (80–3,000 Hz) and digitized at 25 kHz (MC Rack software; Multi Channel Systems). Spikes were recorded on individual electrodes, and if spikes reflected responses from more than one RGC they were sorted with principal components analysis (Offline Sorter; Plexon, Dallas, TX). Sorted units were exported for analysis of their spontaneous and visually evoked responses with NeuroExplorer software (Nex Technologies, Madison, AL). Average poststimulus time histograms (PSTHs) were calculated for all RGCs to 10 presentations of three different full-field luminance levels (Nobles et al. 2012). We defined an excitatory response to light onset or offset when the RGC response exceeded ±3 SE above its mean spontaneous activity over >80% of the trials. From the average PSTH, we calculated the time to peak response, defined as the time from stimulus onset/offset to the RGC peak firing rate. We classified the response of each RGC at each stimulus luminance as ON (excitation at stimulus onset), OFF (excitation at stimulus offset), or ON/OFF (excitation at both stimulus onset and offset). Time to peak of all control RGCs was ≤600 ms, whereas many mutant RGCs had much slower time to peak and these cells were defined as delayed RGCs.
RESULTS
Identification of b-wave mutant in CBA/CaJ mice.
Figure 1 compares the ERG phenotypes of CBA/CaJ and Grm6−/− with C57BL/6J control mice at 6 wk of age. The dark-adapted ERG a-waves in CBA/CaJ and Grm6−/− mice have amplitudes and kinetics that are indistinguishable from control mice (Fig. 1, A and C; P > 0.05). In agreement with a previous ERG study of this mouse line (Xue et al. 2015), the amplitude of the dark-adapted ERG b-wave of CBA/CaJ mice is significantly reduced compared with control (P < 0.0001). The presence of the b-wave in the dark-adapted CBA/CaJ ERG contrasts with the absence of this component in Grm6−/− mice. The CBA/CaJ ERG waveforms also include prominent high-frequency oscillatory potentials. CBA/CaJ light-adapted ERGs show a marked reduction in amplitude compared with control (P < 0.0001) but were not different from cone ERGs of Grm6−/− mice (Fig. 1, B and D; P > 0.05). A similar ERG phenotype was noted when CBA/CaJ mice were examined at 9 mo of age (data not shown).
Fig. 1.

Comparison of dark-adapted (A) and light-adapted (B) ERGs obtained from control (C57BL/6J), CBA/CaJ, and Grm6 −/− mice at 6 wk of age. Dashed lines indicate time of flash presentation. Flash strength (in log cd s/m2) is indicated on left of each set of waveforms. C: amplitude of dark-adapted a-wave and b-wave plotted as a function of flash luminance. D: amplitude of light-adapted ERG b-wave plotted as a function of flash luminance. In C and D, data points indicate average ± SD of 3 mice per genotype. Repeated-measures analysis of variance was used to compare response functions. In comparison to control, CBA/CaJ had comparable dark-adapted ERG a-waves (P > 0.05) and reduced ERG b-waves under dark-adapted (P < 0.0001) and light-adapted (P < 0.0001) conditions. Light-adapted ERG b-waves of CBA/CaJ and Grm6 −/− mice were comparable (P > 0.05).
We first used complementation assays to define the genetic basis for the abnormal ERG b-wave in the CBA/CaJ mice. We excluded Cacna1f as the causative gene, because female F1 offspring generated by crossing CBA/CaJ males and Cacna1fnob2 homozygous females had normal ERGs. In contrast, all F1 offspring generated by crossing CBA/CaJ and Grm6nob3 homozygotes had reduced ERG b-waves, indicating that the CBA/CaJ ERG phenotype reflects a mutation in Grm6. Subsequent sequence analyses identified a mutation with an A to T substitution at position Chr:11:50851337 (build GRCm38), which leads to a missense mutation at codon 66, changing ATG (methionine, Met) to TTG (leucine, Leu) (p.Met66Leu; Fig. 2A). Met66 lies in the glutamate binding region of mGluR6 and is invariant across many mammals, including humans (Fig. 2B). Hereafter we refer to the CBA/CaJ mutant line as Grm6nob8.
Fig. 2.
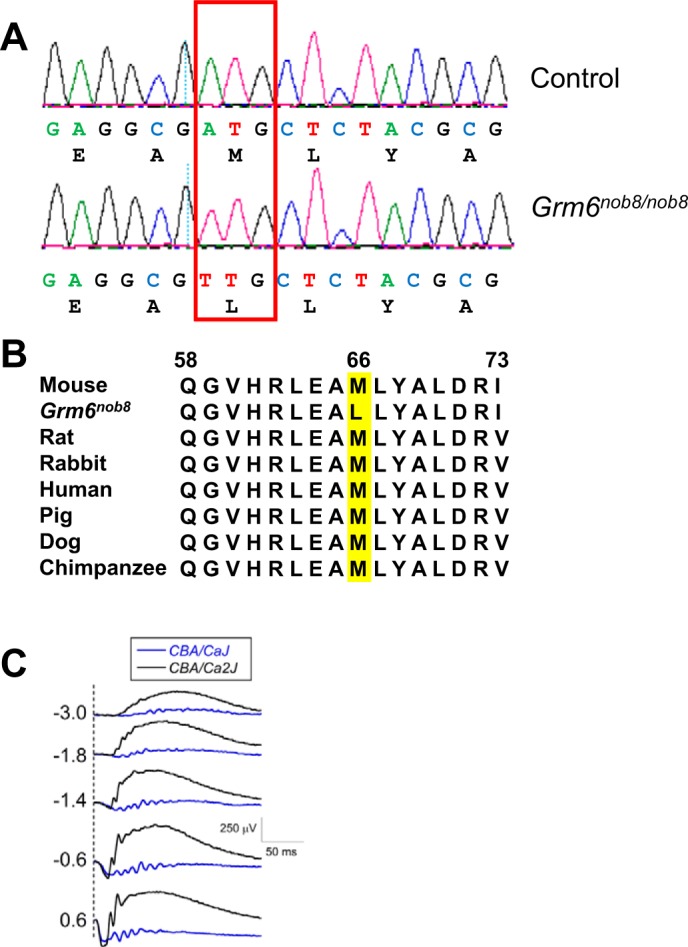
A: sequence reads of fragment of Grm6 gene of control (C57BL/6J) and Grm6nob8 mice. A mutation changes codon 66, ATG to TTG in the Grm6nob8 mice, substituting leucine for methionine (p.Met66Leu). B: species comparison of region containing the Met66 codon (highlighted in yellow) shows that this region of mGluR6 is highly conserved. C: comparison of ERGs obtained from currently available CBA/CaJ (Grm6nob8) mice and the CBA/Ca2J mice recovered from embryos cryopreserved in 2002. Dashed lines indicate time of flash presentation. Flash strength (in log cd s/m2) is indicated on left of each set of waveforms.
Because CBA/CaJ is a standard inbred line provided by The Jackson Laboratory we investigated when the mutation arose. We recovered CBA/Ca2J mice that were cryopreserved in 2002, and these mice had normal ERGs (Fig. 2C) and did not have the A to T transversion. Furthermore, DNA samples obtained in 2006 from CBA/CaJ mice also did not have the A to T transversion. Thus the Grm6nob8 mutation arose recently and was propagated to be present in the CBA/CaJ colony at The Jackson Laboratory.
Because the main difference between the ERGs of the two Grm6 mutants (Grm6nob8 and Grm6–/–) lies in the dark-adapted b-wave, we focused our efforts primarily on the rod pathway.
Expression of mGluR6 transduction proteins is altered in Grm6nob8 retina.
The gross morphology of the Grm6nob8 retina is normal, and all cellular and plexiform layers are present and comparable to controls (Fig. 3A). We examined the expression patterns of proteins that form the pre- and postsynaptic complex at the photoreceptor-to-DBC synapse. In the Grm6nob8 retina, the postsynaptic proteins calbindin (horizontal cells) and PKCα (rod DBCs) were comparable to control and Grm6−/− (Fig. 3B). We also compared the localization of presynaptic structural components (PSD95, ribeye, pikachurin, PNA) in control, Grm6nob8, and Grm6−/− retinas (Fig. 3B). The general distribution of these proteins was comparable across these three models, but ribeye and pikachurin expression were fainter in the Grm6nob8 retina and the cone pedicles identified by PNA labeling show an uneven staining pattern in both Grm6nob8 and Grm6−/− retinas (Fig. 3B).
Fig. 3.

Retinal anatomy. A: by light microscopy, no difference was observed between control and Grm6nob8 retinas. B: by immunohistochemistry, no difference was noted in the cellular localization of calbindin, PKCα, or PSD95 between control, Grm6nob8, and Grm6−/− retinas. Ribeye and pikachurin appeared correctly localized but at reduced levels in the Grm6nob8 retina. PNA was also correctly localized but had an uneven distribution in Grm6nob8 that is similar to that in the Grm6−/− retinas. Scale bars: 100 µm (A) and 5 µm (B). ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer.
mGluR6 expression occurs almost exclusively at the DBC dendritic tips in control retina and is absent in the Grm6−/− retina (Fig. 4A). In contrast, mGluR6 labeling in the Grm6nob8 retina is reduced in the dendritic tips and is present in the cell bodies, suggesting abnormal trafficking of the mutant protein. When we quantified mGluR6 expression with western blotting of whole retina lysates, we found expression is absent in the Grm6−/− retinas, as expected, whereas mGluR6 is present but significantly reduced in the Grm6nob8 retinas compared with control (Fig. 4B, Fig. 5B). Under standard Western blot conditions, the molecular weight of mGluR6 in control retinas was greater than in Grm6nob8. Since the Grm6nob8 mutation occurs near several sites that could undergo posttranslational modification (Nørskov-Lauritsen and Bräuner-Osborne 2015), we tested the idea that this was disrupted in the mutant. We treated the retinal lysates with PNGase to remove N-glycosylation. Consistent with mGluR6 undergoing posttranslational modifications, mGluR6 in control retinas was shifted to a lower molecular weight, similar to the size of the deglycosylated mGluR6 in the Grm6nob8 retinas. This indicates that the proteins are the same size but the extent of glycosylation in the mutant is decreased.
Fig. 4.

mGluR6 is present at reduced levels and is mislocalized in the Grm6nob8 retina. A: immunohistochemistry for mGluR6 in control, Grm6−/−, and Grm6nob8 retinas at 6–8 wk of age. Scale bars: 5 µm. B: Western blots for mGluR6 in control, Grm6−/−, and Grm6nob8 retinas. “+” indicates that homogenates were digested with PNGase F before running gels. β-Actin was used as a loading control.
Fig. 5.

DBC signal transduction proteins are reduced in the Grm6nob8 retina compared with control (C57BL/6J) at 6–8 wk of age. A: TRPM1, GPR179, and LRIT3 are localized to the dendritic tips of DBCs in the Grm6nob8 retinas, albeit at lower expression levels. RGS7 and RGS11 expression are undetectable in the Grm6nob8 retinas. Scale bars: 5 μm. B: Western blots document reduced levels of GPR179, TRPM1, LRIT3, and RGS11 in the Grm6nob8 retina. Bars indicate average (± SE) of 3 independent experiments. **P < 0.01, ***P < 0.001. NS, P > 0.05.
The members of the DBC signal transduction complex are interdependent for correct localization to the DBC dendritic tips (Gregg et al. 2014; Pearring et al. 2011). In Grm6nob8 retinas, the expression patterns of TRPM1, GPR179, and LRIT3 are similar to controls, although their expression levels are lower (Fig. 5). The reduction in expression of RGS7 and RGS11 in the Grm6nob8 retina (Fig. 5B) is consistent with their dependence upon mGluR6 expression (Gregg et al. 2014).
Retinal ganglion cell electrophysiology.
The presence of a small dark-adapted ERG b-wave suggests that some rod DBC function is retained in Grm6nob8 mice. To examine how this signal is transmitted, we studied visually evoked spiking responses of RGCs to full-field flashes using a MEA in control, Grm6−/−, and Grm6nob8 retinas under scotopic conditions (Fig. 6, Ai). Responses across the three control genotypes (C57BL/6J, Grm6nob8/+, and Grm6+/−) were similar and have been combined. In control retinas ~12% of RGCs with spontaneous activity show visual responses at 0.01 cd/m2, increasing to ~40% at 0.03 cd/m2 and ~80% at 1.58 cd/m2 (Fig. 6Aii). In the Grm6nob8 retinas, there are fewer responsive RGCs compared with control at all three stimulus levels, although only the difference at 1.58 cd/m2 was statistically significant. In the Grm6−/− retinas there were even fewer responsive cells at the two lowest flash levels, and the differences at both 0.03 cd/m2 and 1.58 cd/m2 significantly differed from wild type. These data are consistent with the idea that the Grm6nob8 signal is generated in the rod pathway and transmitted to the RGCs via the normal crossover pathway using the AII amacrine cell.
Fig. 6.

Grm6nob8 RGC visual responses reflect a mixture of normal and abnormal ON pathway inputs. Ai: representative PSTHs show responses to light onset for RGCs from the 3 different genotypes with short onset latencies (left) and those in the mutants with slow onset latencies (right). Open bar below x-axis indicates timing of the light flash. Color convention is the same as in Aii. Aii: mean (± SE) % of visually responsive RGCs (those with spontaneous as well as stimulus-evoked spiking) are compared across Grm6nob8, Grm6−/−, and controls across 3 dim flashes under scotopic conditions (n = retinas, cells recorded). B: frequency histograms compare the time to peak of the firing rate of ON (Bi) and OFF (Bii) responses in control, Grm6nob8, and Grm6−/− RGCs. ON responses of both Grm6nob8 and Grm6−/− RGCs are delayed compared with control RGCs (Kruskal-Wallis, P < 0.001; n = cells recorded). Differences in distribution were not observed for OFF responses. C: time to peak for ON (Ci) and OFF (Cii) responses from ON, ON/OFF, and OFF RGCs from control and Grm6nob8. Bars indicate mean, and whiskers represent 5th and 95th percentiles. Individual points indicate measures that fall outside of the 5th to 95th percentile range. These data show that the only difference between genotypes is in the ON responses of both ON and ON/OFF RGCs. In Grm6nob8 mice there are many more RGCs whose ON responses had time to peak within the control range (≤0.7 s) compared with Grm6−/−. Of RGCs with ON responses (which includes both ON and ON/OFF cells), 86 of 164 (52%) were within the control range for Grm6nob8 (n = 8 retinas) while 11 of 35 (31%) were within the control range for Grm6−/− (n = 5 retinas). Differences were not observed in OFF response times to peak across genotypes (Cii). In Bi and Bii, the histograms are binned (0.4 s), as indicated on x-axis. In Ci and Cii, numbers indicate number of cells. **P < 0.01, *** 0.001. NS, P > 0.05.
In previous studies of Grm6 mutant mice, we and others observed that the time to peak of ON RGCs was delayed compared with controls, whereas in OFF RGCs time to peak was normal (Maddox et al. 2008; Rentería et al. 2006). We analyzed this in detail for the responses at the brightest flash luminance (1.58 cd/m2). In control retinas all RGCs (ON, ON/OFF, and OFF) had time to peak <700 ms (Fig. 6Bi) and the mean was 270 ms (median = 250 ms). In contrast, RGCs with responses to luminance onset in Grm6nob8 and Grm6−/− retinas had longer median time to peak, and both mutants differed significantly from control (means 630 and 770 ms, medians 550 and 700 ms for Grm6nob8 and Grm6−/−, respectively; P < 0.0001). Across all three genotypes the time to peak of the OFF responses was similar (~320 ms; Fig. 6Bii). None of the Grm6−/− ON responses had time to peak ≤420 ms, whereas 28% of Grm6nob8 ON responses were between 250 and 420 ms (compared with 97% of control ON responses). We divided Grm6nob8 RGCs into those with delayed ON or delayed ON/OFF responses (Fig. 6Ci), and both classes of cells were significantly delayed compared with ON responses in control RGCs (P = 0.001) but did not differ significantly from each other. The results indicate that there are ON RGCs in Grm6nob8 retinas that are more similar to control than Grm6−/− retinas, which is consistent with both the pattern of expression of mGluR6 and the reduced but not absent ERG b-wave. The distribution of time to peak of OFF responses was similar in control, Grm6nob8, and Grm6−/− (Fig. 6Cii).
DISCUSSION
We report a new mouse model carrying a Grm6 mutation, p.Met66Leu. This mutation is a hypomorphic allele, as it reduces, but does not eliminate, mGluR6 expression at the DBC dendritic tips. This phenotype is distinct from all Grm6 mouse models that have been reported to date, which are null alleles and abolish mGluR6 expression (Maddox et al. 2008; Masu et al. 1995; Pinto et al. 2007; Qian et al. 2015). As a result, the Grm6nob8 mouse provides a model with which to better understand the role of mGluR6 in DBCs.
The Grm6nob8 mouse was initially discovered because of its reduced-amplitude ERG b-wave in the presence of a normal-amplitude a-wave (Xue et al. 2015). This ERG pattern reflects decreased DBC function (Hood and Birch 1996; Kofuji et al. 2000; Robson and Frishman 1995, 1996; Sharma et al. 2005) resulting from either a defect in transmission between photoreceptors and DBCs or defective DBC signaling (Pardue and Peachey 2014).
The overall structure of the Grm6nob8 retina was normal, and many proteins that mark specific structural features of the outer plexiform layer were correctly localized. While the Grm6nob8 ERG signature matches mouse models lacking any one of several presynaptic proteins, including CACNA1F and CABP4, it lacks the ectopic neurites noted in these models (Chang et al. 2006; Haeseleer et al. 2004; Mansergh et al. 2005).
We did observe abnormalities in the levels of several proteins involved in the DBC signal transduction cascade in the Grm6nob8 retina. The expression of TRPM1, GPR179, and LRIT3 appeared reduced, and RGS7 and RGS11 were essentially undetectable. This supports a central role for mGluR6 in the organization of the DBC signaling complex (Gregg et al. 2014) and suggests that a normal complex cannot be maintained in the absence of mGluR6. Consistent with all of these changes, we observed that the proportion of Grm6nob8 visually responsive RGCs as well as of RGCs with short time to peak response lies between control and Grm6−/−.
The data presented support a model in which the p.Met66Leu mutation reduces the ability of mGluR6 to localize to the DBC dendritic tips. As a consequence, mutant mGluR6 is retained within the cell body, where it is unable to participate in the formation of a functional signaling complex. This interpretation matches that made in in vitro studies where wild-type mGluR6 or a series of mGluR6 mutants involving missense changes identified in human cCSNB patients were expressed in cell culture (Beqollari et al. 2009; Zeitz et al. 2007). In these studies, the wild-type protein was trafficked to the cell surface whereas the mutant protein was not. This defect in trafficking to the cell surface was not restricted to mutations in a particular region of the protein. Instead, similar abnormalities have been reported when mutations impacted distinct regions of the mGluR6 protein, including the ligand binding domain, the intracellular domain, and a conserved cysteine-rich domain (Beqollari et al. 2009; Zeitz et al. 2007). We now document that the mouse p.Met66Leu mutation reduces protein glycosylation and note that abnormal glycosylation of other G protein-coupled receptors has been linked to abnormal trafficking (Free et al. 2007; Min et al. 2015; Soto and Trejo 2010). Although the p.Met66Leu mutation does not directly involve a glycosylation site, it is worth noting that rhodopsin glycosylation at Asn15 is impacted by the p.Pro23His mutation (Chen et al. 2014). Our results indicate that the Grm6nob8 mouse presents an opportunity to better understand the processes that govern trafficking of mGluR6 and perhaps other proteins as well. Whether the human mGluR6 mutations impair glycosylation, another posttranslational modification, protein folding, or some other aspect of the process by which mGluR6 is trafficked to the DBC dendritic tips is not known.
Although the dark-adapted ERG b-wave phenotypes of Grm6nob8 and Grm6−/− were distinct, we noted little difference in the cone ERG b-waves of these two genotypes. It is possible that the p.Met66Leu mutation has a greater impact on trafficking to cone compared with rod DBC dendritic tips. Alternatively, it is possible that the impact on trafficking is comparable but the normal density of mGluR6 is normally lower in the dendritic tips of cone compared with rod DBCs. The present data cannot distinguish between these and other potential explanations, and resolution of this issue will require additional work.
While recognizing that direct comparison between in vitro and in vivo expression is difficult, our immunohistochemistry data indicate that some fraction of p.Met66Leu mGluR6 is correctly trafficked to the dendritic tips of rod DBCs. Furthermore, our electrophysiological data indicate that once mGluR6 is correctly trafficked to the DBC dendritic tip the p.Met66Leu mutant protein is able to support the formation of a functional signaling complex and DBC signal transduction. This interpretation is supported by the presence of puncta for other DBC signal transduction proteins with reduced but not absent expression, a reduced but not absent ERG b-wave, and the visual responses of RGCs in Grm6nob8 as compared with Grm6−/− RGCs when dim stimuli are used.
Given the poorly trafficked protein functions, the question is raised of whether the Grm6nob8 phenotype could be normalized by chemical chaperones that improve trafficking of mutant mGluR6 to the DBC dendritic tips. Chemical chaperones have proven effective for a number of disease-causing mutations that result in misfolding of proteins, for example, in RPE65 (Li et al. 2016), LGI1 (Yokoi et al. 2015), and CLRN1 (Alagramam et al. 2016). This question has clinical relevance, as cCSNB patients have been reported with point mutations in the ligand binding domain (Zeitz et al. 2015). As a result, the Grm6nob8 mouse provides a platform for evaluating chemical chaperones and other candidate compounds.
The identification of a mouse mutant for Grm6 in which the b-wave is not abolished provides an exception to the general observation that mutants for DBC proteins lack the ERG b-wave under normal testing conditions (Pardue and Peachey 2014), although a small response can be evoked from some models with long-duration flashes (Ray et al. 2014). With a single exception (Peachey et al. 2012b), all of the reported mutants are thought to produce null alleles due to genetic engineering or the presence of large insertions. While these models are of value for establishing a key role for a given gene product in DBC function, it is likely that there is much to learn from point mutants that involve key functional domains within the protein complex that comprises the DBC signaling cascade.
GRANTS
This work was supported by National Eye Institute Grants R01 EY-12354, R01 EY-19943, and P30 EY-025585; the Department of Veterans Affairs; Foundation Fighting Blindness; and unrestricted grants from Research to Prevent Blindness to Cleveland Clinic Cole Eye Institute and University of Louisville Department of Ophthalmology and Visual Sciences.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.S.P., N.H., R.G.G., M.A.M., and B.C. conceived and designed research; N.S.P., N.H., B.F., S.B., G.P., S.Y.K., L.R., M.L.B., and M.S. performed experiments; N.S.P., N.H., B.F., S.B., G.P., S.Y.K., L.R., M.L.B., M.S., M.A.M., and B.C. analyzed data; N.S.P., N.H., B.F., G.P., S.Y.K., L.R., M.L.B., R.G.G., M.A.M., and B.C. interpreted results of experiments; N.S.P., N.H., R.G.G., M.A.M., and B.C. prepared figures; N.S.P., N.H., G.P., R.G.G., M.A.M., and B.C. drafted manuscript; N.S.P., N.H., R.G.G., M.A.M., and B.C. edited and revised manuscript; N.S.P., N.H., B.F., S.B., G.P., S.Y.K., L.R., M.L.B., M.S., R.G.G., M.A.M., and B.C. approved final version of manuscript.
REFERENCES
- Alagramam KN, Gopal SR, Geng R, Chen DH, Nemet I, Lee R, Tian G, Miyagi M, Malagu KF, Lock CJ, Esmieu WR, Owens AP, Lindsay NA, Ouwehand K, Albertus F, Fischer DF, Bürli RW, MacLeod AM, Harte WE, Palczewski K, Imanishi Y. A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat Chem Biol 12: 444–451, 2016. doi: 10.1038/nchembio.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel LK, Raymond PA. Improved method for obtaining 3-microns cryosections for immunocytochemistry. J Histochem Cytochem 38: 1383–1388, 1990. doi: 10.1177/38.9.2201738. [DOI] [PubMed] [Google Scholar]
- Beqollari D, Betzenhauser MJ, Kammermeier PJ. Altered G-protein coupling in an mGluR6 point mutant associated with congenital stationary night blindness. Mol Pharmacol 76: 992–997, 2009. doi: 10.1124/mol.109.058628. [DOI] [PubMed] [Google Scholar]
- Cao Y, Pahlberg J, Sarria I, Kamasawa N, Sampath AP, Martemyanov KA. Regulators of G protein signaling RGS7 and RGS11 determine the onset of the light response in ON bipolar neurons. Proc Natl Acad Sci USA 109: 7905–7910, 2012. doi: 10.1073/pnas.1202332109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Posokhova E, Martemyanov KA. TRPM1 forms complexes with nyctalopin in vivo and accumulates in postsynaptic compartment of ON-bipolar neurons in mGluR6-dependent manner. J Neurosci 31: 11521–11526, 2011. doi: 10.1523/JNEUROSCI.1682-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT, Nusinowitz S, Rengarajan K, Boyd AP, Sidney SS, Phillips MJ, Stewart RE, Chaudhury R, Nickerson JM, Heckenlively JR, Boatright JH. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res 47: 624–633, 2007. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacna1f: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci 23: 11–24, 2006. doi: 10.1017/S095252380623102X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Jastrzebska B, Cao P, Zhang J, Wang B, Sun W, Yuan Y, Feng Z, Palczewski K. Inherent instability of the retinitis pigmentosa P23H mutant opsin. J Biol Chem 289: 9288–9303, 2014. doi: 10.1074/jbc.M114.551713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Berson EL, Fishman GA, Sandberg MA, Alexander KR, Derlacki DJ, Rajagopalan AS. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci USA 102: 4884–4889, 2005. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen JW, Pangeni G, Pyle IS, McCall MA. Functional changes in Tg P23H-1 rat retinal responses: differences between ON and OFF pathway transmission to the superior colliculus. J Neurophysiol 114: 2368–2375, 2015. doi: 10.1152/jn.00600.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free RB, Hazelwood LA, Cabrera DM, Spalding HN, Namkung Y, Rankin ML, Sibley DR. D1 and D2 dopamine receptor expression is regulated by direct interaction with the chaperone protein calnexin. J Biol Chem 282: 21285–21300, 2007. doi: 10.1074/jbc.M701555200. [DOI] [PubMed] [Google Scholar]
- Gregg RG, Ray TA, Hasan N, McCall MA, Peachey NS. Interdependence among members of the mGluR6 G-protein mediated signalplex of retinal depolarizing bipolar cells. In; G-Protein Signaling Mechanisms in the Retina, Springer Series in Vision Research, edited by Martemyanov KA, Sampath AP. New York: Springer, 2014, p. 67–79. doi: 10.1007/978-1-4939-1218-6_5 [DOI] [Google Scholar]
- Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci 7: 1079–1087, 2004. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan N, Ray TA, Gregg RG. CACNA1S expression in mouse retina: novel isoforms and antibody cross-reactivity with GPR179. Vis Neurosci 33: E009, 2016. doi: 10.1017/S0952523816000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DC, Birch DG. Beta wave of the scotopic (rod) electroretinogram as a measure of the activity of human on-bipolar cells. J Opt Soc Am A Opt Image Sci Vis 13: 623–633, 1996. doi: 10.1364/JOSAA.13.000623. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 20: 5733–5740, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Samardzija M, Yang Z, Grimm C, Jin M. Pharmacological amelioration of cone survival and vision in a mouse model for Leber congenital amaurosis. J Neurosci 36: 5808–5819, 2016. doi: 10.1523/JNEUROSCI.3857-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox DM, Vessey KA, Yarbrough GL, Invergo BM, Cantrell DR, Inayat S, Balannik V, Hicks WL, Hawes NL, Byers S, Smith RS, Hurd R, Howell D, Gregg RG, Chang B, Naggert JK, Troy JB, Pinto LH, Nishina PM, McCall MA. Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J Physiol 586: 4409–4424, 2008. doi: 10.1113/jphysiol.2008.157289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet 14: 3035–3046, 2005. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- Mao CA, Agca C, Mocko-Strand JA, Wang J, Ullrich-Lüter E, Pan P, Wang SW, Arnone MI, Frishman LJ, Klein WH. Substituting mouse transcription factor Pou4f2 with a sea urchin orthologue restores retinal ganglion cell development. Proc Biol Sci 283: 20152978, 2016. doi: 10.1098/rspb.2015.2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R, Takada M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell 80: 757–765, 1995. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- Min C, Zheng M, Zhang X, Guo S, Kwon KJ, Shin CY, Kim HS, Cheon SH, Kim K-M. N-linked glycosylation on the N-terminus of the dopamine D2 and D3 receptors determines receptor association with specific microdomains in the plasma membrane. Biochim Biophys Acta 1853: 41–51, 2015. doi: 10.1016/j.bbamcr.2014.09.024. [DOI] [PubMed] [Google Scholar]
- Nobles RD, Zhang C, Müller U, Betz H, McCall MA. Selective glycine receptor α2 subunit control of crossover inhibition between the on and off retinal pathways. J Neurosci 32: 3321–3332, 2012. doi: 10.1523/JNEUROSCI.5341-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel JM, Fernandez de Castro JP, Demarco PJ Jr, Franco LM, Wang W, Vukmanic EV, Peng X, Sandell JH, Scott PA, Kaplan HJ, McCall MA. Iodoacetic acid, but not sodium iodate, creates an inducible swine model of photoreceptor damage. Exp Eye Res 97: 137–147, 2012. doi: 10.1016/j.exer.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørskov-Lauritsen L, Bräuner-Osborne H. Role of post-translational modifications on structure, function and pharmacology of class C G protein-coupled receptors. Eur J Pharmacol 763: 233–240, 2015. doi: 10.1016/j.ejphar.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Pardue MT, Peachey NS. Mouse b-wave mutants. Doc Ophthalmol 128: 77–89, 2014. doi: 10.1007/s10633-013-9424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peachey NS, Pearring JN, Bojang P Jr, Hirschtritt ME, Sturgill-Short G, Ray TA, Furukawa T, Koike C, Goldberg AF, Shen Y, McCall MA, Nawy S, Nishina PM, Gregg RG. Depolarizing bipolar cell dysfunction due to a Trpm1 point mutation. J Neurophysiol 108: 2442–2451, 2012b. doi: 10.1152/jn.00137.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peachey NS, Ray TA, Florijn R, Rowe LB, Sjoerdsma T, Contreras-Alcantara S, Baba K, Tosini G, Pozdeyev N, Iuvone PM, Bojang P Jr, Pearring JN, Simonsz HJ, van Genderen M, Birch DG, Traboulsi EI, Dorfman A, Lopez I, Ren H, Goldberg AF, Nishina PM, Lachapelle P, McCall MA, Koenekoop RK, Bergen AA, Kamermans M, Gregg RG. GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 90: 331–339, 2012a. doi: 10.1016/j.ajhg.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearring JN, Bojang P Jr, Shen Y, Koike C, Furukawa T, Nawy S, Gregg RG. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J Neurosci 31: 10060–10066, 2011. doi: 10.1523/JNEUROSCI.1014-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto LH, Vitaterna MH, Shimomura K, Siepka SM, Balannik V, McDearmon EL, Omura C, Lumayag S, Invergo BM, Glawe B, Cantrell DR, Inayat S, Olvera MA, Vessey KA, McCall MA, Maddox D, Morgans CW, Young B, Pletcher MT, Mullins RF, Troy JB, Takahashi JS. Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis Neurosci 24: 111–123, 2007. doi: 10.1017/S0952523807070149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Ji R, Gregg RG, Peachey NS. Identification of a new mutant allele, Grm6(nob7), for complete congenital stationary night blindness. Vis Neurosci 32: E004, 2015. doi: 10.1017/S0952523815000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray TA, Heath KM, Hasan N, Noel JM, Samuels IS, Martemyanov KA, Peachey NS, McCall MA, Gregg RG. GPR179 is required for high sensitivity of the mGluR6 signaling cascade in depolarizing bipolar cells. J Neurosci 34: 6334–6343, 2014. doi: 10.1523/JNEUROSCI.4044-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentería RC, Tian N, Cang J, Nakanishi S, Stryker MP, Copenhagen DR. Intrinsic ON responses of the retinal OFF pathway are suppressed by the ON pathway. J Neurosci 26: 11857–11869, 2006. doi: 10.1523/JNEUROSCI.1718-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Response linearity and kinetics of the cat retina: the bipolar cell component of the dark-adapted electroretinogram. Vis Neurosci 12: 837–850, 1995. doi: 10.1017/S0952523800009408. [DOI] [PubMed] [Google Scholar]
- Robson JG, Frishman LJ. Photoreceptor and bipolar cell contributions to the cat electroretinogram: a kinetic model for the early part of the flash response. J Opt Soc Am A Opt Image Sci Vis 13: 613–622, 1996. doi: 10.1364/JOSAA.13.000613. [DOI] [PubMed] [Google Scholar]
- Robson JG, Saszik SM, Ahmed J, Frishman LJ. Rod and cone contributions to the a-wave of the electroretinogram of the macaque. J Physiol 547: 509–530, 2003. doi: 10.1113/jphysiol.2002.030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Ball SL, Peachey NS. Pharmacological studies of the mouse cone electroretinogram. Vis Neurosci 22: 631–636, 2005. doi: 10.1017/S0952523805225129. [DOI] [PubMed] [Google Scholar]
- Shiells RA, Falk G, Naghshineh S. Action of glutamate and aspartate analogues on rod horizontal and bipolar cells. Nature 294: 592–594, 1981. doi: 10.1038/294592a0. [DOI] [PubMed] [Google Scholar]
- Slaughter MM, Miller RF. 2-Amino-4-phosphonobutyric acid: a new pharmacological tool for retina research. Science 211: 182–185, 1981. doi: 10.1126/science.6255566. [DOI] [PubMed] [Google Scholar]
- Soto AG, Trejo J. N-linked glycosylation of protease-activated receptor-1 second extracellular loop: a critical determinant for ligand-induced receptor activation and internalization. J Biol Chem 285: 18781–18793, 2010. doi: 10.1074/jbc.M110.111088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Shen SQ, Corbo JC, Kefalov VJ. Circadian and light-driven regulation of rod dark adaptation. Sci Rep 5: 17616, 2015. doi: 10.1038/srep17616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi N, Fukata Y, Kase D, Miyazaki T, Jaegle M, Ohkawa T, Takahashi N, Iwanari H, Mochizuki Y, Hamakubo T, Imoto K, Meijer D, Watanabe M, Fukata M. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat Med 21: 19–26, 2015. doi: 10.1038/nm.3759. [DOI] [PubMed] [Google Scholar]
- Yu M, Kang K, Bu P, Bell BA, Kaul C, Qiao JB, Sturgill-Short G, Yu X, Tarchick MJ, Beight C, Zhang SX, Peachey NS. Deficiency of CC chemokine ligand 2 and decay-accelerating factor causes retinal degeneration in mice. Exp Eye Res 138: 126–133, 2015. doi: 10.1016/j.exer.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz C, Forster U, Neidhardt J, Feil S, Kälin S, Leifert D, Flor PJ, Berger W. Night blindness-associated mutations in the ligand-binding, cysteine-rich, and intracellular domains of the metabotropic glutamate receptor 6 abolish protein trafficking. Hum Mutat 28: 771–780, 2007. doi: 10.1002/humu.20499. [DOI] [PubMed] [Google Scholar]
- Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res 45: 58–110, 2015. doi: 10.1016/j.preteyeres.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Zeitz C, van Genderen M, Neidhardt J, Luhmann UF, Hoeben F, Forster U, Wycisk K, Mátyás G, Hoyng CB, Riemslag F, Meire F, Cremers FP, Berger W. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci 46: 4328–4335, 2005. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]