

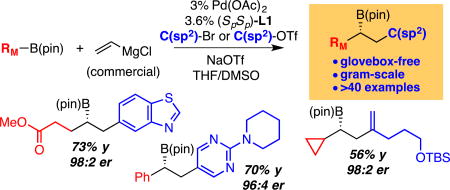
Abstract
Catalytic enantioselective conjunctive cross-couplings that employ Grignard reagents are shown to furnish an array of non-racemic chiral organoboronic esters in an efficient and highly selective fashion. The utility of sodium triflate in facilitating this reaction is two-fold: it enables "ate" complex formation and overcomes catalytic inhibition by halide ions.
Graphical Abstract
INTRODUCTION
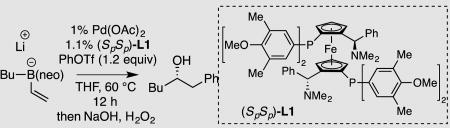
Chiral organoboron compounds are important synthetic intermediates that can be converted to a broad array of chiral materials by mild stereospecific reactions.1 For this reason, intense efforts have been directed towards the construction of these compounds in an enantioselective fashion. While ground breaking work by Brown established the utility of chiral organoboranes,2 organoboronic esters have become the preferred class of boron reagents in contemporary asymmetric synthesis because of their stability and reactivity properties. With these reagents, powerful stoichiometric asymmetric homologation reactions have enabled a broad array of C–C and C–heteroatom bond constructions directly from the organoboronic ester starting materials.3 Similarly, many catalytic processes have been developed that provide access to chiral organoboronic esters from simple building blocks.4 In this vein, our laboratory recently described a catalytic conjunctive cross-coupling reaction (Scheme 1) that operates on alkenyl boronic ester-derived "ate" complexes and produces enantiomericallyenriched alkyl boronic esters in an efficient fashion.5 Since the alkenyl boron "ate" complex is generated in situ from the reaction of appropriate organoboronic esters and organolithium compounds, the conjunctive cross-coupling represents a three-component reaction that merges simple starting materials to generate chiral organoboronic ester products. Because the conjunctive process depicted in Scheme 1 may offer streamlined and convergent routes for the synthesis of chiral compounds, we sought to enhance the operational utility of the reaction by reengineering it to employ readily available and functional-group-tolerant Grignard reagents in place of organolithium compounds. We also aimed to extend the substrate scope from C(sp2) triflate electrophiles to common and inexpensive C(sp2) halide electrophiles.
Scheme 1.

The Pd-Catalyzed Conjunctive Cross-Coupling Reaction.
In this Article, we describe experiments that reveal the mechanistic challenges associated with extending the conjunctive cross-coupling reaction in these directions, and we present an effective solution that enables broadly useful reactions with aryl halides and that also allows use of Grignard-derived "ate" complexes.
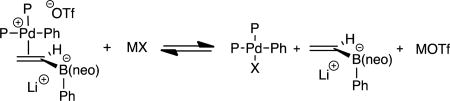
As an operative mechanistic hypothesis to guide development of transition-metal catalyzed conjunctive cross-coupling, it was considered that a π-acidic cationic Pd(II) catalyst is generated by oxidative addition of an LnPd(0) complex to an aryl triflate (Scheme 2). Subsequent association of the Pd(II) adduct with the alkenylboron "ate" complex is proposed to induce a metallate rearrangement; isotope labeling experiments indicate that this elementary reaction occurs with an antiperiplanar relationship between the migrating group (RM) and the Pd center (C). Rearranged complex D then releases the conjunctive coupling product E upon reductive elimination. While the metal-induced metallate rearrangement step (C→D) finds some precedent in the stoichiometric rearrangement reactions of Pt acetylides described by Wrackmeyer6 and in Pd-catalyzed reactions of alkenyl alanes studied by Fillion,7 this elementary transformation has not been used in the catalytic construction of boronic esters. Indeed, in terms of precedent, the catalytic cycle in Scheme 2 is most closely aligned with a reaction studied by Murakami8 involving alkynylborane "ate" complexes, although Murakami proposes alkene carbopalladation followed by invertive reductive displacement of Pd(II) upon 1,2-metallate rearrangement.9 Aside from these important precedents, it should be noted that Deng10 and Ishikura11 have documented relevant catalytic reactions involving outer-sphere addition of alkyne- and indole-derived boron "ate" complexes to palladium allyl and allenyl complexes by a process involving concomitant metallate shift. Similarly, a stoichiometric addition of an alkynyl boronate to an iron(pentadienyl) complex was noted to occur with metallate rearrangement.12
Scheme 2.

Proposed Mechanism for the Pd-Catalyzed Conjunctive Cross-Coupling Reaction.
RESULTS AND DISCUSSION
Challenges in Conjunctive Cross-Coupling: Boron "Ate" Complexes Derived from Grignard Reagents
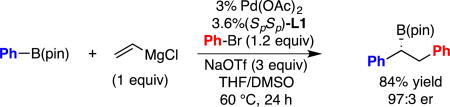
As mentioned above, while the conjunctive cross-coupling is efficient and highly selective, the initial implementation required halide-free organolithium-derived boron "ate" complexes. Because trace amounts of lithium halide salts often arise during the course of lithium-halogen exchange reactions (via elimination), obtaining halide-free organolithium reagents is technically demanding. With a goal of extending the conjunctive cross-coupling reaction to readily accessible starting materials that may have greater functional group compatibility than or-ganolithium reagents, we investigated reactions of "ate" complexes derived from commercially available Grignard reagents (Aldrich). As depicted in Figure 1A, when halide-free vinyllithium was replaced with commercial vinylmagnesium bromide in THF for construction of the "ate" complex from PhB(pin), the conjunctive cross-coupling reaction failed to deliver any product. To learn about underlying reasons for the difference in reactivity between the lithium- and magnesium-based reagents, the reaction of the vinyl metal reagents and PhB(pin) was analyzed by 11B NMR spectroscopy. As depicted in Figure 1B, the reaction between halide-free vinyllithium and PhB(pin) leads to rapid disappearance of the resonance for PhB(pin) (δ = 30.9 ppm) and appearance of a new resonance at δ = 5.8 ppm consistent with efficient formation of a four-coordinate boron species.13 In contrast, the analogous reaction with vinylmagnesium bromide provides ca. 20 % conversion to the putative "ate" complex. While formation of "ate" complexes from Grignard reagents has not been studied in significant detail previously, extant reports describing the reaction between Grignard reagents and α-haloboronates14 imply that access to "ate" complexes as transient reactive intermediates is feasible; however, a report by Blakemore15 suggests that formation of Grignard-derived "ate" complexes may be more difficult than the analogous reactions of lithium derivatives, as is observed here. Thus, one significant challenge to employing Grignard reagents in conjunctive couplings may arise from their diminished nucleophilicity, and hence diminished ability to generate the requisite "ate" complexes, as compared to organolithium reagents.
Figure 1.

A. Comparison of conjunctive cross-coupling with vinyllithium (eq. 1) versus vinylmagnesium bromide (eq. 2). B. 11B NMR analysis of the reaction between vinyl metal reagents and PhB(pin).
Challenges in Conjunctive Cross-Coupling: Inhibition by Halide Salts
While inefficient "ate" complex formation with vinylmagnesium bromide accounts for diminished efficiency of conjunctive cross-coupling with the magnesium-based process, the lack of any conjunctive coupling product at all, even when ca. 20% "ate" complex was generated from the Grignard reagent, suggests other effects might also be operative. In particular, the above-mentioned observation that conjunctive couplings are ineffective with lithium halide-containing "ate" complexes, and that conjunctive couplings are much less effective with aryl and alkenyl halides, suggested that endogenous halide ions might inhibit the catalytic process. To probe the inhibitory effect of halides on the conjunctive coupling, the experiments in Table 1 were conducted. Relative to the standard reaction conditions with 1 mol% catalyst loading and LiI free "ate" complex (entry 1, vinyllithium obtained from tetravinylstannane by Li-Sn exchange16 or from vinyl iodide and n-BuLi, followed by recrystallization from ether17), addition of 1 mol% LiI leads to a significant erosion of conjunctive cross-coupling efficiency (cf. entries 1 and 2), but an otherwise high level of enantioselectivity. The observation that the inhibitory effect of 1 mol % LiI is ameliorated by conducting the reaction with 5 mol % Pd(OAc)2/Mandyphos (L118) (entry 6) suggests that the effect may be due to interaction of LiI and a catalytically active complex. Considering the inhibitory effect of LiI it is unsurprising that conjunctive cross-coupling of aryl halide electrophiles are inefficient even when lithium-halide-free "ate" complexes are employed: the conjunctive cross-coupling itself releases lithium halide as a direct product of the reaction.
Table 1.
Halide Inhibition in Conjunctive Coupling.a
| |||
|---|---|---|---|
|
| |||
| entry | alteration | yieldb | erc |
| 1 | none | 77 | 98:2 |
| 2 | 1% LiI | 13 | 98:2 |
| 3 | 1% LiBr | 41 | 98:2 |
| 4 | 1% LiCl | 40 | 98:2 |
| 5 | 1% (n-Bu)4NCl | 31 | 98:2 |
| 6 | 1% LiI, 5% catalyst | 69 | 98:2 |
| 7 | PhCl instead of PhOTf | <5 | nd |
| 8 | PhBr instead of PhOTf | 9 | 96:4 |
| 9 | PhI instead of PhOTf | 9 | 96:4 |
| 10 | 100% LiBr | 23 | 22:78 |
| 11 | 100% n-Bu4NBr | 19 | 92:8 |
The "ate" complex was prepared by addition of n-butyllithium to vinylB(neo) and the conjunctive coupling was conducted at 0.17 M.
Yield represents isolated yield of purified material.
Enantiomer ratio (er) determined by chiral SFC analysis.
The halide inhibition observed in conjunctive coupling reactions is consonant with the working hypothesis for the reaction mechanism wherein π-bonding between a cationic transition metal complex and the reacting "ate" complex is a necessary prerequisite. In this scenario, it is plausible that halide ions outcompete the olefin for binding to palladium (eq. 3) thereby leading to reaction inhibition. In related stoichiometric processes, the presence of halide ions has been shown to inhibit carbopalladation of alkenes by sequestering cationic palladium complexes.19 Similarly, while catalytic Heck20, Stille,21 and other22 reactions often exhibit acceleration due to the presence of halide ions, this effect is generally traced to an acceleration of oxidative addition; when oxidative addition is not the slow step of catalysis, then halide ions can act as inhibitors.23 Indeed, halide inhibition of catalysis has been documented in the case of catalytic Heck reactions24 and catalytic activation/cross-coupling of cyclopropanes.25
 |
(3) |
Effect of Additives on Boron "Ate" Complex Formation from Grignard Reagents and Subsequent Conjunctive Coupling
The above-described studies suggest that development of a strategy for effective "ate" complex formation and concomitant removal of halide ions from the reaction medium might enable conjunctive cross-coupling reactions with Grignard reagents and also allow the use of organic halide electrophiles. It was reasoned that efficient construction of boron "ate" complexes from Grignard reagents might be facilitated by the addition of appropriate additives that enhance the reactivity of magnesium-based reagents. Along these lines we first examined the capacity for LiCl to facilitate "ate" complex formation. Important studies by Knochel have shown that lithium chloride increases the reactivity of Grignard reagents in Mg-halogen exchange reactions26 and increases the reactivity of Hauser bases27 (in the form of so-called "turbo Grignard reagents" and "turbo Hauser bases"). While we considered that addition of LiCl would likely compound the problem of halide inhibition in catalytic conjunctive coupling reactions, it would nonetheless reveal the capacity for additives to enhance "ate" complex formation. As depicted in Figure 2, when vinylmagnesium chloride was added to PhBpin in THF in the presence of 1 equiv. of LiCl, complete conversion to the "ate" complex was observed by 11B NMR analysis. Unsurprisingly, the resulting complex was unreactive in conjunctive coupling. In an effort to uncover additives that are less likely to inhibit conjunctive coupling reactions, we studied the capacity for other weakly basic metal salts to promote "ate" complex formation (see Supporting Information for complete list). It was found that both LiOTf and NaOTf can effect "ate" complex formation, although the later ion pair is less effective and requires 2 equivalents to achieve >95% conversion to the "ate" complex. In terms of mechanistic features, it is worth noting the effectiveness of Bu4NOTf in facilitating conversion to the "ate" complex. While NaOTf has been proposed to activate Grignard reagents by halide abstraction to generate RMgOTf,28 the lack of Lewis acidity of Bu4NOTf suggests that the ability of additives to facilitate "ate" complex formation stems predominantly from Lewis basicity of the triflate as opposed to either halide abstraction or Lewis acid activation of the boronate through pinacolate O→LA donation.29 Although, it should be noted that NaBPh4, a non-basic Lewis acid, also promotes "ate" complex formation, albeit less effectively than Bu4NOTf; this observation suggests that Lewis acid association may play a beneficial but less significant role in promoting association between the boronic ester and Grignard reagent.
Figure 2.

11B NMR of the reaction between vinylMgCl and PhB(pin) in the presence of additives.
To minimize halide inhibition of catalysis it was considered that cations that are able to ion pair with halide might serve as scavenging agents and facilitate catalysis. While AgOTf was considered to be a reasonable candidate for such a strategy, addition of AgOTf was found to rapidly decompose the halide-free lithium-derived "ate" complex as determined by 11B NMR analysis. We speculated that non-redox active cationic metals whose halide salts are either insoluble or tightly ion-paired in THF solvent might be introduced as metal triflates and, upon anion exchange, act as effective scavengers. In this connection, while the issue of LiCl solubility in THF has received attention,30 the solubility of other metal halides has not been reported. To aid in the interpretation of reaction outcomes, the solubility of a series of metal salts in THF solvent was measured. In these experiments, 1–3 grams of anhydrous salt was stirred in 21 mL anhydrous tetrahydrofuran for 24 hours at room temperature. The solution was allowed to stand undisturbed overnight, the supernatant then filtered to remove remaining non-dissolved salt, and a 10 mL portion of the solution evaporated to constant weight. Using this procedure, the data in Table 2 was collected.
Table 2.
Solubility of metal salts in anhydrous THF solvent.a
| salt | solubility (mg/mL) | solubility (M) |
|---|---|---|
| LiCl | 49.5 | 1.12 |
| LiBr | 388 | 4.47 |
| LiOTf | 473 | 3.03 |
| NaCl | 0.20 | 0.0036 |
| NaCl (1:1 THF:DMSO)b | 0.32 | 0.0055 |
| NaBr | 0.15 | 0.0015 |
| NaOTf | 220 | 1.30 |
| NaOTf (1:1 THF:DMSO)b | 289 | 1.68 |
| KCl | 0.30 | 0.0039 |
| KBr | 0.30 | 0.0022 |
| KOTf | 4.0 | 0.0213 |
| MgCl2 | 40.6 | 0.427 |
| Mg(OTf)2 | 4.4 | 0.014 |
See text and supporting information for procedural details.
For these entries, saturated concentration determined by slowly adding salt to solvent until the solution remained turbid.
Considering the remarkable difference in solubility between NaOTf and NaCl, as well as the ability of NaOTf to promote "ate" complex formation from Grignard reagents, this additive was examined in conjunctive coupling reactions that employ vinylmagnesium chloride. Of note, NaOTf31 is a commercially available and inexpensive salt. After optimization of sol-vent (vide infra for discussion of solvent effects), catalyst loading, stoichiometry, reaction time and temperature (see SI, Table S2), the optimal set of conditions were found to be as depicted in equation 4. As shown, a commercially available solution of vinylmagnesium chloride in THF (Aldrich) was added to a mixture of PhB(pin) and two equivalents of NaOTf in THF/DMSO, followed by Pd(OAc)2, ligand L1, and PhOTf. After reaction at 40 °C for 24 h, the conjunctive coupling product was isolated in 81% yield and 96:4 er. This level of selectivity and reaction efficiency is comparable to that obtained with "ate" complexes prepared from halide-free vinyllithium reagents (eq. 1, Fig 1). It is worth noting that commercial vinylmagnesium bromide could also be utilized with similar levels of yield and selectivity when KOTf (2 equiv) was employed as an additive. In addition to its ability to facilitate the reaction of Grignard-derived "ate" complexes with aryl or alkenyl triflates, the apparent halide-scavenging ability of NaOTf can also enable the reaction of aryl bromide electrophiles: with an additional equivalent of NaOTf added, these electrophiles also engage in efficient and selective conjunctive coupling with Grignard-derived "ate" complexes (eq. 5).32 Aryl iodides behave similarly with PhI reacting in 81% yield and 97:3 er (data not shown).
 |
(4) |
 |
(5) |
Formation and Stability of Boron Ate Complexes: Sol-vent Effects
While NaOTf clearly facilitates formation of boron "ate" complexes from Grignard reagents and can enable conjunctive coupling of Mg-based reagents, two features remained challenging. First, even with NaOTf, formation of "ate" complexes from Grignard reagents and alkyl boronic esters is not efficient (ca. 50% conversion to "ate" complex). Second, whereas the Li-based boron "ate" complexes have good long-term stability, the Mg-based reagents lack stability over a time course comparable to a typical catalytic reaction. These aspects were probed by 11B NMR and the data is depicted in Figure 3. When 3-butenylB(pin) was reacted with vinyllithium, the derived "ate" complex forms immediately (data not shown) and is stable even after 24 hours at room temperature. In contrast, even with NaOTf added, vinyl magnesium chloride only converts ca. 50% of the alkylB(pin) substrate to the derived "ate" complex at 1 h and this complex is not stable: after standing for 24 h, the complex is largely converted back to three-coordinate boron species (mixture of (3-butenyl)Bpin and vinylBpin).
Figure 3.

11B NMR of reactions between vinyl metal reagents and 3-butenylB(pin).
Considering that the polarity and/or coordinating ability of the reaction medium might enhance the stability of "ate" complexes, we examined complexation reactions in different solvents. Most effective was the inclusion of DMSO as a co-solvent: as depicted in Figure 3, the reaction of vinylmagnesium chloride with the alkylB(pin) substrate and NaOTf in THF/DMSO (1:1) solvent mixture proceeds in high conversion and furnishes a boron "ate" complex that persists with little change even after 24 hours at room temperature (data not shown) or 40 °C (Figure 3). This observation is expected to aid in the development of practical conjunctive coupling reactions, and may also prove useful in the design of other boron-based processes.
That the increased stability of Grignard-derived boron "ate" complexes in the presence of DMSO co-solvent translates to increased reaction yield, can be ascertained by examining the data in Table 3. Across a small selection of boronic esters and electrophiles, comparison of conjunctive couplings in either THF solvent and THF/DMSO (1:1) revealed that, while reactions are slower in the solvent mixture, the selectivity and yield are enhanced in the presence of DMSO. Most strikingly, conjunctive couplings with the alkylB(pin) derivative are ineffective in THF solvent, whereas in THF/DMSO a reasonable yield and selectivity are obtained (entries 1 and 2). Notably, even the reaction of arylB(pin) reagents appear to benefit by the inclusion of DMSO co-solvent (cf. entry 3 and 4), although the beneficial effect is less substantial in these cases. Overall, the data presented in the above two sections argues for the use of NaOTf additive and THF/DMSO solvent mixture for effective and general catalytic conjunctive couplings of Grignard-derived complexes. These conditions were surveyed across a range of substrates (vide infra).
Table 3.
Effect of solvent on catalytic conjunctive coupling reactions.a
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | RM | Ar | solvent | yieldb | erc |
| 1 | 3-butenyl | Ph | THF | <5% | n/a |
| 2 | 3-butenyl | Ph | THF/DMSO | 73 | 91:9 |
| 3 | Ph | Ph | THF | 72 | 94:6 |
| 4 | Ph | Ph | THF/DMSO | 81 | 96:4 |
| 5 | Ph | p-MeO-Ph | THF | 78 | 92:8 |
| 6 | Ph | p-MeO-Ph | THF/DMSO | 89 | 98:2 |
| 7 | Ph | p-CF3-Ph | THF | 67 | 75:25 |
| 8 | Ph | p-CF3-Ph | THF/DMSO | 85 | 95:5 |
Reactions conducted as described in the text (see SI for additional details) and the conjunctive coupling was conducted at 0.17 M.
Yield represents isolated yield of purified material.
Enantiomer ratio (er) determined by chiral SFC analysis of boronic ester.
Scope of Catalytic Conjunctive Cross-Coupling with Gri-gnard-Derived "Ate" Complexes
With effective conditions established to employ Grignard-derived "ate" complexes in conjunctive couplings, the scope of the catalytic asymmetric transformation was surveyed. As depicted in Table 4, it was found that the Mg-based system allows conjunctive couplings with a range of aromatic carbocycles, heterocycles and olefinic organotriflate electrophiles. Of note, an aldehyde group attached to the electrophile (product 6) survives the reaction intact, an observation that points to the buffering effect the boron atom imposes on the precursor nucleophilic Grignard reagent. Importantly, labile functional groups such as nitriles, amides, esters, ketones, aldehydes, halides, and unprotected alcohols (employing two equivalents of nucleophile) all survive "ate" complex formation and conjunctive coupling, and are incorporated into non-racemic products selectively and with useful levels of reaction efficiency. It is worth noting that electron-deficient migrating groups such as a para-trifluoromethylphenyl group can be employed successfully (product 25) with the current conditions whereas with the lithium-derive “ate” complex in THF solvent the reaction furnished <5% product. Lastly, it should be pointed out that conjunctive couplings involving vinylB(pin) and alkyl or aryl Grignard reagents (method A) also appear to be effective.
Table 4.
Conjunctive coupling between Grignard-derived boron "ate" complexes and organotriflates.a

|
Conjunctive coupling was conducted at 0.17 M. Yields represent isolated yields of purified material. Both the yield and the enantiomer ratio (er) represent the average value for two experiments.
Product isolated as the derived alcohol.
Reaction conducted at 60 °C.
Solvent = THF.
A survey of the substrate scope involving organobromide electrophiles is depicted in Table 5. Of note, the yield and selectivity with this substrate class parallels that observed when using organotriflate electrophiles so long as an added equivalent of NaOTf is included in the reaction mixture. In these reactions, it was possible to demonstrate compatibility with furan, thiophene, quinoline, pyridine, pyrimidine, indole, benzothiazole and other functionalized organic bromides suggesting that a large collection of targets may ultimately be available from conjunctive couplings, even when the corresponding organotriflate electrophile is not readily available. It should be noted that under the current conditions, alkenyl bromides are significantly less effective than alkenyl triflate electrophiles (i.e. substrate 8 of Table 4 is prepared in 34% yield, 94:6 er from the alkenyl bromide versus 76% yield, 96:4 er from the triflate).
Table 5.
Conjunctive coupling between Grignard-derived boron "ate" complexes and organic bromides.a

|
Conjunctive coupling was conducted at 0.17 M. Yields represent isolated yields of purified material. Both the yield and the enantiomer ratio (er) represent the average value for two experiments.
Reaction conducted at 55 °C.
Conjunctive Cross-Coupling with C(sp2) Triflates and Li-Based Boron "Ate" Complexes Derived from Li-Halogen Exchange
In addition to enabling reactions of Grignard reagents, it was considered that the halide-scavenging ability of NaOTf might enable the direct use of organolithium reagents generated by lithium-halogen exchange (i.e. without taking efforts to remove lithium halide by-products). Thus vinyllith-ium, prepared by treatment of vinyl bromide with 2 equiv. tert-butyllithim was directly added to PhB(pin) in the presence of NaOTf. The derived "ate" complex was then engaged in conjunctive coupling with PhOTf. In this experiment (data not shown) the cross-coupling furnished 1 in 70% yield and 95:5 er. While addition of NaOTf was clearly effective, it was found that KOTf performs somewhat better as a LiBr scavenger for aryl triflate electrophiles whereas NaOTf provided higher selectivity when alkenyl triflate electrophiles were employed (Table 5).
Functional Group Compatibility in Conjunctive Cross-Coupling: Boron-Buffered Nucleophilicity via Kinetic Trapping
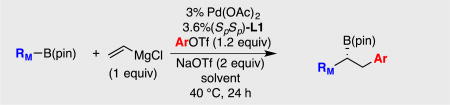
Aspects of the functional group tolerance exhibited during the course of catalytic conjunctive coupling reactions are informative and merit comment. The examples in Tables 4–6 above reveal a range of functional groups – either attached to the electrophile or the "ate" complex – that are compatible with catalytic conjunctive coupling reactions. While compatibility of functional groups attached to the electrophile (i.e. product 6, Table 5) can be anticipated because of the modest basicity and nucleophilicity of "ate" complexes33, the ability to assemble functionalized "ate" complexes by reacting functionalized boronic esters with organometallic compounds (Grignard and organolithium reagents) is less anticipated.34 For example, effective production of compounds 22 – 24 in Table 6 implies that amides, esters, and alkyl halides survive treatment with organolithium reagents. It was considered that this functional group compatibility likely arises from the boronic ester's ability to act as both a kinetic and thermodynamic trap that protects against direct reaction of strong nucleophiles with labile functional groups. It was expected that this feature of "ate" complex formation might ultimately allow conjunctive couplings to be operated as a three-component reaction with-out the need to pre-generate the boron "ate" complex in situ before introduction of the electrophile and the catalyst. To further probe the rapidity of "ate" complex formation relative to reaction of organometallic reagents with other functional groups, more challenging competition experiments were conducted. In the first, a mixture of vinylB(pin) and bromobenzene was treated with tert-butyllithium at −98 °C in ether (eq. 6); the solvent was then removed and the 11B NMR in THF obtained. In this experiment, the 11B NMR resonance corresponding to vinylB(pin) (δ 29.6 ppm) was replaced with a resonance at 7.8 ppm corresponding to "ate" complex 43; a resonance at 5.8 ppm corresponding to PhLi (generated by Li/Br exchange) addition to vinylB(pin) was not observed. Thus addition of tert-butyllithium to vinylB(pin) appears to outcompete Li-Br exchange. In a similar experiment (eq. 7), it was found that addition of tert-butyllithium to vinylB(pin) also outcompetes addition of the alkyllithium to benzaldehyde.
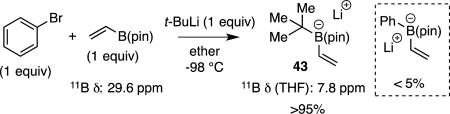 |
(6) |
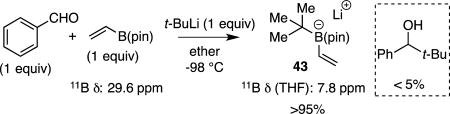 |
(7) |
Table 6.
Conjunctive coupling with halide-containing organolithium-derived boron "ate" complexes.a

|
Conjunctive coupling was conducted at 0.17 M. Yields represent isolated yields of purified material. Both the yield and the enantiomer ratio (er) represent the average value for two experiments.
Product isolated as the derived alcohol.
NaOTf was employed in place of KOTf and the solvent was THF.
Reaction conducted at 60 °C.
Glovebox-Free and Preparative Scale Procedure
To probe the capacity for conjunctive couplings to be conducted without the aid of a glovebox, as a three-component assembly without preformation of "ate" complexes, and on preparative scale, we examined the reactions shown in Figure 4. In these experiments, the solid reagents were weighed in the open atmosphere, combined and transferred to a dried flask, and then the headspace of the reaction vessel purged with dry nitrogen gas. After addition of liquid reagents and solvent, the flask was cooled to 0 °C, the Grignard reagent added, and the reaction then allowed to proceed at the indicated temperature overnight. With this straightforward procedure, the derived conjunctive coupling products can be obtained in good yield and outstanding enantioselectivity. As depicted in equation 8, the reaction can be operated in this manner even on preparative scale and provides functionalized products such as 42 in a practical fashion. Lastly, this procedure applies regardless of whether the substrates are both solids (eq. 8), both liquids (eq. 10) or one of each (eq. 9).
Figure 4.

Conjunctive couplings conducted without the aid of a glovebox.
CONCLUSIONS
We have identified that key challenges associated with the use of Grignard-derived boronic ester "ate" complexes arise from a combination of ineffective "ate" complex formation and inhibition of conjunctive coupling reactions by the presence of halide ions. The latter problem likely contributes to the diminished reactivity of organic halides in conjunctive couplings as well. The addition of NaOTf or KOTf largely counteracts these problems and provides a convenient and broad scoped catalytic conjunctive coupling process. We anticipate that these reactions may find use in organic synthesis and that the utility of alkali metal triflates may find use in development of other catalytic processes involving boron reagents.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-118641).We thank Solvias for supporting this program by providing MandyPhos ligand.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Reviews, see: Brown HS, Singaram B. Acc. Chem. Res. 1988;21:287.Ramachandran PV, Brown HC, editors. Organoboranes for Synthesis. American Chemical Society; Washington, DC: 2001. ACS Symposium Series 783.Matteson DS, editor. Stereodirected Synthesis with Organoboranes. Springer; New York: 1995. Leonori D, Aggarwal VK. Accts. Chem. Res. 2014;47:3174. doi: 10.1021/ar5002473.
- 2.(a) Brown HC, Ramachandran PV. Pure & App. Chem. 1991;63:307. [Google Scholar]; (b) Brown HC, Ramachandran PV. Acct. Chem. Res. 1992;25:16. [Google Scholar]
- 3.(a) Negishi E, Idacavage MJ. Org. React. 1985;33:1. [Google Scholar]; (b) Aggarwal VK, Fang GY, Ginesta X, Howells DM, Zaja M. Pure Appl. Chem. 2006;78:215. [Google Scholar]; (c) Leonori D, Aggarwal VK. Top. Organomet. Chem. 2015;49:271. [Google Scholar]
- 4.Reviews: Hall DG, Lee JCH, Ding J. Pure Appl. Chem. 2012;84:2263.Neeve EC, Geier SJ, Mkhalid IAI, Westcott SA, Marder TB. Chem. Rev. 2016;116:9091. doi: 10.1021/acs.chemrev.6b00193.Ferris GE, Mlynarski SN, Morken JP. Diborane(4) Compounds (Update 2012).Bode JW, editor. Science of Synthesis. Vol. 3. Thieme; 2012. p. 227.
- 5.Zhang L, Lovinger GJ, Edelstein EK, Szymaniak AA, Chierchia MP, Morken JP. Science. 2016;351:70. doi: 10.1126/science.aad6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sebald A, Wrackmeyer B. J. Chem. Soc., Chem. Commun. 1983:309. [Google Scholar]
- 7.(a) Fillion E, Carson RJ, Trépanier VE, Goll JM, Remorova AA. J. Am. Chem. Soc. 2004;126:15354. doi: 10.1021/ja045783t. [DOI] [PubMed] [Google Scholar]; (b) Fillion E, Trépanier VE, Heikkinen JJ, Remorova AA, Carson RJ, Goll JM, Seed A. Organometallics. 2009;28:3518. [Google Scholar]
- 8.(a) Ishida N, Shimamoto Y, Murakami M. Org. Lett. 2009;11:5434. doi: 10.1021/ol902278q. [DOI] [PubMed] [Google Scholar]; (b) Ishida N, Shinmoto T, Sawano S, Miura T, Murakami M. Bull. Chem. Soc. Jpn. 2010;83:1380. [Google Scholar]; (c) Ishida N, Narumi M, Murakami M. Org. Lett. 2008;10:1279. doi: 10.1021/ol8001305. [DOI] [PubMed] [Google Scholar]; (d) Ishida N, Shimamoto Y, Murakami M. Org. Lett. 2010;12:3179. doi: 10.1021/ol1011136. [DOI] [PubMed] [Google Scholar]; (e) Ishida N, Ikemoto W, Narumi M, Murakami M. Org. Lett. 2011;13:3008. doi: 10.1021/ol2008439. [DOI] [PubMed] [Google Scholar]
- 9.Preliminary DFT studies are supportive of the reaction proceeding by the cycle in Scheme 2 as opposed to the carbopalladation-based pathway.
- 10.Chen Y, Li N-S, Deng M-Z. Tetrahedron Lett. 1990;31:2405. [Google Scholar]
- 11.(a) Ishikura M, Terashima M, Okamura K, Date T. J. Chem. Soc. Chem. Commun. 1991:1219. [Google Scholar]; (b) Ishikura M, Agata I. Heterocycles. 1996;43:1591. [Google Scholar]; (c) Ishikura M, Matsuzaki Y, Agata I, Katagiri N. Tetrahedron. 1998;54:13929. [Google Scholar]; (d) Ishikura M, Agata I, Katagiri N. J. Heterocyclic Chem. 1999;36:873. [Google Scholar]; (e) Ishikura M, Hino A, Yaginuma T, Agata I, Katagiri N. Tetrahedron. 2000;56:193. [Google Scholar]; (f) Ishikura M, Kato H. Tetrahedron. 2002;58:9827. [Google Scholar]; (g) Ishiura M, Ida W, Yanada K. Tetrahedron. 2006;62:1015. [Google Scholar]
- 12.Pelter A, Gould KJ. J. Chem. Soc. Chem. Commun. 1974:1029. [Google Scholar]
- 13.For 11B NMR of four-coordinate pinacol boron ate complexes, see: Wrackmeyer B. Ann. Rep. NMR Spectrosc. 1988;20:61.
- 14.Brown HC, De Lue NR, Yamamoto Y, Maruyama K, Kasahara T, Murahashi S, Sonoda A. J. Org. Chem. 1977;42:4088. [Google Scholar]
- 15.Blakemore PR, Marsden SP, Vater HD. Org. Lett. 2006;8:773. doi: 10.1021/ol053055k. [DOI] [PubMed] [Google Scholar]
- 16.Seyferth D, Weiner MA. J. Am. Chem. Soc. 1961;83:3583. [Google Scholar]
- 17.Shinokubo H, Miki H, Yokoo T, Oshima K, Utimoto K. Tetrahedron. 1995;51:11681. [Google Scholar]
- 18.Almena Perea JJ, Lotz M, Knochel P. Tetrahedron Asymm. 1999;10:375. [Google Scholar]
- 19.(a) Kawataka F, Shimizu I, Yamamoto A. Bull. Chem. Soc. Jpn. 1995;68:654. [Google Scholar]; (b) Kayaki Y, Shimizu I, Yamamoto A. Chem. Lett. 1995;24:1089. [Google Scholar]; (c) Amatore C, Carré E, Jutand A, M'Barki MA, Meyer G. Organometallics. 1995;14:5605. [Google Scholar]; (d) Amatore C, Godin B, Jutand A, Lemaître F. Chem. Eur. J. 2007;13:2002. doi: 10.1002/chem.200600153. [DOI] [PubMed] [Google Scholar]; (e) Amatore C, Godin B, Jutand A, Lemaître F. Organometallics. 2007;26:1757. [Google Scholar]
- 20.(a) Jeffrey T. J. Chem. Soc. Chem. Commun. 1984:1287. [Google Scholar]; (b) Jeffrey T. Tetrahedron Lett. 1985;26:2667. [Google Scholar]; (c) Grigg R, Stevenson P, Worakun T. Tetrahedron. 1988;44:2033. [Google Scholar]; (d) Andersson C-M, Hallberg A. J. Org. Chem. 1988;53:2112. [Google Scholar]
- 21.(a) Scott WJ, Crisp GT, Stille JK. J. Am. Chem. Soc. 1984;106:4630. [Google Scholar]; (b) Scott WJ, Stille JK. J. Am. Chem. Soc. 1986;108:3033. doi: 10.1021/ja00263a015. [DOI] [PubMed] [Google Scholar]; (c) Scott WJ, McMurry JE. Acc. Chem. Res. 1988;21:47. [Google Scholar]
- 22.Beletskaya IP. J. Organomet. Chem. 1983;250:511. [Google Scholar]
- 23.Jutand A. Appl. Organomet. Chem. 2004;18:574. [Google Scholar]
- 24.Ozawa F, Kubo A, Hayashi T. J. Am. Chem. Soc. 1991;113:1417. [Google Scholar]
- 25.Aoki S, Fujimura T, Nakamura E, Kuwajima I. J. Am. Chem. Soc. 1988;110:3296. [Google Scholar]
- 26.(a) Krasovskiy A, Knochel P. Angew. Chem. Int. Ed. 2004;43:3333. doi: 10.1002/anie.200454084. [DOI] [PubMed] [Google Scholar]; (b) Barl NM, Werner V, Sämann C, Knochel P. Heterocycles. 2014;88:827. [Google Scholar]; (c) Klatt T, Markiewicz JT, Sämann C, Knochel P. J. Org. Chem. 2014;79:4253. doi: 10.1021/jo500297r. [DOI] [PubMed] [Google Scholar]; (d) Tilly D, Chevallier F, Mongin F, Gros PC. Chem. Rev. 2013;114:1207. doi: 10.1021/cr400367p. [DOI] [PubMed] [Google Scholar]; (e) Bao RL-Y, Zhao R, Shi L. Chem. Commun. 2015;51:6884. doi: 10.1039/c4cc10194d. [DOI] [PubMed] [Google Scholar]
- 27.(a) Krasovskiy A, Krasovskaya V, Knochel P. Angew. Chem. Int. Ed. 2006;45:2958. doi: 10.1002/anie.200504024. [DOI] [PubMed] [Google Scholar]; (b) Lin W, Baron O, Knochel P. Org. Lett. 2006;8:5673. doi: 10.1021/ol0625536. [DOI] [PubMed] [Google Scholar]; (c) Neufed R, Teuteberg TL, Herbst-Irmer R, Mata RA, Stalke D. J. Am. Chem. Soc. 2016;138:4796. doi: 10.1021/jacs.6b00345. [DOI] [PubMed] [Google Scholar]
- 28.Kendall AJ, Salazar CA, Martino PF, Tyler DR. Organometallics. 2014;33:6171.See also, Reetz MT, Harmat N, Mahrwald R. Angew. Chem. Int. Ed. Engl. 1992;31:342.
- 29.For examples of boronate activation by Lewis acid-oxygen binding see: Corey EJ, Barnes-Seeman D, Lee TW. Tetrahedron Asymm. 1997;8:3711.Midland M. J. Org. Chem. 1998;63:914.Rauniyar V, Hall DG. J. Am. Chem. Soc. 2004;126:4518. doi: 10.1021/ja049446w.
- 30.Krasovskiy A, Knochel P. Synthesis. 2006:890. [Google Scholar]
- 31.Surya Prakash GK, Mathew T. Sodium 1,1,1-Trifluoromethanesulfonate. 2010 e-EROS Encyclopedia of Reagents for Organic Synthesis. [Google Scholar]
- 32.For examples of halide abstraction by NaOTf, see: From LnRuCl Quebatte L, Scopelliti R, Severin K. Eur. J. Inorg. Chem. 2005:3353.From LnI(III)Cl, Brantley JN, Samant AV, Toste FD. ACS Cent. Sci. 2016;2:341. doi: 10.1021/acscentsci.6b00119.LnAuCl, Serra D, Moret M-E, Chen P. J. Am. Chem. Soc. 2011;133:8914. doi: 10.1021/ja110405q.LnPdCl, Delis JGP, Groen JH, Vrieze K, van Leeuwen PWNM, Veldman N, Spek AL. Organometallics. 1997;16:551.
- 33.(a) Feeney K, Berionni G, Mayr H, Aggarwal VK. Org. Lett. 2016;17:2614. doi: 10.1021/acs.orglett.5b00918. [DOI] [PubMed] [Google Scholar]; (b) Berionni G, Leonov AI, Mayer P, Ofial AR, Mayr H. Angew. Chem. Int. Ed. 2015;54:2780. doi: 10.1002/anie.201410562. [DOI] [PubMed] [Google Scholar]
- 34.For addition of alkyllithium reagents to B(pin) derivatives bearing esters and amides see: Odachowski M, Bonet A, Essafi S, Conti-Ramsden P, Harvey JN, Leonori D, Aggarwal VK. J. Am. Chem. Soc. 2016;138:9521. doi: 10.1021/jacs.6b03963.Armstrong RJ, García-Ruiz C, Myers EL, Aggarwal VK. Angew. Chem. Int. Ed. 2017;56:786. doi: 10.1002/anie.201610387.Wang Y, Noble A, Myers EL, Aggarwal VK. Angew. Chem. Int. Ed. 2016;55:4270. doi: 10.1002/anie.201600599.Review: Matteson DJ. J. Organomet. Chem. 1999;581:51.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.