

Abstract
Organic cation (OC) transporter 2 (OCT2) mediates the first step in the renal secretion of many cationic drugs: basolateral uptake from blood into proximal tubule cells. The impact of this process on the pharmacokinetics of drug clearance as estimated using a physiologically-based pharmacokinetic approach relies on an accurate understanding of the kinetics of transport because the ratio of the maximal rate of transport to the Michaelis constant (i.e., Jmax/ Kt) provides an estimate of the intrinsic clearance (Clint) used in in vitro–in vivo extrapolation of experimentally determined transport data. Although the multispecificity of renal OC secretion, including that of the OCT2 transporter, is widely acknowledged, the possible relationship between relative affinity of the transporter for its diverse substrates and the maximal rates of their transport has received little attention. In this study, we determined the Jmax and apparent Michaelis constant (Ktapp) values for six structurally distinct OCT2 substrates and found a strong correlation between Jmax and Ktapp; high-affinity substrates [Ktapp values <50 µM, including 1-methyl-4-phenylpyridinium, or 1-methyl-4-phenylpyridinium (MPP), and cimetidine] displayed systematically lower Jmax values (<50 pmol cm−2 min−1) than did low-affinity substrates (Ktapp >200 µM, including choline and metformin). Similarly, preloading OCT2-expressing cells with low-affinity substrates resulted in systematically larger trans-stimulated rates of MPP uptake than did preloading with high-affinity substrates. The data are quantitatively consistent with the hypothesis that dissociation of bound substrate from the transporter is rate limiting in establishing maximal rates of OCT2-mediated transport. This systematic relationship may provide a means to estimate Clint for drugs for which transport data are lacking.
Introduction
At physiologic pH, about 40% of prescribed drugs carry a net positive charge (Neuhoff et al., 2003; Ahlin et al., 2008), and the kidney is the principal site of clearance of these organic cations (OCs) (Hagenbuch, 2010). Renal OC clearance is supported by the two-step process of active secretion that occurs in the proximal tubule: uptake of OC from the blood into proximal tubule cells across the peritubular (basolateral) membrane, followed by efflux into the tubular filtrate across the luminal (apical) membrane. The sequential activity of these two processes effectively defines the pharmacokinetics of many cationic drugs. In humans, basolateral OC transport is dominated by OC transporter, OCT2, whereas apical OC transport involves the combined influence of the multidrug and toxin extruder, MATE1 and MATE2/2-K (Pelis and Wright, 2011; Motohashi and Inui, 2013). Although MATE-mediated OC/H+ exchange represents the active step in net OC secretion, the process is initiated by the electrogenic, albeit passive, accumulation of OCs by the uniporter, OCT2 (Pelis and Wright, 2011).
Efforts to predict pharmacokinetic profiles of drug clearance increasingly rely on physiologically-based pharmacokinetic (PBPK) models that take into account the contribution of the individual metabolic events that influence drug clearance (Harwood et al., 2013), including renal transport (Posada et al., 2015; Burt et al., 2016). Mechanistically-based PBPK models require knowledge of the kinetic parameters of the underlying mediated transport processes (Harwood et al., 2013), including the Michaelis–Menten parameters, Jmax (maximal rate of transport) and Kt (concentration of substrate resulting in half-maximal transport). Importantly, the scaling of transport activity determined in vitro to that occurring in vivo often uses the ratio Jmax/Kt to provide an estimate of the intrinsic clearance (Clint) supported by a particular transporter (Sjögren et al., 2009; Harwood et al., 2013). However, despite insights into the contribution of a transport process to clearance of a target drug that are provided by the relationship between Jmax and Kt, little attention has been given to the basis for different rates of transport that a multidrug transporter, such as OCT2, can exhibit for its structurally diverse suite of substrates.
In this study, we determined the kinetics of OCT2-mediated transport for a structurally diverse set of known substrates and found a strong positive correlation between Jmax and apparent Kt; in other words, substrates for which the transporter had low apparent affinity (i.e., high Ktapp) systematically displayed the highest maximal rates of transport. We also noted a similar correlation between IC50 values for inhibition of OCT2-mediated transport and the degree of trans stimulation arising from preincubating cells in the inhibitory test agent; whereas preloading cells with low-affinity substrates resulted in high levels of trans stimulation, high-affinity ligands (i.e., low IC50 values) produced no stimulation or even a trans inhibition of transport activity. In light of these results, we suggest that dissociation of a substrate from OCT2 is most likely the predominant influence in defining its maximal rate of flux.
Materials and Methods
Chemicals and Reagents.
Chinese hamster ovary (CHO) cells containing the Flp-In target site, as well as hygromyocin and zeocin, were acquired from Invitrogen (Carlsbad, CA). [3H]Tetraethylammonium ([3H]TEA; 54 Ci/mmol) and [3H]cimetidine (80 Ci/mmol) were purchased from American Radiolabeled Chemicals (St. Louis, MO). [3H]1-Methyl-4-phenylpyridinium ([3H]MPP; 80 Ci/mmol), [3H]N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium ([3H]NBD-MTMA; 80 Ci/mmol) (Belzer et al., 2013) and [3H]choline (66.7 Ci/mmol), as well as unlabeled MPP and NBD-MTMA (Aavula et al., 2006), were prepared by the Synthesis Core of the Southwest Environmental Health Sciences Center/Department of Chemistry of the University of Arizona (Tucson, AZ). Additional [3H]MPP (80 Ci/mmol) was purchased from Perkin-Elmer (Waltham, MA). [14C]Metformin was purchased from Moravek Biochemicals (Brea, CA). Diphenidol was purchased from Santa Cruz Biotechnology (Dallas, TX), and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise specified.
Cell Culture.
CHO cells were stably transfected at a single Flp-In recombinase site with the open reading frame for OCT2 in pcDNA5/FRT/V5-His TOPO (Pelis et al., 2007). Cells were cultured at 37°C with 5% CO2. Culture medium contained Ham's F-12 nutrient mixture with 10% fetal bovine serum (Fisher Scientific, Pittsburgh, PA). Hygromyocin (100 µg/ml) or zeocin (100 µg/ml) was added to the culture medium of CHO–OCT2 cells or CHO–wild-type cells, respectively, to maintain cell line expression profiles. Cells were passed every 2–4 days. When seeded into 96-well plates (Greiner; VWR International, Arlington Heights, IL) for transport assays, they were grown to confluence in antibiotic-free media.
Transport Assays.
Wild-type CHO cells and cells expressing human OCT2 were typically plated at densities sufficient to reach confluence within 24 hours (50,000 cells in 96-well cell culture plates), after which they were used in transport experiments. For time course and kinetics experiments, plates containing culture media were placed in an automatic fluid aspirator/dispenser (model 406; BioTek, Winooski, VT) and automatically rinsed/aspirated three times with Waymouth’s buffer (WB; in mM: 135 NaCl, 13 HEPES-NaOH, 28 D-glucose, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, and 0.8 MgSO4), pH 7.4, at room temperature, after which transport buffer (60 μl) containing substrate and any additional required test agents was automatically introduced into each well. Following the experimental incubation, the transport reaction was stopped by the rapid addition (and simultaneous aspiration) of ∼750 μl cold (4°C) WB. Following final aspiration of the cold stop, 200 μl scintillation cocktail (Microscint 20; Perkin-Elmer, Waltham, MA) was added to each well and the plates were sealed (Topseal-A; Perkin-Elmer) and allowed to sit for at least 2 hours before radioactivity was assessed in a 12-channel, multiwell scintillation counter (Wallac Trilux 1450 Microbeta; Perkin-Elmer). Accumulated substrate is expressed as picomoles per cm2 of nominal cell surface or, in some cases, as a clearance in µl cm−2. For the purpose of comparison with literature values that express rates of transport per mg cell protein, we find the conversion factor of 0.035 mg protein cm−2 to be reasonably accurate (Schömig et al., 2006).
To determine the effect on rates of substrate uptake of preloading cells with a test agent, cells were rinsed and then exposed for 20 minutes to WB containing the test agent (or just WB as a control). Following this preincubation, uptake of radiolabeled substrate was measured (in the absence of the preloaded compound), as outlined above.
Data Analysis.
Uptake in OCT2-expressing cells was corrected for transport measured in wild-type CHO cells determined under the same experimental conditions and reported as a mean ± S.E. or mean ± S.D., as indicated, for each data set. The significance of observed differences used simple t tests or analysis of variance (with Tukey's multiple comparisons test), as appropriate, with differences at the 0.05 level considered to be significant. Analyses used either Excel 2007 (Microsoft, Redmond, WA) or Prism 6 (GraphPad, La Jolla, CA).
Results
Figure 1 shows the time course of uptake (expressed as clearance) of three probe substrates for OCT2 (MPP, TEA, and NBD-MTMA), two commonly used drugs (cimetidine and metformin) and one endogenous OC (choline). Over the course of 2 minutes, uptake of all six substrates was adequately described as an exponential increase toward a constant, i.e., steady state accumulation. Extrapolated rates of transport at time zero for each compound were calculated from their first-order rate constants and plateau (steady state) values. For five of the six substrates, these estimates of the initial rate were within 30% of the rates of transport estimated from net accumulation at 30 seconds, whereas the rate of MPP transport was within 45%. Consequently, 30-second uptakes were used for subsequent kinetic experiments.
Fig. 1.

Time course of OCT2-mediated transport of six structurally diverse substrates, expressed as clearance. Each point is the mean (±S.E.) of two separate experiments, each performed in quintuplicate. The substrate concentrations of [3H]labeled MPP, TEA, cimetidine, NBD-MTMA, and choline ranged from 10 to 12 nM; [14C]metformin concentration was ∼12 µM. Each uptake was corrected for substrate accumulation measured in non-OCT2–expressing cells. Lines reflect best fits of first-order association (Prism; GraphPad, San Diego, CA).
Figure 2 shows representative examples of the kinetics of OCT2-mediated transport of the six test substrates. Mediated transport of each was a saturable function of increasing substrate concentration that was adequately described by the Michaelis–Menten equation:
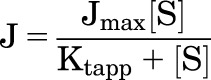 |
(1) |
where J is the rate of mediated transport from a substrate concentration of [S], Jmax is the maximal rate of mediated substrate transport, and Ktapp is the apparent Michaelis constant, i.e., the substrate concentration in the bulk medium that resulted in half-maximal mediated transport. The kinetic values determined in five to nine separate experiments for each substrate are summarized in Table 1. Jmax values ranged from ∼30 pmol cm−2 min−1 (for MPP, cimetidine, and NBD-MTMA) to as high as ∼1000 pmol cm−2 min−1 (for metformin).
Fig. 2.

Kinetics of OCT2-mediated uptake of the six structurally distinct test substrates. Each plot shows data from a representative experiment (see Table 1 for compiled data). The data points reflect mean uptake (±S.E.) from five to six replicate wells. The lines reflect best fits of the Michaelis–Menten equation (Prism).
TABLE 1.
Kinetics of OCT2-mediated transport of the indicated substrates
TE reflects the ratio Jmax/Ktapp.
| Substrate | Jmax (±S.E.) (pmol cm−2 min−1) | Ktapp (±S.E.) (µM) | TE (10−3 cm min−1) | n |
|---|---|---|---|---|
| MPP | 35.2 ± 9.1 | 5.1 ± 0.8 | 6.5 ± 1.4 | 9 |
| NBD-MTMA | 36.2 ± 5.5 | 14.5 ± 4.0 | 3.0 ± 0.3 | 9 |
| Cimetidine | 27.7 ± 7.5 | 20.8 ± 2.8 | 1.3 ± 0.3 | 6 |
| TEA | 276 ± 125 | 71.9 ± 9.9 | 4.7 ± 2.8 | 6 |
| Choline | 673 ± 116 | 465 ± 116 | 1.6 ± 0.1 | 5 |
| Metformin | 1046 ± 171 | 518 ± 45 | 2.0 ± 0.2 | 8 |
In addition to reflecting Clint (Sjögren et al., 2009), the ratio of Jmax to Ktapp has been used as a measure of transport efficiency (TE) (Schömig et al., 2006) because it is approximately analogous to enzymatic catalytic efficiency (kcat/Km), and, as such, it provides a useful means to compare the relative influence of a transporter to the flux of its structurally distinct substrates (Eisenthal et al., 2007). Cimetidine had the lowest TE at 1.3 × 10−3 cm min−1 (or expressed as a clearance, ∼40 µl mg−1 min−1), whereas MPP had the highest at 6.5 × 10−3 cm min−1 (∼200 µl mg−1 min−1) (Table 1). Indeed, Schömig et al. (2006) found that OCT1, OCT2, and OCT3/EMT all display higher TEs for MPP transport than the other substrates they reviewed. But it was noteworthy that of the six substrates studied in this work, the only TE values found to be different from one another were those for MPP and cimetidine (P < 0.05); the other values were neither different from one another nor from the other two substrates. In other words, in our cells the average TE of OCT2 for its substrates was ∼3.2 (±0.8) × 10−3 cm min−1.
The similarity of TE over a broad range of substrate structure and absolute rates of transport is evident in the relationship between increases in Jmax and corresponding increases in the apparent Kt for transport, shown in Fig. 3. For the six test substrates, there was a strong correlation (r = 0.93) between a decrease in the apparent affinity of OCT2 for a substrate and the maximal rate of transport of that compound (expressed as the log of each parameter). At one end of the range, the Jmax for cimetidine transport of ∼28 pmol cm−2 min−1 was associated with a Ktapp of ∼21 µM, whereas, at the other extreme, metformin’s Jmax of ∼1000 pmol cm−2 min−1 was associated with a Ktapp of 500 µM.
Fig. 3.

Relationship between Jmax and apparent Kt for the six test substrates. Each point is the mean value (±S.E.) for five to nine separate determinations of the kinetics of OCT2-mediated transport.
The correlation between low affinity of substrate for OCT2 and high rate of substrate uptake proved to be true for efflux, as well. As a uniporter, OCT2 can support an electrogenic flux of OCs (its normal mode of operation) or, alternatively, an electroneutral exchange of these charged substrates (Budiman et al., 2000). When operating as an exchanger, trans-oriented substrate gradients can stimulate transport from the cis compartment, resulting in classic trans stimulation, or counterflow (e.g., Stein, 1986). In light of the correlation between maximum transport rate and apparent affinity of OCT2 for its substrates evident in Fig. 3, we reasoned that oppositely oriented substrate gradients would stimulate transport of a probe substrate to a degree that was proportional to the trans compound’s affinity for the transporter. To test this, we assembled eight known substrates of OCT2, and four inhibitors of OCT2-mediated transport, and assessed their relative affinity for the transporter by determining their IC50 values for inhibition of MPP transport. As expected, increasing concentrations of each compound resulted in increasing inhibition of OCT2-mediated MPP transport (Fig. 4), which was described by the relationship:
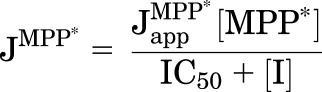 |
(2) |
where [I] is the concentration of inhibitor; JMPP* is the rate of OCT2-mediated transport of [3H]MPP from a concentration equal to [MPP*] (∼10 nM); IC50 is the concentration of inhibitor that reduces mediated (i.e., inhibitable) substrate transport by 50% (note: MPP uptake at each inhibitor concentration was corrected for uptake measured in wild-type CHO cells); and JMPP*app is a constant that includes the maximal rate of MPP transport times the ratio of the inhibitor IC50 and the Ktapp for transport of MPP (Groves et al., 1994). Because the nM concentration of [3H]MPP was much less than its Ktapp (6.5 µM), the IC50 values represent a reasonable approximation of each inhibitor’s Ki. Resulting IC50 values were as low as 0.8 µM (for ethidium) to as large as 435 µM (for choline) (Table 2).
Fig. 4.

Determination of IC50 values for inhibition of OCT2-mediated MPP transport produced by eight OCT2 substrates and four inhibitors of OCT2-mediated transport. Each figure shows inhibition of [3H]MPP transport (∼10 nM; 30-second uptake), expressed as percentage of control uptake, averaged for two to six separate experiments for each inhibitor (see Table 2). The listed IC50 values reflect the average of the values obtained in the individual experiments with each test agent.
TABLE 2.
IC50 values for inhibition of OCT2-mediated transport of MPP
In each experiment, rates of transport were based on 30-second uptakes of 10–20 nM 3H]MPP in the presence of increasing concentrations (typically eight concentrations) of test inhibitor (n = 4–6 wells of a 96-well plate for each concentration). n refers to the number of separate experiments performed.
| Inhibitor | IC50 (µM) | n |
|---|---|---|
| Ethidium | 0.8 ± 0.1 | 2 |
| MPP | 7. | 1 |
| Imipramine | 28.9 ± 15.7 | 3 |
| Diphenidol | 34.3 ± 7.9 | 5 |
| NBD-MTMA | 46 ± 20 | 2 |
| TEA | 77 ± 3 | 2 |
| Quinidine | 92.6 ± 29.1 | 6 |
| Cimetidine | 113 ± 53 | 2 |
| Tramadol | 140.1 ± 31.9 | 3 |
| Metformin | 223 ± 30 | 4 |
| TMA | 301 ± 15 | 2 |
| Choline | 435 ± 153 | 2 |
We then determined the effect on [3H]MPP uptake of preincubating the cells in concentrations of each compound at least twofold, and generally >fivefold, larger than its IC50 (Fig. 5). There was a significant correlation between apparent affinity for OCT2 (i.e., IC50) and the rate of [3H]MPP uptake produced by preloading the cells with the six test substrates; the degree of trans stimulation increased as apparent affinity for the transporter decreased (black circles in Fig. 5). This correlation was extended by the influence on MPP uptake of preloading the cells with two other known OCT substrates: ethidium and tetramethylammonium. Whereas the high-affinity substrate ethidium (IC50 of 0.8 µM and Ktapp of ∼2 µM) (Lee et al., 2009) had comparatively little effect on the rate of MPP uptake, the low-affinity substrate tetramethylammonium (Dresser et al., 2000) supported a marked stimulation of MPP transport (gray filled circles in Fig. 5). Two compounds shown previously to be nontransported inhibitors of OCT2, quinidine (Arndt et al., 2001) and diphenidol (Harper and Wright, 2013), and the known OCT2 inhibitor imipramine (Zolk et al., 2009), trans-inhibited MPP uptake (open circles in Fig. 5). Preincubating cells with tramadol, a substrate for OCT1 (Stamer et al., 2016), resulted in a modest, although not significant, stimulation of MPP transport.
Fig. 5.

Relationship between IC50 values for inhibition of OCT2-mediated MPP transport and the degree of stimulation of MPP uptake resulting from preloading cells with test compound. Each point is the average (±S.E.) of trans stimulation measured in four to seven separate experiments with the test compounds, plotted as a function of their respective IC50 values (from Table 2). *Indicates a difference (relative to control uptake) significant at P < 0.05.
Discussion
The multisubstrate specificity of renal OC secretion was established by Ullrich (1994), and, since his seminal studies (Ullrich et al., 1991, 1992; David et al., 1995; Ullrich and Rumrich, 1996), the ability of the underlying molecular processes, i.e., the OCTs and MATEs, to transport compounds of broadly diverse structure has been clearly established (Nies et al., 2011; Pelis and Wright, 2011). The first step in renal secretion—substrate uptake from blood into proximal tubule cells—is mediated by OCTs, and in humans OCT2 is the dominant process. Over the last 20 years, the kinetics of OCT2-mediated transport has been reported for a structurally diverse range of compounds (Nies et al., 2011). But interstudy comparisons of kinetic parameters are rife with caveats. Indeed, marked differences in kinetic values obtained by different laboratory groups (that often use different experimental and analytical methodologies) have plagued the study of membrane transport (e.g., Bentz et al., 2013). Nevertheless, the comparatively large database for such studies (see Nies et al., 2011 and Supplemental Table 1) invites examination of the relationship between reported values for Jmax for different OCT2 substrates and their associated apparent Kt values. As shown in Fig. 6, these data suggest that there is an inverse correlation between the apparent affinity of OCT2 for a substrate and its maximal rate of transport. The results of the current study confirmed this general observation (the gray shaded data points in Fig. 6) and support the hypothesis that there is a systematic relationship between the affinity of OCT2 for a substrate and the maximal rate of that substrate’s transport.
Fig. 6.

Relationship between Jmax and apparent Kt values for OCT2-mediated transport of 23 distinct substrates. Solid black circles show values reported in the literature (see Supplemental Table 1 for a list of these values and the corresponding references). Shaded gray circles show the values for the six substrates used in the present study. The line is simple linear regression of these data (Prism).
However, it should be noted that increases in apparent Kt values are an expected consequence of increases in Jmax simply owing to the influence of unstirred water layers (UWLs) (Winne, 1973; Barry and Diamond, 1984). For carrier-mediated uptake into cells, transport activity reduces the concentration of substrate in the layer of effectively static fluid immediately adjacent to the extracellular face of the membrane, the UWL, thereby establishing a gradient of substrate concentration between the surface of the membrane and the (well-stirred) bulk medium. Consequently, the concentration of substrate in the experimental solution is greater than that actually available to the transporter, so apparent Kt values (Ktapp) determined experimentally overestimate the actual concentration of substrate required to half-saturate the transporter. The bias that an UWL introduces into measured values of Ktapp is directly proportional to the Jmax of the transport process (Winne, 1973). We recently showed that simply changing the number of transporters expressed in the plasma membrane, thereby changing Jmax, can result in 5- to 10-fold changes in the measured Ktapp values for OCT2-mediated transport of TEA and MPP (Shibayama et al., 2015). At steady state, the influence of UWLs and Jmax on an experimentally determined Kt value is given by:
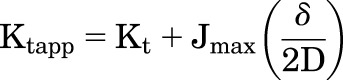 |
(3) |
where Ktapp is the experimentally determined Michaelis constant, Kt is the true Michaelis constant of the transporter, Jmax is the maximal rate of mediated transport, D is the diffusion coefficient of the transported substrate (∼6 × 10−6 cm2 sec−1 for molecules the size of MPP or other small OCs) (Avdeef et al., 2004), and δ is the thickness of the UWL. The effective thickness of UWLs in unstirred multiwell plates has been measured using a variety of techniques with average values being on the order of 1500 μm (Korjamo et al., 2009). Using that value and a Jmax of 1000 pmol cm−2 min−1 (the value we measured for metformin transport and the upper end of Jmax values reported in the literature for OCT2-mediated transport; Fig. 6), the bias introduced to steady state estimates of Kt is on the order of 200 µM. By this reasoning, our measured value of ∼500 µM for the Ktapp for metformin transport could reflect a true Kt of ∼300 µM. By similar logic, the smaller Jmax for MPP transport (∼35 pmol cm−2 min−1) suggests that measured Kt values should overestimate the true value by ∼7 µM. However, these calculations assume the concentration of substrate at the membrane is at steady state during the entire course of measurement, reflecting the impact of the UWL on establishing a constant gradient in concentration between the membrane surface and the bulk medium. In fact, this is not the case. The substrate concentration at the membrane immediately following addition of an experimental solution is actually zero, owing to the presence of substrate-free medium at the membrane surface that remains even after thorough aspiration of preincubation media. Although substrate quickly diffuses through this layer, the ensuing activity of the transporter begins to influence the concentration. The steady state condition was assumed in all previous assessments of the impact of UWLs on transport kinetics (Winne, 1973; Thomson and Dietschy, 1977; Balakrishnan et al., 2007), but it takes several minutes of unabated transport activity before the steady state condition reflected in the relationship derived by Winne (1973) is established. Our modeling of this effect indicates that, during the course of the 30-second uptakes we used in our studies, the concentration of substrate at the membrane was both constantly changing and, on average, higher than expected at steady state, thereby blunting the influence of UWLs on the measured Kt (unpublished observations). Consequently, at the low end of the Jmax range (e.g., MPP, cimetidine), Kt values may have been overestimated by 1–3 µM (rather than 7 µM), whereas, at the high end (metformin, choline), the overestimate was likely on the order of 100 µM (rather than 200 µM). Thus, although the ubiquitous presence of UWLs will contribute to the relationship between Jmax and Ktapp apparent in Figs. 3 and 6, it cannot account for the bulk of this correlative effect. Supporting this conclusion, we point to the trans stimulation of MPP uptake produced by preloading cells with OCT2 substrates. Despite the fact that there would have been no substantive UWL at the cytoplasmic face of the protein to influence the interaction of the preloaded substrate with the transporter, the correlation between maximal rate of transport and the apparent affinity of substrate for the transporter was observed.
It is also worth commenting on the influence of preloading cells with nontransported inhibitors of OCT2 on the rate of substrate uptake. Diphenidol was an effective inhibitor of OCT2 (IC50 of 34 µM), but is not itself transported by OCT2 (Harper and Wright, 2013). As a weak base (pKa of 9.2), a modest rate of diffusion of the uncharged species would, however, be expected to occur, leading to its gradual intracellular accumulation during the 20-minute preincubation in 500 µM diphenidol. Intracellular diphenidol would likely bind to the cytoplasmic face of the transporter, and its failure to support the conformational changes required for translocation would be expected to result in a trans inhibition of MPP uptake, as noted in Fig. 5. Although the transportability of quinidine by OCT2 has not been clearly established, its enantiomer, quinine, is a nontransported inhibitor of OCT2 (Arndt et al., 2001), so the trans inhibition produced by preincubating cells in a 500 µM concentration of this weak base was also expected.
The Jmax of substrate transport into a cell is the product of the number of functional transporters expressed at the membrane and the turnover number (TN) of each transporter (the number of substrate molecules it can translocate from one side of the membrane to the other per unit time under saturating conditions). Assuming that the transport process is adequately described by the alternating access model (Kaback et al., 2011), the TN is influenced by the rate of interconversion of the outward- and inward-facing forms of the carrier and/or the rate of dissociation of substrate from the carrier (Stein, 1986). Our observation that the line of stably expressing CHO cells that supported OCT2-mediated transport of different substrates at maximal rates that varied by almost 40-fold suggests that the TN of OCT2 can vary by that amount, depending on substrate identity. That TNs varied inversely with apparent affinity of OCT2 for substrate invites the suggestion that substrate dissociation may be rate-limiting to the rate of transport. However, identifying the basis for transporter TN is difficult because we lack information on rates of substrate association (kon; M−1 sec−1) with, and dissociation (koff; sec−1) from, the carrier. However, Stein (1986) described a method that permits a tentative test of the hypothesis that substrate dissociation is the rate-limiting element of transporter turnover. The argument makes the temporary assumption that TN is given by koff. Given that the dissociation constant for substrate binding to the transporter, KD, is equal to koff/kon, one calculates an estimate for kon from koff/KD, identified tentatively as TN/Kt, and then compares the estimated kon to a range of likely values for that parameter. This comparison takes advantage of the existing database for small ligand interactions with their binding partners. Rates of association for ligands can range from 104 to 109 M−1 sec−1. However, the applicable range can be narrowed markedly by considering a range of measured kon values for low molecular weight ligands with their protein-binding partners: from about 1 × 105 M−1 sec−1 to about 1 × 107 M−1 sec−1 (e.g., Stein, 1986; Bloom et al., 1987; Wright et al., 1992; Kapur and Seeman, 2000). Moreover, there is evidence that large differences in KD values observed for a range of structurally distinct ligands generally reflect changes in koff, rather than kon, which tend to be relatively constant (Tummino and Copeland, 2008). For the current purpose, we use 105 to 107 M−1 sec−1 for the range of likely kon values for OCT2 substrates. The TN of OCT2 for transport of atenolol at 37°C has been estimated from the kinetics of its transport and measurement of the number of (total) OCT2 transporters expressed in the HEK 293 expression system used in the study (Yin et al., 2015); the resulting TN was ∼3 seconds−1. The range of TN values for transporters is generally reported as 10–2000 seconds−1 (Stein and Litman, 2015), and for the current purpose we will use the range 1–1000 seconds−1. As shown in Fig. 6, Michaelis constants reported in the literature for OCT2-mediated transport range from ∼1 µM to ∼1000 µM, and we will use this range. Figure 7 shows a matrix of estimated kon values for physiologically relevant pairs of TN and Kt values, and values lying within the likely range of association rates are highlighted. In fact, those highlighted values represent the majority of those in the matrix and, as expected, show a clear correlation between high TN (i.e., high Jmax) and high Kt (i.e., low affinity), consistent with the observed correlation between those parameters and supporting the hypothesis that dissociation of substrate from the carrier is rate-limiting for establishing maximal rates of OCT2-mediated transport.
Fig. 7.

Calculated association rates (kon; M−1 sec−1) for a range of likely Kt (µM) and turnover number (sec−1) values for substrate interaction with OCT2. The bold/highlighted values represent kon values considered most likely relevant for low molecular weight interactions of the type anticipated to occur between OCT2 and its substrates. (See text for discussion.)
Jmax and Kt values for drug transporters determined from in vitro experiments are playing an increasingly important role in PBPK (Harwood et al., 2013; Burt et al., 2016). When interpreted as a measure of Clint, the ratio of Jmax to Kt is an element of the scaling factors used for the in vitro–in vivo extrapolation required for mechanistic PBPK (Harwood et al., 2013). If future work confirms the rate-limiting impact of apparent affinity of OCT2 for substrate on the maximal rate of transport, then establishing Clint of an in vitro system for several substrates may provide scaling factors that are common for a wide range of OCT2 substrates for which direct transport data are not available.
In summary, maximal rates of OCT2-mediated uptake for an array of structurally distinct substrates were inversely correlated with the apparent affinity of the transporter for these compounds. Similarly, the rate of trans stimulation of transport activity produced by preloading cells with a structurally diverse range of known OCT2 substrates was inversely correlated with the apparent affinity of the transporter for the preloaded compound. The correlation between affinity of OCT2 for a substrate and the maximal rate of transport of that compound was consistent with the hypothesis that the rate-limiting step in OCT2-mediated transport is dissociation of substrate from the transporter (rather than differences in the rate of conformational changes associated with translocation of structurally distinct substrates). These observations may provide useful insights for applying scaling factors for OCT2 activity required for mechanistic PBPK modeling of novel compounds for which direct transport data are lacking.
Abbreviations
- CHO
Chinese hamster ovary
- Clint
intrinsic clearance
- MATE
multidrug and toxin extruder
- MPP
1-methyl-4-phenylpyridinium
- NBD-MTMA
N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][l,2,5]oxadiazol-4-yl)amino]ethanaminium iodide
- OC
organic cation
- OCT
OC transporter
- PBPK
physiologically-based pharmacokinetic
- TE
transport efficiency
- TEA
tetraethylammonium
- TN
turnover number
- UWL
unstirred water layer
- WB
Waymouth’s buffer
Authorship Contributions
Participated in research design: Severance, Wright.
Conducted experiments: Severance, Sandoval.
Performed data analysis: Severance, Sandoval, Wright.
Wrote or contributed to the writing of the manuscript: Severance, Sandoval, Wright.
Footnotes
This work was supported by National Institutes of Health [Grants 5R01DK58251/DK058251-S1, 5T32HL07249, and 5P30ES006694].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Aavula BR, Ali MA, Mash EA, Bednarczyk D, Wright SH. (2006) Synthesis and fluorescence of n, n, n-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium iodide, a pH-insensitive reporter of organic cation transport. Synth Commun 36:701–705. [Google Scholar]
- Ahlin G, Karlsson J, Pedersen JM, Gustavsson L, Larsson R, Matsson P, Norinder U, Bergström CA, Artursson P. (2008) Structural requirements for drug inhibition of the liver specific human organic cation transport protein 1. J Med Chem 51:5932–5942. [DOI] [PubMed] [Google Scholar]
- Arndt P, Volk C, Gorboulev V, Budiman T, Popp C, Ulzheimer-Teuber I, Akhoundova A, Koppatz S, Bamberg E, Nagel G, et al. (2001) Interaction of cations, anions, and weak base quinine with rat renal cation transporter rOCT2 compared with rOCT1. Am J Physiol Renal Physiol 281:F454–F468. [DOI] [PubMed] [Google Scholar]
- Avdeef A, Nielsen PE, Tsinman O. (2004) PAMPA--a drug absorption in vitro model 11: matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur J Pharm Sci 22:365–374. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Hussainzada N, Gonzalez P, Bermejo M, Swaan PW, Polli JE. (2007) Bias in estimation of transporter kinetic parameters from overexpression systems: interplay of transporter expression level and substrate affinity. J Pharmacol Exp Ther 320:133–144. [DOI] [PubMed] [Google Scholar]
- Barry PH, Diamond JM. (1984) Effects of unstirred layers on membrane phenomena. Physiol Rev 64:763–872. [DOI] [PubMed] [Google Scholar]
- Belzer M, Morales M, Jagadish B, Mash EA, Wright SH. (2013) Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J Pharmacol Exp Ther 346:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz J, O’Connor MP, Bednarczyk D, Coleman J, Lee C, Palm J, Pak YA, Perloff ES, Reyner E, Balimane P, et al. (2013) Variability in P-glycoprotein inhibitory potency (IC50) using various in vitro experimental systems: implications for universal digoxin drug-drug interaction risk assessment decision criteria. Drug Metab Dispos 41:1347–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JW, Halonen M, Lawrence LJ, Rould E, Seaver NA, Yamamura HI. (1987) Characterization of high affinity [3H]pirenzepine and (-)-[3H] quinuclidinyl benzilate binding to muscarinic cholinergic receptors in rabbit peripheral lung. J Pharmacol Exp Ther 240:51–58. [PubMed] [Google Scholar]
- Budiman T, Bamberg E, Koepsell H, Nagel G. (2000) Mechanism of electrogenic cation transport by the cloned organic cation transporter 2 from rat. J Biol Chem 275:29413–29420. [DOI] [PubMed] [Google Scholar]
- Burt HJ, Neuhoff S, Almond L, Gaohua L, Harwood MD, Jamei M, Rostami-Hodjegan A, Tucker GT, Rowland-Yeo K. (2016) Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur J Pharm Sci 88:70–82. [DOI] [PubMed] [Google Scholar]
- David C, Rumrich G, Ullrich KJ. (1995) Luminal transport system for H+/organic cations in the rat proximal tubule: kinetics, dependence on pH; specificity as compared with the contraluminal organic cation-transport system. Pflugers Arch 430:477–492. [DOI] [PubMed] [Google Scholar]
- Dresser MJ, Gray AT, Giacomini KM. (2000) Kinetic and selectivity differences between rodent, rabbit, and human organic cation transporters (OCT1). J Pharmacol Exp Ther 292:1146–1152. [PubMed] [Google Scholar]
- Eisenthal R, Danson MJ, Hough DW. (2007) Catalytic efficiency and kcat/KM: a useful comparator? Trends Biotechnol 25:247–249. [DOI] [PubMed] [Google Scholar]
- Groves CE, Evans KK, Dantzler WH, Wright SH. (1994) Peritubular organic cation transport in isolated rabbit proximal tubules. Am J Physiol 266:F450–F458. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B. (2010) Drug uptake systems in liver and kidney: a historic perspective. Clin Pharmacol Ther 87:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JN, Wright SH. (2013) Multiple mechanisms of ligand interaction with the human organic cation transporter, OCT2. Am J Physiol Renal Physiol 304:F56–F67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood MD, Neuhoff S, Carlson GL, Warhurst G, Rostami-Hodjegan A. (2013) Absolute abundance and function of intestinal drug transporters: a prerequisite for fully mechanistic in vitro-in vivo extrapolation of oral drug absorption. Biopharm Drug Dispos 34:2–28. [DOI] [PubMed] [Google Scholar]
- Kaback HR, Smirnova I, Kasho V, Nie Y, Zhou Y. (2011) The alternating access transport mechanism in LacY. J Membr Biol 239:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Seeman P. (2000) Antipsychotic agents differ in how fast they come off the dopamine D2 receptors: implications for atypical antipsychotic action. J Psychiatry Neurosci 25:161–166. [PMC free article] [PubMed] [Google Scholar]
- Korjamo T, Heikkinen AT, Mönkkönen J. (2009) Analysis of unstirred water layer in in vitro permeability experiments. J Pharm Sci 98:4469–4479. [DOI] [PubMed] [Google Scholar]
- Lee WK, Reichold M, Edemir B, Ciarimboli G, Warth R, Koepsell H, Thévenod F. (2009) Organic cation transporters OCT1, 2, and 3 mediate high-affinity transport of the mutagenic vital dye ethidium in the kidney proximal tubule. Am J Physiol Renal Physiol 296:F1504–F1513. [DOI] [PubMed] [Google Scholar]
- Motohashi H and Inui K (2013) Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J 15:581-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhoff S, Ungell AL, Zamora I, Artursson P. (2003) pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug-drug interactions. Pharm Res 20:1141–1148. [DOI] [PubMed] [Google Scholar]
- Nies AT, Koepsell H, Damme K, Schwab M. (2011) Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol 201:105–167. [DOI] [PubMed] [Google Scholar]
- Pelis RM, Dangprapai Y, Wunz TM, Wright SH. (2007) Inorganic mercury interacts with cysteine residues (C451 and C474) of hOCT2 to reduce its transport activity. Am J Physiol Renal Physiol 292:F1583–F1591. [DOI] [PubMed] [Google Scholar]
- Pelis RM, Wright SH. (2011) Renal transport of organic anions and cations. Compr Physiol 1:1795–1835. [DOI] [PubMed] [Google Scholar]
- Posada MM, Bacon JA, Schneck KB, Tirona RG, Kim RB, Higgins JW, Pak YA, Hall SD, Hillgren KM. (2015) Prediction of renal transporter mediated drug-drug interactions for pemetrexed using physiologically based pharmacokinetic modeling. Drug Metab Dispos 43:325–334. [DOI] [PubMed] [Google Scholar]
- Schömig E, Lazar A, Gründemann D. (2006) Extraneuronal monoamine transporter and organic cation transporters 1 and 2: a review of transport efficiency. Handb Exp Pharmacol 175:151–180. [DOI] [PubMed] [Google Scholar]
- Shibayama T, Morales M, Zhang X, Martínez-Guerrero LJ, Berteloot A, Secomb TW, Wright SH. (2015) Unstirred water layers and the kinetics of organic cation transport. Pharm Res 32:2937–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjögren E, Lennernäs H, Andersson TB, Gråsjö J, Bredberg U. (2009) The multiple depletion curves method provides accurate estimates of intrinsic clearance (CLint), maximum velocity of the metabolic reaction (Vmax), and Michaelis constant (Km): accuracy and robustness evaluated through experimental data and Monte Carlo simulations. Drug Metab Dispos 37:47–58. [DOI] [PubMed] [Google Scholar]
- Stamer UM, Musshoff F, Stüber F, Brockmöller J, Steffens M, Tzvetkov MV. (2016) Loss-of-function polymorphisms in the organic cation transporter OCT1 are associated with reduced postoperative tramadol consumption. Pain 157:2467–2475. [DOI] [PubMed] [Google Scholar]
- Stein WD. (1986) Transport and Diffusion across Cell Membranes, Academic Press, New York. [Google Scholar]
- Stein WD, Litman T. (2015) Channels, Carriers, and Pumps: An Introduction to Membrane Transport, Academic Press, San Diego, CA. [Google Scholar]
- Thomson AB, Dietschy JM. (1977) Derivation of the equations that describe the effects of unstirred water layers on the kinetic parameters of active transport processes in the intestine. J Theor Biol 64:277–294. [DOI] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. (2008) Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 47:5481–5492. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ. (1994) Specificity of transporters for ‘organic anions’ and ‘organic cations’ in the kidney. Biochim Biophys Acta 1197:45–62. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Papavassiliou F, David C, Rumrich G, Fritzsch G. (1991) Contraluminal transport of organic cations in the proximal tubule of the rat kidney. I. Kinetics of N1-methylnicotinamide and tetraethylammonium, influence of K+, HCO3-, pH; inhibition by aliphatic primary, secondary and tertiary amines and mono- and bisquaternary compounds. Pflugers Arch 419:84–92. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Rumrich G. (1996) Luminal transport system for choline+ in relation to the other organic cation transport systems in the rat proximal tubule: kinetics, specificity: alkyl/arylamines, alkylamines with OH, O, SH, NH2, ROCO, RSCO and H2PO4-groups, methylaminostyryl, rhodamine, acridine, phenanthrene and cyanine compounds. Pflugers Arch 432:471–485. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Rumrich G, Neiteler K, Fritzsch G. (1992) Contraluminal transport of organic cations in the proximal tubule of the rat kidney. II. Specificity: anilines, phenylalkylamines (catecholamines), heterocyclic compounds (pyridines, quinolines, acridines). Pflugers Arch 420:29–38. [DOI] [PubMed] [Google Scholar]
- Winne D. (1973) Unstirred layer, source of biased Michaelis constant in membrane transport. Biochim Biophys Acta 298:27–31. [DOI] [PubMed] [Google Scholar]
- Wright SH, Pajor AM, Moon DA, Wunz TM. (1992) High-affinity phlorizin binding in Mytilus gill. Biochim Biophys Acta 1103:212–218. [DOI] [PubMed] [Google Scholar]
- Yin J, Duan H, Shirasaka Y, Prasad B, Wang J. (2015) Atenolol renal secretion is mediated by human organic cation transporter 2 and multidrug and toxin extrusion proteins. Drug Metab Dispos 43:1872–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF. (2009) Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2). Naunyn Schmiedebergs Arch Pharmacol 379:337–348. [DOI] [PubMed] [Google Scholar]