

Abstract
Objective:
To determine the relationship between sleep quality and CSF markers of Alzheimer disease (AD) pathology in late midlife.
Methods:
We investigated the relationship between sleep quality and CSF AD biomarkers in a cohort enriched for parental history of sporadic AD, the Wisconsin Registry for Alzheimer's Prevention. A total of 101 participants (mean age 62.9 ± 6.2 years, 65.3% female) completed sleep assessments and CSF collection and were cognitively normal. Sleep quality was measured with the Medical Outcomes Study Sleep Scale. CSF was assayed for biomarkers of amyloid metabolism and plaques (β-amyloid 42 [Aβ42]), tau pathology (phosphorylated tau [p-tau] 181), neuronal/axonal degeneration (total tau [t-tau], neurofilament light [NFL]), neuroinflammation/astroglial activation (monocyte chemoattractant protein–1 [MCP-1], chitinase-3-like protein 1 [YKL-40]), and synaptic dysfunction/degeneration (neurogranin). To adjust for individual differences in total amyloid production, Aβ42 was expressed relative to Aβ40. To assess cumulative pathology, CSF biomarkers were expressed in ratio to Aβ42. Relationships among sleep scores and CSF biomarkers were assessed with multiple regression, controlling for age, sex, time between sleep and CSF measurements, and CSF assay batch.
Results:
Worse subjective sleep quality, more sleep problems, and daytime somnolence were associated with greater AD pathology, indicated by lower CSF Aβ42/Aβ40 and higher t-tau/Aβ42, p-tau/Aβ42, MCP-1/Aβ42, and YKL-40/Aβ42. There were no significant associations between sleep and NFL or neurogranin.
Conclusions:
Self-report of poor sleep was associated with greater AD-related pathology in cognitively healthy adults at risk for AD. Effective strategies exist for improving sleep; therefore sleep health may be a tractable target for early intervention to attenuate AD pathogenesis.
To delay or prevent dementia due to Alzheimer disease (AD), it is critical to identify modifiable risk factors. Sleep quality is a promising target for intervention during the preclinical phase of AD, when pathogenesis has begun but cognition is still intact.1 Sleep is associated with AD brain pathophysiology, including amyloid mis-metabolism and deposition, tau hyperphosphorylation and aggregation, and neuronal and synaptic dysfunction and degeneration. We previously found that self-report of poor sleep was associated with greater brain amyloid burden, as measured by PET with Pittsburgh compound B (PiB).2 In young men, sleep deprivation diminished the diurnal fluctuation of CSF amyloid levels.3 In rodent models of AD, sleep restriction alters amyloid metabolism and tau phosphorylation,4–6 and tau-deficient mice show disturbed sleep architecture.7 Maintenance of axons8 and synapses occurs during sleep,9 and sleep loss elevates inflammation and microglial activation.10
Preclinical AD brain pathology can be detected via CSF biomarkers; however, little is known of their relationship with sleep. CSF β-amyloid 42 (Aβ42, a marker of amyloid deposition) may be associated with sleep-disordered breathing11 and sleep fragmentation.12 However, less is known about other markers of AD pathophysiology. Furthermore, CSF biomarkers combined in ratios have superior diagnostic and prognostic power compared to single biomarkers.13,14 Therefore we examined the relationship between self-reported sleep and CSF biomarkers of amyloid deposition and plaque formation (Aβ42), tau phosphorylation state and tau pathology (phosphorylated tau [p-tau]), axonal degeneration (total tau [t-tau], neurofilament light [NFL]), neuroinflammation (monocyte chemoattractant protein–1 [MCP-1], chitinase-3-like protein 1 [YKL-40]), and synaptic dysfunction/degeneration (neurogranin) in cognitively healthy adults in late middle age.
METHODS
Participants and study design.
Participants were drawn from a longitudinal cohort enriched with parental history of AD, the Wisconsin Registry for Alzheimer's Prevention (WRAP).15 Beginning in 2001, 1,500+ WRAP participants were recruited from the community via advertisements and word of mouth and were aged 40–65 years at study entry. Participants underwent extensive cognitive testing and medical history assessment at baseline, 4 years later, and every 2 years subsequently at the University of Wisconsin–Madison. Beginning in 2010, participants were sequentially recruited by letter and telephone into substudies that included CSF collection. Recruitment into CSF substudies was based on temporal proximity to recent or upcoming evaluations for the parent study (average interval was 0.84 years), as well as self-reported interest in substudies that were advertised at WRAP participant events. All WRAP participants were included in the present analysis who had assayed CSF samples, completed sleep questionnaires, and were cognitively normal. When CSF was available from multiple time points, the data from the CSF sample collected closest to the sleep questionnaire were used.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the University of Wisconsin–Madison Health Sciences Institutional Review Board and participants provided informed written consent.
CSF collection and quantification.
CSF was collected in the morning (mean 10:22 am ± 1 hour 12 minutes SD) following a 12-hour fast. Lumbar puncture was performed with a Sprotte 25- or 24-gauge spinal needle at L3/4 or L4/5, using gentle extraction into polypropylene syringes. Approximately 22 mL of CSF was gently mixed to remove gradient effects and centrifuged at 2,000 g for 10 minutes. A total of 0.5 mL aliquots of supernatants was frozen in polypropylene tubes and stored at −80°C. The samples were sent in 2 batches to the University of Gothenburg in Sweden to be assayed. For the Aβ42/Aβ40 ratio, CSF Aβ42 and Aβ40 were measured by electrochemiluminescence using an Aβ triplex assay (MSD Human Aβ peptide Ultra-Sensitive Kit, Meso Scale Discovery, Gaithersburg, MD). For all other ratios, CSF Aβ42 was quantified with a sandwich ELISA, as were t-tau and p-tau181 (INNOTEST β-amyloid1-42, hTAU-Ag, and phospho-tau[181P], respectively; Fujirebio Europe, Ghent, Belgium). Sandwich ELISAs were also used to assay CSF YKL-40 (R&D Systems, Minneapolis, MN) and NFL (NF-light ELISA kit, UmanDiagnostics AB, Umeå, Sweden). MCP-1 was measured using the Meso Scale Discovery technique (MSD Human MCP-1; Meso Scale Discovery). All assays were conducted by board-certified laboratory technicians blinded to participant clinical characteristics. Assays were completed in a single round of analyses using one batch of reagents, yielding intra-assay coefficients of variation below 10%.
CSF measures were selected for this analysis based on their ability to predict subsequent amyloid accumulation, as we have reported previously in an overlapping sample.13 For statistical analyses, Aβ42 was expressed in ratio to Aβ40 (the more abundant, nontoxic fragment), to assess the pathologic species (Aβ42) while accounting for individual differences in amyloid production. T-tau, p-tau181, NFL, MCP-1, YKL-40, and neurogranin were expressed in ratios to Aβ42 to reflect coincident pathologies and because these ratios have better diagnostic and prognostic power than each species expressed alone.13,14 CSF Aβ42 decreases as plaque burden increases, whereas the other CSF markers analyzed are elevated when pathology is greater. Therefore lower Aβ42 and Aβ42/Aβ40 indicate greater pathology, while ratios of other CSF markers to Aβ42 indicate greater pathology when they are elevated.13
Sleep assessment.
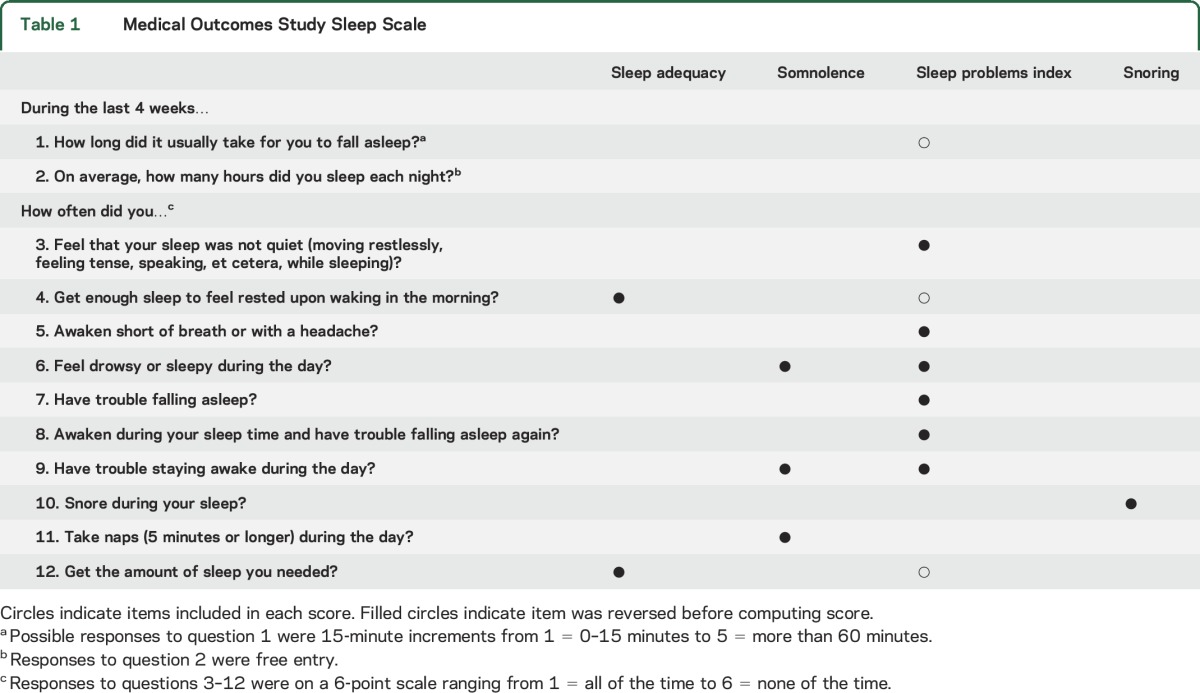
Sleep was assessed with the Medical Outcomes Study (MOS) Sleep Scale and the Epworth Sleepiness Scale (ESS). The MOS Sleep Scale was selected because it assesses multiple sleep domains with low participant burden and good internal consistency.16 It gives scores in 6 sleep domains, derived from 12 questions (table 1). Responses were elicited for the last 4 weeks using a 6-point scale, then converted to a 0–100 scale before being summed to give sleep scores. Primary analyses focused on scores we previously found to be associated with brain amyloid burden measured by PiB-PET: sleep adequacy, somnolence, and sleep problems index.2 Secondary analyses of symptoms of sleep-disordered breathing (snoring and waking short of breath) were conducted to better understand the relationship between sleep and CSF biomarkers. Higher scores indicate more of the construct being measured, such that worse sleep is reflected by lower sleep adequacy and higher somnolence and sleep problems index. The ESS was selected because it is a widely used tool in clinical sleep research for assessing daytime sleepiness and sleep propensity, with good internal validity and test–retest reliability.17 Participants use a 4-point scale to rate their likelihood of dozing off in 8 common situations, with higher scores indicating greater sleepiness (range 0–24).
Table 1.
Medical Outcomes Study Sleep Scale
APOE, family history, and cognitive assessment.
Participants were classified as carriers or noncarriers of one or more APOE ε4 alleles, determined by standard PCR techniques. Positive parental history of AD was defined as having one or both parents with AD as determined by a validated interview18 or autopsy-confirmed or probable AD as outlined by research criteria.19 Detailed medical history and phone interviews were conducted to confirm family history negative participants. Global cognitive function was assessed with the Mini-Mental State Examination,20 and learning and memory with the Rey Auditory Verbal Learning Test.21 Clinical diagnosis of cognitively normal was determined by consensus of a multidisciplinary panel, based on physical examination, medical and social history, neuropsychological testing, and self-reports and informant reports of cognitive and functional status.
Statistical analysis.
Separate multiple regression models were run for each combination of CSF and sleep measures, with CSF as the dependent variable and sleep as the independent variable, with covariates sex, age at CSF sample, CSF assay batch, and time between CSF sample and sleep assessment (all mean centered). Due to missing data, the number of participants included in each analysis ranged from 94 to 101. Ratios of CSF values to Aβ42 were log-transformed to achieve normal distributions. Casewise diagnostics did not reveal any outliers (±3 SD from the mean) that warranted removal, based on influence (Cook distance <0.039) or leverage (DFBETA <0.197). To account for multiple comparisons, a false discovery rate approach was used, which controls error while preserving power by adjusting the p value criterion for significance based on the number of tests performed.22 All analyses were conducted with IBM SPSS 23 (SPSS Inc., Chicago, IL).
RESULTS
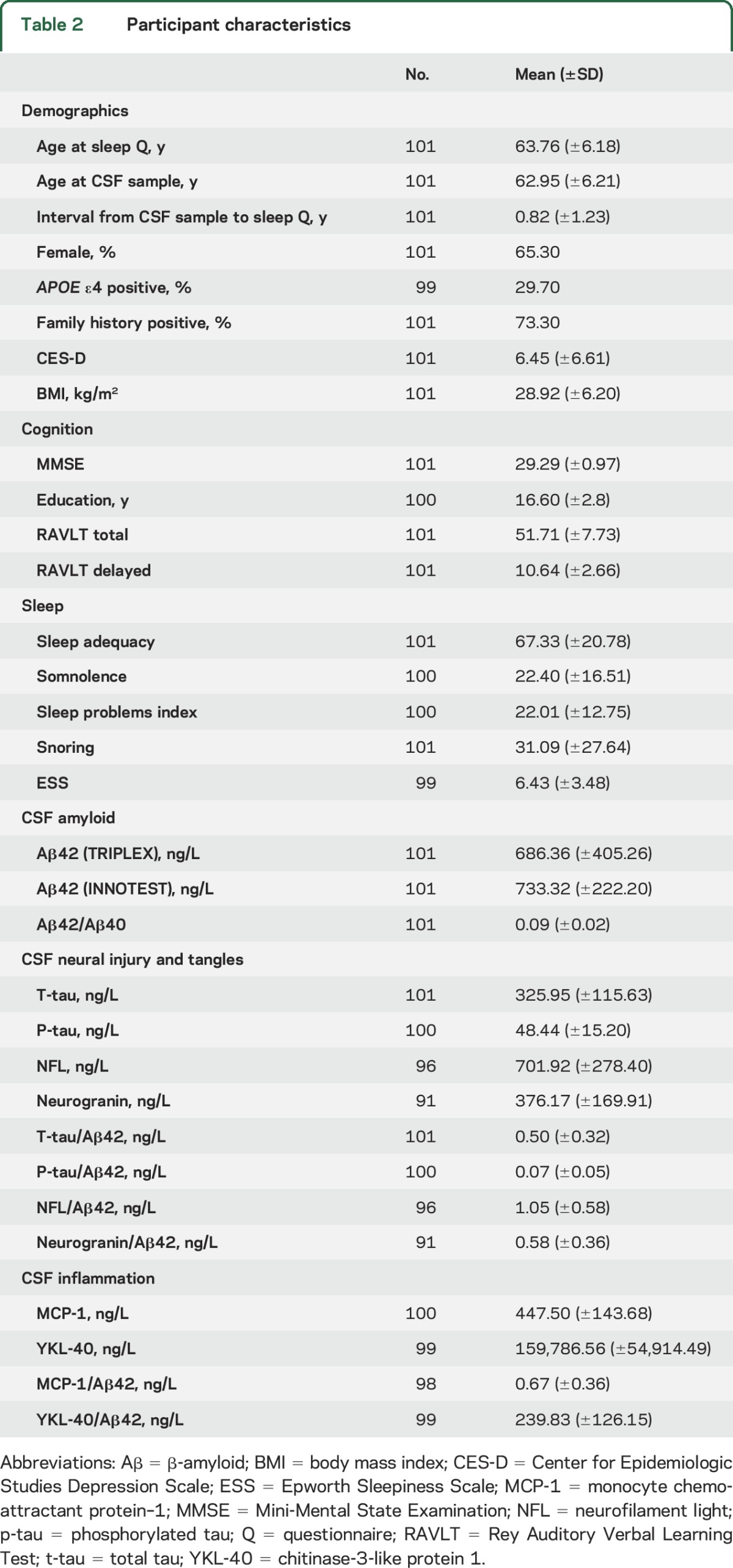
A total of 101 members of the WRAP cohort met criteria for inclusion (completed CSF assays, MOS Sleep Scale, and ESS). Participant characteristics are summarized in table 2. The sample was in late midlife (age 63.76 ± 6.18 years at time of sleep questionnaires), cognitively normal, and enriched with AD risk (73.3% with family history of AD, 29.7% APOE ε4+).
Table 2.
Participant characteristics
Regression results are summarized in table 3 and significant relationships between CSF biomarkers and sleep scores are plotted in the figure. Lower Aβ42/Aβ40 was associated with lower sleep adequacy (figure, A), indicating that less adequate sleep was associated with greater amyloid pathology. Aβ42/Aβ40 was not significantly associated with somnolence, sleep problems index, or ESS. Elevated NFL/Aβ42 was associated with greater somnolence (figure, B). There was no significant relationship between NFL/Aβ42 and the other sleep measures examined here. T-tau/Aβ42 and p-tau/Aβ42 were significantly higher with lower sleep adequacy and greater somnolence and sleep problems index (figure, C–H). Neither t-tau/Aβ42 nor p-tau/Aβ42 was related to ESS. Higher YKL-40/Aβ42 was associated with lower sleep adequacy and greater sleep problems index (figure, I and J). There was no significant relationship between YKL-40/Aβ42 and daytime sleepiness (somnolence or ESS). Higher MCP-1/Aβ42 was associated with greater somnolence (figure, K) but no other sleep measures. Neurogranin/Aβ42 was not associated with any of the sleep measures. Greater tau was associated with lower sleep adequacy. There were no other significant relationships between sleep and CSF biomarkers expressed alone (table e-1 at Neurology.org). There were no significant interactions with APOE ε4 status or age. There was no significant association between CSF biomarkers and self-report of sleep-disordered breathing symptoms or sleep duration. Depression, body mass index (BMI), education, cardiovascular disease, and sleep-affecting medications (table e-2) did not explain any additional variance in CSF biomarkers (table e-3), except for BMI with log(NFL/Aβ42). The relationship between somnolence and log(NFL/Aβ42) remained statistically significant and effect size (model R2) did not change substantially after controlling for BMI (supplemental data).
Table 3.
Results of multiple regression of CSF biomarkers on sleep measures, controlling for sex, age at CSF sample, CSF assay batch, and time between CSF sample and sleep assessment (all mean centered)
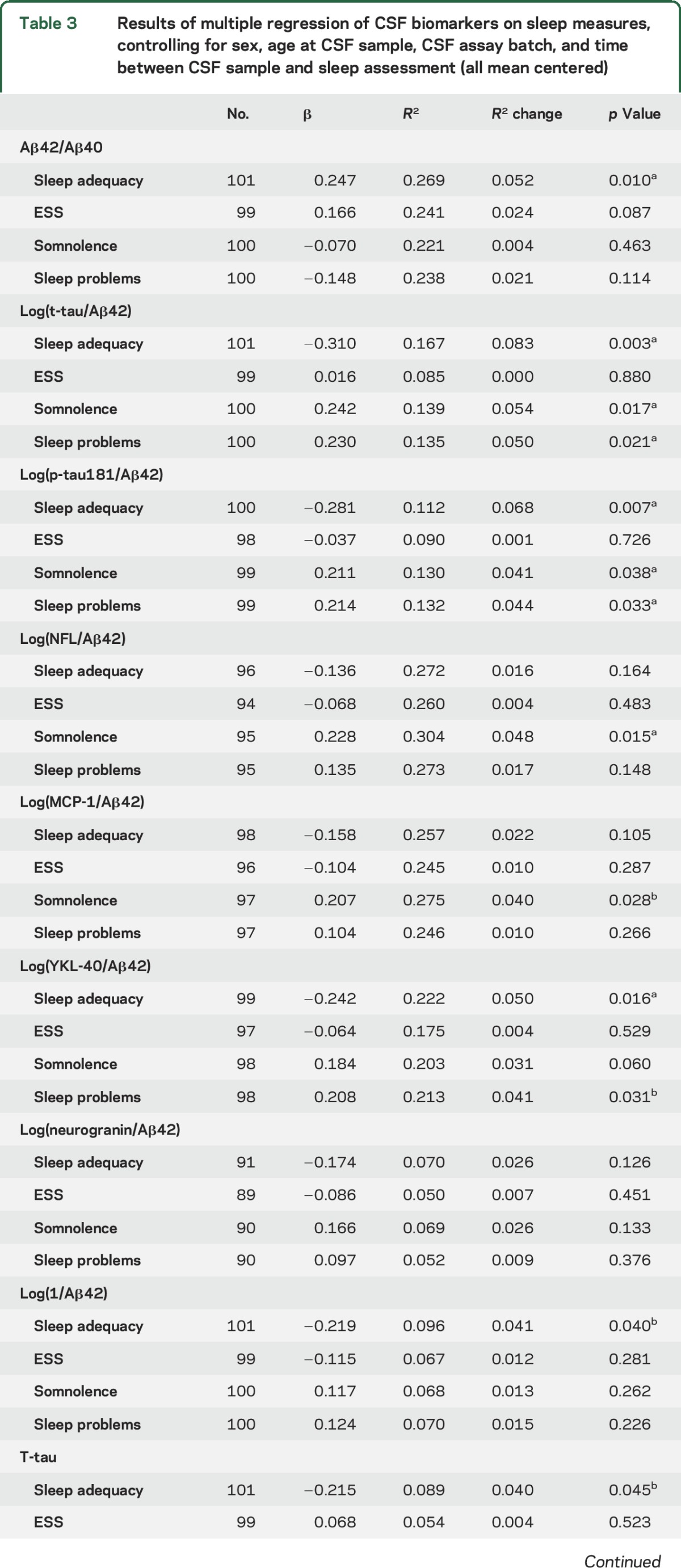
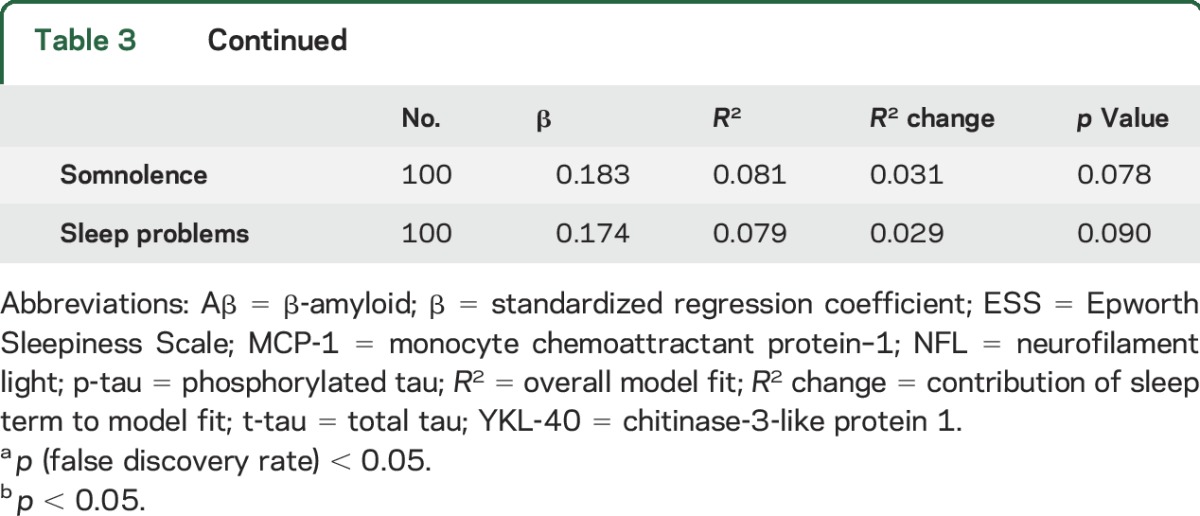
Figure. Association of CSF biomarkers with sleep scores.

(A–K) Regression results and 95% confidence intervals are plotted for the significant relationships between CSF biomarkers and sleep scores. CSF measures are adjusted for age, sex, CSF batch, and time between CSF sample and sleep questionnaire. Higher sleep scores indicate more of the construct being measured. Aβ = β-amyloid; MCP-1 = monocyte chemoattractant protein–1; NFL = neurofilament light; p-tau = phosphorylated tau; t-tau = total tau; YKL-40 = chitinase-3-like protein 1.
DISCUSSION
Self-report of less adequate sleep, greater daytime sleepiness, and more sleep problems were associated with CSF biomarkers of amyloid deposition in combination with tau pathology, axonal degeneration, and neuroinflammation.
We have previously shown that self-report of poor sleep was associated with greater brain amyloid burden, measured with PiB-PET.2 The CSF results reported here mirror the PiB-PET findings, and reveal further relationships between sleep and CSF biomarkers of cumulative AD pathology. CSF biomarkers give complimentary information to amyloid PET scans. First, abnormalities may become apparent in CSF before PET.23 Second, CSF can be collected and assayed using widely available methods, whereas PET remains more expensive and requires substantial infrastructure. Finally, whereas a PET scan assesses only a single form of pathology, CSF can be assayed for multiple pathologic species simultaneously.
CSF biomarker ratios indexing multiple pathologies capture the temporal relationship between pathologies, which is a better indicator of disease stage than absolute levels of individual pathologies because AD is a progressive and multifactorial neurodegenerative disease.1 Multipathology ratios have superior predictive power for development of neuropathologic features of AD13 and appear to provide superior diagnostic markers of AD.14 Our findings confirm their utility in the context of sleep, given that multipathology biomarkers were associated with more sleep domains than single-pathology markers. In particular, the only single-pathology markers associated with sleep were amyloid and tau, which were associated with lower sleep adequacy. Yet when expressed in ratio to each other, they were also associated with daytime somnolence and sleep problems. The value 1/Aβ42 was associated with sleep scores in a similar but not identical pattern to the ratios, confirming that the ratios capture information that is different from single markers and that our results were not driven by Aβ42 in the denominator. This is further supported by effect sizes, which were greater for CSF ratios than 1/Aβ42.
There is evidence that sleep affects AD pathology through multiple pathways. Sleep disturbance may promote amyloid plaque formation through increased amyloid production or reduced amyloid clearance. Orexin, an important regulator of the sleep–wake cycle, has been shown to drive production and deposition of Aβ in transgenic mice, possibly by promoting wakefulness.3 It has also been shown in mice that clearance of exogenous amyloid is greater during sleep, as a result of increased glymphatic flow.4 Thus sleep disruption could promote a buildup of soluble amyloid, leading to aggregation into plaques. These hypotheses remain to be tested in humans.
Interestingly, CSF biomarkers of AD pathology were associated with daytime sleepiness indexed by MOS somnolence but not ESS. We previously observed the same distinction with PET-measured amyloid burden.2 The difference could be due to the nature of the daytime sleepiness: ESS indexes irresistible sleepiness (falling asleep in inappropriate situations such as driving) whereas somnolence may index resistible sleepiness (feeling drowsy and deliberate napping). Orexin, a wake promoter, is elevated in patients with mild cognitive impairment and associated with tau pathology.24 It is possible that the sleep disturbance reported here in conjunction with resistible, but not irresistible, sleepiness reflects an abnormal upregulation of the orexin system.
YKL-40 and MCP-1 in CSF are indicative of neuroinflammation and astroglial activation associated with amyloid plaques, with diagnostic and prognostic utility for AD.25–27 Sleep restriction broadly promotes proinflammatory cytokines, possibly by disrupting diurnal fluctuations of growth and stress hormone release, and their modulation by slow-wave sleep.10 Both neurotoxic and neuroprotective roles have been proposed for YKL-40 and MCP-1 during early stages of amyloid plaque formation27 and further study is required to interpret their relationship with poor sleep.
Degeneration of synapses is an early feature of AD indexed by neurogranin, a postsynaptic protein with diagnostic utility for AD.28 Maintenance of membranes and synapses is upregulated during sleep8,9; therefore we hypothesized that sleep disturbance would increase synaptic injury. However, we found no relationship between neurogranin and sleep. Synaptic degeneration in AD may be secondary to amyloid and tau pathology29; therefore it may be that a relationship between synaptic injury and sleep is not yet detectable in this relatively young healthy cohort.
We did not find a relationship between CSF biomarkers and symptoms of obstructive sleep apnea (OSA). This lack of relationship is surprising, given consistent findings that OSA is a risk factor for dementia, possibly promoting AD pathogenesis via sleep fragmentation30 or hypoxia.31 It is possible, however, that OSA impedes the transfer of proteins into the CSF, altering the relationship between CSF concentrations and CNS pathology in patients with OSA.11 Alternatively, OSA severity in this sample may have been too low to detect a relationship with CSF, as on average participants rated their snoring frequency as low. Sleep-disordered breathing often goes undetected by patients, and subjective reports are not reliable in detecting sleep apnea. Neither snoring nor waking short of breath was significantly correlated with sleep adequacy, somnolence, ESS, or each other. This fits with literature showing that snoring and daytime sleepiness add distinct information to predict SDB.32 Given that we did not directly test for OSA, further studies measuring sleep and breathing are needed to understand the contribution of sleep-disordered breathing to the relationship between poor sleep and CSF biomarkers of AD.
Sleep disturbances may have an effect on AD pathology, but on the other hand, AD pathology may affect sleep quality. Animal models show that increasing amyloid plaque burden is accompanied by increasingly fragmented sleep, which is rescued by elimination of Aβ deposits in the mouse brain by active immunization.33 In humans, there is evidence that amyloid plaques disrupt slow-wave sleep, impairing memory consolidation.34 Slow-wave sleep is important for feeling refreshed35; therefore our finding that perceptions of inadequate sleep were related to more AD pathology could reflect an inability to obtain sufficient slow-wave sleep as a result of amyloid plaques or possibly elevated presence of oligomeric forms of Aβ.
Limitations of this study include the cross-sectional design and use of subjective sleep measures. There is evidence of bidirectional relationships between sleep and amyloid3,33 that cannot be disentangled by this cross-sectional study design. Longitudinal follow-up within individuals will be important to determine the course of sleep and brain pathology in preclinical AD. Variability in CSF collection time may have contributed some unaccounted for variance in amyloid levels, and mean collection time was 10:22 am, close to a 10 am nadir in CSF amyloid reported by one study.36 However, the diurnal fluctuation was smaller in older adults, and we found no correlation between CSF collection time and amyloid levels.
This study measured sleep through self-report. Objective sleep measures such as actigraphy and polysomnography would clarify the contribution of sleeping brain activity, breathing, and sleep-wake rhythmicity to AD pathogenesis. However, the investigation of self-reported sleep quality is itself an important clinical question. There is substantial interindividual variability in the effect of sleep disturbance on cognitive and physiologic functioning.37 Furthermore, sleep health is multidimensional,38 and current objective techniques (e.g., polysomnography and actigraphy) do not fully capture “adequate” sleep.39 Thus self-report provides important complementary information on functional outcomes and perceptions.
These results add to a growing body of evidence linking sleep quality with AD neuropathology. Our findings demonstrate that the relationship between sleep and AD pathology is present in late midlife in the absence of cognitive impairment. Furthermore, we show that the relationship is detectable using self-report and CSF biomarkers, tools that are relatively inexpensive and accessible, making them appealing for further research in large cohorts and clinical trials. ESS is a simple and widely used measure of sleep dysfunction in clinical sleep research. Given the lack of associations shown here with CSF, and previously with PET biomarkers of AD,2 we recommend that other sleep measures be included in studies of the relationship between sleep and AD. Findings were not altered by race, education, depressive symptoms, BMI, or use of sleep medications.
Sleep may be a modifiable risk factor for AD during the earliest stages of the disease, before dementia symptoms appear. Although the effect sizes reported here are modest, it is estimated that delaying AD onset by a mere 5 years would reduce AD cases by 5.7 million and save $367 billion in health care spending in the United States.40 Many effective pharmaceuticals, devices, and behavioral interventions are already available in the clinic for improving sleep quality. Follow-up studies are needed to identify the aspects of sleep that are most amenable to modification and most effective in affecting AD pathology, to ultimately delay AD or diminish AD symptoms.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Caitlin A. Cleary, BSc, Sandra Harding, MS, Nancy Davenport-Sis, BSc, LeAnn DeRungs, NP, and the WRAP study team for assistance with data collection; and the participants in the Wisconsin Registry for Alzheimer's Prevention for their participation.
GLOSSARY
- Aβ42
β-amyloid 42
- AD
Alzheimer disease
- BMI
body mass index
- ESS
Epworth Sleepiness Scale
- MCP-1
monocyte chemoattractant protein–1
- MOS
Medical Outcomes Study
- NFL
neurofilament light
- OSA
obstructive sleep apnea
- p-tau
phosphorylated tau
- PiB
Pittsburgh compound B
- t-tau
total tau
- WRAP
Wisconsin Registry for Alzheimer's Prevention
- YKL-40
chitinase-3-like protein 1
Footnotes
Supplemental data at Neurology.org
Editorial, page 419
AUTHOR CONTRIBUTIONS
Kate Sprecher: design of the study, analysis and interpretation of the data, drafting and revising the manuscript. Rebecca L. Koscik: design of the study, interpretation of the data, revising the manuscript. Cynthia M. Carlsson: interpretation of the data, revising the manuscript. Henrik Zetterberg: analysis and interpretation of the data, revising the manuscript. Kaj Blennow: analysis and interpretation of the data, revising the manuscript. Ozioma C. Okonkwo: interpretation of the data, revising the manuscript. Mark A. Sager: design of the study, revising the manuscript. Sanjay Asthana: revising the manuscript. Sterling C. Johnson: interpretation of the data, revising the manuscript. Ruth M. Benca: design of the study, interpretation of the data, revising the manuscript. Barbara B. Bendlin: design of the study, interpretation of the data, revising the manuscript.
STUDY FUNDING
This research was funded by grants R01 AG027161, R01 AG021155, ADRC P50 AG033514, R01 AG037639, and National Research Service Award F31 AG048732 from the National Institute on Aging, and by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR000427.
DISCLOSURE
K. Sprecher, R. Koscik, and C. Carlsson report no disclosures relevant to the manuscript. H. Zetterberg is co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. K. Blennow served as a consultant or at advisory boards for Eli Lilly, Fujirebio Europe, IBL International, Novartis, and Roche Diagnostics, and is co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. O. Okonkwo, M. Sager, S. Asthana, and S. Johnson report no disclosures relevant to the manuscript. R. Benca has served as a consultant to Merck, Janssen, and Jazz, and receives grant support from Merck. B. Bendlin reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sprecher KE, Bendlin BB, Racine AM, et al. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol Aging 2015;36:2568–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol 2014;71:971–977. [DOI] [PubMed] [Google Scholar]
- 4.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science 2013;342:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Meco A, Joshi YB, Praticò D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer's disease with plaques and tangles. Neurobiol Aging 2014;35:1813–1820. [DOI] [PubMed] [Google Scholar]
- 6.Kang J-E, Lim MM, Bateman RJ, et al. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 2009;326:1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantero JL, Hita-Yañez E, Moreno-Lopez B, Portillo F, Rubio A, Avila J. Tau protein role in sleep-wake cycle. J Alzheimers Dis 2010;21:411–421. [DOI] [PubMed] [Google Scholar]
- 8.Cirelli C. The genetic and molecular regulation of sleep: from fruit flies to humans. Nat Rev Neurosci 2009;10:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maret S, Faraguna U, Nelson AB, Cirelli C, Tononi G. Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nat Neurosci 2011;14:1418–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faraut B, Boudjeltia KZ, Vanhamme L, Kerkhofs M. Immune, inflammatory and cardiovascular consequences of sleep restriction and recovery. Sleep Med Rev 2012;16:137–149. [DOI] [PubMed] [Google Scholar]
- 11.Ju YES, Finn MB, Sutphen CL, et al. Obstructive sleep apnea decreases central nervous system-derived proteins in the cerebrospinal fluid. Ann Neurol 2016;80:154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ju Y-ES, McLeland JS, Toedebusch CD, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol 2013;70:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Racine AM, Koscik RL, Nicholas CR, et al. Cerebrospinal fluid ratios with Aβ42 predict preclinical brain β-amyloid accumulation. Alzheimers Dement 2016;2:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janelidze S, Zetterberg H, Mattsson N, et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol 2016;3:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer's disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer's Prevention. J Geriatr Psychiatry Neurol 2005;18:245–249. [DOI] [PubMed] [Google Scholar]
- 16.Viala-Danten M, Martin S, Guillemin I, Hays RD. Evaluation of the reliability and validity of the Medical Outcomes Study sleep scale in patients with painful diabetic peripheral neuropathy during an international clinical trial. Health Qual Life Outcomes 2008;6:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johns MW. Reliability and factor analysis of the Epworth Sleepiness Scale. Sleep 1992;15:376–381. [DOI] [PubMed] [Google Scholar]
- 18.Kawas C, Segal J, Stewart WF, Corrada M, Thal LJ. A validation study of the Dementia Questionnaire. Arch Neurol 1994;51:901–906. [DOI] [PubMed] [Google Scholar]
- 19.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt M. Rey Auditory Verbal Learning Test: A Handbook. Los Angeles: Western Psychological Services; 1996. [Google Scholar]
- 22.Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol 2000;279:R1–R8. [DOI] [PubMed] [Google Scholar]
- 23.Palmqvist S, Mattsson N, Hansson O. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain 2016;139:1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liguori C, Nuccetelli M, Izzi F, et al. Rapid eye movement sleep disruption and sleep fragmentation are associated with increased orexin-A cerebrospinal-fluid levels in mild cognitive impairment due to Alzheimer's disease. Neurobiol Aging 2016;40:120–126. [DOI] [PubMed] [Google Scholar]
- 25.Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS One 2012;7:e30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Craig-Schapiro R, Perrin RJ, Roe CM, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry 2010;68:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer's disease. Biomark Med 2012;6:455–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tarawneh R, D'Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol 2016;73:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorostkar MM, Zou C, Blazquez-Llorca L, Herms J. Analyzing dendritic spine pathology in Alzheimer's disease: problems and opportunities. Acta Neuropathol 2015;130:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaffe K, Laffan AM, Harrison SL, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 2011;306:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiota S, Takekawa H, Matsumoto SE, et al. Chronic intermittent hypoxia/reoxygenation facilitate amyloid-β generation in mice. J Alzheimers Dis 2013;37:325–333. [DOI] [PubMed] [Google Scholar]
- 32.Netzer NC, Stoohs RA, Netzer CM, Clark K, Strohl KP. Using the Berlin Questionnaire to identify patients at risk for the sleep apnea syndrome. Ann Intern Med 1999;131:485–491. [DOI] [PubMed] [Google Scholar]
- 33.Roh JH, Huang Y, Bero AW, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med 2012;4:150ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mander BA, Marks SM, Vogel JW, et al. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci 2015;18:1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh JK. Enhancement of slow wave sleep: implications for insomnia. J Clin Sleep Med 2009;5:S27–S32. [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y, Potter R, Sigurdson W, et al. Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch Neurol 2012;69:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Dongen HPA, Baynard MD, Maislin G, Dinges DF. Systematic interindividual differences in neurobehavioral impairment from sleep loss: evidence of trait-like differential vulnerability. Sleep 2004;27:423–433. [PubMed] [Google Scholar]
- 38.Buysse DJ. Sleep health: can we define it? Does it matter? Sleep 2014;37:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krystal AD, Edinger JD. Measuring sleep quality. Sleep Med 2008;9(suppl 1):S10–S17. [DOI] [PubMed] [Google Scholar]
- 40.Alzheimer's Association. Changing the Trajectory of Alzheimer's Disease: How a Treatment by 2025 Saves Lives and Dollars. Chicago: Alzheimer's Association; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.