

SUMMARY
Global transcriptomic imbalance is a ubiquitous feature associated with cancer, including hepatocellular carcinoma (HCC). Analyses of 1,225 clinical HCC samples revealed that a large numbers of RNA binding proteins (RBPs) are dysregulated and that RBP dysregulation is associated with poor prognosis. We further identified that oncogenic activation of a top candidate RBP, negative elongation factor E (NELFE), via somatic copy number alterations enhanced MYC signaling and promoted HCC progression. Interestingly, NELFE induces a unique tumor transcriptome by selectively regulating MYC-associated genes. Thus, our results revealed NELFE as an oncogenic protein that may contribute to transcriptome imbalance in HCC through the regulation of MYC signaling.
Graphical abstract
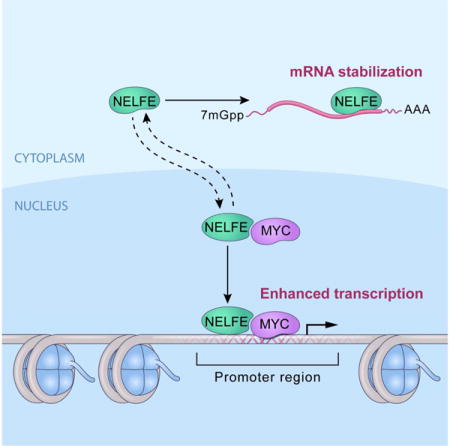
INTRODUCTION
Cancer development centers on the concept that cancer cells acquire multiple cellular properties known as hallmarks of cancer via genetic and epigenetic –mechanisms (Hanahan and Weinberg, 2011). A reasonably small number of oncogenes and tumor suppressor genes are believed to be required for the maintenance of malignant features. However, one feature observed in many cancer transcriptomic studies, e.g. the Cancer Genome Atlas (TCGA), is that cancer cells, including hepatocellular carcinoma (HCC) cells, often contain alterations of thousands of seemingly unrelated coding and non-coding RNA transcripts. These cancer-associated transcriptomes may represent fitness traits in tumor evolution as they have been demonstrated to be stable across different datasets (Lee et al., 2006; Roessler et al., 2010; Teufel et al., 2012; Wang et al., 2012). Whether this trait is acquired stochastically or via a specific mechanism is still unresolved. It is possible that RNA binding proteins (RBPs), because of their ability to regulate the abundances and functions of RNA transcripts at multiple levels (including transcription, RNA localization, biogenesis, RNA stability and translation (Kechavarzi and Janga, 2014)), may contribute to these oncogenic fitness traits.
Given the importance of RBPs in many cellular processes, defects in their functions in cancer are unsurprising. In fact, RBP dysregulation has been linked to several human diseases, including muscular atrophies, neurological disorders, and cancer (Castello et al., 2013; Chen et al., 2013; Kim et al., 2013). Currently, more than 1,500 RBPs have been curated and more than 800 mRNA RBPs have been identified (Castello et al., 2012; Gerstberger et al., 2014; Kechavarzi and Janga, 2014; Lukong et al., 2008). An interesting hypothesis is that dysregulation of members of the RBP community collectively contribute to the transcriptomic imbalance in tumor cells and thus drive tumorigenicity, including HCC. While several studies indicate that RBPs are important in regulating gene expression in cellular development, homeostasis and disease states, how and to what extent RBPs modulate the cancer transcriptome is largely unexplored.
HCC represents the second most common cause of cancer-related deaths worldwide (Theise, 2014) and is on the rise in the United States (El-Serag, 2011). HCC is an aggressive tumor type with poor prognosis due to the diverse etiological factors implicated during tumor development, heterogeneity of the tumor, and the late stage at which HCC is generally diagnosed. Despite many potential therapeutic targets, the overall survival is poor (Theise, 2014). Like other solid tumors, one of the genomic hallmarks of HCC is global dysregulation of the transcriptome of both coding and non-coding RNAs (Lee et al., 2006; Roessler et al., 2010; Teufel et al., 2012; Wang et al., 2012). In fact, the tumor specific transcriptome of HCC is associated with clinical characteristics, suggesting that changes in the transcriptome drive tumorigenesis (Boyault et al., 2007; Lee et al., 2006). These observations led us to hypothesize that RBPs are key mediators of oncogenic transcriptomic changes in HCC.
RESULTS
Global alterations of RNA binding proteins in HCC
To assess the role of mRNA binding proteins (mRBPs) in HCC, we analyzed tumor-associated transcriptome and somatic copy number alterations (SCNA) of more than 1,200 clinical samples (Figure 1A). We first determined global gene expression patterns of all known mRBPs in 241 matched pair of HCC and non-tumor tissues microarray dataset in the well-established LCI datasets. From a total of 13,101 genes analyzed, we identified 8,608 differentially expressed genes, including 526 out of 672 RBPs (78%) between tumor and paired non-tumor tissues (paired t-test, p<0.001). Among these 526 RBPs, approximately 86% of RBPs were preferentially upregulated in HCC (452/526, one-sided Fisher’s exact test, p=2.2×10−16) compared to non-tumor (Table S1). Using compound covariate predictor, nearest neighbor, and support vector machine algorithms, we identified 474 RBPs that are differentially expressed and predicted tumor and non-tumor classes with more than 93% accuracy (Permutation p<0.001, Tables S1). We next performed the same multivariate class prediction analysis described above with TCGA-LIHC datasets of 418 samples. The 474 RBP gene classifier significantly discriminated tumor from non-tumor samples in the TCGA-LIHC dataset with at least 91% accuracy (Table S1). Hierarchal clustering analysis revealed that 474 RBPs separate HCC from non-tumor in both TCGA-LIHC and Stanford datasets (Chen et al., 2002) (Figure 1B and Figure S1A), suggesting that these RBPs are dysregulated in a diverse group of HCCs. We found that global expression of 474 RBPs is significantly associated with patient prognosis in both the LCI and LEC datasets (Figure 1C). In contrast, global expression patterns of transcription factors (TFs) (629/1558) are not associated with HCC prognosis (Figure S1B).
Figure 1.
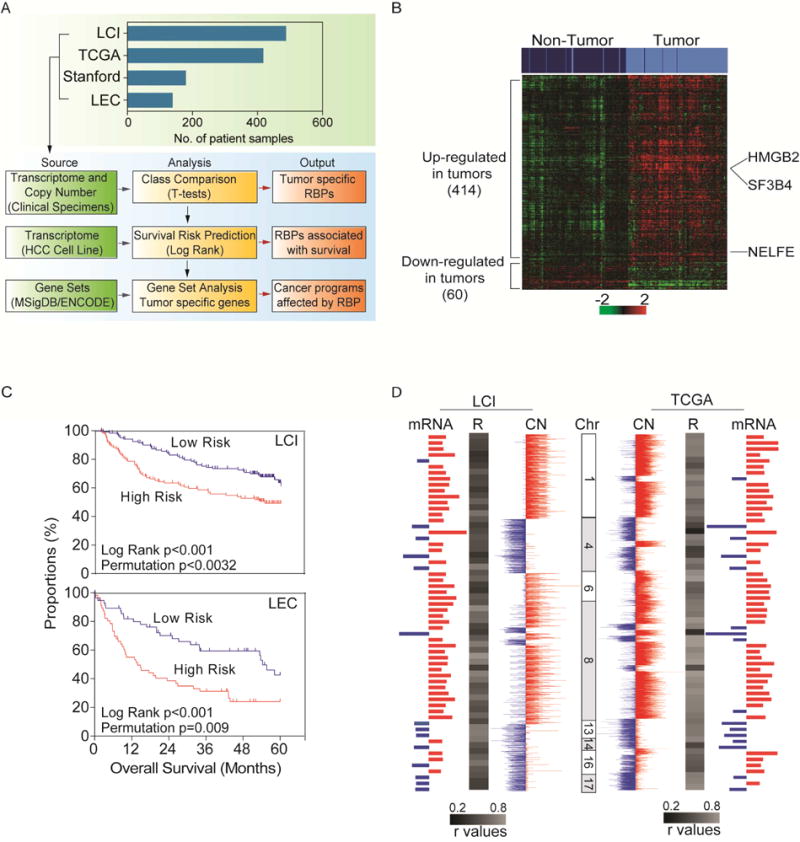
Alterations of RNA binding proteins in hepatocellular carcinoma (HCC). (A) Schematic overview of the study design. Clinical HCC microarray were used to determine differentially expressed RBPs in the LCI datasets (n=488) and validated in the TCGA-LIHC (n=418), Stanford (n=180) and LEC (n=139) datasets using class comparison and survival prediction analysis. (B) Hierarchical clustering and heatmap of LCI datasets of 474 RBPs. Top bars represent sample clustering. Dark blue is non-tumor and light blue is tumor. (C) Kaplan-Meier survival analysis of LCI (High n=120, Low n=121) and LEC (High n=56, Low n=57) datasets based on predictive survival analysis using the 474 RBP gene signatures by gene expression. (D) Integrative analysis of arrayCGH and gene expression microarray of the LCI dataset (on left) and SNParray data and RNASeq TCGA datasets (right). Red represents increase in somatic copy number (SCN) or elevated gene expression in tumors compared to non-tumors. Blue represents loss of SCN or decreased gene expression in tumors compared to non-tumors. See also Figure S1 and Table S1 and S2.
Among the 474 dysregulated RBPs, 414 RBPs (87%) were preferentially upregulated in HCC compared to non-tumor, indicating that there is a preferential enrichment for activated RBPs in HCC (one-sided Fisher’s exact test, p<0.001, Figure 1B). In order to search for potential HCC drivers, we performed integrative analysis of 64 HCC cases from the LCI dataset that contains matched transcriptome and SCNA (Roessler et al., 2012). Integrative global analysis of 13,101 genes yielded 975 whose expression was positively correlated with SCNA (Pearson Correlation ≥ 0.2, p<0.05). Among these correlated genes, 88 were RBPs with a mean Pearson coefficient of 0.43 (95% [CI]: 0.40–0.46) and 60 were significantly differentially expressed RBPs (Table S2). Of the 60 differentially expressed RBPs, there was preferential enrichment for RBPs with elevated gene expression and increased SCNAs (one-sided Fisher’s Exact test, p<0.001) (Figure 1D, Table S2). Consistent data were obtained in the TCGA-LIHC data set (Mean Pearson coefficient: 0.5869 and 95% [CI]: 0.5425–0.6312) (Figure 1D). In contrast, TFs were not significantly enriched (one-sided Fisher’s exact test, p≥0.05) in both the LCI and TCGA-LIHC datasets (Table S2), suggesting that RBPs are preferentially selected in HCC. Taken together, these data reveal that RBP dysregulation is functionally linked to HCC biology and severity.
NELFE is important for the progression of hepatocellular carcinoma
In HCC, the top three RBP differentially expressed between tumor and non-tumor were HMGB2, SF3B4 and NELFE (Table S2). HMGB2 is a high mobility group box protein while SF3B4 is splicing factor 3b, subunit 4 and NELFE, also known as RDBP, encodes negative elongation factor E. Moreover, high NELFE levels have been demonstrated to be associated with HCC (Iida et al., 2012). We selected NELFE for further analysis due to its elevated gene expression in HCC, high correlation between mRNA and copy number with a low FDR in both the TCGA and LCI datasets (Figure S1C–D and Table S2). We validated the LCI dataset using quantitative polymerase chain reaction (q-PCR) of the NELFE gene that showed high correlation with arrayCGH data in the 64 HCC samples (Figure S1E).
To investigate whether NELFE is associated with overall survival, we analyzed the SCNA data of 76 HCC samples using log 2>0.2 as a cutoff and mRNA levels (1/3 vs 2/3) to increase the statistical power. In both SCNA and mRNA analyses, we found that NELFE was associated with overall survival in three independent datasets either significantly or displaying a trend that did not achieve statistical significance (Figure 2A, Figure S2A). To determine if NELFE copy number was associated with HCC cell growth, we performed q-PCR using 10 HCC cell lines and the telomerase immortalized normal human hepatocyte line HHT4 (Jiang et al., 2010), which exhibits a near diploid karyotype and expresses many hepatocyte-specific genes. Notably, HHT4 cells have one NELFE copy (Figure S2B), consistent with previous karyotyping analysis showing a loss of one copy of chromosome 6 (Jiang et al., 2010). We found that some HCC cell lines showed an increased NELFE somatic copy number gain compared to normal diploid cells (male gDNA) and an abundant expression of the NELFE protein (Figure S2B–C). Tumor cells with an increased NELFE had increased cell proliferation (Figure S2D). Thus, the pathophysiological levels of NELFE were associated with HCC cell growth.
Figure 2.
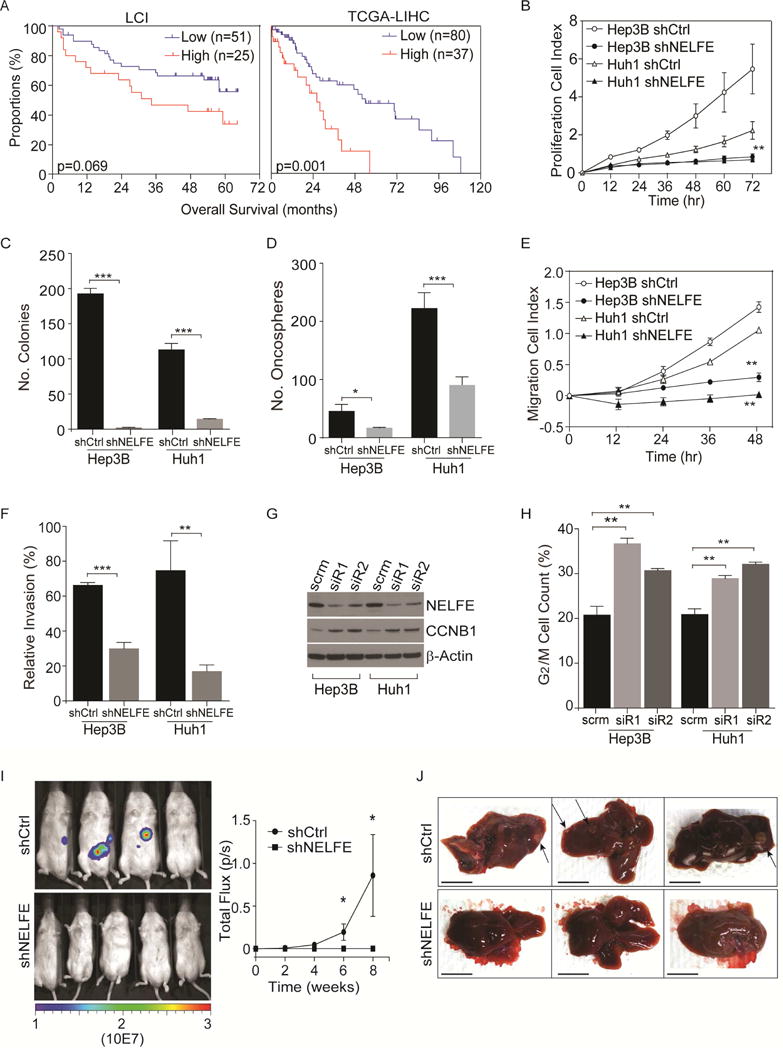
Role of the RNA binding protein NELFE in hepatocellular carcinoma cells. (A) Kaplan-Meier survival analysis of LCI and TCGA-LIHC datasets based on segmentation values of NELFE (high: log2 >0.2, low: log2<0.2). (B–H) Cell proliferation rates measured by xCELLigence (B), colony formation (C), oncosphere formation measured by Algimatrix 3D assay (D), cell migration (E), cell invasion (F), cropped immunoblot (G), and proportions of cells in during G2/M phase measured by 7′AAD staining using flow cytometry (H) of Hep3B and Huh1 HCC cells after shNELFE or shCtrl via lentivirus. Statistical significance for the proliferation rate (B) is measured at time-point 72 hr, results shown in (E–H) were measured at 48 hr. For invasion assay (with matrigel), relative invasion index is calculated by normalizing to cells that have migrated (no matrigel). (I) Bioluminescence of NOD/SCID mice at eight weeks (middle panel). On the right panel, mean signal from shCtrl (n=4) or shNELFE (n=6). (J) Representative livers of three mice in each group. Arrows are pointing at tumor nodules. Scale bars, 1 cm. *p<0.05, **p<0.01, ***p<0.001. All data are mean ± SD. See also Figure S2.
To further analyze the relevance of NELFE in HCC, we knocked down NELFE in Hep3B, Huh1 and SMMC-7721 HCC cell lines using a lentivirus expressing NELFE shRNA (shNELFE) and performed cell proliferation, colony- and oncospheroid formation assays compared to vector control (shCtrl). We used these cells for knockdown studies due to their higher copy number relative to male gDNA and increased proliferative rates (Figures S2B, S2D). The knockdown of NELFE reduced the protein levels of NELFE (Figure S2E), decreased cell proliferation as measured over time by xCELLigence compared to shCtrl (Figure 2B, Figure S2F) in these HCC cell lines, and decreased the number of colonies significantly (Figure 2C, Figure S2G). Additionally, we performed recently developed oncospheroid assay (Takai et al., 2016) and cell migration and invasion assays. HCC cells transduced with NELFE shRNA lentivirus had a significant reduction of oncosphere formation (Figure 2D and Figure S2H) and lost their ability to migrate or invade the matrigel (Figures 2E–F and Figure S2I–J). No significant effect after NELFE inhibition on apoptosis was found using Annexin-V flow cytometry analysis and Caspase 3/7 fluorometric assay on both Hep3B and Huh1 cells compared to control (Figures S2K–L). Interestingly, NELFE knockdown increased Cyclin B1 protein expression (Figure 2G), which is associated with G2/M arrest. Indeed, the percentage of G2/M cells was significantly higher in HCC cells transfected with NELFE siRNA compared to scrambled control (scrm) as determined by FACS (Figure 2H).
To determine the effect of NELFE on HCC tumorigenicity in vivo, we orthotopically injected Huh1-Luc cells (stably expressing luciferase) cells that have been transduced with shNELFE or shCtrl into the livers of NOD/SCID mice (Figure S2M). At weeks eight, luciferase bioluminescence demonstrated that the knockdown of NELFE suppressed orthotopic tumor formation in mice (Figure 2I). Further examination revealed that the shCtrl group had large tumor nodules detectable in the mouse liver, whereas there were no visible nodules in the shNELFE group (Figure 2J). To determine whether the tumors are of Huh1 origin, we immunohistochemically analyzed mouse livers using an antibody specific to human HLA-A. Accordingly, shCtrl mice liver showed high levels of HLA-A staining in the tumor but not the surrounding liver tissues. In contrast, in the shNELFE group, HLA-A staining was not evident, suggesting NELFE knockdown reduced colonization of HCC cells in vivo (Figure S2N). Similar results regarding the effect of NELFE on HCC tumorigenicity were also observed in Hep3B cells using a subcutaneous model (Figure S2M, S2O). Overexpression of NELFE in HCC cells with two copies of NELFE (Figure S2P) resulted in enhanced cell proliferation, colony formation, oncosphere formation and cell migration in both Huh7 and MHCC97 cells (Figure S2Q–T). Taken together, the results presented here suggest that NELFE plays a significant role in HCC progression and tumorigenicity.
NELFE preferentially enhances MYC signaling
To further analyze the role of NELFE in HCC, we analyzed differentially expressed transcripts in Hep3B cells comparing siRNA-mediated NELFE knockdown and a scrm siRNA using HTA 2.0 arrays (~40K probe sets consisting of multiple species of RNAs including mRNAs, lncRNAs, and miRNAs) (Figure 3A). We identified 3,070 transcripts that were significantly altered upon NELFE knockdown and, of the more than nine different categories of RNAs represented on the array, mRNAs were most significantly affected (Figure 3B, one-sided Fisher’s exact test, p=4.49×10−165). This is consistent with previous results indicating that NELFE primarily binds mRNAs (Castello et al., 2012). Interestingly, NELFE knockdown also has an effect on lncRNAs and snoRNAs, suggesting that NELFE regulates multiple RNA species.
Figure 3.
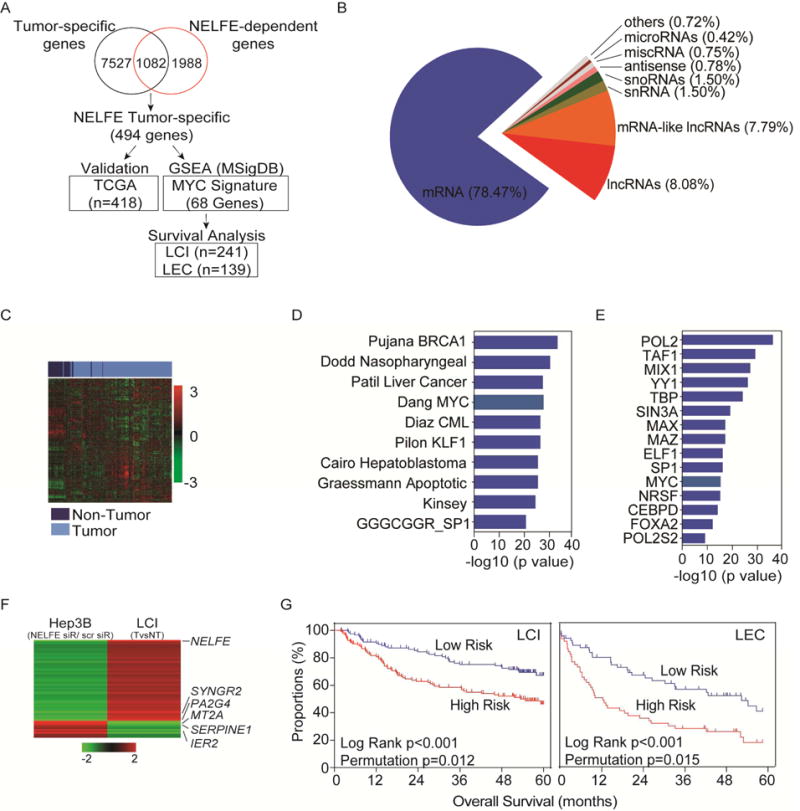
NELFE enhances MYC signaling in HCC. (A) Schematic of overview of microarray analysis to identify correlative genes that are both differentially expressed in clinical samples and in Hep3B cells. (B) Differentially expressed genes were used to identify RNA species affected by NELFE siRNA in Hep3B HCC cells. (C) Hierarchical clustering analysis of the TCGA-LIHC dataset using 494 NELFE-dependent gene list. (D) GSEA analysis of 494 NELFE-dependent genes showing the top 10 most enriched genesets. (E) ENCODE analysis of 494 NELFE-dependent genes showing the top 15 most enriched proteins. 343/494 genes are MYC-related genes within 500 bp from the transcription start site. (F) Heatmap of 68 NELFE-dependent MYC targets (RDMTs) in the LCI datasets and Hep3B siRNA-mediated knockdown of NELFE. Data represents fold change between tumor vs non-tumor for the LCI datasets. For Hep3B, data is fold change between NELFE siRNA vs scrm. (G) Kaplan-Meier curve using the 68 NELFE-dependent MYC-related genes. Predictive survival analysis was performed on the LCI or the LEC datasets with log rank and permutation p values. See also Figure S3 and Table S3.
Among the 3,070 NELFE-dependent transcripts, 78.5% were mRNAs (Figure 3B), 1,082 of which were differentially expressed between HCC and matched non-tumor samples in the LCI dataset (Figure 3A). While the remaining 1,988 transcripts were considered to be affected by NELFE knockdown in vitro, they did not overlap with the differentially expressed genes identified using the LCI dataset and were not used for further analysis. When we compared the distribution of fold change differences in both gene lists, there were a similar degree of changes between overlapping and non-overlapping genes (i.e., an average of 1.4 fold change), indicating that genes that changed in HCC were similarily affected by NELFE knockdown. Of the 1,082 NELFE-dependent mRNAs, 494 show concordant expression upon NELFE knockdown in HCC cells and in HCC samples with a reciprocal expression pattern (i.e., up-regulated in HCC tissues but down-regulated in HCC cells treated with NELFE siRNA or vice versa) (Table S3). We reasoned that by performing such a stringent filter, we may increase the likelihood of identifying NELFE associated genes that are physiologically relevant, accurately represent the true tumor biology, and act as key drivers of HCC. Indeed, these 494 NELFE-dependent genes could accurately discriminate tumor from non-tumor with more than 91% accuracy in the TCGA-LIHC dataset (Figure 3C, Table S3), confirming the clinical relevance of NELFE-dependent genes that are HCC-associated.
To further analyze the functional importance of the 494 NELFE-dependent gene set, we performed gene enrichment analysis using the Molecular Signatures Database. We found an enrichment for liver cancer associated signatures and the Dang_Bound_by_Myc (68/494) signature, a list made up of 1,103 MYC responsive genes according to the MYC Target Gene Database (MTGD) (Figure 3D and Table S3). We further identified genes that are bound by MYC in more than 90 cell lines but may not be represented on the MTGD using the ENCODE ChIP-Seq Significance Tool (Auerbach et al., 2013). Surprisingly, more than 70% (343/494) of the 494 genes were MYC-associated genes (Figure 3E, q-value=6.46×10−29). Furthermore, when analyzing Huh1 cells treated with NELFE siRNA microarray data, we also found that NELFE-dependent mRNAs were enriched for MYC signaling (Figure S3A–D).
Since siRNA mediated NELFE knockdown affects a significant number of MYC-related genes, we hypothesized that NELFE may affect MYC directly. We first investigated the relationship between NELFE and MYC in the HCC clinical samples. We reason that if NELFE co-amplifies with MYC in HCC samples then these NELFE-dependent mRNAs may be directly regulated by MYC. Approximately 12.5% or 15% of HCC samples have amplifications of both NELFE and MYC (log2>0.32) in the LCI or TCGA dataset, respectively. Whereas 32.8% or 38% of HCC samples had MYC amplification, only 17 or 11% of HCC samples had NELFE amplification (Figure S3E). We tested for a correlation between NELFE and MYC gene copy number and found no significant correlation in the LCI or TCGA dataset (Figures S3F). Whereas the mRNA levels between the two genes were correlated in TCGA-LIHC dataset (Pearson, p=0.03), this trend was not observed in the LCI dataset (Figures S3F). These data indicate a weak mutual relationship between NELFE and MYC expression in HCC clinical samples, and that NELFE may enhance MYC signaling independent of the MYC gene amplification.
We also performed a Student’s t-tests to determine differentially expressed genes between NELFE-high and NELFE-low expressing HCC samples in the LCI and TCGA-LIHC datasets (Figure S3G). We reason that if NELFE preferentially affects MYC-associated genes, MYC signaling would be significantly enriched in NELFE-high HCC compared to NELFE-low HCC. This analysis resulted in 683 genes that overlapped in both datasets. We found a significant enrichment of genes in liver cancer associated signatures, including the Patil Liver Cancer and Cairo Hepatoblastoma signatures and other signatures, such as E2F Hallmark Markers and Cell Cycle, all of which are consistent with NELFE’s role in promoting cell proliferation and HCC progression as described above. There was also significant enrichment for MYC-associated genes in both analyses (GSEA q-value: 6.61×10−12, ENCODE q-value: 1.96×10−25) (Figures S3H–I), suggesting that high levels of NELFE in HCC is correlated with MYC signaling. To further confirm specificity, we performed the same analysis with Argonuate 2 (AGO2), another top ranking RBP candidate, and found that AGO2-related genes are significantly different than NELFE-associated genes (data not shown).
Since NELFE-related genes are likely associated with HCC and MYC-related genes, we determined if the 68 NELFE-dependent MYC targets (referred to as RDMT), which are correlated with NELFE expression in HCC samples from the Dang_Bound_by_Myc cancer signature, are associated with HCC prognosis in the LCI and LEC datasets (Table S3, Figure 3F). Consistently, the survival risk prediction based on 10-fold cross-validation with 1000 permutations classified patients into low- and high-risk groups with a significant difference in survival as analyzed by Kaplan–Meier plot in both the LCI and LEC datasets (Figure 3G). These data suggest that NELFE may enhance MYC signaling to promote HCC progression.
NELFE enhances MYC-induced HCC
We determined whether NELFE functionally interacts with and modulates MYC by investigating whether the knockdown of NELFE via siRNA can affect MYC mRNA or protein expression. Upon siRNA mediated NELFE knockdown, we observed no significant changes in either the mRNA or protein expression of MYC (Figure S4A), which was consistent with our clinical analysis of HCC tumors. Using TaqMan q-PCR gene copy number analysis in several HCC cell lines, we determined that Hep3B, SMMC-7721 and Huh7 cells had two copies of MYC, whereas Huh1 and MHCC97 cells had more than four copies of MYC when compared to male gDNA or HHT4 cells (Figure S4B). These data suggest that MYC amplification is not the main driver of HCC with activated MYC signaling.
To test the hypothesis that NELFE can enhance MYC-induced HCC, we first overexpressed lenti-mGFP-NELFE in HHT4 cells, an hTERT immortalized normal human hepatocyte cell line. Interestingly, overexpression of NELFE resulted in reduced cell proliferation but no significant changes in colony formation of HHT4 cells (Figures S4C–D). This is consistent with the known phenomenon that aberrant activation of oncogenes results in growth arrest or apoptosis in normal cells (Lowe et al., 2004), as multiple alterations in oncogenes and tumor suppressor genes are required for the transformation of normal cells into tumorigenic cells (Hahn and Weinberg, 2002). We thus tested if simian virus 40 T-antigen (SV40) could override NELFE-induced growth arrest of HHT4 cells in the presence or absence of MYC overexpression (Figure S4E–F). We found that SV40-NELFE had an increase in cell colony formation compared to SV40 or SV40-vector (SV40-Ctrl) (Figure 4A). Although SV40-NELFE cells had no significant effect on oncosphere formation (Figure 4B), SV40-NELFE cells demonstrate enhanced cell proliferation (Figure 4C). Similarly, SV40-MYC cells showed enhanced colony formation, oncosphere formation and cell proliferation compared to SV40-Ctrl (Figures 4A–C). Consistently, SV40-MYC+NELFE cells showed enhanced cell proliferation, colony formation and oncosphere formation compared to SV40-Ctrl, SV40-NELFE or SV40-MYC (Figures 4A–C). Consistent with MYC-associated gene expression in HCC tumor specimens and in NELFE siRNA expressing HCC cells, we found that NELFE altered MYC-associated genes, such as PA2G4, CCNE2, IER2 and EGR1, randomly selected from the 68 MYC gene set (Figure 4D). RT-PCR analysis of SV40-Ctrl, SV40-NELFE, SV40-MYC or MYC+NELFE demonstrate that NELFE enhances MYC-associated genes such as PA2G4 and CCNE2 and suppresses IER2 or EGR1 expression in SV40-MYC+NELFE compared to SV40-Ctrl cells. Furthermore, NELFE overexpression in SV40-MYC cells enhanced the induction of PA2G4 and repression IER2 expression (Figure 4D), suggesting that the increased levels of NELFE in a MYC background can affect MYC-associated genes.
Figure 4.
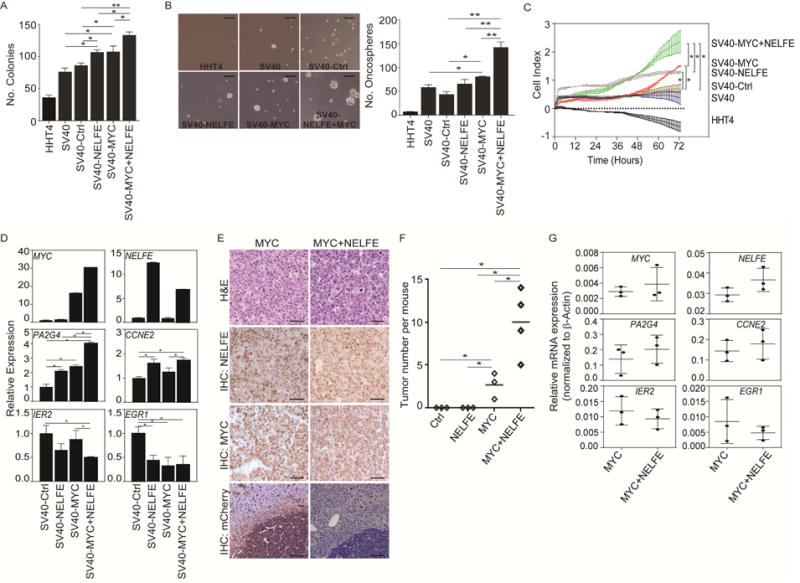
NELFE enhances MYC tumorigenicity. (A) Bar graph of colony formation assay of HHT4 cells or HHT4 cells ectopically expressing the indicated proteins at day 10. (B) Representatie image and quantifiation of oncosphere formation assay at day 7 measured by Algimatrix 3D assay. Scale bar, 200 μM. (C) Proliferation rates of different cell lines up to 72 hr. (D) RT-PCR analysis of relative mRNA expression of MYC-related genes. (E) Hematoxylin and eosin and immunohistochemical staining of indicated tumors. Scale bars, 40 μM. (F) Number of tumor nodules four weeks aftr the injection of indicated cells. Short horizontal lines represent the mean. (G) RT-PCR analysis of relative mRNA expression of MYC-related genes in MYC or MYC+NELFE tumor tissues. Data were first normalized to β–actin to get dCt. Relative mRNA was then calculated using 2dCt. *p<0.05, **p<0.01. All data are mean ± SD. See also Figure S4.
We further examined whether NELFE can enhance MYC-induced HCC in vivo. We cloned the NELFE cDNA into the Sleeping Beauty (SB) vector (Ctrl) (Figure S4G) and hydrodynamically injected different combinations of pT2.EF1α.NELFE.PGK.mCherry, pT3-EF1α-Myc, and CMV-SB10 transposase plasmids via tail vein into Trp53flox/flox;albumin-Cre mice (Figure S4H) and sacrificed them four weeks post-injection. As shown in Figure 4E, MYC+NELFE IHC staining of liver sections showed mCherry in both tumor cells and non-tumor cells, indicating that NELFE was effectively delivered into mice livers. As expected, MYC was able to induce tumor nodules, consistent with other MYC only models (Beer et al., 2004; Shachaf et al., 2004). While NELFE alone did not induce HCC, it enhanced MYC-induced HCC development (Figure 4F). H&E and IHC analysis confirmed that NELFE-induced tumors present HCC characteristics and stained for high levels of NELFE and MYC protein expression (Figure 4E). Consistent with cell line-based data described above, we found that NELFE, PA2G4 and CCNE2 mRNA levels were increased, whereas IER2 and EGR1 expression is decreased in MYC+NELFE compared to MYC alone (Figure 4G). Taken together, these results indicate that NELFE enhances MYC-induced HCC tumor formation in vivo.
NELFE interacts with and regulates the stability of the mRNA of MYC-associated genes
Although NELFE has been previously identified as an mRBP (Castello et al., 2012), its mRNA targets are largely undiscovered. Thus, we determined the RNA transcript sequence preference for NELFE binding using the RNAcompete assay, which represents direct binding in vitro, as only RNA and protein are present in the binding reaction (Ray et al., 2009; Ray et al., 2013). RNAs associated with NELFE were interrogated by microarray and computational analysis, producing both Z (intensity) scores for each individual 7-mer along with consensus RNA binding sequences (the predicted RNA binding motif), resulting in the International Union of Pure and Applied Chemistry (IUPAC) nomenclature AGAGWWW (Figure 5A, left). We then scanned coding genes containing the RNA consensus motif to determine the NELFE associated genes. We considered a gene to be an NELFE associated gene if it has a log odds score ≥ 8, which we considered as a part of the NELFE-hits list. To increase the stringency and decrease the number of false positives, we overlapped the NELFE-hits list with 3,070 differentially expressed genes (DEGs) list described in Figure 3A, focusing on only mRNAs due to our current findings that more than 75% of DEGs are mRNAs (Figure 3B) (Castello et al., 2012). Notably, there is significant enrichment for NELFE-hits in DEGs compared to undifferentially expressed genes (uDEGs) (one-sided Fisher’s exact test, p=03.72×10−49) (Figure 5A, left, Figure S5A) and NELFE-hits have more motifs per gene in DEGs than uDEGs (Figure S5B), indicating that genes affected by NELFE siRNA are more likely to contain NELFE binding sequences than uDEGs.
Figure 5.
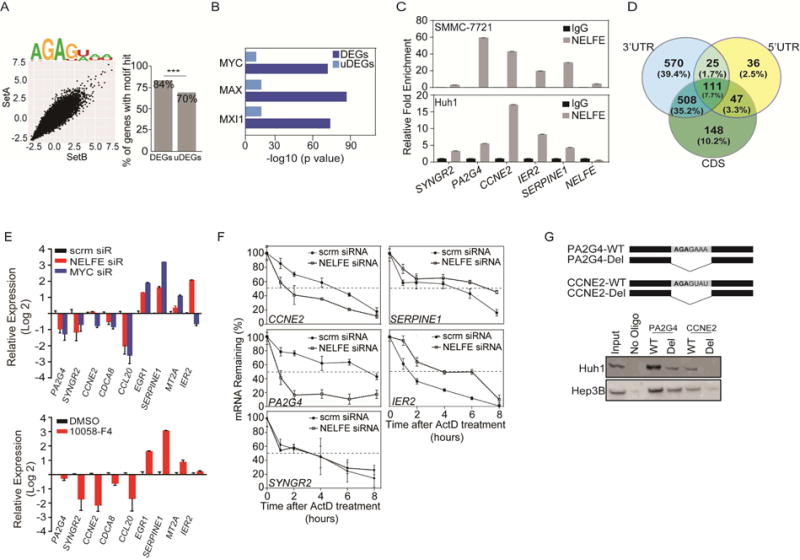
NELFE preferentially interacts with MYC-related genes. (A) 7-mer Z scores and motifs for the two probe sets for NELFE (left). Differentially (DEGs) and undifferentially (uDEGs) expressed genes were scanned for NELFE motifs (right) (***p=3.7×10−49). (B) ChIP-Seq ENCODE analysis of 1,836 DEGs that were predicted as NELFE associated genes from (A). Showing only the MYC and its family members. (C) RNA immunoprecipitation followed by RT-PCR analysis of MYC-related and NELFE associated genes (CCL20, PA2G4, CCNE2, IER2 and SERPINE1) and MYC-related genes that is not an NELFE predicted gene (SYNGR2). (D) Venn diagram of 1,445 genes whose NELFE RNA consensus sequence was found in the 3′UTR, 5′UTR or CDS. (E) RT-PCR of Huh1 cells after 72 hr of specified siRNA (top) or 16 hr of 10058-F4 treatment (bottom). (F) RT-PCR of MYC-related genes in Huh1 cells after NELFE siRNA followed by Actinomycin D treatment (10 μg/ml) over time. Dotted lines are at 50% mRNA remaining. (G) Synthetic RNA oligos with the indicated NELFE consensus sequences for PA2G4 or CCNE2 (Top) were examined via RNA pull down and blotted for NELFE (Bottom: cropped immunoblot). All data are mean ± SD. See also Figure S5 and Table S4.
Using ENCODE ChIP-Seq data, we investigated whether there is a significant enrichment for MYC-related genes in the NELFE-hits list and found 1,164/1,839 genes were MYC-related genes (ENCODE q-value:1.25×10−72) (Table S4). There was also a significant enrichment for NELFE-associated genes that were also bound by MYC partners, including MAX (q=8.52×10−87) and MXI1 (q=2.41×10−74) (Figure 5B, dark blue). To increase the confidence that DEGs were enriched with MYC-related genes, we performed the same analysis using uDEGs as a control. Accordingly, uDEGs were not as enriched with MYC, MAX, or MXI1 related genes compared to DEGs (Figure 5B, light blue). We next performed RNA-immunoprecipitation (RIP) followed by RT-PCR to confirm that NELFE interacts with MYC-related genes. We selected genes that were predicted to be bound by NELFE (i.e., PA2G4, CCNE2, IER2 and SERPINE1) and also found on the RDMT gene list for validation. We also performed PCR on SYNGR2, a MYC-associated gene from the RDMT gene list that was not predicted to be bound by NELFE as a negative control. Accordingly, CCNE2, PA2G4, and IER2 were at least 5-fold enriched compared to IgG control, whereas SYNGR2 showed negligible differences in enrichment compared to IgG in three HCC cell lines (Figure 5C and Figure S5C).
To determine how NELFE affects MYC-related genes, we first examined the location of the RNAcompete derived consensus motif in the 1,839 NELFE-hits list. Of the 1,839 NELFE hits, we were able to map 1,445 genes accordingly to either the 3′UTR, 5′UTR or CDS. We found 570 genes had an NELFE motif in the 3′UTR (one-sided Fisher’s exact test, p=0.017), 36 in the 5′UTR, and 148 genes had at least one NELFE consensus motif in the protein coding sequences (CDS) (Figure 5D). Interestingly, there was a large proportion (35.2%, one-sided Fisher’s exact test, p<0.01) of genes with the consensus motif in both the 3′UTR and 5′UTR, suggesting that NELFE may regulate multiple post-transcriptional processes. Notably, approximately 7% of genes did not have at least one NELFE binding motif found throughout the mRNA sequence, suggesting that NELFE may indirectly affect the expression of some genes.
We next determined whether MYC-associated genes are affected by siRNA mediated knockdown of NELFE or MYC in HCC cells by performing RT-PCR. We selected five RDMTs whose gene expression were among the most affected from the 68 gene list in both our microarray and LCI datasets analyses (i.e., PA2G4, SYNGR2, MT2A, SERPINE1 and IER2 (Figure 5E and Table S3) and selected MYC-associated genes found on the ENCODE analysis including CCNE2, CDCA8 and CCL20. Consistent with our microarray data (Figure 3G), NELFE or MYC knockdown decreased the expression of CCNE2, CDCA8 and CCL20 and induced the expression of EGR1, SERPINE1, MT2A and IER2 (Figure 5E and Figure S5D, top panels) in HCC cells. To determine whether MYC regulates RDMTs, we used a small molecule MYC/Max inhibitor, 10053-F4 in HCC cells. 10058-F4 inhibits MYC/Max heterodimerzation both in vitro and in vivo (Wang et al., 2007; Yin et al., 2003). The treatment of both HCC cell lines with 10058-F4 (200 μM for 16 hr) altered the expression RDMTs and NELFE-dependent genes, consistent with both our microarray and siRNA-mediated NELFE or MYC inhibition (Figure 5E and Figure S5D, bottom panels).
Since there was a significant enrichment for genes with an NELFE consensus RNA motif in the 3′UTR, we hypothesize that these mRNA/NELFE interactions play a role in the degradation, stability and/or translation of its targets. Accordingly, about 64.4% (367/570) of the genes with a consensus motif in the 3′UTRs are also MYC-related genes (one-sided Fisher’s exact test, p=1.57×10−23), suggesting that NELFE may regulate its target’s stability. We thus blocked transcription with Actinomycin D or 5,6-Dichlorobenzimidazole 1-b-D-ribofuranoside (DRB) in HCC cells that have been transfected with scrm or NELFE siRNA and measured the relative change of MYC-related gene’s mRNA levels over time. We investigated SERPINE1, PA2G4 and IER2, as they are predicted to have an NELFE RNA consensus motif in the 3′UTR region only. We also investigated CCNE2, which is predicted to have NELFE consensus sequence motifs found throughout the gene (5′UTR, 3′UTR, and CDS). Compared to scrm, NELFE knockdown significantly reduced the stability of CCNE2 and PA2G4 and increased the stability of IER2 and SERPINE1 mRNAs (Figure 5F and Figure S5E). However, NELFE knockdown did not affect SYNGR2 mRNA stability, a MYC-related gene without the presence of NELFE consensus sequence motifs. To confirm that NELFE interacts with the 3′UTR of MYC targets, we deleted the NELFE consensus motif in two MYC-related genes, PA2G4 and CCNE2, and examined their binding by NELFE via RNA pulldown (Figure 5G, top panel). We found that both PA2G4-WT and CCNE2-WT RNA binds to NELFE. However, the deletion of the RNA consensus sequence significantly reduced the NELFE binding (Figure 5G, bottom panel). These data suggest that NELFE affects the mRNA stability of some MYC-related genes that was predicted to bind to.
NELFE modulates MYC-related genes
Since NELFE knockdown did not affect the mRNA stability of some RDMTs tested (Figure 5F), we wanted to determine whether NELFE knockdown decreased MYC’s binding ability to promoter regions. We reason that NELFE may play a direct role in regulating MYC activity without regulating its mRNA or protein expression. To determine whether NELFE knockdown affects MYC activity, we performed a MYC binding ELISA, which determines the binding activity of MYC to oligonucleotides with the consensus binding site CACGTG. We found that the MYC binding in Hep3B and Huh1 cells significantly decreased upon inhibition of NELFE (Figure 6A). We further investigated whether downregulation of NELFE affects MYC binding to RDMT promoters via ChIP assay in HCC cells. We selected several RDMT genes in which MYC was predicted to directly bind and regulate, such as CCL20, CCNE2, PA2G4, SERPINE1, SYNGR2 and HBB (the Hemoglobin beta gene) as a negative control. It should be noted that SYNGR2 is not predicted to be bound by NELFE. We found a significant reduction of MYC binding to CCNE2, PA2G4, SERPINE1 and SYNGR2, and an appreciable but not statistically significant reduction in CCL20 upon NELFE knockdown (Figure 6B). In contrast, overexpression of NELFE enhanced MYC binding to these RDMT genes in HHT4 cells (Figure 6C). Interestingly, overexpression of NELFE in SV40-MYC cells did not have an additive effect in enhancing MYC binding to SYNGR2’s promoter region compared to SV40-NELFE cells. We also determined whether the inhibition of MYC may alter the binding of NELFE to the RDMT gene promoters by ChIP with an anti-NELFE antibody. We found a significant reduction of NELFE binding to PA2G4 and SYNGR2 but not in other genes which were tested upon MYC knockdown (Figure 6D). Moreover, we found evidence of a direct interaction between NELFE and MYC using co-immunoprecipitation analysis in Hep3B and Huh1 cells overexrepessed with GFP-NELFE upon micrococcal nuclease digestion (Figure 6E and Fig S6A). Consistently, we found evidence of interactions between endogenous NELFE and MYC by immunoprecipitating anti-NELFE and immunoblotting for MYC (Figure S6B). Together, our data reveal that NELFE may bind directly to some MYC-related genes and/or interact with MYC itself to regulate MYC signaling in HCC cells.
Figure 6.
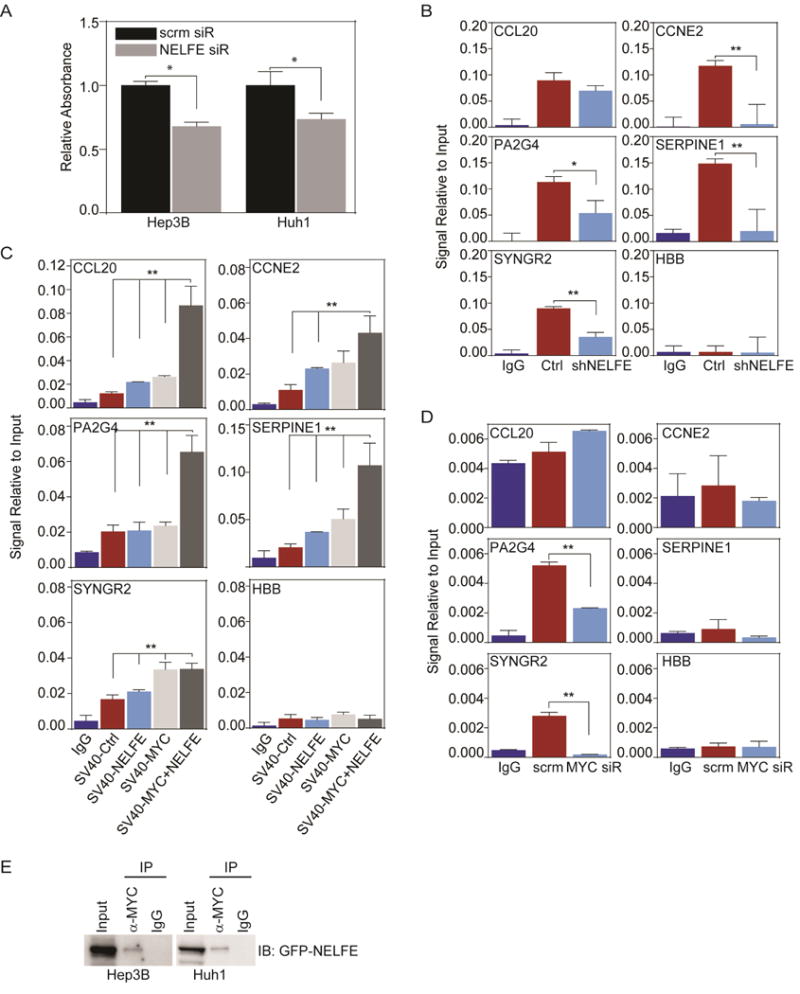
NELFE affects MYC-related genes by modulating MYC binding. (A) MYC transam assay on HCC cells treated with 48 hr of NELFE siRNA compared to scrm (*p<0.01). (B) ChIP-PCR of MYC-related genes of HCC cells after shNELFE compared shCtrl. Data is relative to 2% input. (C) ChIP-PCR of MYC-related genes of HHT4-SV40 cells overexpressed with Ctrl, NELFE, MYC or MYC+NELFE. Anti-MYC was used for IP. (D) ChIP-PCR of MYC-related genes in Hep3B cells after 48 hr of MYC siRNA or scrm. Anti-NELFE was used for IP. (E) Represented immunoblot of co-IP assay in HCC cells. MYC or Rabbit IgG was used for IP. All data are mean ± SD. See also Figure S6.
DISCUSSION
Carcinogenesis is partially dictated by aberrant transcriptional events that consist of several oncogenic signaling pathways (Hanahan and Weinberg, 2011), possibly through the activation of RBPs due to their key roles in maintaining RNA homeostasis. Several RBPs are highly expressed in solid tumors and have been demonstrated to be drivers of carcinogenesis (Busa et al., 2007; Chen et al., 2013; Han et al., 2013; Nguyen et al., 2014; Ortiz-Zapater et al., 2012). As the role of RBPs in cancer emerges, the ability of RBPs to interact with thousands of RNAs makes it an appropriate group of proteins to be selectively dysregulated in cancer. Furthermore, these proteins are highly conserved, stable, translationally efficient, tightly regulated, and abundantly expressed in cells; the idea that alterations of RBPs may bring large scale changes in global gene expression is conceivable (Mittal et al., 2009). Thus, the dysregulation (i.e., activation) of RBP in tumors may lead to significant transcriptomic imbalance.
Our work provide several lines of evidence supporting the hypothesis that the activation of some RBPs contribute to the HCC transcriptome and oncogenicity. We show that more than 70% of RBPs are significantly altered, whose transcriptomic changes are attributed by increased gene copy numbers, suggesting that members of the RBP gene family can be oncogenic. Interestingly, we did not observe the same degree of alterations with TFs, suggesting that a preferential global alteration of RBPs is evident during HCC progression. Moreover, the differentially expressed RBPs are associated with HCC prognosis, suggesting the importance of RBPs in tumor biology of a subset of HCC. Furthermore, by demonstrating that NELFE activation enhanced HCC progression through MYC signaling, we provide evidence of one of multipile mechanisms through which changes in an RBP could have an oncogenic effect on the HCC transcriptome.
MYC, a proto-oncogenic TF that regulates cell growth, proliferation, apoptosis, and metabolic pathways, cooperates with many other oncogenic events to initiate tumorigenesis (Gabay et al., 2014). While chromosome 8q24, containing the MYC locus, gains are observed during hepatocarcinogenesis, an activated MYC signaling without detectable concomitant overexpression or amplification of MYC has been demonstrated to be strongly associated with HCC malignancy (Chan et al., 2004; Kaposi-Novak et al., 2009; Thorgeirsson and Grisham, 2002). The exact mechanism leading to these aberrant changes, however, remains unclear. Consistent with the above view, our study provides insight into how MYC signaling can drive hepatocarcinogenesis without MYC amplification, probably through the activation of NELFE. Considering that MYC regulates 15% of all genes, it is foreseeable that deregulation of its signaling is an important driver of hepatocarcinogenesis (Dang, 2012). However, while MYC inhibitors have been exploited as potential molecular targets for tumors with MYC amplification (Delmore et al., 2011), their potential as an effective cancer therapeutic agents are still uncertain. By inhibiting NELFE in tumors with high MYC signaling (i.e. using our 68 MYC signature to stratify patients) may prove to be a viable therapeutic target for many tumor types, including HCC.
Our results reveal that NELFE contributes to HCC progression in concert with MYC through multiple scenarios of NELFE-mediated regulation of MYC-related genes. We found that NELFE affected the mRNA stability and MYC occupancies at promoter regions of some genes, such as CCNE2, PA2G4 and SERPINE1. These findings suggest that the regulation of the same genes could be affected by two different mechanisms. Moreover, for some genes such as SYNGR2, its regulation is mostly by NELFE or MYC occupancy at its promoter regions. Interestingly, we also found that NELFE can occupy the promoter region of some genes, such as PA2G4 and SYNGR2. To complicate the matter, it is known that NELFE is a part of the NELF-mediated Pol II pausing complex, which may contribute to the regulation of MYC-related genes (Rahl et al., 2010). NELFE has one RNA recognition motif, which directly interacts with nascent RNAs to promote Pol II pausing, stalling elongation on more than 30% of genes (Adelman and Lis, 2012; Castello et al., 2012; Rahl et al., 2010). Our analysis of NELFE-targets in HCC cells showed that only about 16% of genes are bound by NELFE either in the 5′UTR or CDS compared to 39.4% at the 3′UTR. Taken together, NELFE may regulate MYC-related genes through different mechanisms depending on the binding sites, specificity of the target or the availability of MYC at the promoter region. Thus, additional mechanistic studies detailing the regulatory circuitry between NELFE and MYC will be further investigated.
The role of RBPs in cancer have been described (Wurth, 2012). Indeed, many studies have demonstrated that certain elevated RBPs affect RNA metabolism to promote tumorigenesis in different cancer types (Chen et al., 2013; Han et al., 2013; Nguyen et al., 2014; Ortiz-Zapater et al., 2012). The observation that aberrant global transcriptomic alterations occur in tumors suggests the presence of a common mechanism that drives tumorigenesis. One interesting observation from the current study is that while a substantial amount of RBPs show increased gene amplification and transcription, the average changes in gene expression levels among most RBPs between HCC and non-tumors are small (~1.5 fold change). It should be noted that this phenomenon is fairly stable as it was observed in three independent datasets. This is in contrary to the observation that some studies, including this study, demonstrate the activation of a few RBPs can promote tumorigenesis by tilting the transcriptome balance. One plausible scenario is that significant changes of most RBPs might be incompatible for tumor cell growth, but the collective subtle changes of RBPs may be required to achieve a tumor-related transcriptome during tumor evolution. It is interesting to speculate that few RBPs may have a dominant feature to drive transcriptome imbalance while many others may work cooperatively in HCC. This leads to the possibility that the ubiquitous dysregulation of RBPs observed in HCC acts as a cancer driver. This idea is conceivable since not many RBPs have been identified as oncogenes or tumor suppressors. In fact, approximately 8% known oncogenes or tumor suppressors are RBPs (Vogelstein et al., 2013). It is worth noting that the loss of function of some RBPs have been identified in human diseases (Lukong et al., 2008) (14% of tumor-related RBPs are down-regulated in HCC in our transcriptomic analysis). It is likely that cancer may largely require the activation of RBPs to achieve tumor-specific transcriptome to drive tumorigenesis.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xin Wei Wang (xw3u@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Study
Four-weeks-old male NOD/SCID mice (NOD.CB17-Prkdcscid/NcrCrl) and athymic nude mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA). The animal study protocol was approved by the National Cancer Institute-Bethesda Animal Care and Use Committee. For subcutaneous tumorigenic studies, Hep3B cells were transfected with shCtrl or shNELFE lentivirus with 8 μg/ml polybrene for 24 hr. Media was changed and 2 μg/ml puromycin was supplemented for selection for 7 days. Cells were then trypan blue counted and 5×106 Hep3B cells were subcutaneously injected into bilateral regions of the nude mice. Tumor volume were then measured weekly until sacrifice at weeks 12, where tumors were weighed and fixed with 10% formalin, followed by paraffin embedding. For orthotopic studies, Huh1 cells were stably transfected with the luciferase and GFP expression vector. Cells were suspended with Hank’s Balanced Salt Solution (1×105 cells/50 μl) and injected into the left hepatic lobe of NOD/SCID mice. Every 2 weeks, luciferase signals were measured using IVIS Lumina Series III (PerkinElmer, Waltham, MA) after intraperitoneal injection of 3mg D-Luciferin (PerkinElmer). Luciferase signals were quantified using Living Image Software (PerkinElmer). Mice were sacrificed 8 weeks after injection and the livers were fixed with 10% formalin overnight and transferred to 70% ethanol before embedding with paraffin. For subcutaneous injection, Hep3B cells were transfected with an NELFE lentivirus or vector and selected for 7 days. 5×106/100 μl cells were subcutaneously injected bilaterally into athymic nude mice and tumor development was observed weekly. At 12 weeks, mice were sacrificed and tumors were weighed, collected and fixed as described above. Trp53flox/flox mice were crossed with albumin-Cre mice (Jackson Laboratories) (Katz et al., 2012). For the Sleeping beauty animal studies, pT2.EF1a.NELFE.PGK.mCherry plasmid was cloned by Gibson assembly. Plasmid DNA was purified by EndoFree Maxiprep DNA Kit (Qiagen). pT2 (20 g), pT3-EF1α-Myc (5 g, a kind gift of Dr. Xin Chen, UCSF) and CMV-SB10 transposase (1 g) plasmids were delivered to ~8 weeks-old mice by hydrodynamic tail vein injection. Mice were humanely euthanized by CO2 asphyxiation after 4–8 weeks of injection. All animal protocols were approved by the UMass Medical School Institutional Animal Care and Use Committee.
Cell lines
Huh1, HLE, and HuH7 cells were cultured in Dulbecco’s modified Eagle Medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin and L-glutamine. MHCC97-H cells were cultured in Dulbecco’s modified Eagle Medium (Life Technologies) supplemented with 10% defined FBS, penicillin, streptomycin and L-glutamine. Hep3B cells were cultured in Minimum Essential Medium (Life Technologies) supplemented with 10% FBS, penicillin, streptomycin and L-glutamine, non-essential amino acids and sodium pyruvate. SMMC-7721 cells were cultured in RPMI Media 1640 (Life Technologies) supplemented with 10% defined FBS, penicillin, streptomycin and L-glutamine. SNU-398 and SNU-449 cells were cultured in Minimum Essential Medium (Life Technologies) supplemented with heat inactivated 10% FBS, penicillin, streptomycin and L-glutamine, non-essential amino acids and sodium pyruvate. PLC/PRF/5 cells were cultured in Minimum Essential Medium (Life Technologies) supplemented with 10% FBS, penicillin, streptomycin and L-glutamine. HHT4 cells were cultured in HBM medium supplemented with 20% Knockout serum replacement and SingleQuots (Lonza), penicillin, streptomycin and L-glutamine (Jiang et al., 2010). HHT4 cells were cultured on fibronectin (BD Biosciences) coated plates. See also Key Resources Table for cell lines used in the study and sources.
METHOD DETAILS
Plasmids Details
pLKO.1 shNELFE and pLKO.1 shCtrl were purchased from OpenBiosystems (TRC lentivirus). Lenti-C-NELFE mGFP tagged ORF or vector were purchased from Origene. pBABE-zeo-largeTgenomic (Addgene #1778) and pWzl-Blast-Myc (Addgene #10674) were transfected into HHT4 cells followed by selection with puromycin and blasticidin, respectively to generate stable cell lines. HHT4-SV40 cells were transfected with NELFE or vector lentivirus at MOI 5 (NELFE mGFP tagged ORF, Origene). See also Key Resources Table for list of plasmids used.
Immuno-blotting
Protein lysates were separated on Bis-tris 4–12% or 10% tris-glycine SDS-polyacrylamide gels (Life Technologies) and transferred to a nitrocellulose membrane (Life Technologies). Protein detection was performed using anti-NELFE (Abcam, cat# ab170104), anti-β–actin (Sigma-Aldrich, cat#A5316), anti-Cyclin B1 (Cell Signaling Technology, cat #4138), anti-HLA-A (Abcam, cat#52922), anti-HRAS (Santa Cruz, cat#sc-520) and anti-SV40 T Ag (Santa Cruz, cat#sc-147). See also Key Resources Table for more product information for different assays. All immunoblots are cropped for viewing. For full immunoblots of this manuscript, please contact the lead author.
Quantitative RT-PCR
RNA isolation via Trizol is performed and quality of RNA was assessed using the Nanochip on the Bioanalyzer (Agilent Technologies). cDNA was prepared using the ABI High Capacity with 2 μg of total RNA to generate cDNA via Reverse Transcription Kit (Applied Biosystems), and PCR reactions were carried out with TaqMan Gene Expression assay probes (Applied Biosystems) for NELFE, PA2G4, SYNGR2, CCNE2, CDCA8, CCL20, EGR1, SERPINE1, MT2A and IER2 mRNA (See TaqMan Assays in the Key Resources Table). The data were acquired using the ABI SDS 2.4 Software Package (Applied Biosystems) and analyzed using the 2−ΔΔ Ct method. The Ct value of these genes was normalized by subtracting the Ct of the endogenous control 18S mRNA. See also Key Resources Table for list of taqman assay sources.
mRNA Stability Analysis
Hep3B or Huh1 cells were treated with scrm or NELFE siRNA for 48 hr followed by treatment of Actinomycin D (10 μg/ml) or 5,6-Dichlorobenzimidazole (DRB) (50 μM, Sigma-Aldrich) for 0, 1, 2, 4, 6 or 8 hr followed by Trizol RNA extraction. Quantitative RT-PCR analysis was performed and relative mRNA analysis was performed using the 2 −ΔΔ Ct method with 18S as endogenous control. mRNA levels were calibrated to the 0 timepoint.
Apoptosis analysis: Caspase-Glo 3/7 Assay and Annexin V/7-AAD
Apoptosis was assayed with the Hep3B or Huh1cell lines transfected with two NELFE siRNA or scrambled control. After 48 hr of transfection, apoptosis was monitored with the Caspase-Glo 3/7 Assay Reagent (Promega). Luciferase measurements were acquired after 2 hr of incubation with the Omega Reader (MBG Labtech). For the 7-AAD/Annexin V staining, cells were washed with 1×PBS twice and trypsinized followed by addition of ice cold 1×PBS. Cells were then passed through a 40 μM (BD Biosciences) cell strainer into a 15 ml conical tube and centrifuged at 4 °C for 5 min. Cells were washed in FACs buffer followed by a second centrifugation at 4 °C. 100ul of 1× Annexin V binding buffer per 1×105 cells were used to resuspend cell pellets followed by FITC Annexin V and 7-AAD staining in the dark for 15 min at room temperature and analyzed on the BD FACSCanto Flow Cytometry Machine (BD Biosciences). Flow cytometry data were analyzed using FlowJo to calculate Annexin V and 7-AAD distribution.
ChIP-PCR
ChIP assays were performed as per the manufacturer’s protocol using a SimpleChIP Plus Enzymatic IP Kit (Cell Signaling Technology, Danvers, MA, USA). ChIP grade antibodies specific to MYC (Santa Cruz, cat. sc-40) were used for immunoprecipitations with rabbit immunoglobulin G (IgG, Cell Signaling) as control. Purified chromatin was examined by quantitative reverse transcriptase–PCR. Primer sequences are shown in Key Resources Table. Cycle threshold (Ct) values were normalized to the 2% input sample (Percentage of input=2% × 2(C[T] 2%Input sample–C[T] IP sample)).
RNA Pulldown Assay
Hep3B or Huh1 HCC cells were cultured on 15 cm plates until 80% confluent. Cells were then washed 2× with ice cold 1× PBS on ice. 1mL of RIPA buffer supplemented with 1mM of PMSF was then added onto plates and incubated on ice for 10 min. Cells were then scraped and collected followed by a 10 min incubation on ice. Lysates were centrifuge for 10 min at 13K RPM for 10 min at 4C. The supernatant was then transferred into a new microcentrifuge tube. For IP, 50 μl of neutravidin slurry (Thermo Scientific) were washed twice with 1× PBS. The slurry was then centrifuged for 1 min at 1500 RPM and the supernatant was removed via pipetting. 8 μl of 100 μM mRNA oligos (HPLC grade generated by IDT) were then incubated with the slurry and 100ul of 1× TBS for 2 hr, washed 3× with 1×TBS and incubated with 150ul of whole lysates freshly prepared and incubated overnight in 4 °C with rotation. 4 μl (~2.5%) of the lysates were used as input. The slurry was then washed and immuno-blotting was performed to test for binding (NELFE from Abcam (cat. ab170104)). For oligo sequence, see Supplemental Materials.
MYC TransAm DNA binding ELISA
HCC cells treated with NELFE siRNA or scrm siRNA in 10 cm plates for 48 hr followed by nuclear extraction using NE-PER (Thermo Scientific, cat. 78833). 2.5 μg of nuclear extracts was used for MYC Binding assay using a 96-well format per manufacturer’s instructions (Active Motif, cat. 43396).
Cell proliferation and Migration/Invasion Assays
XCELLigence (ACEA Biosciences) assays were performed for cell proliferation and cell migration and invasion assays. 1×105 HCC cells were plated in triplicates in 6-well plates and incubated overnight. Cells were then transfected with either shCtrl or shNELFE lentivirus with 8 μg/ml polybrene supplementation and incubated overnight. 3×103–5×103 cells were plated onto 8-well E-Plates with appropriate medium supplemented with on with 2 μg/ml of puromycin for Huh1 cells and 1 μg/ml for Hep3B and SMMC-7721 cells. For migration/invasion assays, 3×104–4×104 cells were plated on 8-well CIM Plates (ACEA Biosciences) with or without matrigel in quadruplicates.
Cell colony formation
1×105 HCC cells were plated onto a 6-well plate overnight. Cells were then transfected with shNELFE or shCtrl lentivirus with 8 μg/ml polybrene supplementation and incubated overnight at 37° C. Cells were then trypsinized and 5×103 cells were plated in triplicates using 6-well plates with 2 μg/ml puromycin selection for Huh1 cells or 1 μg/ml puromycin for Hep3B cells and cultured for 10 days. Cells were then washed with ice cold 1× PBS followed by ice cold fixation with methanol for 30 min. After fixation, cells are washed with dH2O followed by 2 hr of 0.05% crystal violet staining and 2× washes with dH2O. Colonies were then manually counted. For overexpression studies, 5×103 cells were plated in triplicates using 6-well plates with antibiotic selection.
Cell-cycle analysis
For the cell cycle analysis, cells were washed with 1×PBS twice and trypsinized followed by addition of ice cold 1×PBS. Cells were then passed through a 40 μM (BD Biosciences) cell strainer into a 15ml conical tube and centrifuged at 4° C for 5 min and washed with FACs buffer followed by a second centrifugation at 4° C. Cells were then stained with 7-AAD in the dark for 15 min at room temperature and analyzed on the BD FACSCanto Flow Cytometry Machine (BD Biosciences). Flow cytometry data were analyzed using FlowJo to calculate cell-cycle distribution.
Immunoprecipitation
Hep3B and Huh1 cells overexpressed with p-c-mGFP-NELFE lentivirus were cultured on 15 cm plates until 90% confluency. Cells were washed with 20 ml of ice cold 1×PBS 3× followed by addition of 1 ml of IP lysis buffer (ThermoScientific, Cat. 87787) supplemented with PMSF (1 mM final concentration). Cells were scraped off plates and incubated on ice for 10 min. Whole cell lysates were then centrifuged at 13K RPM for 10 min. A third of the supernatant was then treated with micrococcal nuclease (Cell Signaling Cat# 10011, 0.5 μl/4×106 cells) for 20 min at 37 °C followed by immuno-blotting. The IP was performed using the IP Kit Dynabeads Protein G per manufacturers protocol (ThermoScientific, cat#10007D). 2.5 μg of MYC (Abcam, Cat: ab32072) antibody was used for the pull down and Rabbit IgG (Millipore) as control, which were incubated overnight. IP products were then resuspended with 2 μl of reducing buffer (ThermoScientific), 5 μl of LDS buffer (ThermoScientific), and 13 ul of elution buffer (from kit) and heated at 70 °C for 10 min. IP products without beads were then loaded onto 10% Tris-glycine gels and ran for 2.5 hr at 140 volts. Gel was then transferred onto nitrocellulose membrane (iBlot 7 min at 15 volts) and blotted using NELFE (sc377052).
For endogenous MYC/NELFE interaction, Hep3B and Huh1 cells were cultured on 15 cm plates until 70% confluency. Cells were washed with 20 ml of ice cold 1×PBS 3× followed by addition of 1ml of IP lysis buffer supplemented with PMSF (1 mM final concentration). Cells were scraped off plates and incubated on ice for 10 min. Whole cell lysates were then centrifuged at 12K for 20 min. The IP was performed using the IP Kit Dynabeads Protein G per manufacturers protocol (ThermoScientific, cat#10007D). 5 μg of NELFE (Santa Cruz, Cat: sc377052) was used for the pull down and Mouse IgG (Millipore) as control. The IP were then eluted with 15 μL of elution buffer and 5 μl of loading dye buffer (ThermoScientific). Lysates were then loaded onto 10% tris-glycine gel and run on ice for 2.5 hr and transferred onto a nitrocellulose membrane (iBlot 7 min at 15volts). MYC (Cat. ab32072) blotted in non-reducing/non-denaturing conditions.
Organotypic culture
AlgiMatrix™ 3D Culture System (6-well plate) (Life Technologies) was used for the organotypic cell culture as developed by our group. 1×105 cells were plated in triplicates in 6-well plates for 24 hr followed by lentiviral transfection with 8 μg/ml polybrene supplementation and incubated overnight. 1.5×105 cells were seeded to Algimatrix 3D 6-well plates and cultured in the regular cell culture medium for 10 days. Media was changed every 3–4 days. Spheres were collected by dissolving the matrix with 5ml of AlgiMatrix Dissolving Buffer (Life Technologies) followed by centrifugation at 300g for 5min. Pellets were then resuspended with 2 ml serum free medium and plated on fibronectin coated 6-well plates and incubated at 37° C for 10 min. Media were aspirated and cells were then fixed using 100% cold methanol followed by ice cold 1×PBS wash and 2 hr staining with 0.5% crystal violet staining. Colonies were then manually counted.
Histology and immunohistochemistry
The paraffin blocks were sectioned at 5 μm and stained with haematoxylin and eosin. Anti-HLA-A antibody (Abcam), anti-MYC (Santa Cruz), anti-NELFE (Santa Cruz), and and Envision™+ Kit (Dako, Glostrup, Denmark) were used for detection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Clinical Liver Samples and Array Data
The data for the LCI datasets (N = 488) are available on Gene Expression Omnibus (GEO) GSE14520 (http://www.ncbi.nlm.nih.gov/geo). The data for the Laboratory of Experimental Carcinogenesis (LEC, N = 139) are available on GEO with accession numbers GSE1898 and GSE4024 (microarray platform GPL1528). For the Stanford data set see http://genome-www.stanford.edu/hcc/Figures/ArrayInformation.htm (Chen et al., 2002). For clinical information and preliminary processing of the LCI or LEC dataset see the following references (Lee et al., 2004; Lee et al., 2006). 64 HCC samples from the LCI datasets were also analyzed using the arrayCGH data (Agilent Human-Genome-CGH-105A Oligo Microarrays G4412A). These data were downloaded from GEO GSE14322. For more information on the dataset see the following references (Roessler et al., 2012). See also Key Resources Table for complete data sets used.
TCGA Data processing and integration
LIHC RNASeqV2 and Copy Number datasets from TCGA (Downloaded 03-27-2015) was downloaded using the R (v3.12 package TCGA Assembler, http://www.compgenome.org/TCGA-Assembler) (Zhu et al., 2014). TCGA Assembler was used to integrate copy number data with RNASeq V2 data (n=366 samples with both data types) along with clinical data. RNASeqV2 data were log2 transformed and imported into BRB Array (v4.40) for class comparison analysis [n=418, (Non-Tumor=50, Tumor=368)]. Pearson correlation analysis was performed between mRNA levels and copy number with r > 0.2 to be correlated using R (v3.12) with Bonferroni adjustment.
Gene Expression Microarrays and arrayCGH
3×105 Hep3B or Huh1 cells are plated in quadruplicates on 10 cm plates and transfected with scrambled or NELFE siRNA for 48 hr. RNA isolation via Trizol is performed and quality of RNA is assessed using the Nanochip on the Bioanalyzer (Agilent Technologies). Gene expression profiles using the Affymetrix HTA2.0 gene chip were then performed. Quality control using principal components and Pearson correlation of signal intensities were performed to exclude outlier replicates followed by RMA normalization using Expression Console (Affymetrix) resulting in triplicates for each group. Differentially expressed genes were determined using Transcriptome Analysis Console TAC (Affymetrix) with triplicates per group using p<0.005. The data are available on GSE73219. The arrayCGH data were imported into BRB-ArrayTools (v4.3), http://linus.nci.nih.gov/BRB-ArrayTools.html, along with the corresponding gene expression arrays for integrative analysis using the BRB-CGHTools (v1.3.2). Briefly, the Circular Binary Segmentation (CBS) method is used in the CGHTools package to identify regions in each chromosome followed by the mergeLevels algorithm is used to merge two segments if the distribution of the log2 ratios of the probes is not significantly different. Pearson correlation analysis between expression and copy number of 64 samples (clinical samples with both mRNA and arrayCGH data) was performed with R>0.2 as positively correlated. See also Key Resources Table for complete table of datasets used.
Analysis of NELFE RNA-binding Using RNAcompete
The RNA pool generation, RNAcompete pulldown assays, and microarray hybridizations were performed as previously described (Ray et al., 2009; Ray et al., 2013). Briefly, GST-tagged NELFE (NELFE) (20 pmoles) and RNA pool (1.5 nmoles) were incubated in 1 mL of Binding Buffer (20 mM Hepes pH 7.8, 80 mM KCl, 20 mM NaCl, 10% glycerol, 2 mM DTT, 0.1 μg/μL BSA) containing 20 μL L-glutathione sepharose 4B (GE Healthcare) beads (pre-washed 3 times in Binding Buffer) for 30 minutes at 4° C, and subsequently washed four times for two minutes with Binding Buffer at 4° C. One-sided Z-scores were calculated for the motifs as described previously (Ray et al., 2013). RNAcompete data has been uploaded onto Geo Omnibus GSE93949 (to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=wzirmocuflihrkd&acc=GSE93949) and the International Union of Pure and Applied Chemistry (IUPAC) nomenclature for the NELFE consensus sequence is AGAGWWW.
Scanning for NELFE motifs
Human RefSeq gene sequences (hg19 assembly) corresponding to differentially and non-differentially regulated mRNAs from NELFE knockdown experiments (DEG and uDEG, respectively), were downloaded from the UCSC Table Browser (https://genome.ucsc.edu/) (Karolchik et al., 2004). NELFE motif occurrences were identified using a position weight matrix (PWM) generated by previously described methods (Ray et al., 2013) and scored by log odds using the Bio.motifs module in Biopython version 1.64 (Cock et al., 2009). For a given substring in a sequence, the log odds score is the sum of each position-specific score defined as:
Where pij is the probability of observing nucleotide j at position i, and bj is the background probability of nucleotide j. The background nucleotide frequencies were computed from the DEG and uDEG sequences. Subsequences with log odds score greater than or equal to eight were considered motif hits. NELFE motif enrichment was assessed, in DEG versus uDEG sequences with one or more motif hit, by performing a one-sided Fisher’s exact test using the R (version 3.2.3; https://www.r-project.org/) function fisher.test with parameter alternative=“greater”. Additionally, a permutation test was performed by shuffling the DEG/uDEG labels of each gene and repeating the above enrichment test to exclude the possibility that the observed enrichment was due to chance. After 1000 iterations, the observed odds ratios were significantly higher than the odds ratios in the randomized sets.
Other Statistical analysis
Statistical analysis was performed using Graphpad Prism (v6) and R (v3.12). Enrichment analysis was used to test whether a specific gene list (observed) is different from a gene list randomly selected from all genes in the analysis (expected) using Chi-square or Fisher’s Exact test followed by Bonferroni correction testing. Student’s t-tests were performed when there are two groups. One-way ANOVA was performed amongst different groups followed by Tukey’s posthoc test. All data analysis is performed with at least 3 replicates and are presented as mean ± SD of replicates. Microarray analysis of HCC samples was performed using the paired t-test to look for differentially expressed genes with a p<0.001 as a cutoff. For class prediction analysis, we randomly partitioned all 488 HCC and NT samples of the LCI datasets into a training (NT=113, T=116) and test set (NT=118, T=229). Class prediction analysis (tumor vs. non-tumor) was performed using BRB-ArrayTools (v4.3) to test whether a specific gene signature can classify experiments into phenotype classes based on gene expression. Briefly, three methods of prediction are used: Compound Covariate Predictor (CCP), 1-Nearest Neighbors (1NN), and Support Vector Machines (SVM) with 1000 permutation to determine whether the cross-validated misclassification rate is lower than expected by chance. One-way ANOVA with Tukey’s post hoc testing when comparing multiple groups. Kaplan-Meier curves are calculated using the Cox’s proportional hazards model. Survival risk group prediction analysis was performed using BRBArray (v4.3) with 1000 permutation using specific gene lists as specified according to the analysis described above (http://linus.nci.nih.gov/BRB-ArrayTools.html). For NELFE survival analysis, we used 1/3 vs 2/3 in our LCI and TCGA-LIHC datasets to separate into two groups. This is due to the low number of samples (n=72) in the arrayCGH dataset from LCI datasets, which was then also performed in the TCGA-LIHC datasets for consistency. Briefly, each gene signature is used to assess whether the association of the expression data to survival data is significant. A log-rank statistic is computed for the cross-validated Kaplan-Meier curve and random reshuffling is performed to generate a permutation p-value. Hierarchal clustering analysis was performed using Genesis (v1.7.6). Log 2 data were mean centered to genes and experiments before hierarchal clustering analysis were performed using Pearson complete linkage distance parameters. GSEA analysis was performed using GSEA/MSigDB (Broad Institute, http://www.broadinstitute.org/gsea/msigdb/index.jsp). ENCODE ChIP-Seq Significance tool (http://encodeqt.simple-encode.org/) was used to investigate the number of genes bound by specific ENCODE ChIP data with the following parameters: 500bp upstream and downstream from the TSS and Hg19 GRch37 assembly.
Supplementary Material
Table S1, related to Figure 1. 526 Differentially expressed RBPs found in HCC patient samples. Fishers exact test of elevated RBPs in HCC. 474 Tumor specific RBPs in HCC patient samples. Multivariate class prediction analysis using three different prediction analyses.
Table S2, related to Figure 1. Up-regulated tumor-associated RBPs in HCC clinical samples from two different cohorts (LCI and TCGA). Transcription factors whose mRNA is correlated with Copy number.
Table S3, related to Figure 3. 494 NELFE-dependent genes that are differentially expressed between Hep3B siRNA and the LCI cohort analysis. 68 NELFE-dependent MYC related genes. Highlighted are also NELFE predicted hits.
Table S4, related to Figure 5. NELFE predicted genes from RNACompete experiment. 1,164 NELFE hits that are also MYC related genes per ENCODE.
SIGNIFICANCE.
Tumor initiation and maintenance require the selection of a unique tumor-promoting transcriptome induced by driver genes. Our study indicates that RNA binding proteins can mediate cancer-associated transcriptomic changes in hepatocellular carcinoma (HCC). We show that negative elongation factor E (NELFE) is oncogenic and can selectively regulate MYC-associated genes in HCC with poor prognosis. Our results are consistent with the hypotheses that NELFE activation drives hepatocarcinogenesis and that the NELFE-MYC axis may be exploited as a viable therapeutic target for HCC.
HIGHLIGHTS.
mRNA binding proteins (RBPs) are globally dysregulated in HCC
Tumor associated RBPs are predictive of HCC patient survival
NELFE is oncogenic and enhances MYC signaling
NELFE regulates MYC signaling by binding directly to MYC or its targets
Acknowledgments
We thank Zhipeng Yu for his aid in the immunoblots and Snorri Thorgeirsson for proofreading this manuscript and providing valuable scientific advice for this project. We thank Zhi-Ming Zheng and Michael Bustin for their valuable scientific advice. We also thank Xiaolin Wu at the Microarray Core Facility and Dominic Espinosa at the Protein Expression Laboratory of the Frederick National Laboratory for Cancer Research for microarray and virus production, respectively. We thank the NIH Fellows Editorial Board for their editorial work. This work was supported by grants (Z01 BC 010877 and Z01 BC 010876) from the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, Bethesda, MD. W.X. was supported by grants from the NIH (P01HL131471) and the American Cancer Society (129056-RSG-16-093).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.D. and X.W.W. conceived the idea. H.D. carried out most of the in vitro and in vivo experiments and data analysis. A.T. conducted in vivo experiments and some data analysis. H.M. and W.X. carried out the MYC+NELFE animal experiments. D.R. carried out the RNAcompete experiments. K.H. performed bioinformatics analysis for RNAcompete experiments and Y.P. performed further bioinformatics analysis for RNAcompete gene list. Q.M. and T.R.H. conceived of RNAcompete, helped interpret the data, and provided collaborative support. M.F. carried out the immunohistochemistry experiment. H.D. and X.W.W. interpreted data and co-wrote the manuscript. X.W.W provided overall support for this work.
DATA AND SOFTWARE AVAILABILITY
All data have been deposited. All gene expression and SCNA data used in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) as noted in the Key Resourcees Table and Methods section. Software used in this study are noted in the Method Details section above and the Key Resources Table.
Dang et al. show that a large numbers of RNA binding proteins (RBPs) are dysregulated in hepatocellular carcinoma (HCC) and that NELFE, a RBP, enhances MYC-induced HCC development by regulating the binding of MYC to target promoters and the mRNA stability of several MYC-regulated genes.
References
- Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nature reviews Genetics. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach RK, Chen B, Butte AJ. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics. 2013;29:1922–1924. doi: 10.1093/bioinformatics/btt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, Arvanitis C, Attardi LD, Feng S, Ruebner B, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2:e332. doi: 10.1371/journal.pbio.0020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, Saric J, Belghiti J, Franco D, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- Busa R, Paronetto MP, Farini D, Pierantozzi E, Botti F, Angelini DF, Attisani F, Vespasiani G, Sette C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene. 2007;26:4372–4382. doi: 10.1038/sj.onc.1210224. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Hentze MW, Preiss T. RNA-binding proteins in Mendelian disease. Trends Genet. 2013;29:318–327. doi: 10.1016/j.tig.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Chan KL, Guan XY, Ng IO. High-throughput tissue microarray analysis of c-myc activation in chronic liver diseases and hepatocellular carcinoma. HumPathol. 2004;35:1324–1331. doi: 10.1016/j.humpath.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19:209–216. doi: 10.1038/nm.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, Lai KM, Ji J, Dudoit S, Ng IO, et al. Gene expression patterns in human liver cancers. MolBiolCell. 2002;13:1929–1939. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nature reviews Genetics. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593–1603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- Han W, Xin Z, Zhao Z, Bao W, Lin X, Yin B, Zhao J, Yuan J, Qiang B, Peng X. RNA-binding protein PCBP2 modulates glioma growth by regulating FHL3. J Clin Invest. 2013;123:2103–2118. doi: 10.1172/JCI61820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Iida M, Iizuka N, Tsunedomi R, Tsutsui M, Yoshida S, Maeda Y, Tokuhisa Y, Sakamoto K, Yoshimura K, Tamesa T, Oka M. Overexpression of the RD RNA binding protein in hepatitis C virus-related hepatocellular carcinoma. Oncol Rep. 2012;28:728–734. doi: 10.3892/or.2012.1821. [DOI] [PubMed] [Google Scholar]
- Jiang W, Wang XW, Unger T, Forgues M, Kim JW, Hussain SP, Bowman E, Spillare EA, Lipsky MM, Meck JM, et al. Cooperation of tumor-derived HBx mutants and p53–249(ser) mutant in regulating cell proliferation, anchorage-independent growth and aneuploidy in a telomerase-immortalized normal human hepatocyte-derived cell line. Int J Cancer. 2010;127:1011–1020. doi: 10.1002/ijc.25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaposi-Novak P, Libbrecht L, Woo HG, Lee YH, Sears NC, Conner EA, Factor VM, Roskams T, Thorgeirsson SS. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009;69:2775–2782. doi: 10.1158/0008-5472.CAN-08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SF, Lechel A, Obenauf AC, Begus-Nahrmann Y, Kraus JM, Hoffmann EM, Duda J, Eshraghi P, Hartmann D, Liss B, et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology. 2012;142:1229–1239. doi: 10.1053/j.gastro.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014;15:R14. doi: 10.1186/gb-2014-15-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, Demetris AJ, Thorgeirsson SS. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. NatMed. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Mittal N, Roy N, Babu MM, Janga SC. Dissecting the expression dynamics of RNA-binding proteins in posttranscriptional regulatory networks. Proc Natl Acad Sci U S A. 2009;106:20300–20305. doi: 10.1073/pnas.0906940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell. 2014;26:248–261. doi: 10.1016/j.ccr.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Zapater E, Pineda D, Martinez-Bosch N, Fernandez-Miranda G, Iglesias M, Alameda F, Moreno M, Eliscovich C, Eyras E, Real FX, et al. Key contribution of CPEB4-mediated translational control to cancer progression. Nat Med. 2012;18:83–90. doi: 10.1038/nm.2540. [DOI] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Chan ET, Pena Castillo L, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nature biotechnology. 2009;27:667–670. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, Wang XW. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Research. 2010;70:10202–10212. doi: 10.1158/0008-5472.CAN-10-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012;142:957–966. doi: 10.1053/j.gastro.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Takai A, Fako V, Dang H, Forgues M, Yu Z, Budhu A, Wang XW. Three-dimensional Organotypic Culture Models of Human Hepatocellular Carcinoma. Scientific reports. 2016;6:21174. doi: 10.1038/srep21174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel A, Marquardt JU, Staib F, Galle PR. Snapshot liver transcriptome in hepatocellular carcinoma. Journal of hepatology. 2012;56:990–992. doi: 10.1016/j.jhep.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Theise ND. Liver cancer. Lyon, France: International Agency for Research on Cancer; 2014. [Google Scholar]
- Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. NatGenet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Prochownik EV. Improved low molecular weight Myc-Max inhibitors. Molecular cancer therapeutics. 2007;6:2399–2408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
- Wang XW, Heegaard NH, Orum H. MicroRNAs in liver disease. Gastroenterology. 2012;142:1431–1443. doi: 10.1053/j.gastro.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurth L. Versatility of RNA-Binding Proteins in Cancer. Comparative and functional genomics. 2012;2012:178525. doi: 10.1155/2012/178525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Qiu P, Ji Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nature methods. 2014;11:599–600. doi: 10.1038/nmeth.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure 1. 526 Differentially expressed RBPs found in HCC patient samples. Fishers exact test of elevated RBPs in HCC. 474 Tumor specific RBPs in HCC patient samples. Multivariate class prediction analysis using three different prediction analyses.
Table S2, related to Figure 1. Up-regulated tumor-associated RBPs in HCC clinical samples from two different cohorts (LCI and TCGA). Transcription factors whose mRNA is correlated with Copy number.
Table S3, related to Figure 3. 494 NELFE-dependent genes that are differentially expressed between Hep3B siRNA and the LCI cohort analysis. 68 NELFE-dependent MYC related genes. Highlighted are also NELFE predicted hits.
Table S4, related to Figure 5. NELFE predicted genes from RNACompete experiment. 1,164 NELFE hits that are also MYC related genes per ENCODE.