

Abstract
Peripheral inflammation induces sensitization of nociceptive spinal cord neurons. Both spinal tumor necrosis factor (TNF) and neuronal membrane insertion of Ca2+ permeable AMPA receptor (AMPAr) contribute to spinal sensitization and resultant pain behavior, molecular mechanisms connecting these two events have not been studied in detail. Intrathecal (i.t.) injection of TNF-blockers attenuated paw carrageenan-induced mechanical and thermal hypersensitivity. Levels of GluA1 and GluA4 from dorsal spinal membrane fractions increased in carrageenan-injected rats compared to controls. In the same tissue, GluA2 levels were not altered. Inflammation-induced increases in membrane GluA1 were prevented by i.t. pre-treatment with antagonists to TNF, PI3K, PKA and NMDA. Interestingly, administration of TNF or PI3K inhibitors followed by carrageenan caused a marked reduction in plasma membrane GluA2 levels, despite the fact that membrane GluA2 levels were stable following inhibitor administration in the absence of carrageenan. TNF pre-incubation induced increased numbers of Co2+ labeled dorsal horn neurons, indicating more neurons with Ca2+ permeable AMPAr. In parallel to Western blot results, this increase was blocked by antagonism of PI3K and PKA. In addition, spinal slices from GluA1 transgenic mice, which had a single alanine replacement at GluA1 ser 845 or ser 831 that prevented phosphorylation, were resistant to TNF-induced increases in Co2+ labeling. However, behavioral responses following intraplantar carrageenan and formalin in the mutant mice were no different from littermate controls, suggesting a more complex regulation of nociception. Co-localization of GluA1, GluA2 and GluA4 with synaptophysin on identified spinoparabrachial neurons and their relative ratios were used to assess inflammation-induced trafficking of AMPAr to synapses. Inflammation induced an increase in synaptic GluA1, but not GluA2. Although total GluA4 also increased with inflammation, co-localization of GluA4 with synaptophysin, fell short of significance. Taken together these data suggest that peripheral inflammation induces a PI3K and PKA dependent TNFR1 activated pathway that culminates with trafficking of calcium permeable AMPAr into synapses of nociceptive dorsal horn projection neurons.
Introduction
Peripheral inflammation elicits a state of central or spinal sensitization that, in part, underlies development of the resultant pain behavior (Latremoliere and Woolf, 2009). This spinal plasticity encompasses a plethora of mechanisms and transduction pathways, many working in parallel. We have focused our efforts on delineating one such pathway initiated by spinal tumor necrosis factor (TNF), which is presumably released from activated glia, and culminates with trafficking of GluA1 and GluA4 subunit enriched, GluA2 lacking, α-amino-3-hydroxy-5-methyl-4-isoxazol proprionic acid (AMPA) receptors into plasma membranes (Beattie et al., 2002; Choi et al., 2010). These newly-inserted receptors are Ca2+ permeable AMPA receptors (Katano et al., 2008; Park et al., 2009; Vikman et al., 2008).
All AMPA receptors are comprised of four subunits (GluA1-4) that form a tetramer when assembled (Keinänen et al., 1990). The majority of AMPA receptors in spinal cord of adult rats contain GluA2 paired with either GluA1 or GluA3 and are Ca2+ impermeable. Receptors lacking GluA2 subunits are Ca2+ permeant (Burnashev et al., 1992; Hollmann et al., 1991) and, in naïve animals, are found in low levels throughout laminae I–IV (Engelman et al., 1999; Kopach et al., 2011). Significantly, there is a small population of nociceptive lamina I projection neurons that lack GluA1, but instead contain GluA4, (Polgár et al., 2008; 2010). Other nociceptive neurons that develop Ca2+ permeable AMPA receptors with GluA4 have more recently been identified in deep dorsal horn (Cabañero et al., 2013). Plasmalemmal AMPA receptors are normally in dynamic equilibrium with those in the cytosolic compartment (Scannevin and Huganir, 2000). Equilibrium is altered by changes in afferent drive. Acutely, increased nociceptive drive favors membrane insertion of GluA1 enriched and GluA2 lacking AMPA receptors and removal of GluA2 containing receptors from the membrane of some cell types (Choi et al., 2010; 2012; Galan et al., 2004; Park et al., 2008; Pezet et al., 2008). Analogous membrane trafficking of GluA4 subunit enriched, GluA2 lacking AMPA receptors occurs in deep dorsal horn during morphine-induced hypersensitivity (Cabañero et al., 2013). Following endocytosis, AMPA receptors are either recycled or degraded depending, among other things, on the proportion of AMPA and NMDA receptor activation and the phosphorylation state of GluA1 (Ehlers, 2000; Fernández-Monreal et al., 2012).
Levels of spinal cord TNF increase during the acute phase of inflammation (Christianson et al., 2012; Raghavendra et al., 2004) and pharmacological blockade of spinal TNF attenuates inflammation-induced pain behavior (Choi et al., 2012; Christianson et al., 2012). In cortical neurons, acute application of TNF increases the magnitude of AMPA evoked miniature excitatory post-synaptic currants (EPSCs) (Steinmetz and Turrigiano, 2010), while in hippocampal neurons (Leonoudakis et al., 2008; Ogoshi et al., 2005; Stellwagen et al., 2005) and spinal motor neurons (Ferguson et al., 2008; Yin et al., 2012), TNF is known to elicit membrane insertion of GluA2 lacking AMPA receptors.
A small body of evidence implicates phosphatidylinositol 3 kinase (PI3K) as an intermediary between acute peripheral inflammation (intraplantar formalin and intracolonic capsaicin) and activity driven increases in GluA1 in membrane fractions obtained from dorsal horn (Galan et al., 2004; Pezet et al., 2008). Other data exist linking protein kinase A (PKA) with spinal AMPA receptor trafficking down stream of NMDA receptor activation (Ehlers, 2000) and many studies have shown that peripheral injury elicits phosphorylation of GluA1 ser 845 by PKA (Choi et al., 2010; Fang et al., 2003; Peng et al., 2011) and of GluA1 ser 831 by PKC and/or CaMKII. Spinal blockade of either PKA or PKC activity prevents peripheral inflammation induced hyperalgesia (Jones and Sorkin, 2005; Peng et al., 2011; Sluka, 2002; Willis, 2001).
In this study, using an animal model of intraplantar carrageenan, combined with subcellular fractionation techniques, we demonstrate peripheral inflammation induced increases in dorsal spinal cord plasma membrane GluA1 and GluA4 as well as a small, simultaneous loss of GluA2. Furthermore, we show that this trafficking is dependent on spinal TNF, NMDA, PI3K and PKA. Immunohistochemistry and confocal microscopy indicate that a significant portion of the increased membrane GluA1 on identified spinal projection neurons is synaptic. In vitro studies conducted in parallel, which used kainate induced cobalt labeling of Ca2+ permeable AMPA receptors showed that TNF induces a PI3K and PKA dependent insertion of Ca2+ permeable AMPA receptors in dorsal horn neurons. In a follow up study, using knock-in mice with point mutations on GluA1 that do not permit phosphorylation at ser 831 (CaMKII/PKC site) or ser 845 (PKA site), we demonstrated that TNF induced trafficking of Ca2+ permeable AMPA receptors is both PKC and PKA dependent.
Materials and Methods
Animals and surgery
Male Holtzman rats (Harlan Industries, Indianapolis, IN), weighing 250–350 g were used for behavioral experiments and for in vivo inflammation followed by tissue harvest and either Western blots or immunohistochemistry. Female Sprague Dawley rats (175–250 g; Charles River Laboratories, Wilmington, MA) provided spinal cord slices for in vitro cobalt labeling experiments. Transgenic mice, which lacked the ability to phosphorylate GluA1 at ser 845 or at ser 831 due to single alanine substitutions were generated (Johns Hopkins University, (H.-K. Lee et al., 2003)) and bred to C57BL/6 mice (Harlan Industries) to produce heterozygous mating pairs at University of California, San Diego. This mutation prevents insertion of GluA1-containing Ca2+ permeable AMPAr into the plasma membrane. These mice were bred and homozygous knock-ins and wild type littermates used for behavioral (both sexes) and in vitro cobalt labeling (female) experiments. Animals were maintained on a 12-h light/dark cycle with free access to food and water; all experiments were performed during the light cycle. Experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committees of the University of California, San Diego and the University of California, Irvine (under the Guide for Care and Use of Laboratory Animals, National Institutes of Health publication 85–23, Bethesda, MD, USA).
To permit bolus intrathecal (i.t.) drug delivery in the in vivo rat studies, lumbar i.t. catheters (PE-5, 8.5 cm in length, Spectranetics, Colorado Springs, CO, USA) were implanted via cisternal exposure under isoflurane anesthesia (2–4%) and externalized as described elsewhere (Yaksh and Rudy, 1976). Following catheter implantation rats were housed singly. Experiments were performed 5–10 days following catheter implantation.
Drugs
In Vivo
Etanercept (Enbrel, 30 and 100 μg, TNF antagonist, Amgen, Thousand Oaks, CA, USA) and a receptor-neutralizing antibody specific for TNFR1 (125 μg, R&D Systems, Minneapolis, MN, USA) were used for behavioral experiments. The following agents were used prior to tissue harvest and subsequent immunoblotting. Etanercept (ETA), 100 μg; LY294002 (2-morpholin-4-yl-8-phenylchromen-4-one), 50 μg, a non-selective PI3K inhibitor; H-89 (dihydrochloride hydrate, 26 μg, a PKA inhibitor), Calbiochem, La Jolla, CA); AKAP st-Ht31(2.8 μg, Promega, Madison, WI, USA, inhibitor protein for PKA anchoring onto AKAPs); and MK-801 hydrogen maleate, (MK-801, 30 μg, a non-competitive NMDA receptor antagonist) were delivered i.t. in 10 μl vehicle followed by a 10 μl saline flush. LY294002 was obtained from Sigma Aldrich, St. Louis, MO. All agents were dissolved in physiological saline, except for LY294002 which was dissolved in 5% DMSO, 2.5% EtOH and 92.5% saline.
In vitro
Wortmannin (100 μM, a non-selective PI3K antagonist; Tocris Bioscience, Ellisville, MI) and H89 (10 μM) were used as pretreatments prior to adding TNF (20 nM Rats; 12 nM Mice), to slices in the bath.
Induction of inflammation
Rats
To induce a state of local inflammation 100 μl of a 2 % λ carrageenan solution (Sigma, St Louis, MO, USA; v/w in physiological saline) was injected subcutaneously into the ventral aspect of the left hind paw under brief isoflurane anesthesia.
Mice
The injection volume of carrageenan was reduced to 20 μl. After injection, animals were immediately placed in their test compartments or home cages for behavioral testing or tissue harvest, respectively. In other experiments, restrained mice were injected with 20 μl of a 2.5% formalin solution into the dorsal surface of the left hindpaw.
Behavioral assessment of pain behavior
Rats
For measurement of tactile thresholds, rats were placed in individual compartments with wire mesh bottoms. Animals were acclimated for periods of 60 min in the testing room and 30 min in the test cages prior to assessment of baseline thresholds, for two consecutive days prior to carrageenan injection. Von Frey filaments were used to measure tactile allodynia following the Dixon up-down method (Chaplan et al., 1994). Briefly, calibrated filaments (Stoelting, Wood Dale, IL, USA) with buckling forces between 0.41 and 15.2 g were applied perpendicularly to the mid-paw plantar surface (L4 dermatome) until the filament was slightly bent. Testing was started with the 2.0 g hair and the next filament was applied after a few seconds or when the animal was calm with both hind paws placed on the grid. A positive response was noted when the rat displayed a brisk withdrawal from the stimulus. The 50% probability withdrawal threshold was determined and plotted versus time. To assess thermal hypersensitivity, a modified Hargreaves type device was employed (Dirig et al., 1997). Rats were placed in individual Plexiglas cubicles on top of a glass surface and after habituation, a radiant heat stimulus was applied to each paw and the latency defined as the time required for paw withdrawal. After determination of basal thresholds, ETA or neutralizing antibody (mechanical only) was injected through the catheter and the rat returned to the testing chamber. Animals were lightly anesthetized 57 min after the i.t. injection and interplantar injection of carrageenan performed 60 min post-i.t. injection. Mechanical and thermal withdrawal thresholds were measured at 1, 2, 3 and 4 h post carrageenan injection.
Mice
Mice were treated similar to the rats with the following modifications. Testing took place in a dedicated mouse room, prior to the experiment mice were acclimated to the testing room and apparatus for two hours each, on three successive days. Animals of either sex were used, but on any given day, only male or female mice were used and equipment was washed thoroughly between uses. Knock-in mice and wild-type littermates were tested in the same session. Acute mechanical and thermal withdrawal thresholds were determined prior to carrageenan and formalin testing. Thermal escape latencies were measured using a modified Hargreaves device (Dirig et al., 1997), mechanical thresholds were performed as for rats, but with less stiff von Frey filaments (buckling forces from 0.2 to 2.0 gm). Each mouse was tested three times/day on each hindpaw, this was repeated for two days and all responses were averaged. Carrageenan: Following carrageenan injection, mice were placed in testing compartments and mechanical thresholds were re-determined at 1 hr intervals for 4 hours. At the end of this period, mice were placed back in the Hargreaves device and thermal escape latencies were again measured. Formalin: Flinches were counted in 1 min bins for the first hour post-injection in an automated device (Yaksh et al., 2001). Responses were divided into phase 1 (min 1–9), phase 2 (min 10–60). In all behavioral experiments, the experimenter was blinded to drug treatment (rats) or genotype (mice).
Tissue harvest for Western blot
One hour after i.t. ETA/saline vehicle or 5 min after LY294002, H89, AKAP st-Ht31, or the appropriate vehicle, carrageenan was injected into the paw under light isoflurane anaesthesia. Control animals were anesthetized, but received no injection. Animals recovered in warm chambers and then returned to their home cages. One hour after carrageenan, rats were re-anesthetized, decapitated and the spinal cord extruded with cold saline. After removal of a 1 cm length of lumbar enlargement (L2-L5), the left dorsal quadrant was dissected and immediately frozen with dry ice and stored at −70°C. Samples were shipped on dry ice to Sweden for analysis.
Subcellular fractionation
Lumbar spinal cord tissue (20–25 mg) was homogenized in 1 ml hypotonic buffer containing 10 μM NaHCO3, Phosphatase Inhibitor Cocktail I (Sigma P2850 1:1000), Phosphatase Cocktail II (Sigma P5726 1:1000) and Protease Inhibitor (Sigma P8340 1:1000) at pH 7.4, using a dounce homogenizer. The homogenate was incubated on ice for 10 min and centrifuged at 4°C for 10 min at 1200 RCF using a 50.4-Ti rotor (Beckman Coulter). The pellet containing large cell debris was discarded and the supernatant was collected and centrifuged at 21600 RCF for 30 min. The resulting second pellet contains internal membranes and was discarded. The second supernatant was collected and centrifuged for 2h at 150000 RCF creating a third pellet containing external membranes. Hypotonic buffer (2 ml, pH 7.4) was added to wash this pellet. After an additional centrifugation at 150000 RCF for 2 h the supernatant was discarded and the fourth pellet enriched in external membranes was re-suspended in 50 μl extraction buffer and the samples stored in −70°C until analysis. Method adapted from (Lehel et al., 1995).
Western blots
Samples containing the external membrane were denatured at 65°C for 30 min and proteins separated by gel electrophoresis (NuPAGE 3–8% Tris Acetate gels, Invitrogen) and transferred to nitrocellulose membranes (Invitrogen). Non-specific binding was blocked using Odyssey blocking buffer (Licor, Lincoln, NE, USA) for 1 h at room temperature and membranes were then incubated with primary antibody GluA1, GluA2, or GluA4 (1:1000, Millipore, Billerica, MA, USA) and β-actin (1:10 000, Cell Signaling Technology, Beverly, MA, USA) in blocking buffer with Tween-20 (0.1%) overnight at 4°C. After washing, membranes were incubated with secondary antibody (goat anti-rabbit 800CW and goat anti-mouse 680 (1:10 000 Licor) dissolved in TBS with 0.1% Tween and conjugated to IRDye-680 or IRDye-800CW for 1 h, washed and scanned using Odyssey Imager (Odyssey CLx, Licor). Image Studio Analysis software (Licor) was used for quantification of band intensity and signals for the GluAs were normalized to β-actin. Tissue from control animals (i.t. vehicle and no carrageenan) was run on every gel.
Co-localization of GluA subunits and synaptophysin on spinal nociceptive projection neurons
Animals
Eight rats were anesthetized with an intraperitoneal ketamine (50 mg/kg)/xylazine (5mg/kg)/acepromazine (1mg/kg) cocktail, supplemented as necessary. Animals were positioned in a stereotaxic device and a Hamilton syringe with a 30 gauge needle, used to pressure inject each rat bilaterally with 1ul of 10% Fluororuby (Millipore #AG335). Injections were aimed at the lateral parabrachial area, using the co-ordinates of AP: −9.0 mm, ML: ± 2.3 mm, D: −7.2 mm relative to Bregma. The injection needle was angled 16° posteriorally in order to enter the brain without piercing the sagittal sinus. The needle was left in place for 10 min after each injection, following skin suturing, infiltration of the incision with lidocaine and subcutaneous injection of Carprofen in 5 ml lactated Ringers, animals were returned to their home cages for 14 days. On the day of the experiment, all animals were lightly anesthetized with isoflurane and four were injected bilaterally into the ventral surface of the hindpaws with λ-carrageenan (100 μl, Wako Pure Chemical Industries, Japan), Animals were allowed to wake up and were re-anesthetized with 5% isoflurane for 8 min starting 1h after paw injection at which time they were transcardially perfused with cold heparinized 0.9% saline followed by chilled 4% paraformaldahyde in 0.1 M phosphate buffer. Spinal cords were removed and post-fixed for 6 h and transferred to sucrose for cryoprotection. Horizontal sections (20 μm) of the spinal cord, to allow the best view of lamina I dendritic trees, were cut using a cryostat and placed directly on slides. Serial sections were collected until Fluororuby staining was no longer observed with epi-flourescent illumination. Brains were removed, brainstems were blocked and post-fixed overnight, then transferred to 30% sucrose and 30 μm sections cut to confirm injection sites.
Immunohistochemistry
Tissue from control and carrageenan animals was processed in parallel and neither the histologist nor those doing the analysis of the stained tissue were aware of treatment. Sections from the lumbar enlargement with Fluororuby staining were initially incubated for one hour with 10% normal goat serum (NGS, Vector Laboratories) at room temperature (RT) followed by an overnight incubation with rabbit anti-synaptophysin (Abcam #14692) (1:2000; diluted in Signal Stain, Cell Signaling #8112L) at RT. A biotinylated goat anti-rabbit secondary (Vector Laboratories; Burlingame, CA.) was applied at 1:500 for one hour at room temperature in a 1% BSA-PBS dilution buffer. Following PBS rinses, Avidin Biotin Complex-Horse Radish Peroxidase (ABC-HRP, Vector Laboratories) was prepared per manufacturers instructions and slides were incubated for another hour followed by a two min incubation in Cy5 Tyramide Signal Amplification reagent (TSA, Perkin Elmer). Slides were then rinsed in PBS and placed in fresh PBS for 30 minutes in a 37°C oven and exposed to freshly prepared pepsin solution (Promega) at 1 mg/ml in 0.2 M HCl for two min. This limited digestion process served to ‘open up’ the synapses and expose the synaptic glutamate receptors. This procedure is apparently necessary in the dorsal horn (Nagy et al., 2004), although surprisingly it is not necessary for motor neurons (Ferguson et al., 2008).
Following three PBS rinses and a one hour incubation in 10% NGS, slides were incubated in Signal Stain dilution buffer for three days at 4°C with either rabbit monoclonal anti-GluA1 (1:300, Millipore #04-855), rabbit polyclonal anti-GluA2 (1:300, Chemicon #AB1768), or rabbit polyclonal anti-GluA4 (1:200, Millipore #1508). Confocal Microscopy: Lamina 1 Fluroruby-labeled projection neurons were scanned with a Leica TCS SP5 confocal laser microscope utilizing the Argon laser for 488nm GluA immunoreactivity, a 543 HeNe laser for Fluororuby and an HeNe laser to detect synaptophysin. Scans, with a 63× oil immersion lens, were carried out sequentially to avoid fluorescent bleedthrough. Z stacks were system optimized with a 0.13 μm step size. Digital files were sent to UCSF for analysis.
Confocal image processing and automated quantification of synaptic AMPAR subunits: Confocal stacks were first batch processed by channel and deblurred using 3D blind iterative deconvolution (AutoQuant, Media Cybernetics, Rockville, MD). To ensure that synaptophysin and GluA expression were quantified only around cells that projected to the parabrachial area, a masking technique was used to isolate Fluororuby-labeled cells. Metamorph software (Molecular Devices, Sunnyvale, CA) was used to detect Fluororuby signal above a predetermined threshold, from which a binary image was created. A mask was then created using an integrated morphometry application, followed by a 15 pixel dilation of this mask to ensure that synaptophysin surrounding the cell was included. This mask was then laid over the synaptophysin/GluA image to isolate only that region that was determined to be positive for Fluororuby. Total GluA (green) and synaptophysin (red) were quantified, and yellow pixels produced by the colocalization of the synaptophysin (presynaptic) and GluA (postsynaptic) signified synaptic GluA. The number of red, green, and yellow pixels above a predetermined threshold was then automatically quantified by the Metamorph software. Values for each color were normalized by the total Fluorouby-masked area within each plane. This process was carried out for each optical plane of each confocal image. In total, 239 confocal stacks were analyzed, with an average 40 optical planes per stack.
TNF-induced Co2+ labeling
Animals were deeply anesthetized with sodium pentobarbital (40 mg/kg) and decapitated. A laminectomy was then performed and lumbosacral spinal cord dissected out and immediately immersed in ice-cold aerated “stabilization buffer” (SB; containing in mM: 139 sucrose, 32.5 NaCL, 2.5 KCl, 10 MgSO4, 12 Glucose, 24 NaHCO3, 1 NaH2PO4, and 0.5 CaCl2) for 5 min. The dura was removed and the spinal cord embedded on end in 2.5% agar, and transverse slices (500 μm) cut on a vibratome (Leica VT 1200, Nusslock, Germany). These thick slices were returned to the aerated SB for 25 min (at 4°C) and then brought up to room temperature for another 20 min before transfer to “uptake buffer” (UB; containing, in mM: 139 Sucrose, 57.5 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 12 Glucose). Slices from rat spinal cord were divided into four groups, pre-incubation with H89, wortmannin or vehicle for 10 min, followed by addition of TNF for 10 more min. The fourth group was pre-incubated with vehicle followed by addition of saline instead of TNF (control). Slices were taken from each animal for the vehicle/saline, vehicle/TNF and at least one of the antagonists given as a pretreatment before TNF and processed simultaneously to reduce experimental variability. Pretreatment/treatment paradigms were followed by the addition of a small volume of concentrated Co2+ and kainate to a final bath concentration of 2.5 mM Co2+, 100 μM kainate for a final 20 min. The pretreatment agents and TNF were not removed during the kainate stimulation period. Cobalt loading was terminated by a UB wash containing 2 mM Ca-EDTA to remove extracellular Co2+ followed by treatment with (NH4)2S (0.067%, 5 min) to precipitate intracellular Co2+ and tissue was then fixed overnight in 4% PFA. Slices were cryoprotected (30% sucrose for 24 hours), OCT embedded and frozen sections cut at 30 μm. Thin sections were then processed in the dark for 60 min with AgNO3 enhancement of the Co2+ stain, as described (Yin et al., 1999) before re-washing to terminate the process.
Nonspecific Co2+ uptake occurs in the top- and bottom-most thin sections in each slice due to tissue damage resulting from vibratome sectioning, staining in the center of each slice is only faintly stained due to difficulty in reagent penetration. Thus, analyzed sections were confined to those 50–150 μm from each end of the thick slice (stack). Sections from the same stack were mounted together and analyzed. Images were acquired by light microscopy (Olympus model BX51, Olympus Japan) using a digital camera (20×). For quantification, photographs were captured using identical exposure parameters and imported into an image analysis program (ImagePro Plus software, MediaCybernetics, Silver Springs, MD) for background subtraction and analysis. Cells with black or dark brown somata of at least twice background intensity were classified as Co2+ positive. Neurons with markedly atrophic or irregular somata were not counted. Care was taken to exclude areas of artifact (obvious non-cellular punctate labeling) as well as capillaries where Co2+ occasionally accumulated. Cobalt positive neurons were counted and divided into those in superficial (laminae I–III) and deep (laminae IV–V) dorsal horn. Frequency of Co2+ labeled neurons within each stack (3–12 sections/stack) was averaged. Stacks from the same animal, subjected to the same treatment were also averaged to obtain an animal mean.
Analysis and counting was performed blinded by investigators with no knowledge of treatment group. Mouse tissue was treated equivalently. Tissue was taken from GluA1 ser 831 and ser 845 knock in mice and their respective wild type littermates. Tissue from each animal was subjected to control and TNF conditions.
Statistics
Graph Pad Prism 4.0 (GPP 4.0, GraphPad Software, Inc., San Diego, CA) was used for statistical analyses and graphs. One-way ANOVAs with post-hoc Bonferroni corrected multiple comparison tests were used to compare behavioral responses and Co2+ positive cell counts among experimental groups. Unpaired t-tests were used to analyze differences between Western blot results. Percent area for GluA, synaptophysin, and colocalization was averaged across planes for each confocal stack, and a linear mixed model was used to test for significant differences between treatment conditions, with confocal stacks for each animal treated as a repeated, random measure. All data is depicted as means ± SEM in graphs. P≤ 0.05 was considered significant.
Results
Inflammation induces allodynia and increases dorsal horn Ca2+–permeable AMPAr: both are blocked by spinal TNF inhibition
In order to confirm the role of spinal TNF in inflammation-induced pain behavior, we pretreated animals intrathecally with the TNF sequestering agent ETA or a TNFR1 neutralizing antibody. Prior to the intraplantar carrageenan-injection, mean 50% mechanical withdrawal thresholds were at cutoff (15.2 g) for all groups. When carrageenan was injected into the plantar surface of the paw, characteristic inflammation and mechanical allodynia were generated in the saline pre-treated animals. Withdrawal thresholds fell within the first hour after injection and continued to fall throughout the experiment. Compared to saline control, intrathecal pre-treatment with ETA, reduced mechanical sensitization (fig. 1A) for the first h for both doses. Anti-allodynia completely dissipated by 3h for the 30 μg dose while the 100 μg dose was efficacious for the remainder of the experiment (4h). The neutralizing antibody blocked the mechanical allodynia to a similar degree as the higher dose of ETA (fig. 1C). Intrathecal ETA at 100 μg also delayed development of thermal hyperalgesia by several h (fig. 1B).
Figure 1.
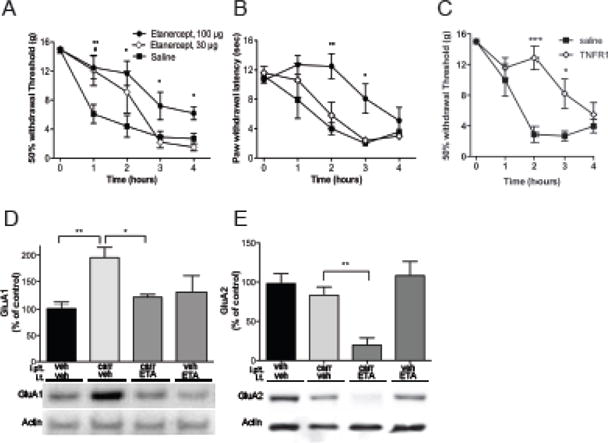
Carrageenan induced hyperalgesia and AMPAr trafficking are mediated by endogenous TNF release and TNFR1 receptor activation. Pretreatment with i.t. etanercept (ETA, # refers to 30 μg and * refers to 100 μg dose) reduces paw carrageenan-induced (A) mechanical allodynia and (B) thermal hyperalgesia. (C) Pretreatment with i.t. TNFR1 neutralizing antibody reduces paw carrageenan-induced mechanical allodynia. (D) Paw carrageenan elicits an increase in GluA1 levels in the plasma membrane fraction of the ipsilateral dorsal spinal cord, which is blocked by i.t. pretreatment with ETA. (E) While paw carrageenan induces a modest tendency towards less GluA2 in the plasma membrane fraction, pretreatment with i.t. ETA results in significant reduction in plasmalemmal GluA2. Data is presented as mean±SEM, n = 5–8 per group, *, # p < 0.05, ** p < 0.01, *** p < 0.001.
Following peripheral inflammation, AMPAr trafficking was assessed by differential centrifugation followed by Western blots for GluA subunits. An increased membrane GluA1/GluA2 ratio is indicative of increased numbers of Ca2+-permeable AMPAr. Previously, we used a total membrane preparation and found that the levels of the GluA subunit were increased in the lumbar spinal cord 1 h after intraplantar injection of carrageenan. Here we performed a more rigorous subcellular fractionation and confirmed that the levels of GluA1 were increased in the plasma membrane fraction generated from the dorsal horn of the lumbar spinal cords collected 1 h after injection of carrageenan, compared to controls (195±21% vs. 100±36% p<0.01) (fig. 1D). In some experiments, we also measured GluA4 subunits in ipsilateral spinal cord of carrageenan injected animals and saw a similar, albeit smaller, increase of this subunit in membrane fractions of injected animals compared to control (160±18% vs, 100±19% p<0.034)(fig. 2O). The increases in both GluA1/GluA2 and GluA4/GluA2 ratios as shown in the Western blots could represent changes in either or both the superficial and deep laminae of the dorsal horn. In an attempt to more closely examine these changes, we used immunostaining to examine lamina 1 projection neurons that were putative nociceptors.
Figure 2.
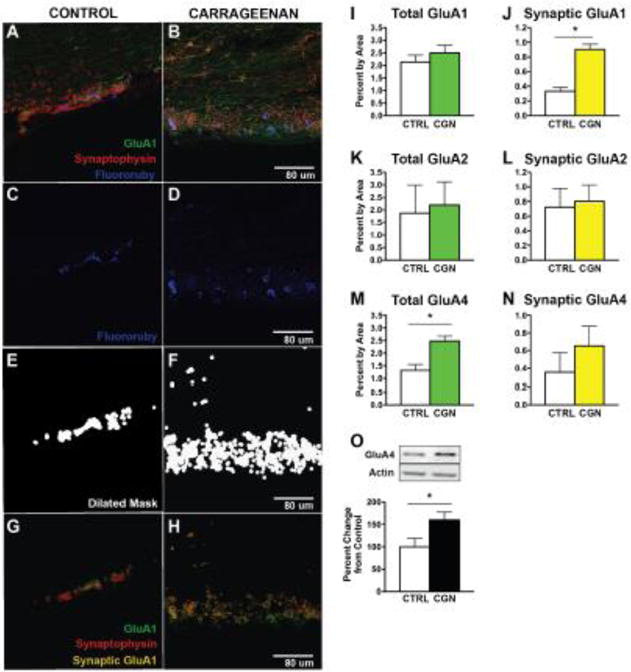
Carrageenan increases GluA1 and GLuA4 AMPAR subunit expression on dorsal horn neurons, but not GluA2. (A–H) Masking technique to isolate backfilled cells and calculate membrane percentage containing each AMPAR subunit (GluA1, GluA2 or GluA4). (A–B) Representative confocal images of dorsal horn neurons labeled for AMPAR subunit (GluA1, green), synaptophysin (red), and Fluororuby tracer back-labeled from parabrachial nucleus (blue). (C–F) To quantify AMPAR/synaptophysin colocalization, Fluororuby channel was first isolated (C–D), and automated imaging software produced a dilated binary mask of Fluororuby signal above threshold (E–F). (G–H) Puncta for GluA1 (green), synaptophysin (red), and synaptic AMPAR subunit (yellow) around the Fluororuby-masked area were then quantified for each optical plane. Total GluA1 (I) was unchanged after carrageenan (p > 0.05) while synaptic GluA1 (J) was significantly increased in response to carrageenan (*p < 0.0001, n=3 per group). Carrageenan had no significant effect on total (K) or synaptic (L) GluA2 (p > 0.05). Total GluA4 (M) was significantly increased in response to carrageenan (*p < 0.05), but synaptic GluA4 (N) was not significantly changed (*p > 0.05, n=3 per group). (O) Western blot analysis confirmed the significant increase of GluA4 in the plasma membrane fraction, in response to carrageenan (*p < 0.05, n = 9–10 per group).
Our confocal examination of synaptic GluA subunits on lamina 1 neurons before and after paw carrageenan supported the idea that some portion of these changes were occurring in the superficial dorsal horn (fig 2A–H). As expected, 1 h post injection, total GluA1 and GluA2 were unchanged in the Fluroruby labeled cells (fig 2I,K). Importantly, the area occupied by GluA1 in the synapse as indicated by co-localization with synaptophysin almost tripled from 0.334 ±0.05 to 0.9 ±0.07, while synaptic GluA2 remained stable 0.73 ±0.25 vs 0.81 ± 0.22 (fig. 2J,L). Surprisingly, total area staining for GluA4 almost doubled at this time (fig. 2M; p=0.028). This could represent a post-translational change in the subunit or an unmasking that allowed it to be recognized by the antibody. Synaptic GluA4 showed trend to increase but this change fell short of significance 0.354± 0.22 vs 0.654±0.22, p=0.393 (fig. 2N).
In the Western blot experiments, the increase in membrane GluA1 was prevented by intrathecal pretreatment with the TNF-blocker (ETA, 100 μg) (p<0.05). While there was a tendency for GluA2 levels in the spinal plasma membrane fractions from rats subjected to intraplantar injection of carrageenan in this set of experiments to decrease, it was not significant. However, when vehicle controls and vehicle/carrageenan results across all of the experiments were combined (fig. 8), carrageenan elicited a reduction of GluA2 (p≤0.03). Unexpectedly, intrathecal injection of 100 μg i.t. ETA, in combination with the carrageenan, massively reduced dorsal GluA2 levels in membrane fractions from spinal cord compared to controls (19±22% vs. 100±48%, p<0.01, fig. 1D). Importantly, this only occurred when carrageenan followed ETA pretreatment. Intrathecal injection of ETA in animals without peripheral inflammation had no effect on spinal membrane levels of GluA1 or GluA2 (fig. 1D, E).
Figure 8.
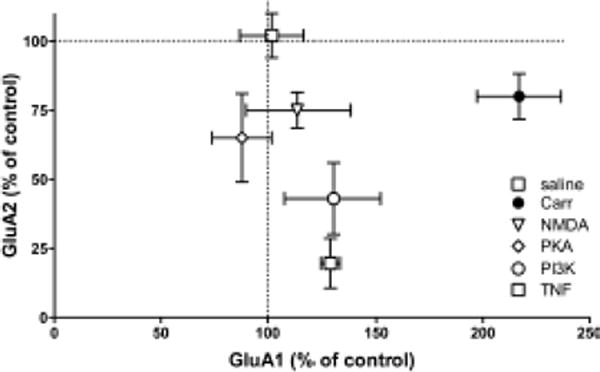
Summary of plasma membrane levels of GluA1 and GluA2 after treatment. Rats were treated with saline, carrageenan, or carrageenan and i.t. inhibitors of NMDAR, PKA, PI3K, or TNF. Mean ± SEM from each treatment group is graphed as a percentage of saline/vehicle controls indicated as the origin on graph.
Inhibition of spinal PI3K, PKA, and NMDAR prevents inflammation-induced dorsal horn trafficking of membrane GluA1
To further investigate the intracellular pathways involved in AMPA subunit trafficking, the effects of intrathecal pretreatment with the PI3K-inhibitor LY294002, PKA-inhibitor H89, PKA anchoring inhibitor St-Ht31 and NMDAR inhibitor MK801 were examined. Intrathecal injection of LY294002 (50 μg) prior to paw injection completely prevented carrageenan-induced increase of GluA1 levels (130±55% vs. 229±79%, p<0.05, fig. 3A) and strongly reduced GluA2 levels compared to control (43±36% vs. 100±25%, p<0.05, fig. 3B) in lumbar dorsal horn membrane fractions. Thus, LY294002 and ETA had similar effects on GluA1 and GluA2 plasma membrane levels compared to carrageenan-injected animals that received vehicle (fig. 8). Intrathecal injection of H89 (26 μg) and St-Ht31 (2.8 μg) also prevented carrageenan-induced increase of GluA1 levels (81±42% and 98±43%, vs. 197±80%, p<0.05, fig. 4A), but did not alter levels of GluA2 in the plasma membranes compared to vehicle/carrageenan-injected animals. Pretreatment with i.t. MK801 (30 μg) prevented carrageenan-induced increase of GluA1 protein levels in the spinal plasma membrane fraction (114±49%, vs. 213±80%, p<0.05, fig. 5A), while it did not produce changes in GluA2 protein levels (figs 5B). Thus PKA inhibition and NMDA receptor antagonism generated similar degrees of blockade of carrageenan-induced trafficking of GluA1 and GluA2 into and out of the spinal plasma membrane, respectively (fig 8). Intrathecal injection of PI3K, PKA or NMDAR inhibitors, in the absence of intraplantar carrageenan, did not alter membrane levels of either GluA1 or GluA2 (fig 3–5).
Figure 3.
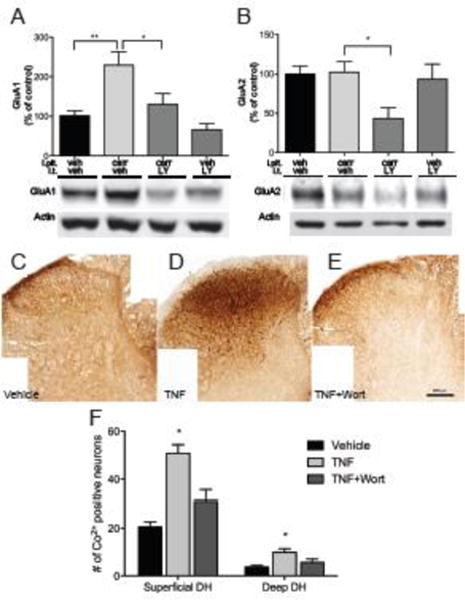
Carrageenan-induced AMPAr trafficking and membrane insertion of Ca2+ permeable AMPAr is PI3K dependent. (A) Pretreatment with i.t. LY294002 (LY) blocks carrageenan-induced trafficking of GluA1 to the plasmalemma in ipsilateral dorsal horn. (B) Injection of LY prior to paw injection leads to a pronounced reduction in plasmalemma GluA2, which is not observed in non-inflamed animals. (C–E) Representative examples of Co2+ uptake induced by kainate in lumbar spinal cord sections incubated with vehicle, TNF, or Wortmannin (Wort), a non-selective PI3K antagonist. (F) Quantification of cells with positive Co2+ staining shows increased Co2+ uptake in both superficial and deep lumbar dorsal horn sections after TNF incubation, which is prevented Wort. Data is presented as mean±SEM, n=3, * p < 0.05, ** p < 0.01
Figure 4.
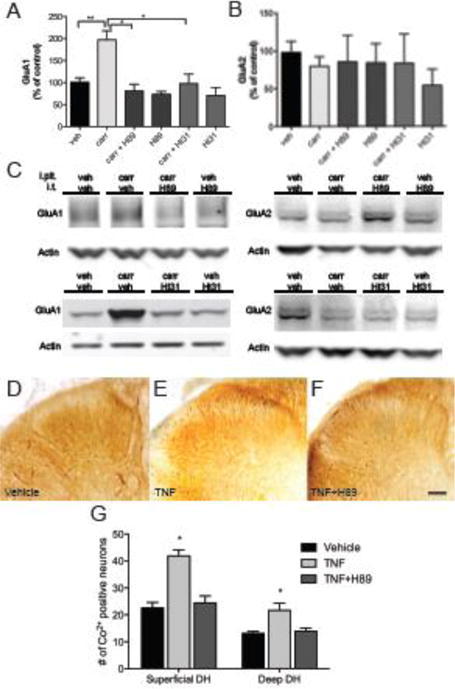
Carrageenan-induced AMPAr trafficking and insertion of Ca2+ permeable AMPAr into neuronal membranes is PKA dependent. (A) Pretreatment with H89, a PKA inhibitor, or St-Ht31, which blocks the interaction of PKA with A-Kinase anchoring proteins, blocks carrageenan-induced trafficking of GluA1 to the plasmalemma in ipsilateral dorsal horn, (B) while levels of GluA2 are unchanged. (C) Representative Western blots. (D–F) Examples of Co2+ uptake induced by kainate in lumbar spinal cord sections incubated with vehicle, TNF, or H89. (G) Quantification of cells with positive Co2+ staining shows an increase of Co2+ uptake in both superficial and deep lumbar dorsal horn sections after incubation with TNF, which is prevented by H89. Data is presented as mean±SEM, n=3, * p < 0.05, ** p < 0.01
Figure 5.
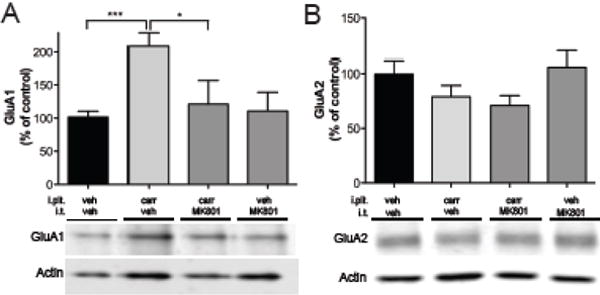
Carrageenan-induced AMPAr trafficking is NMDA receptor dependent. (A) Pretreatment with MK801, an NMDA receptor antagonist, blocks inflammation –induced trafficking of GluA1 to the plasma membrane. (B) MK801 treatment had no affect on GluA2 at this time. Data is presented as mean±SEM, * n=4, p < 0.05, and *** p < 0.001
To measure changes in numbers of calcium-permeable AMPAr in cells, spinal cord slices were treated with Co2+ in the presence of kainite to visualize differences between treatments. We first examined basal numbers of Co2+-positive neurons throughout the dorsal horn after kainate-induced Co2+ -staining. Inspection of the tissue indicated that while all of the tissue analyzed for the wortmannin studies came from the lumbar enlargement (L4-6), much of the tissue used for the H89 experiments (which were performed first) came from more rostral lumbar cord. This difference in rostro-caudal level had no effect on basal numbers of stained neurons in the superficial dorsal horn (22.6 ±2.0 neurons for set 1 H89 and 20.6 ±2.1 neurons for set 2, wortmannin; p≤0.56) (fig 3F, 4G). There was, however, a difference in basal staining in laminae IV–V with more neurons stained in the more rostral segments (means 13.0 ±0.7, set 1 and 3.4 ±0.6, set 2; p≤0.001). In all cases, the Co2+-positive cells encompassed small to large neurons with no particular anatomical distribution. The general pattern of basal staining is quite similar to that reported by Kopach et al. (Kopach et al., 2011) who observed just over 20 Co2+ positive cells/section in laminae I–II and approximately 10 Co2+ positive cells/section in laminae III–V in control rats. Variations in number of stained cells in deep dorsal horn could result from some combination of absolute rostro-causal level as shown here or age of the animals as older rats exhibit less kainate induced cobalt staining in the spinal cord (Engelman et al., 1999).
Irrespective of level or lamina, exposure to TNF consistently produced a significant increase in the number of neurons that exhibited kainate –induced Co2+ -staining (fig. 3F, 3G) indicating that TNF induced trafficking of functional GluA1 enriched AMPA receptors to the plasmalemma is a common neuronal event. In the 2nd set of experiments, when all tissues were in the lumbar enlargement this was seen as a 2.5 to 3 fold increase in Co2+-positive neurons in superficial and deep dorsal horn, respectively. Representative images of superficial laminae neurons stained after TNF exposure are shown in figures 3D and 4E. In comparison, rats injected with complete Freund’s adjuvant 24 h before the experiment where the number of stained cells in superficial (I–II) and deep (laminae III–VII) of the lumbar enlargement increased by 2.77 and 3.73 fold, respectively (Kopach et al., 2011). Given the differences in inflammatory stimuli (TNF incubation vs intraplantar CFA) as well as differences in delimiting the deep dorsal horn, the results are remarkable similar. In more rostral levels of the lumbar spinal cord, percentage increases in TNF-induced staining fell to 85 and 65% (laminae IV and V) over basal, it must be noted that for the deep dorsal horn in this tissue, despite the smaller percentage increase, the increase in absolute numbers of stained neurons was considerable and significant. Pretreatment with wortmannin reduced the TNF-induced increase in stained neurons in the superficial laminae (p≤0.016, fig 3E–F) and although there was a tendency for the levels to be above those seen in the control slices, this difference was not significant. Pretreatment with the PKA antagonist H89 blocked TNF-induced staining in both superficial (p≤0.001) and deep dorsal horns (p≤0.01, fig. 4F–G). Importantly, in the presence of TNF and wortmannin, numbers were no different than those seen under control conditions. The difference between the number of stained neurons in the deeper laminae under basal conditions could be due to the different levels of the spinal cord examined, to technical issues, e.g. different types/numbers of neurons or smaller diameter allowing for easier diffusion of the kainate, or alternatively, it could be due to evolving changes in our techniques over time. Given the strong similarity in the superficial laminae, it is more likely due to the spinal level sampled.
Mice unable to phosphorylate GluA1 do not differ from WT in mechanical or thermal thresholds
Phosphorylation of ser 831 and ser 845 on GluA1 are important events in the regulation of GluA1 subunit trafficking into neuronal membranes. In order to examine whether this affects pain behavior in our paradigm, we used mice in which GluA1 ser 831 or ser 845 had been substituted with alanine throughout the body. Changes in acute nociceptive thresholds were assessed by measuring acute thermal and mechanical withdrawal thresholds. Male mice with an alanine substitution on GluA1 for ser 831 had acute thermal withdrawal latencies of 5.61 s ±0.29, their male wild type littermates had latencies of 6.01 s ±0.29. Male mice with an alanine substituted for serine at the 845 residue of GluA1 and their male wild type littermates were also tested. The mean withdrawal latency for the male ser 845 knock in mice was 6.17 s ±0.32 and that for the wild type mice was 5.73 s ±0.34. There was no difference between the two mouse lines (p>0.05) (fig. 6A, B). We then looked at basal 50% probability mechanical withdrawal thresholds of mice. Male ser 831 mutant mice had thresholds of 1.83 g ±0.06 while their wild type counterparts had thresholds of 1.56 g ±0.12. The ser 845 males had thresholds of 1.4 g ±0.15 and their wild type littermates had mean thresholds of thresholds of 1.6 g ±0.15. (p>0.05; figs 6C,D). No difference in the mean mechanical thresholds in female ser 845 knock in mice (1.89 g ±0.06) and the female wild type controls (1.94 g ±0.05), was observed (fig. 6E; p>0.05). Following basal mechanical threshold determination, mutant and wild type mice were treated with intraplantar carrageenan. Injection precipitated a largely identical inflammatory response and drop in mechanical withdrawal threshold in both strains over 4 h of observation (p>0.05; figs 6C, D).
Figure 6.
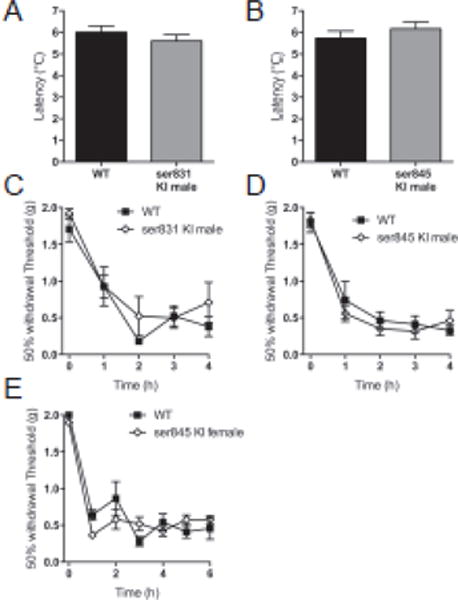
Ability of mice to phosphorylate GluA1 at ser 831 or ser 845 has no effect on pain behavior. Acute thermal thresholds of (A) male ser 831 mice that cannot phosphorylate GluA1 at the PKC site, and (B) male ser 845 mice that cannot phosphorylate GluA1 at the PKA site, are unchanged from their wild type littermates. (C–E) Development of post-carrageenan mechanical hyperalgesia in male and female ser 831 mice and male ser 845 mice is identical to their wild type littermates.
Loss of the PKA or PKC GluA1 phosphorylation site has no effect in phase 2 of the formalin test
Following formalin injection, flinches were counted in one-min bins for 60 min and summed for phase 1 and 2. Consistent with the lack of difference seen for acute thermal latencies and mechanical thresholds, ser 831 mice have identical phase 1 responses as their wild type littermates (Table I). Counter to our expectations, they also showed no difference in their phase 2 responses. Ser 845 mice (lacking the PKA phosphorylation site showed a diminished phase 1 response, but were no different than control with respect to their phase 2 response. Unusually, the phase 1 response of both the ser 845 knock in and wild type mice were substantially less than that seen for the 831 mice, while the phase 2 responses of all four strains were roughly equivalent.
Table I.
Phosphorylation of GluA1 at ser 845 or ser 831 does not affect the second phase of the formalin test. Number of flinches after formalin injection in the hind paw in mice unable to phosphorylate GluA1 at either ser 831 or ser 845 showed that the mice responded that same as their respective wild type in the second phase, while ser 845 had reduced flinching in the first phase. Data is presented as mean±SEM,
| Phase 1 | Phase 2 | |
|---|---|---|
| Ser 831 knock in mice | 231.1 ±25.6 | 1079 ±64.6 |
| Ser 831 wild type | 225.0 ± 41.8 | 930.7 ±125.0 |
| Ser 845 knock in mice | 72.4 ±12.1* | 938.2 ±77.6 |
| Ser 845 wild type | 148.9±27.1 | 1088.0 ±127.4 |
represents p < 0.05
Loss of the PKA or PKC GluA1 phosphorylation site prevents TNF-induced AMPAr insertion in slices
Given the lack of expected behavioral differences in facilitated pain states, we decided to look at TNF induced insertion of Ca2+ permeable AMPA receptors in dorsal horn neurons. This would bypass both the peripheral nervous system and higher cognitive centers and focus exclusively on dorsal horn neuronal changes. Similar to results observed in rats, incubation with TNF resulted in increased numbers of Co2+ stained neurons throughout the dorsal horn compared to control conditions for wild type littermates for both knock-in strains. This was more prominent for littermates of the ser 845 mice (p≤0.01). Paired testing of spinal sections treated with vehicle and TNF from the same mouse indicated that it was also true for the wild type mice derived from the ser 831 strain (p≤0.05). Representative images of superficial dorsal horn neurons, stained after TNF exposure, are shown in figure 7A,C. No difference, paired or otherwise, was present in tissue from either homozygous knock-in strain, between vehicle and TNF-treated conditions. This indicated that although we were not able to observe behavioral changes between the knock-ins and the wild types, there are physiological differences readily apparent at the level of the spinal dorsal horn. Thus, phosphorylation at both ser 831 (PKC binding site) and ser 845 (PKA binding site) is necessary for the TNF activity-induced trafficking of Ca2+ permeable AMPAr to the plasma membrane.
Figure 7.
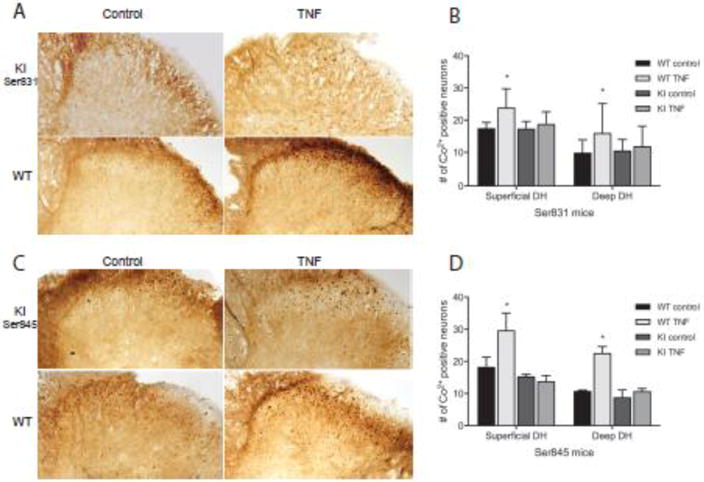
Ability of mice to phosphorylate GluA1 at ser 831 or ser 845 controls TNF insertion of Ca2+ permeable AMPAr into neuronal membranes. (A,C) Representative examples of Co2+ uptake induced by kainate in lumbar spinal cord sections incubated with vehicle or TNF, taken from knock in and wild type mice. (B, D) Quantification of cells with positive Co2+ staining shows an increase in Ca2+ permeable AMPAr in both superficial and deep lumbar dorsal horn sections after incubation with TNF in wild type mice. This effect was lost in slices from both ser 831 and ser 845 knockin mice. Data is presented as mean±SEM, n=3, * p < 0.05
Discussion
The present study demonstrates that peripheral inflammation induced a rapid increase in plasma membrane GluA1, consistent with previous work (Choi et al., 2010; Tao, 2012) and goes further to show that there is an increase in the GluA1/GluA2 ratio at the synapse. Increases of membrane GluA4 were also demonstrated within the same timeframe, while the synaptic increase did not reach significance this could be due to the tissue harvest time or other variables that were optimized for GluA1 changes. While inflammation-induced movement of GluA2 out of the membrane did not reach significance for all individual experiments, summing the Western blot data across all of the experiments indicated that intraplantar carrageenan also induced a small, yet significant decrease. The resulting increases in membrane GluA1/GluA2 and GluA4/GluA2 ratios imply an increase in Ca2+ permeable AMPA receptors and consequently increased synaptic strength (Bredt and Nicoll, 2003; Gu et al., 1996; Hartmann et al., 2004). While Ca2+ permeable (GluA2-containing) AMPA receptors mediate the majority of acute pain sensations, AMPA receptor subunit trafficking resulting in an increased GluA1/GluA2 ratio comes into prominence in a number of painful conditions, including tissue and nerve injury (Katano et al., 2008; Park et al., 2009; Vikman et al., 2008; Wang et al., 2011). The trafficking of GluA4 enriched and GluA2 lacking receptors in spinal cord has been less studied, but the relationship of membrane GluA4 to spinal sensitization and nociception is starting to emerge (Cabañero et al., 2013; Polgár et al., 2010). Interestingly, the nature of the injury may govern the final pattern of the GluA1/GluA2 trafficking and possibly that of GluA4 (Tao, 2012). In some acute pain models, notably skin incision (but see (Atianjoh et al., 2010)), capsaicin injection and acute inflammation, insertion of GluA1 enriched AMPA receptors into the membrane occurs with little to no decrease in GluA2 (Galan et al., 2004; Larsson and Broman, 2008; Pezet et al., 2008). In models of longer lasting inflammation, a decrease in GluA2 appears to be the more consistent change observed at the period of peak nociception (Park et al., 2009; Tao, 2010). The mechanisms of insertion and deletion are beginning to be elucidated, e.g. GluA2 removal from the membrane is NMDA receptor dependent and requires phosphorylation of GluA2 at ser 880 (Beattie et al., 2000; D. Z. Lee et al., 2012; Malinow and Malenka, 2002; Park et al., 2009; Seidenman et al., 2003). Morphine-induced hypersensitivity appears to be a unique pain state thus far, in that it is associated with trafficking of GluA4, but not GluA1 homomeric AMPA receptors to synaptic membranes in laminae III–V (Cabañero et al., 2013). The present results indicate a predominant inflammation/injury-induced insertion of Ca2+ permeable AMPA receptors and deletion of GluA2 containing Ca2+ impermeable AMPA receptors and are thought to reflect the sum of the constitutive balance of AMPA receptors in the plasma membranes plus the activity driven insertion and deletion of GluA1 (or GluA4) enriched and GluA2 containing AMPA receptors, respectively. Our results demonstrating that paw carrageenan produces reliable increases in synaptic membrane GluA1, with a less prominent decrease in membrane GluA2 in the dorsal horn confirm our previous work and fit with intraplantar carrageenan being a transitional model between the very acute and more chronic models. The increase in non-synaptic membrane GluA4 that we observed is harder to put in context. Given that (Polgár et al., 2010) and (Cabañero et al., 2013) looked at GluA4 in membranes of superficial and deep dorsal horn nociceptive neurons, respectively, confidence is increased in our supposition that we are looking at GluA4 trafficking into the plasma membrane. The GluA4 positive neurons in lamina 1 are associated with a pronounced input from peptidergic afferents, which may strongly link them to changes resulting from inflammation (Polgár et al., 2010). As the Cabañero data from the deep dorsal horn indicates a synaptic localization of the GluA4-enriched receptors, it is highly possible that harvesting at a later time point may have given us that result as GluA1-enriched receptors are inserted first into the extrasynaptic membrane and only then move laterally to the synapse (Ferguson et al., 2008). Indeed, Bredt and Nicoll (2003) have speculated that in cultured hippocampal neurons, extrasynaptic AMPA receptors “serve as a reservoir for synaptic receptors”.
The reasons for injury specific patterns and relative amount of insertion and deletion are still being explored and may depend on the balance of signal transduction cascades engaged as agents as varied as insulin and reactive oxygen species, in addition to NMDA and pro-inflammatory cytokines, are postulated to play a role in varying conditions.
Convergence of signals to regulate GluA1 insertion
Spinal antagonism of TNF or TNFR1 blocked the carrageenan-induced GluA1 behavioral hypersensitivity in the present study. Others, including ourselves, have previously published that spinal antagonism of NMDA receptors (D. Z. Lee et al., 2012; K. Ren et al., 1992) and PI3K (Choi et al., 2010; Leinders et al., 2014; Pezet et al., 2008; Xu et al., 2011) and PKA activity (Jones and Sorkin, 2005; Peng, 2010; Sluka, 2002) all block peripheral injury/inflammation induced pain behavior. Similarly, activity driven receptor trafficking observed in this study was influenced by antagonism of spinal TNF and NMDA receptors and blockade of PI3K and PKA activity. Indeed, antagonism of TNF, NMDA, PI3K or PKA activity prior to carrageenan resulted in membrane GluA1 levels no different from control (fig. 8). In parallel studies, using kainate induced cobalt labeling, we showed in vitro that exposure to TNF increases the number and/or activity of Ca2+-permeable AMPAr in dorsal horn neurons and that this increase is also dependent on both PI3K and PKA activity. These data are complementary to our Western blot findings. It is unknown what percentage of these TNF-induced results reflects GluA4 containing Ca2+ permeable AMPA receptors, but if it were to be significant, we speculate that their trafficking is also PI3K and PKA dependent. Moreover, we have recently shown that TNF-induced insertion of functional Ca2+ permeable AMPA receptors into the plasma membrane of motor neurons is both PI3K and PKA dependent (Yin et al., 2012). PI3K is thought to exert its effects following formation of a complex with GluA1. Activation of the chemokine receptor CXCR2 results in its forming a complex together with PI3K and GluA1 and elicits a PKA dependent phosphorylation of GluA1 ser 845 in hippocampal neurons, (Catalano et al., 2008), a similar mechanism may exist in dorsal horn nociceptive neurons downstream of the TNFR1 receptor. PI3K also forms a complex with an atypical isoform of PKC and GluA1 in hippocampal neurons, formation of this complex leads to phosphorylation of GluA1 ser818 and insertion of the AMPA receptor into the synaptic membrane (S.-Q. Ren et al., 2013). In addition, PIP3, a product of PI3K activation, is thought by many to be involved in AMPA receptor trafficking to and clustering at the post-synaptic density (Arendt et al., 2010).
Selective blockade of the TNF pathway in the face of maintained afferent drive results in uncoupling of Glu subunits and a massive loss of membrane GluA2
We have previously shown that following paw carrageenan, phosphorylation of GluA1 at ser 845, the PKA phosphorylation site is downstream of TNF (Choi et al., 2010). Spinal NMDAr activation also induces PKA dependent phosphorylation at ser 845 with subsequent AMPA receptor insertion into the membrane (Ehlers, 2000; Esteban et al., 2003; Lu et al., 2001; Peng et al., 2011). These data imply that TNF and glutamate acting on TNF and NMDA receptors respectively, presumably working in parallel, participate in and are required for carrageenan-induced sensitization. Furthermore, they imply that PKA is downstream of both mediators. Interestingly, inhibition of PKA activity prior to carrageenan injection has effects on membrane GluA2 no different than that of NMDA receptor inhibition, i.e. no different than control animals that received vehicle (fig. 8). However, additional removal of GluA2 was observed in the presence of carrageenan following intrathecal pretreatment with either ETA or the PI3K inhibitors, the portion of the cascade that is uniquely downstream of TNF and not the NMDAr.
The detailed mechanisms regulating GluA2 insertion and removal from membranes are not fully understood. Following NMDAr activation, the initial removal of GluA2 – containing AMPAr from spinal membranes in pain models is frequently attributed to phosphorylation of GluA2 at ser 880 by PKC, this phosphorylation disrupts the anchoring of the receptor to the post synaptic density, (Kopach et al., 2013; Park et al., 2009). However, it has been proposed that the phosphorylation state of GluA2 ser 880 is more involved with later recycling of GluA2 back to the membrane than the initial internalization (Lin and Huganir, 2007).
The massive decrease in membrane GluA2 containing AMPAr appears to occur only when the NMDAr/PKC pathway is activated by normal afferent drive initiated by the peripheral carrageenan, while the parallel TNF/PI3K pathway is inhibited. This outcome may depend on a specific characteristic of the adaptor protein, PICK1 (protein interacting with C kinase-1). PICK1 is a Ca2+ sensing protein that binds the PZD domain on the GluA2, but not the GluA1 AMPAr subunit (Hanley, 2008; Xia et al., 1999). The strength of the PICK1-GluA2 bond may be either increased, thus restricting the bound GluA2 to the endosomal compartment or decreased, resulting in liberating the previously bound GluA2 from the endosomes (Hanley and Henley, 2005; Terashima et al., 2004). Mice lacking the PICK1 protein or the GluA2 PZD domain have normal protein levels of GluA2, but fail to induce LTD in cerebellar neurons (Steinberg et al., 2006). Following peripheral stimulation with carrageenan, increased cytosolic Ca2+, possibly through the ionotropic NMDAr, increases the strength of the PICK1-GluA2 interaction in the cytosol; this restricts GluA2 access to the plasma membrane (Terashima et al., 2004) (fig. 9). Under normal conditions with no antagonism of the TNF/PI3K pathway, subsequent insertion of Ca2+-permeable AMPAr into the plasma membrane, causes further Ca2+ influx and increased intracellular concentration of Ca2+. This disrupts the PICK1-GluA2 interaction, creating a negative feedback loop to restore normal GluA2 trafficking (Jaafari et al., 2012). Blocking the insertion of Ca2+-permeable AMPAr with either ETA or inhibition of PI3K could alter this negative feedback loop, leaving the GluA2 sequestered with PICK1 in the endosomal compartment and shifting the set point of the remaining “free” GluA2 containing AMPAr trafficking towards the cytosol (fig. 9). This would be consistent with our Western blot data where ETA or LY294002 pretreatment before carrageenan blocked insertion of GluA1 into the plasma membrane while keeping GluA2 in endosomes. This is manifested as a pronounced decrease in membrane GluA2 that is not seen in the absence of carrageenan (low afferent drive) or in the absence of TNF or PI3K (negative feedback system intact.
Figure 9.
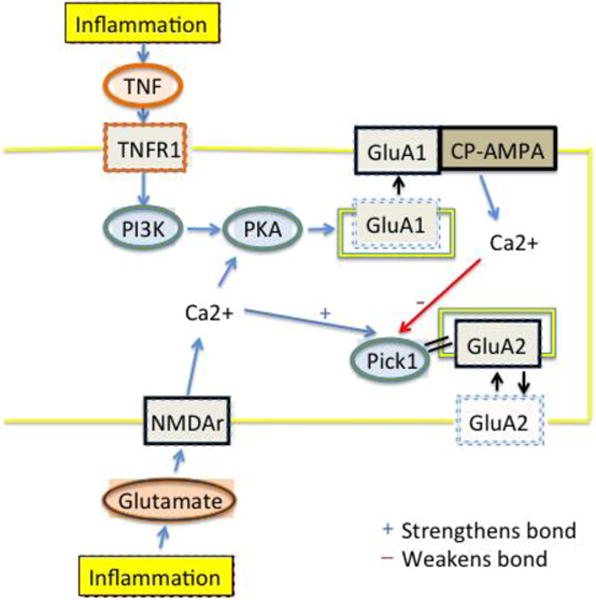
Cartoon indicating that paw carrageenan induces spinal release of TNF acting at TNFR1 and glutamate acting on NMDAr. Activation of NMDAr allows Ca2+ influx, which strengthens the PICK-1/GluA2 bond and sequesters the receptor within cytosolic endosomes. Any GluA2 involved in constitutive trafficking or actively released from the membrane by phosphorylation at ser 880 will be unable to recycle back to the membrane. Activation of TNFR1, PI3K and PKA induces phosphorylation of GluA1 and subsequent insertion of GluA1 containing and GluA2 subunit lacking AMPA receptors into the plasma membrane-these form Ca2+ permeable AMPA receptors (CP-AMPA). NMDAr activation activates this pathway less directly. This hypothesis rests on the fact that local Ca2+ concentrations at the endosomes will vary as a function of differing amounts of Ca2+ influx through the different ionophores and the distance of each ionophore to the PICK1. We postulate that given the known steep gradients of cytosolic Ca2+, that activation of each of the receptor types could result in different local Ca2+ concentrations able to strengthen and weaken the PICK-1/GluA2 bond.
Dissociation of physiological change in DH neurons from behavioral outcome
Posttranslational modification of the GluA1 subunit consisting of phosphorylation at ser 845 is necessary for full enhancement of synaptic AMPA receptors. Phosphorylation at ser 845 is necessary, but not sufficient for plasma membrane insertion of GluA1 containing AMPA receptors and long term potentiation (LTP) (Barry and Ziff, 2002; Esteban et al., 2003; Malinow and Malenka, 2002) and also potentiates the response to glutamate (Banke and Traynelis, 2003). In an attempt to further demonstrate that PKA phosphorylation of the GluA1 AMPA receptor subunit was part of the pathway or if PKA was acting at another site, we bred knock in mice that had an alanine substitution at ser 845. In earlier experiments, mice from this line and other knock in mice that could not phosphorylate ser 831 showed a deficit in hippocampal long-term depression (LTD) and LTP, (H.-K. Lee et al., 2003) and both lines showed deficits in spatial learning. Furthermore, while mechanical hypersensitivity following plantar incision was not prevented in the ser 831 phospho-deficient mice, stress-induced prolongation of the incisional pain was inhibited (Li et al., 2014). In a nerve ligation model of neuropathic pain, knock in mice for ser 845, but not ser 831 displayed a lack of mechanical allodynia (Qiu et al., 2014). In our study, behavior of knock in mice of both strains was indistinguishable from that of wild type littermates in two paradigms used to measure hypersensitivity (carrageenan and formalin). However, spinal cord slices taken from the knock in animals and subjected to the TNF-induced trafficking/kainate induced Co2+ labeling paradigm, demonstrated a lack of measurable TNF effect in the knock in animals, but not their wild-type littermates. It must be noted that Co2+ staining would label Ca2+ permeable AMPAr with either homomeric GluA1or GluA4 subunits. Given that these were not conditional knock in animals, it is possible that the remaining intact GluA4 system ascending nociceptive system developed compensatory actions in lieu of a normally functioning GluA1 system, despite the continued larger number of dorsal horn neurons with GluA1 than GluA4 (Polgár et al., 2010). Taken together these in vitro experiments demonstrate that within the relevant dorsal horn neurons, TNF-induced trafficking of Ca2+ permeable AMPA receptors is dependent on phosphorylation of GluA1 by PKA, but that compensatory actions at other levels of the neuraxis were still permissive for pain behavior.
In summary, carrageenan in the paw induced pain behavior and altered GluA1 and GluA4 trafficking in the plasma membrane in the dorsal horn. These affects were mediated by spinal TNF release through PI3K pathway with involvement of the NMDA pathway. Blocking these pathways reduced GluA1 insertion and subsequent increase in Ca2+-permeability in dorsal horn neurons. TNF seems to be a major player in modulating synaptic strength and inducing LTP, which is important in many acute and chronic pain states.
Acknowledgments
We would like to thank Dr. W.M. Campana for reading an earlier copy of this manuscript
This work was supported by NIH RO1 (LSS), Fullbright Foundation (LSS), Wennergren Fellowship (LSS), Swedish Research Council (CIS), Swedish Foundation for Strategic Research (CIS), Ragnar Söderberg Foundation (CIS) and Willian K. Bowes Jr. Foundation (CIS).
Abbreviations
- AMPA
α-amino-3-hydroy-5-methyl-4-isoxazolepropionic acid
- AMPAr
AMPA receptor
- EPSC
excitatory post-synaptic current
- ETA
Etanercept
- i.t.
intrathecal
- LTD
long term depression
- LTP
long term potentiation
- NMDA
N-methyl-D-aspartic acid
- NMDAr
NMDA receptor
- PI3K
phosphatidylinositol 3 kinase
- PKA
protein kinase A
- PKC
protein kinase C
- TNF
tumor necrosis factor
- TNFR1
TNF receptor 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arendt KL, Royo M, Fernández-Monreal M, Knafo S, Petrok CN, Martens JR, Esteban JA. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2010;13:36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atianjoh FE, Yaster M, Zhao X, Takamiya K, Xia J, Gauda EB, Huganir RL, Tao YX. Spinal cord protein interacting with C kinase 1 is required for the maintenance of complete Freund’s adjuvant-induced inflammatory pain but not for incision-induced post-operative pain. 2010;151:226–234. doi: 10.1016/j.pain.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Traynelis SF. Activation of NR1/NR2B NMDA receptors. Nat Neurosci. 2003;6:144–152. doi: 10.1038/nn1000. [DOI] [PubMed] [Google Scholar]
- Barry MF, Ziff EB. Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol. 2002;12:279–286. doi: 10.1016/s0959-4388(02)00329-x. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Cabañero D, Baker A, Zhou S, Hargett GL, Irie T, Xia Y, Beaudry H, Gendron L, Melyan Z, Carlton SM, Morón JA. Pain after discontinuation of morphine treatment is associated with synaptic increase of GluA4-containing AMPAR in the dorsal horn of the spinal cord. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2013;38:1472–1484. doi: 10.1038/npp.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano M, Trettel F, Cipriani R, Lauro C, Sobrero F, Eusebi F, Limatola C. Chemokine CXCL8 modulates GluR1 phosphorylation. J Neuroimmunol. 2008;198:75–81. doi: 10.1016/j.jneuroim.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Choi JI, Svensson CI, Koehrn FJ, Bhuskute A, Sorkin LS. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. 2010;149:243–253. doi: 10.1016/j.pain.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JIL, Koehrn FJ, Sorkin LS. Carrageenan induced phosphorylation of Akt is dependent on neurokinin-1 expressing neurons in the superficial dorsal horn. Molecular pain. 2012;8:4. doi: 10.1186/1744-8069-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson CA, Fitzsimmons BL, Shim JH, Agrawal A, Cohen SM, Hua XY, Yaksh TL. Spinal matrix metalloproteinase 3 mediates inflammatory hyperalgesia via a tumor necrosis factor-dependent mechanism. Neuroscience. 2012;200:199–210. doi: 10.1016/j.neuroscience.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. Journal of neuroscience methods. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Engelman HS, Allen TB, MacDermott AB. The distribution of neurons expressing calcium-permeable AMPA receptors in the superficial laminae of the spinal cord dorsal horn. J Neurosci. 1999;19:2081–2089. doi: 10.1523/JNEUROSCI.19-06-02081.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD. Protein kinases regulate the phosphorylation of the GluR1 subunit of AMPA receptors of spinal cord in rats following noxious stimulation. Brain Res Mol Brain Res. 2003;118:160–165. doi: 10.1016/j.molbrainres.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. Journal of Neuroscience. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Monreal M, Brown TC, Royo M, Esteban JA. The balance between receptor recycling and trafficking toward lysosomes determines synaptic strength during long-term depression. Journal of Neuroscience. 2012;32:13200–13205. doi: 10.1523/JNEUROSCI.0061-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan A, Laird JMA, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Gu JG, Albuquerque C, Lee CJ, MacDermott AB. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381:793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- Hanley JG. ICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. The EMBO Journal. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann B, Ahmadi S, Heppenstall PA, Lewin GR, Schott C, Borchardt T, Seeburg PH, Zeilhofer HU, Sprengel R, Kuner R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–650. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Jaafari N, Henley JM, Hanley JG. PICK1 mediates transient synaptic expression of GluA2-lacking AMPA receptors during glycine-induced AMPA receptor trafficking. Journal of Neuroscience. 2012;32:11618–11630. doi: 10.1523/JNEUROSCI.5068-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TL, Sorkin LS. Activated PKA and PKC, but not CaMKIIalpha, are required for AMPA/Kainate-mediated pain behavior in the thermal stimulus model. 2005;117:259–270. doi: 10.1016/j.pain.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Katano T, Furue H, Okuda-Ashitaka E, Tagaya M, Watanabe M, Yoshimura M, Ito S. N-ethylmaleimide-sensitive fusion protein (NSF) is involved in central sensitization in the spinal cord through GluR2 subunit composition switch after inflammation. European Journal of Neuroscience. 2008;27:3161–3170. doi: 10.1111/j.1460-9568.2008.06293.x. [DOI] [PubMed] [Google Scholar]
- Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- Kopach O, Kao SC, Petralia RS, Belan P, Tao YX, Voitenko N. Inflammation alters trafficking of extrasynaptic AMPA receptors in tonically firing lamina II neurons of the rat spinal dorsal horn. 2011;152:912–923. doi: 10.1016/j.pain.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopach O, Viatchenko-Karpinski V, Atianjoh FE, Belan P, Tao YX, Voitenko N. PKCα is required for inflammation-induced trafficking of extrasynaptic AMPA receptors in tonically firing lamina II dorsal horn neurons during the maintenance of persistent inflammatory pain. J Pain. 2013;14:182–192. doi: 10.1016/j.jpain.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson M, Broman J. Translocation of GluR1-containing AMPA receptors to a spinal nociceptive synapse during acute noxious stimulation. Journal of Neuroscience. 2008;28:7084–7090. doi: 10.1523/JNEUROSCI.5749-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DZ, Chung JM, Chung K, Kang MG. Reactive oxygen species (ROS) modulate AMPA receptor phosphorylation and cell-surface localization in concert with pain-related behavior. 2012;153:1905–1915. doi: 10.1016/j.pain.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lehel C, Oláh Z, Jakab G, Szállási Z, Petrovics G, Harta G, Blumberg PM, Anderson WB. Protein kinase C epsilon subcellular localization domains and proteolytic degradation sites. A model for protein kinase C conformational changes. J Biol Chem. 1995;270:19651–19658. doi: 10.1074/jbc.270.33.19651. [DOI] [PubMed] [Google Scholar]
- Leinders M, Koehrn FJ, Bartok B, Boyle DL, Shubayev V, Kalcheva I, Yu NK, Park J, Kaang BK, Hefferan MP, Firestein GS, Sorkin LS. Differential distribution of I3Kisoforms in spinal cord and dorsal root ganglia: Potential roles in acute inflammatory pain. 2014 doi: 10.1016/j.pain.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. Journal of Neuroscience. 2008;28:2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Yang Y, Liu S, Fang H, Zhang Y, Furmanski O, Skinner J, Xing Y, Johns RA, Huganir RL, Tao F. Stress induces pain transition by potentiation of AMPA receptor phosphorylation. Journal of Neuroscience. 2014;34:13737–13746. doi: 10.1523/JNEUROSCI.2130-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. Journal of Neuroscience. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Nagy GG, Al-Ayyan M, Andrew D, Fukaya M, Watanabe M, Todd AJ. Widespread expression of the AMPA receptor GluR2 subunit at glutamatergic synapses in the rat spinal cord and phosphorylation of GluR1 in response to noxious stimulation revealed with an antigen-unmasking method. Journal of Neuroscience. 2004;24:5766–5777. doi: 10.1523/JNEUROSCI.1237-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol. 2005;193:384–393. doi: 10.1016/j.expneurol.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Park JS, Voitenko N, Petralia RS, Guan X, Xu JT, Steinberg JP, Takamiya K, Sotnik A, Kopach O, Huganir RL, Tao YX. Persistent inflammation induces GluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. Journal of Neuroscience. 2009;29:3206–3219. doi: 10.1523/JNEUROSCI.4514-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Yaster M, Guan X, Xu JT, Shih MH, Guan Y, Raja SN, Tao YX. Role of spinal cord alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in complete Freund’s adjuvant-induced inflammatory pain. Molecular pain. 2008;4:67. doi: 10.1186/1744-8069-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HY. PKA-dependent spinal AMPA receptor trafficking mediates the capsaicin-induced colon-urethra cross-organ reflex sensitization. PKA-dependent spinal AMPA receptor trafficking mediates the capsaicin-induced colon-urethra cross-organ reflex sensitization. 2010:1–42. doi: 10.1097/ALN.0b013e3181fe4204. [DOI] [PubMed] [Google Scholar]
- Peng HY, Chang CH, Tsai SJ, Lai CY, Tung KC, Wu HC, Lin TB. Protein kinase A-dependent spinal α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate-receptor trafficking mediates capsaicin-induced colon-urethra cross-organ reflex sensitization. Anesthesiology. 2011;114:70–83. doi: 10.1097/ALN.0b013e3181fe4204. [DOI] [PubMed] [Google Scholar]
- Pezet S, Marchand F, D’Mello R, Grist J, Clark AK, Malcangio M, Dickenson AH, Williams RJ, McMahon SB. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. Journal of Neuroscience. 2008;28:4261–4270. doi: 10.1523/JNEUROSCI.5392-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgár E, Al-Khater KM, Shehab S, Watanabe M, Todd AJ. Large projection neurons in lamina I of the rat spinal cord that lack the neurokinin 1 receptor are densely innervated by VGLUT2-containing axons and possess GluR4-containing AMPA receptors. Journal of Neuroscience. 2008;28:13150–13160. doi: 10.1523/JNEUROSCI.4053-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgár E, al Ghamdi KS, Todd AJ. Two populations of neurokinin 1 receptor-expressing projection neurons in lamina I of the rat spinal cord that differ in AMPA receptor subunit composition and density of excitatory synaptic input. Neuroscience. 2010;167:1192–1204. doi: 10.1016/j.neuroscience.2010.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zhang M, Liu Y, Guo Y, Zhao H, Song Q, Zhao M, Huganir RL, Luo J, Xu H, Zhuo M. GluA1 phosphorylation contributes to postsynaptic amplification of neuropathic pain in the insular cortex. Journal of Neuroscience. 2014;34:13505–13515. doi: 10.1523/JNEUROSCI.1431-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ren K, Hylden JL, Williams GM, Ruda MA, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain. 1992;50:331–344. doi: 10.1016/0304-3959(92)90039-E. [DOI] [PubMed] [Google Scholar]
- Ren SQ, Yan JZ, Zhang XY, Bu YF, Pan WW, Yao W, Tian T, Lu W. PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. The EMBO Journal. 2013;32:1365–1380. doi: 10.1038/emboj.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 Serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. Journal of Neuroscience. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA. Stimulation of deep somatic tissue with capsaicin produces long-lasting mechanical allodynia and heat hypoalgesia that depends on early activation of the cAMP pathway. Journal of Neuroscience. 2002;22:5687–5693. doi: 10.1523/JNEUROSCI.22-13-05687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. Journal of Neuroscience. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. Journal of Neuroscience. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX. AMPA receptor trafficking in inflammation-induced dorsal horn central sensitization. Neurosci Bull. 2012;28:111–120. doi: 10.1007/s12264-012-1204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao YX. Dorsal horn alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking in inflammatory pain. Anesthesiology. 2010;112:1259–1265. doi: 10.1097/ALN.0b013e3181d3e1ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JTR. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. Journal of Neuroscience. 2004;24:5381–5390. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikman KS, Rycroft BK, Christie MJ. Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J Physiol (Lond) 2008;586:515–527. doi: 10.1113/jphysiol.2007.145581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mu X, Wu J, Wu A, Fang L, Li J, Yue Y. Differential roles of phosphorylated AMPA receptor GluR1 subunits at Serine-831 and Serine-845 sites in spinal cord dorsal horn in a rat model of post-operative pain. Neurochemical research. 2011;36:170–176. doi: 10.1007/s11064-010-0288-y. [DOI] [PubMed] [Google Scholar]
- Willis WD. Role of neurotransmitters in sensitization of pain responses. Ann N Y Acad Sci. 2001;933:142–156. doi: 10.1111/j.1749-6632.2001.tb05821.x. [DOI] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Xu Q, Fitzsimmons B, Steinauer J, O’Neill A, Newton AC, Hua X-Y, Yaksh TL. Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. Journal of Neuroscience. 2011;31:2113–2124. doi: 10.1523/JNEUROSCI.2139-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Ozaki G, McCumber D, Rathbun M, Svensson C, Malkmus S, Yaksh MC. An automated flinch detecting system for use in the formalin nociceptive bioassay. J Appl Physiol. 2001;90:2386–2402. doi: 10.1152/jappl.2001.90.6.2386. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Hsu CI, Yu S, Rao SD, Sorkin LS, Weiss JH. TNF-α triggers rapid membrane insertion of Ca(2+) permeable AMPA receptors into adult motor neurons and enhances their susceptibility to slow excitotoxic injury. Exp Neurol. 2012;238:93–102. doi: 10.1016/j.expneurol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HZ, Sensi SL, Carriedo SG, Weiss JH. Dendritic localization of Ca(2+)-permeable AMPA/kainate channels in hippocampal pyramidal neurons. J Comp Neurol. 1999;409:250–260. doi: 10.1002/(SICI)1096-9861(19990628)409:2<250∷AID-CNE6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]