

Abstract
The capabilities of the polarizable force fields for alchemical free energy calculations have been limited by the high computational cost and complexity of the underlying potential energy functions. In this work, we present a GPU based general alchemical free energy simulation platform for polarizable potential AMOEBA. Tinker-OpenMM, the OpenMM implementation of the AMOEBA simulation engine has been modified to enable both absolute and relative alchemical simulations on GPUs, which leads to a ~200-fold improvement in simulation speed over a single CPU core. We show that free energy values calculated using this platform agree with the results of Tinker simulations for the hydration of organic compounds and binding of host-guest systems within the statistical errors. In addition to absolute binding, we designed a relative alchemical approach for computing relative binding affinities of ligands to the same host, where a special path was applied to avoid numerical instability due to polarization between the different ligands that bind to the same site. This scheme is general and does not require ligands to have similar scaffolds. We show that relative hydration and binding free energy calculated using this approach match those computed from the absolute free energy approach.
Keywords: AMOEBA, Free Energy Calculation, Graphics Processing Units, Tinker, OpenMM
Graphical Abstract
We have developed Tinker-OpenMM, a computational platform for the calculation of free energies on Graphics Processing Units. This allows for free energy simulations to be run 200 times faster than is possible on a single CPU core. In addition, we have developed a platform for the calculation of relative free energies.
INTRODUCTION
Free energy is the driving force for spontaneous molecular processes and accurate alchemical free energy calculations can benefit a broad range of chemical and biomedical applications1–5. The accurate prediction of the binding affinities for ligands to their target proteins has been a great challenge in the computational drug development process6. Today, it is common to utilize empirical docking algorithms in the identification of potential lead compounds7–11. However, in order to screen large ligand libraries in a short amount of time, empirical docking typically relies on incomplete physics models12, and only account for limited system dynamics (such as loop flexibility) when predicting ligand affinity13. These limitations result in a lack of the accuracy necessary for lead optimization14,15. The calculation of ligand binding free energies from elaborated molecular simulations has also been limited by a combination of underlying force fields and sampling algorithms16,17.
One pathway for the calculation of binding free energies is the double decoupling approach. In this approach, one includes a parameter (lambda) that controls the interaction of a ligand with its environment. When transitioning from lambda=1 (full ligand intermolecular interaction) to lambda=0 (no ligand intermolecular interaction), a ligand’s interaction with its environment is evaluated. Simulations of the system are conducted with the solvated ligand and the protein-ligand complex, and the binding free energy is calculated as the complexation energy minus the solvation energy, plus standard state and other corrections18. In this methodology restraints19 are often used to keep the ligand bound to the protein complex throughout the decoupling process. The magnitude of this restraint term is then analytically corrected for.
Another major class of approaches of binding free energy involve the calculation of the potential of mean force. In these approaches, pioneered by the Roux lab20, one calculates the average force needed to maintain a system in a given configuration (e.g. the distance and orientation between a ligand and the active site). Free energy is then calculated by calculating the work integral from the starting to ending distances. In order to obtain energy data on all relevant distances, a biasing process such as steered MD21,22 or umbrella sampling20,23 is often used. The advantage of this technique is that it allows for the collection of free energy profiles, including information about the energy barriers to binding. The main challenge of this approach is the difficulty in defining an appropriate reaction coordinate for the biasing process. Therefore, this technique has been mostly applied to systems such as channel proteins24,25 that have an obvious pulling dimension. However, this technique can also be applied to general protein-ligand binding26–28.
The free energy between the bound and unbound states in either approach can be sampled by using various techniques such as free energy perturbation (FEP)3, thermodynamic integration (TI)29, metadynamics30–32 or Orthogonal Space Random Walk (OSRW)33,34. A common method for calculating the free energy between neighboring states in alchemical perturbation is the Bennett acceptance ratio (BAR)35. The free energy of binding can then be calculated as the difference between the ligand-host interaction energy and the ligand-water interaction energy. In thermodynamic integration, one utilizes lambda much like in setting up a simulation for BAR and calculate the numerical integration of <∂H/∂λ)>λ from lambda=0 to lambda=129. Compared to BAR, it can be difficult to determine which discrete values of lambda should be used, as convergence can be difficult in regions of high curvature of <∂H/∂λ)>λ. Due to this, comparison studies36 have suggested that TI simulations may require more states than BAR to reach converged free energies. However, TI simulations require less post-simulation processing than BAR based approaches.
The second ingredient of free energy simulations is the choice of force field. Popular force fields include CHARMM37–40 and AMBER41–44. More recent advances have resulted in the development of force fields with more complex electrostatics models, particularly incorporation of polarization. General polarizable force fields include polarizable multipole based AMOEBA,45–47 polarizable OPLS48–50, fluctuating charge51,52 and Drude-Oscillator53–55 based CHARMM force fields. The defining feature of the AMOEBA force field we have been developing is its electrostatic model based on permanent atomic multipoles, as well as many-body polarization through induced dipoles. These added terms, while computationally expensive, allow for a more rigorous modeling of ligand-protein interaction, particularly at short range, than is possible using a fixed-charge based force field.
Previous work using AMOEBA force field has shown an accurate recapitulation of experimental free energies in small molecules hydration,56,57–59 metal ion hydration60–62, as well as ligand binding in synthetic hosts63, and protein systems64–70. The inclusion of a complex electrostatic force leads to increasing computational cost, so that potential it can benefit even more from parallel computing of protein-scale systems consisting of tens of thousands of atoms. Earlier implementations of AMOEBA in Tinker have utilized OpenMP71, which allows for limited parallelism on commercially available CPUs. Massively parallel computation using AMOEBA is possible on supercomputers using the Tinker-HP package72–74. In addition, AMOEBA has been previously implemented in OpenMM, enabling massively parallel molecular dynamics simulations on GPUs75,76. In order to enable alchemical free energy calculations in OpenMM, we have incorporated “lambda” into force and energy calculation via a soft-core approach,77 which is necessary to remove the singularities in van der Waals (vdW) interactions that occurs when atoms are in close contacts.78 In addition, we modified the tinker-OpenMM interface to allow for perturbation of the electrostatic force via the scaling of electrostatic parameters. Another feature of OpenMM that is now supported by the Tinker-OpenMM interface is the addition of support for the CustomCentroidBondForce. This addition enables the coupling of a two groups of atoms (such as a ligand and its binding site).
Compared to the state of CPU alchemical free energy calculations, GPU alchemical free energy calculations is still in its infancy. It is possible to perform MD simulations on GPUs using a few software, including AMBER79, NAMD80, and OpenMM75. However, very few GPU platforms have yet supported alchemical simulations. In addition to the work with OpenMM-AMOEBA described here, the YANK package81 for the use of OpenMM to simulate AMBER force fields is currently in development. Therefore, the AMOEBA on GPU implementation described here(Tinker-OpenMM) constitutes the first available platform for free energy perturbation simulations on GPUs using a polarizable force field.
It is not always necessary to compute the absolute alchemical free energy, and binding or solvation energies relative to a reference ligand are sufficient. In those cases, it may be advantageous to calculate relative energies instead of absolute energies. Many previous relative binding free energy calculation use a “dummy atom” single topology approach82–86 where a pair of ligands are simulated as a common core of atoms connected to a set of atoms sufficient to describe both desired molecules. This dummy atom approach has been used to calculate a number of molecular properties, including binding free energies83–87 Previous work with the AMOEBA force fields on CPUs have accurately calculated the relative binding free energies of ligands to trypsin using a single topology approach66,67,69. The weakness of this scheme is that it is not general; it is more suitable for pairs of molecules with significant chemical similarity. A different approach is that of dual topology free energy calculation, where two ligands are always present in the binding pocket. Relative complexation free energy is calculated via a path starting in a state with fully ligand 1-environmental interaction, and ending at a state of fully ligand 2- environmental interaction. Dual topology free energy calculations have been possible in CHARMM since the late 80’s88 and have more recently been implemented in AMBER79. However, this dual topology scheme is more difficult to implement in a polarizable force field due to the complexity of the electrostatics making it difficult to selectively “scale” the polarization between two ligands. By utilizing a pathway where only one ligand is charged during any perturbation step, we were able to avoid this complication.
Currently, the ability to perform GPU based platform alchemical simulations, particularly for polarizable force fields, has been limited. In this work, we created Tinker-OpenMM, an OpenMM implementation of AMOEBA that enables alchemical free energy calculations on GPUs, while also adding the capability to perform dual topology simulations to both the Tinker89 and OpenMM75,76 platforms. We then proceed to test the GPU based free energy calculations for hydration free energies of aromatic systems90, absolute and relative binding free energies of the sampl4 host-guest systems91.
IMPLEMENTATION DETAILS
Tinker-OpenMM interface
Tinker-OpenMM is built using an interface to pass tinker coordinates and parameters to OpenMM. Tinker reads in the input key and coordinate files, and passes the relevant variables in to a C++ script. This script then uses the OpenMM C API to create the relevant OpenMM parameters and forces, and initiates GPU Molecular Dynamics simulation. Coordinate saving is then managed by occasionally transferring atomic coordinates and velocities from the GPU to main system memory. Tinker then saves these outputs in Tinker coordinate and velocity files, enabling post-processing by Tinker commands (eg. BAR). This interface was originally created by Mark Friedrichs, Lee-Ping Wang, Kailong Mao, and Chao Lu.
Absolute binding free energy
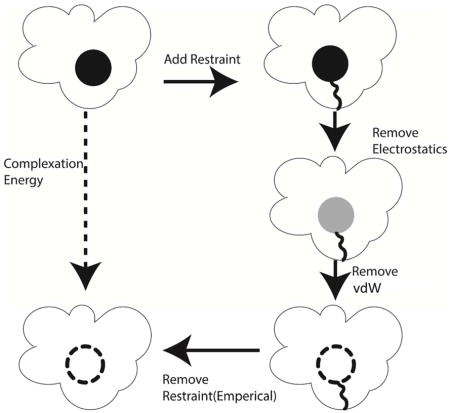
In this work, we employ double-decoupling and alchemical perturbation to compute free energy of binding. First, the electrostatic interactions between the ligand and its environment (water or protein/water) are scaled from 0 to 100% in a series of simulations. With no electrostatic interaction between ligand and surroundings, a series of simulations are run where the (softcore) vdW interactions between ligand and environment are scaled. The path utilized for absolute complexation simulations is shown in Figure 1. This process is also repeated in an aqueous environment to account for hydration free energy.
Figure 1.

Thermodynamic path used to calculate the absolute complexation energy of a ligand using a double-decoupling approach.
After running these simulations, the Bennett Acceptance Ratio (BAR) method is used to calculate the free energy difference between each pair of neighboring states. Since energy is a state function, we can calculate the total complexation energy as the sum of many small perturbations in ligand-environmental interaction strengths. The binding energy is calculated as the complexation free energy, minus the hydration free energy, with the addition of several corrections explained below.
When conducting alchemical perturbation, it is necessary to denote which atoms belong to the ligand. In the simulation system, the ligand atom indices are identified by using the ligand keyword in the key file (e.g. “ligand −1 14” denotes that atoms 1 through 14 belong to a ligand).
Alteration of the electrostatic interactions between the ligand and its environment is accomplished via the scaling of the electrostatic parameters passed from the Tinker interface to OpenMM. The atomic charge, dipole, quadrupole, and polarizability of all ligand atoms are each multiplied by the current simulation electrostatic lambda value (between 0 and 1), which is denoted by the ele-lambda keyword. This results in no electrostatic interaction between the ligand and its environment when ele-lambda=0, and full interaction strength when ele-lambda=1. This methodology also “turns off” the intra-ligand electrostatic interactions. When calculating hydration free energy, the intra-ligand/solute electrostatic contributions are added back by “growing” the electrostatic parameters for ligand alone (in gas phase). However, when calculating binding free energy, this contribution is exactly canceled by an equal omission in the ligand-solvent interaction.
When conducting alchemical perturbation simulations, the change in energy and structure that results from each perturbation needs to be relatively small. To avoid the numerical instability of the standard vdW function when the ligand-environment interaction approaches zero, a softcore buffered 14-7 vdW (energy equation shown below) has been used to calculate the forces and energies.69
| (1) |
Here εij is the well depth, and ρij represents the current interatomic distance divided by rmin, the interatomic distance that results in the lowest vdW energy. In order to use this softcore vdW force, we need to assign the appropriate value of the lambda parameter λij. In this implementation, each ligand atom is assigned a lambda value equal to the vdW-lambda keyword value in the simulation input key file. Each non-ligand atom is assigned a lambda value of 1. When calculating a pairwise vdW interaction, it is necessary to have a set of combining rules to convert two atomic vdW lambdas into a combined, λij. For a pair of atom i and j, λij is determined as the lesser of λi and λj. If the two lambda values are identical (as is the case in an intra-ligand or water-water interaction), λij = 1.
In order to ensure that the ligand stays in the binding pocket even when intramolecular interactions are weak, a distance restraint (k(r − r0)2) is applied between the centers of mass of the ligand and the center of the binding pocket. The bias introduced by the restraint is corrected for at the start and end of our thermodynamic path. The restraint correction at the end of simulation where no intermolecular interaction between ligand and environment is given by92
| (2) |
Here C0 represents standard state concentration (1 mol/L). In this work, we use a force constant (k) of 15 kcal/mol/Å2, and this correction amounts to 6.25 kcal/mol.
In order to remove the ligand restraint from the system with full ligand-protein interaction, we repeat the simulation but with the restraint off. The free energy difference between the two simulations is then calculated using BAR. Alternatively, one could also gradually turn off the restraint while the interaction strength between ligand and protein increases so that no additional correction is needed.
Dual-topology based relative free energy
Relative binding free energy can potentially be calculated more reliably as it avoids simulation of the non-ligand bound form of the protein. In this implementation of the calculation of relative binding free energies, we take a thermodynamic path where we first reduce ligand 1’s electrostatic parameters to zero magnitude. We then proceed to reduce the vdW interactions between ligand 1 and environment, while simultaneously increasing the vdW interactions between ligand 2 and environment. Finally, we increase ligand 2’s electrostatic parameters from zero to full. The path we used to calculate relative complexation energy (ligand binding to receptor in water) is shown in Figure 2. Since the two ligands are never charged at the same perturbation step, ligand 1 and 2 never interact with each other (the vdW interactions are also turned off via the soft-core formula), which requires minimal changes to the electrostatic force in the existing OpenMM code.
Figure 2.

Path used to determine the relative complexation interaction energy of two ligands using a dual topological approach.
In order to run the simulations in our thermodynamic path, we require independent (ligand1 and ligand2) keywords to denote the indices of ligand 1 and ligand 2, respectively. The electrostatic perturbation segments of our path require that we independently control the electrostatic interaction of ligand 1 and ligand2. This is accomplished by having two electrostatic lambda keywords (ele-lambda1 and ele-lambda2, respectively). The charge, dipole, quadrupole and polarizability of each ligand is multiplied by the appropriate ele-lambda variable.
When perturbing the vdW force, we need to assign each ligand atom the correct lambda value. The vdW-lambda of all ligand 1 atoms is equal to the value specified by the vdW-lambda keyword, and vdW-lambda of all ligand 2 atoms is equal to 1 minus vdW-lambda. Therefore, changing the vdw-lambda keyword from 1.0 to 0.0 results in removing all ligand1-environment interactions while setting all ligand2 atoms to full vdW interaction with the environment.
When conducting relative binding simulations or BAR energy calculations, we need to ensure that the two ligands do not interact via the vdW force. Therefore, we need a way for our vdW force and energy calculations kernels to know which ligand each atom belongs to. This is accomplished by adding an internal variable to the vdW force used to designate which ligand (if any) an atom belongs to. This variable is equal to 0 for environmental (non-ligand) atoms, 1 for ligand 1, and 2 for ligand 2. Each pairwise vdW interaction is checked to ensure that ligand1- ligand2 interactions are omitted.
The relative binding free energy is calculated as the relative complexation energy minus the relative hydration energy. Note that if one uses the same force constant for ligand-receptor restraint for all simulations, the restraint correction discussed above is identical for both ligands and drops out in the relative binding free energy.
METHODS
Simulation setup
Prior to all simulation, the system energy was minimized to 1 kcal/mol/Å in order to avoid close atomic contacts. All simulations were run under OpenMM mixed precision mode. Ewald cutoff was set to 7.0 Å, with a 12 Å vdW cutoff in both simulations. All simulations converge the induced dipole moments between iterations to <0.00001 D. Sampl4 and aromatic simulations use a cubic box of 40 Å an Ewald grid of 48 × 48 × 48, while the larger bench7 dataset uses an Ewald grid of 64 × 64 × 64 and a cubic box of 62.23 Å. Example Tinker key files are included in the Supplementary Materials.
Molecular dynamics
Perturbation steps for absolute binding and solvation simulations were conducted with a stepwise reduction of the ele-lambda keyword, followed by a stepwise reduction of the vdw-lambda keyword at 0 ele-lambda. MD used a RESPA integrator, and a BUSSI thermostat. Information on what perturbation steps were used is included in the Supplementary Materials.
Relative binding and solvation simulations were conducting starting with the ele-lambda1 and vdw-lambda keywords at 1.0, and the ele-lambda2 keyword at 0.0. In a series of simulations, the ele-lambda1 keyword is then gradually reduced to 0.0. This is followed by simulations with a stepwise reduction of vdw-lambda1 to 0.0, then a stepwise increase of ele-lambda2 to 1.0.
All CPU simulations were conducted using Tinker dynamic.x for 1ns with a 2fs time step and snapshots saved every 1ps. Each GPU perturbation simulation was conducted using dynamic_omm for 5ns, with a 2fs time-step and snapshots saved every 2ps (except for relative free energy simulations, which had snapshots saved every 1ps). All simulations were conducted at 298K.
Bennett Acceptance Ratio
Bar was computed using Tinker’s BAR program. This program iterates between the two equations below until convergence:
| (3) |
For all CPU based trajectories, BAR used frames 400 to 1000 for calculation, with the initial 400ps equilibration discarded. For absolute free energy trajectories generated on the GPU, BAR used frames 1 to 2500(0–5ns) for calculation. For the relative free energy trajectories generated on the GPU, BAR used frames 1 to 5000(0–5ns) due to more frequently saved snapshots.
Hydration of aromatic compounds
Parameters for the aromatic molecules were previously generated.90 Structures of the 10 compounds are shown in Figure 3. Initial simulation systems were generated by solvating each ligand in water boxes using the Tinker commands solvate and crystal. Initial structures for relative HFE simulations were generated by concatenating ligand 2’s coordinates to the solvated ligand 1 pose.
Figure 3.

Structures of the 12 sampl4 molecules utilized in this study.
In order to calculate the absolute hydration free energy, it is necessary to correct for the contribution of intramolecular electrostatics as we scale the solute electrostatic parameters in “disappearing” or “growing” the solute molecule. The intra-solute electrostatic energy was calculated by running simulations on CPU (this same value was used for both the CPU and GPU simulations). Each molecule was simulated alone in a non-periodic system at ele-lambda values of 0, 0.1, … and 1.0. Simulations were run for 1ns using a time step of 0.1 fs, with structures saved every 0.5 ps at constant volume of 40.0 Å with temperature at 298K. The intra-solute electrostatic energy was then calculated using BAR.
Sampl4 binding simulations
Parameters and starting poses for 12 molecules of the sampl4 dataset were generated as described previously63. Structures of the sampl4 ligands utilized in this study are shown in Figure 4. Relative binding poses were generated as in the relative aromatic simulations.
Figure 4.

Structures of the 10 aromatic compounds used in this study.
The final absolute binding energy was calculated as ΔG of complexation (from no interaction to full interaction) − ΔG of solvation (from no interaction to full interaction) + ΔG of going from no restraint to full restraint at 0 interaction lambda + ΔG of removing the restraint at full interaction energy.
The latest version of Tinker is available at https://github.com/jayponder/tinker. Tinker-OpenMM is available at https://github.com/pren/tinker-openmm. Note that Tinker only works using the modified Tinker-OpenMM, not the main OpenMM release.
RESULTS
Force agreement
Correct simulation of molecular systems requires an accurate calculation of both force and energy. However, since energy is only utilized by Tinker in the BAR process, and isn’t used during OpenMM molecular dynamics, we focused our initial analysis of Tinker-OpenMM on agreement of OpenMM forces with those of Tinker. To ensure that lambda was working in the Tinker-OpenMM implementation, we tested molecule 1 of the sampl4 dataset bound to the host at a range of lambda values, and compared the resulting static forces to those of Tinker. The Tinker-OpenMM platform was able to closely match that of Tinker for all tested lambda values, with a root mean squared error of approximately 8.6*10−4 kcal/mol/Å, and a maximal atomic force deviation of approximately 4.7*10−3 kcal/mol/Å (Table 1). These degrees of deviation are negligible when considering that the RMS force is 31 kcal/mol/Å. The force deviation is partially due to the single precision used in GPU force evaluation.
Table 1.
Force comparison between the Tinker-AMOEBA CPU and Tinker-OpenMM-AMOEBA GPU platforms for Sampl4 molecule 1 at a range of lambda values.
| VDW lambda/ele-lambda | RMSE force (10− 4 kcal/mol/Å) | Max force deviation (10− 3 kcal/mol/Å) |
|---|---|---|
| 1/1 | 8.58 | 4.69 |
| 1/0.5 | 8.59 | 4.66 |
| 1/0.0 | 8.58 | 4.71 |
| 0.5/0.0 | 8.58 | 4.72 |
| 0.0/0.0 | 8.58 | 4.72 |
Computational efficiency
In order to test the speed and scalability of the Tinker-OpenMM platform, we ran 1000 steps of MD on sampl4 molecule 1 (6417 atoms), and the bench7 test case distributed with Tinker (a protein system of 23,558 atoms). For both test systems, the NVidia GTX1070 and GTX 970 were approximately 66-fold and 40-fold faster than an 8 core CPU simulation, respectively (Table 2). A single CPU core is approximately 200-fold slower than simulation on a GTX1070 due to the poor core scalability of Tinker utilizing OpenMP. The GPU platform shows better than linear scaling with respect to system size, with a 3.7-fold increase in particle number resulting in a 2.4-fold or 2.5-fold decrease in speed on the GTX1070 and GTX970 platforms, respectively. This better then linear scaling is likely a result of the smaller sampl4 systems being unable to saturate GPU core utilization, as verified by profiling GPU core utilization during simulations. The change of the vdW force to the softcore 14-7 force resulted in no observable difference in speed compared to the kernel used in OpenMM. This was confirmed by running simulations using a version of Tinker-OpenMM that had been modified to utilize a standard, non-softcore 14-7 vdW force without the presence of the lambda parameter in the codebase.
Table 2.
Performance of Tinker-OpenMM on Nvidia GTX1070 and GTX970 GPUs without the relative binding calculations compared to Tinker CPU running on 8 OpenMP threads (4X of single CPU speed). Values are in nanoseconds/day
| GTX1070 | GTX970 | CPU | |
|---|---|---|---|
| mol01(6417 atoms) | 20.0 | 12.2 | 0.3 |
| bench7(23558 atoms) | 8.3 | 4.8 | 0.16 |
In order to test the cost of utilization of relative vdW, tests were run on bench7 with the relative VDW activated by using two waters (atoms 9000–9002 and 9003–9005) as “ligands” for the alchemical dual topology process. Both of these waters had their ele-lambda values set at 0.0, with a utilized vdW-lambda of 1.0. This allowed for the activation of dual topology kernels without introducing extra costs. This system was minimized, and a speed test was run as above. This resulted in a speed of 4.68 ns/day on a GTX 970, an approximately 2.5% speed reduction when compared to the absolute simulations. This small cost is only present when doing relative free energy calculations; when no ligand2 parameter is set, the cheaper absolute vdW kernel is used for force and energy calculation.
Tinker-OpenMM defaults to a utilizing a “mixed” precision mode in all calculations. This mixed precision mode uses 32-bit floating point calculation for all forces, and integrates using 64-bit floating point precision. Due to the poor double floating point calculation of the consumer GeForce line of graphics cards, the use of double precision for both integration and force calculation results in an 18.1-fold reduction in performance on a GTX 970.
GPU/CPU absolute free energy agreement
As a test of the ability of the Tinker-OpenMM platform to reproduce the results of the Tinker CPU implementation, we performed hydration free energy calculation on a dataset of 10 aromatic compounds, as well as binding free energies on 12 ligands of the sampl4 dataset (9). Both the solvation (Figure 5) and sampl4 datasets (Figure 6) show agreement within the uncertainty of BAR, with R2 values of (0.9924) and (0.9987), respectively. This, along with the static force calculations provides strong evidence that the GPU and CPU implementations of the AMOEBA force field produce comparable results. The fact that a high degree of agreement is possible despite the fact that the GPU simulations were run for 5 times longer (5ns vs 1ns at each perturbation step) is an indication that the tested systems converge relatively rapidly.
Figure 5.

Comparison between the sampl4 binding free energies of 12 sampl4 compounds computed by the Tinker-OpenMM GPU and Tinker CPU platforms. GPU simulations were run for 5ns at each perturbation step, while CPU simulations were run for 1ns.
Figure 6.

Comparison between the calculated solvation free energies for the 10 molecule aromatic compound dataset on the Tinker-OpenMM GPU and Tinker CPU platforms.
GPU/CPU relative free energy agreement
We then proceeded to test the capability of the dual-topology based relative free energy platform by computing the relative solvation values for the aromatic dataset. For all tested aromatic pairs, the relative hydration free energy values computed from the dual-topology approach and the absolute HFE showed an agreement within 0.3 kcal/mol, with an R2 value of 0.999 (Table 3). The observed deviation is likely a result of random, non-systematic statistical error.
Table 3.
Comparison between the Tinker-OpenMM absolute and relative platform calculation of the solvation energy between pairs of aromatic compounds. Values are in kcal/mol.
| Relative from Dual-Topology | Difference by Absolute | |
|---|---|---|
| Aniline/Benzene | 4.2±0.1 | 4.0±0.1 |
| Adenine/Pyrrole | 11.4±0.1 | 11.3±0.1 |
| Aniline/Adenine | −10.2±0.1 | −10.2±0.1 |
| Benzene/3-Methylimidizole | −9.0±0.1 | −8.7±0.1 |
| 3-Methylpytidine/pyridine | −0.1±0.1 | 0.0±0.1 |
Finally, we tested the relative binding prediction of two pairs of sampl4 compounds. The first set of compounds, mol05 and mol06 share similar scaffolds, and show agreement in both complexation and solvation to within the uncertainty of BAR(Table 4).
Table 4.
Comparison between the Tinker-OpenMM absolute and relative platform calculations of the relative binding free energy between pairs of sampl4 compounds. Values are in kcal/mol.
| mol05-mol06 | mol09-mol10 | |||
|---|---|---|---|---|
| Relative from absolute GPU | Relative from dual topology | Relative from absolute GPU | Relative from dual topology | |
| Complexation energy | 44.3±0.1 | 44.3±0.1 | −56.3±0.1 | −56.0±0.1 |
| solvation energy | 47.3±0.1 | 47.3±0.1 | −68.0±0.1 | −68.0±0.1 |
| total ΔΔG | −2.9±0.1 | −2.9±0.1 | 10.4±0.2 | 10.7±0.1 |
The relative binding between molecules 9 and 10 constitutes a more challenging case that cannot be handled using the dummy atom based approach due to the lack of a shared scaffold. In addition, this dissimilarity between the ligands may theoretically make convergence more difficult in the intermediate vdW transitions. Nonetheless, the relative binding platform was still able to agree with the absolute platform to within 0.3 kcal/mol, demonstrating the advantage of dual-topology platform.
DISCUSSION AND CONCLUSIONS
This work reports a GPU implementation of alchemical free energy simulation for polarizable force field AMOEBA. The enhanced speed of GPU over CPU will be valuable for applications such as lead optimization. We have shown that the Tinker-OpenMM GPU platform is capable of reproducing the results of Tinker CPU platform, with an approximately 200-fold improvement in computational performance over what is possible on a single CPU core. This usage of GPU computation greatly improved sampling, which should allow for accounting for slow dynamics such as induced fit effects and other local changes in protein structure. Therefore, we expect the better sampling afforded by the GPU based platform will potentially lead to improved accuracy in ligand binding free energy prediction.
In addition to raw performance, one of the biggest challenges facing the free energy calculation field is the application of techniques to improve sampling of flexible systems to enable convergence with lesser simulation times. One methodology to achieve this increase in sampling efficiency is the calculation of relative binding free energies. Unlike previously utilized dummy atom based approaches 82–86, the framework presented here is general and doesn’t require a shared set of atoms to be utilized effectively. A special path has been designed to avoid unstable ligand-ligand polarization in the dual-topology approach. We expect that for flexible protein systems, the dual-topology approach will be more efficient and reduce the time needed for convergence in comparison with absolute free energy approaches.
Supplementary Material
Acknowledgments
The authors are grateful for support by the National Institutes of Health (R01GM106137 and R01GM114237), CPRIT (RP160657) and the Robert A. Welch Foundation (F-1691).
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1610403. Funding from French CNRS through a PICS grant between UPMC and UT Austin is acknowledged. This work was supported in part by French state funds managed by CalSimLab and the ANR within the Investissements d’Avenir program under reference ANR-11-IDEX-0004-02.
References
- 1.Sanz E, Vega C. Solubility of KF and NaCl in water by molecular simulation. The Journal of chemical physics. 2007;126(1):014507. doi: 10.1063/1.2397683. [DOI] [PubMed] [Google Scholar]
- 2.Seeliger D, De Groot BL. Protein thermostability calculations using alchemical free energy simulations. Biophys J. 2010;98(10):2309–2316. doi: 10.1016/j.bpj.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aqvist J. Ion-water interaction potentials derived from free energy perturbation simulations. The Journal of Physical Chemistry. 1990;94(21):8021–8024. [Google Scholar]
- 4.Garrido NM, Queimada AJ, Jorge M, Macedo EA, Economou IG. 1-Octanol/water partition coefficients of n-alkanes from molecular simulations of absolute solvation free energies. J Chem Theory Comput. 2009;5(9):2436–2446. doi: 10.1021/ct900214y. [DOI] [PubMed] [Google Scholar]
- 5.Jayaraman S, Maginn EJ. Computing the melting point and thermodynamic stability of the orthorhombic and monoclinic crystalline polymorphs of the ionic liquid 1-n-butyl-3-methylimidazolium chloride. The Journal of chemical physics. 2007;127(21):214504. doi: 10.1063/1.2801539. [DOI] [PubMed] [Google Scholar]
- 6.Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, Pande VS. Alchemical free energy methods for drug discovery: progress and challenges. Curr Opin Struct Biol. 2011;21(2):150–160. doi: 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira LG, dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20(10–11):601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 9.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1 Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 10.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52(4):609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 11.Tanrikulu Y, Kruger B, Proschak E. The holistic integration of virtual screening in drug discovery. Drug Discov Today. 2013;18(7–8):358–364. doi: 10.1016/j.drudis.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49(20):5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 13.Fischer M, Coleman RG, Fraser JS, Shoichet BK. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat Chem. 2014;6(7):575–583. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enyedy IJ, Egan WJ. Can we use docking and scoring for hit-to-lead optimization? J Comput Aided Mol Des. 2008;22(3–4):161–168. doi: 10.1007/s10822-007-9165-4. [DOI] [PubMed] [Google Scholar]
- 15.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303(5665):1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 16.Mobley D, Gilson MK. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu Rev Biophysics. 2017:46. doi: 10.1146/annurev-biophys-070816-033654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Buckner JK, Boudon S, Tiradorives J. Efficient Computation of Absolute Free-Energies of Binding by Computer-Simulations - Application to the Methane Dimer in Water. J Chem Phys. 1988;89(6):3742–3746. [Google Scholar]
- 19.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys J. 1997;72(3):1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput Phys Commun. 1995;91(1–3):275–282. [Google Scholar]
- 21.Izrailev S, Stepaniants S, Isralewitz B, Kosztin D, Lu H, Molnar F, Wriggers W, Schulten K. Computational molecular dynamics: challenges, methods, ideas. Springer; 1999. pp. 39–65. [Google Scholar]
- 22.Gullingsrud JR, Braun R, Schulten K. Reconstructing potentials of mean force through time series analysis of steered molecular dynamics simulations. J Comput Phys. 1999;151(1):190–211. [Google Scholar]
- 23.Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J Comput Phys. 1977;23(2):187–199. [Google Scholar]
- 24.Allen TW, Andersen OS, Roux B. Molecular dynamics - potential of mean force calculations as a tool for understanding ion permeation and selectivity in narrow channels. Biophys Chem. 2006;124(3):251–267. doi: 10.1016/j.bpc.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 25.Zhang D, Gullingsrud J, McCammon JA. Potentials of mean force for acetylcholine unbinding from the alpha7 nicotinic acetylcholine receptor ligand-binding domain. J Am Chem Soc. 2006;128(9):3019–3026. doi: 10.1021/ja057292u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woo HJ, Roux B. Calculation of absolute protein–ligand binding free energy from computer simulations. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(19):6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau AY, Roux B. The hidden energetics of ligand binding and activation in a glutamate receptor. Nature structural & molecular biology. 2011;18(3):283–287. doi: 10.1038/nsmb.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doudou S, Burton NA, Henchman RH. Standard free energy of binding from a one-dimensional potential of mean force. J Chem Theory Comput. 2009;5(4):909–918. doi: 10.1021/ct8002354. [DOI] [PubMed] [Google Scholar]
- 29.Straatsma TP, Berendsen HJC. Free-Energy of Ionic Hydration - Analysis of a Thermodynamic Integration Technique to Evaluate Free-Energy Differences by Molecular-Dynamics Simulations. J Chem Phys. 1988;89(9):5876–5886. [Google Scholar]
- 30.Barducci A, Bussi G, Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett. 2008;100(2):020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 31.Laio A, Gervasio FL. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Reports on Progress in Physics. 2008;71(12):126601. [Google Scholar]
- 32.Bussi G, Laio A, Parrinello M. Equilibrium free energies from nonequilibrium metadynamics. Physical review letters. 2006;96(9):090601. doi: 10.1103/PhysRevLett.96.090601. [DOI] [PubMed] [Google Scholar]
- 33.Zheng L, Chen M, Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc Natl Acad Sci U S A. 2008;105(51):20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng L, Chen M, Yang W. Simultaneous escaping of explicit and hidden free energy barriers: Application of the orthogonal space random walk strategy in generalized ensemble based conformational sampling. The Journal of chemical physics. 2009;130(23):06B618. doi: 10.1063/1.3153841. [DOI] [PubMed] [Google Scholar]
- 35.Bennett CH. Efficient Estimation of Free-Energy Differences from Monte-Carlo Data. J Comput Phys. 1976;22(2):245–268. [Google Scholar]
- 36.Shirts MR, Pande VS. Comparison of efficiency and bias of free energies computed by exponential averaging, the Bennett acceptance ratio, and thermodynamic integration (vol 122, art no 144107, 2005) J Chem Phys. 2008;129(22) doi: 10.1063/1.1873592. [DOI] [PubMed] [Google Scholar]
- 37.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 38.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I. CHARMM general force field: A force field for drug - like molecules compatible with the CHARMM all - atom additive biological force fields. J Comput Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. The Journal of Physical Chemistry B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacKerell AD, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56(4):257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 41.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Amber, a Package of Computer-Programs for Applying Molecular Mechanics, Normal-Mode Analysis, Molecular-Dynamics and Free-Energy Calculations to Simulate the Structural and Energetic. Properties of Molecules Comput Phys Commun. 1995;91(1–3):1–41. [Google Scholar]
- 42.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 43.Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J. 2007;92(11):3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meagher KL, Redman LT, Carlson HA. Development of polyphosphate parameters for use with the AMBER force field. J Comput Chem. 2003;24(9):1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 45.Ren PY, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107(24):5933–5947. [Google Scholar]
- 46.Wu JC, Chattree G, Ren P. Automation of AMOEBA polarizable force field parameterization for small molecules. Theoretical chemistry accounts. 2012;131(3):1138. doi: 10.1007/s00214-012-1138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P. Polarizable atomic multipole-based AMOEBA force field for proteins. J Chem Theory Comput. 2013;9(9):4046–4063. doi: 10.1021/ct4003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maple JR, Cao Y, Damm W, Halgren TA, Kaminski GA, Zhang LY, Friesner RA. A polarizable force field and continuum solvation methodology for modeling of protein– ligand interactions. J Chem Theory Comput. 2005;1(4):694–715. doi: 10.1021/ct049855i. [DOI] [PubMed] [Google Scholar]
- 49.Stern HA, Kaminski GA, Banks JL, Zhou R, Berne B, Friesner RA. Fluctuating charge, polarizable dipole, and combined models: parameterization from ab initio quantum chemistry. The Journal of Physical Chemistry B. 1999;103(22):4730–4737. [Google Scholar]
- 50.Kaminski GA, Stern HA, Berne BJ, Friesner RA. Development of an accurate and robust polarizable molecular mechanics force field from ab initio quantum chemistry. The Journal of Physical Chemistry A. 2004;108(4):621–627. [Google Scholar]
- 51.Patel S, Brooks CL. CHARMM fluctuating charge force field for proteins: I parameterization and application to bulk organic liquid simulations. J Comput Chem. 2004;25(1):1–16. doi: 10.1002/jcc.10355. [DOI] [PubMed] [Google Scholar]
- 52.Patel S, Mackerell AD, Brooks CL. CHARMM fluctuating charge force field for proteins: II protein/solvent properties from molecular dynamics simulations using a nonadditive electrostatic model. J Comput Chem. 2004;25(12):1504–1514. doi: 10.1002/jcc.20077. [DOI] [PubMed] [Google Scholar]
- 53.Baker CM, Anisimov VM, MacKerell AD., Jr Development of CHARMM polarizable force field for nucleic acid bases based on the classical Drude oscillator model. The Journal of Physical Chemistry B. 2010;115(3):580–596. doi: 10.1021/jp1092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopes PE, Lamoureux G, Roux B, MacKerell AD. Polarizable empirical force field for aromatic compounds based on the classical drude oscillator. The Journal of Physical Chemistry B. 2007;111(11):2873–2885. doi: 10.1021/jp0663614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baker CM, Lopes PE, Zhu X, Roux B, MacKerell AD., Jr Accurate calculation of hydration free energies using pair-specific Lennard-Jones parameters in the CHARMM Drude polarizable force field. J Chem Theory Comput. 2010;6(4):1181–1198. doi: 10.1021/ct9005773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ren PY, Wu CJ, Ponder JW. Polarizable Atomic Multipole-Based Molecular Mechanics for Organic Molecules. J Chem Theory Comput. 2011;7(10):3143–3161. doi: 10.1021/ct200304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi Y, Wu CJ, Ponder JW, Ren PY. Multipole Electrostatics in Hydration Free Energy Calculations. J Comput Chem. 2011;32(5):967–977. doi: 10.1002/jcc.21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abella JR, Cheng SY, Wang Q, Yang W, Ren P. Hydration Free Energy from Orthogonal Space Random Walk and Polarizable Force Field. J Chem Theory Comput. 2014;10(7):2792–2801. doi: 10.1021/ct500202q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schnieders MJ, Baltrusaitis J, Shi Y, Chattree G, Zheng L, Yang W, Ren P. The Structure, Thermodynamics and Solubility of Organic Crystals from Simulation with a Polarizable Force Field. J Chem Theory Comput. 2012;8(5):1721–1736. doi: 10.1021/ct300035u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiao D, King C, Grossfield A, Darden TA, Ren PY. Simulation of Ca2+ and Mg2+ solvation using polarizable atomic multipole potential. J Phys Chem B. 2006;110(37):18553–18559. doi: 10.1021/jp062230r. [DOI] [PubMed] [Google Scholar]
- 61.Wu JC, Piquemal JP, Chaudret R, Reinhardt P, Ren PY. Polarizable Molecular Dynamics Simulation of Zn(II) in Water Using the AMOEBA Force Field. J Chem Theory Comput. 2010;6(7):2059–2070. doi: 10.1021/ct100091j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grossfield A, Ren P, Ponder JW. Ion solvation thermodynamics from simulation with a polarizable force field. J Am Chem Soc. 2003;125(50):15671–15682. doi: 10.1021/ja037005r. [DOI] [PubMed] [Google Scholar]
- 63.Bell DR, Qi R, Jing ZF, Xiang JY, Mejias C, Schnieders MJ, Ponderc JW, Ren PY. Calculating binding free energies of host-guest systems using the AMOEBA polarizable force field. Phys Chem Chem Phys. 2016;18(44):30261–30269. doi: 10.1039/c6cp02509a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, Shi Y, Ren P. Polarizable Force Fields for Scoring Protein–Ligand Interactions Protein-Ligand Interactions. 1. 2012. pp. 99–120. [Google Scholar]
- 65.Shi Y, Zhu CZ, Martin SF, Ren P. Probing the effect of conformational constraint on phosphorylated ligand binding to an SH2 domain using polarizable force field simulations. J Phys Chem B. 2012;116(5):1716–1727. doi: 10.1021/jp210265d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiao D, Zhang J, Duke RE, Li G, Schnieders MJ, Ren P. Trypsin - ligand binding free energies from explicit and implicit solvent simulations with polarizable potential. J Comput Chem. 2009;30(11):1701–1711. doi: 10.1002/jcc.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiao D, Zhang JJ, Duke RE, Li GH, Schnieders MJ, Ren PY. Trypsin-Ligand Binding Free Energies from Explicit and Implicit Solvent Simulations with Polarizable Potential. J Comput Chem. 2009;30(11):1701–1711. doi: 10.1002/jcc.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Y, Xia Z, Zhang JJ, Best R, Wu CJ, Ponder JW, Ren PY. Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J Chem Theory Comput. 2013;9(9):4046–4063. doi: 10.1021/ct4003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiao D, Golubkov PA, Darden TA, Ren P. Calculation of protein–ligand binding free energy by using a polarizable potential. Proceedings of the National Academy of Sciences. 2008;105(17):6290–6295. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Yang W, Piquemal JP, Ren P. Modeling Structural Coordination and Ligand Binding in Zinc Proteins with a Polarizable Potential. J Chem Theory Comput. 2012;8(4):1314–1324. doi: 10.1021/ct200812y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dagum L, Menon R. OpenMP: an industry standard API for shared-memory programming. Ieee Comput Sci Eng. 1998;5(1):9. [Google Scholar]
- 72.Narth C, Lagardère L, Polack E, Gresh N, Wang Q, Bell DR, Rackers JA, Ponder JW, Ren PY, Piquemal JP. Scalable improvement of SPME multipolar electrostatics in anisotropic polarizable molecular mechanics using a general short - range penetration correction up to quadrupoles. J Comput Chem. 2016;37(5):494–506. doi: 10.1002/jcc.24257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lagardere L, Lipparini F, Polack E, Stamm B, Cances E, Schnieders M, Ren P, Maday Y, Piquemal JP. Scalable Evaluation of Polarization Energy and Associated Forces in Polarizable Molecular Dynamics: II. Towards Massively Parallel Computations using Smooth Particle Mesh Ewald. J Chem Theory Comput. 2014;10(4):1638–1651. doi: 10.1021/ct401096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lipparini F, Lagardère L, Stamm B, Cancès E, Schnieders M, Ren P, Maday Y, Piquemal JP. Scalable evaluation of polarization energy and associated forces in polarizable molecular dynamics: I. toward massively parallel direct space computations. J Chem Theory Comput. 2014;10(4):1638–1651. doi: 10.1021/ct401096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eastman P, Friedrichs MS, Chodera JD, Radmer RJ, Bruns CM, Ku JP, Beauchamp KA, Lane TJ, Wang LP, Shukla D, Tye T, Houston M, Stich T, Klein C, Shirts MR, Pande VS. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J Chem Theory Comput. 2013;9(1):461–469. doi: 10.1021/ct300857j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang L-P, Simmonett AC, Harrigan MP, Brooks BR. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics bioRxiv. 2016:091801. doi: 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levitt M. Protein Folding by Restrained Energy Minimization and Molecular-Dynamics. J Mol Biol. 1983;170(3):723–764. doi: 10.1016/s0022-2836(83)80129-6. [DOI] [PubMed] [Google Scholar]
- 78.Hornak V, Simmerling C. Development of softcore potential functions for overcoming steric barriers in molecular dynamics simulations. J Mol Graph Model. 2004;22(5):405–413. doi: 10.1016/j.jmgm.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 79.Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. Wires Comput Mol Sci. 2013;3(2):198–210. [Google Scholar]
- 80.Phillips JC, Stone JE, Schultent K. Adapting a Message-Driven Parallel Application to GPU-Accelerated Clusters International Conference for High Performance Computing. Networking, Storage and Analysis. 2008:444–452. [Google Scholar]
- 81.81
- 82.Jorgensen WL, Nguyen TB. Monte Carlo simulations of the hydration of substituted benzenes with OPLS potential functions. J Comput Chem. 1993;14(2):195–205. [Google Scholar]
- 83.Ota N, Stroupe C, Ferreira-da-Silva J, Shah SA, Mares-Guia M, Brunger AT. Non - Boltzmann thermodynamic integration (NBTI) for macromolecular systems: Relative free energy of binding of trypsin to benzamidine and benzylamine. Proteins: Structure, Function, and Bioinformatics. 1999;37(4):641–653. doi: 10.1002/(sici)1097-0134(19991201)37:4<641::aid-prot14>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 84.Miyamoto S, Kollman PA. Absolute and Relative Binding Free-Energy Calculations of the Interaction of Biotin and Its Analogs with Streptavidin Using Molecular-Dynamics Free-Energy Perturbation. Approaches Proteins. 1993;16(3):226–245. doi: 10.1002/prot.340160303. [DOI] [PubMed] [Google Scholar]
- 85.Reddy MR, Erion MD. Calculation of relative binding free energy differences for fructose 1, 6-bisphosphatase inhibitors using the thermodynamic cycle perturbation approach. Journal of the American Chemical Society. 2001;123(26):6246–6252. doi: 10.1021/ja0103288. [DOI] [PubMed] [Google Scholar]
- 86.Reddy MR, Viswanadhan VN, Weinstein JN. Relative differences in the binding free energies of human immunodeficiency virus 1 protease inhibitors: a thermodynamic cycle-perturbation approach. Proceedings of the National Academy of Sciences. 1991;88(22):10287–10291. doi: 10.1073/pnas.88.22.10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reddy MR, Erion MD. Calculation of relative binding free energy differences for fructose 1,6-bisphosphatase inhibitors using the thermodynamic cycle perturbation approach. J Am Chem Soc. 2001;123(26):6246–6252. doi: 10.1021/ja0103288. [DOI] [PubMed] [Google Scholar]
- 88.Fleischman SH, Brooks CL. Thermodynamics of Aqueous Solvation - Solution Properties of Alcohols and Alkanes. J Chem Phys. 1987;87(5):3029–3037. [Google Scholar]
- 89.Ponder JW, Richards FM. TINKER molecular modeling package. J Comput Chem. 1987;(8):1016–1024. [Google Scholar]
- 90.Zhang C, Bell DR, Harger M, Ren PY. Polarizable Multipole-Based Force Field for Aromatic Molecules and nucleobases. J Chem Theory Comput. 2017 doi: 10.1021/acs.jctc.6b00918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Muddana HS, Fenley AT, Mobley DL, Gilson MK. The SAMPL4 host-guest blind prediction challenge: an overview. J Comput Aid Mol Des. 2014;28(4):305–317. doi: 10.1007/s10822-014-9735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roux B, Nina M, Pomès R, Smith JC. Thermodynamic stability of water molecules in the bacteriorhodopsin proton channel: a molecular dynamics free energy perturbation study. Biophys J. 1996;71(2):670–681. doi: 10.1016/S0006-3495(96)79267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.