

Abstract
Background
Obstructive sleep apnea syndrome (OSAS) is characterized by chronic intermittent episodes of upper-airway obstruction with hypoxia and is associated with increased risk of cardiovascular diseases, including myocardial hypertrophy. Chronic intermittent hypoxia (CIH) has been shown to induce apoptosis in cardiomyocytes. However, the mechanisms of cardiomyocytes apoptosis under CIH largely remain unclear.
Material/Methods
We used male Sprague-Dawley rats and human cardiomyocyte cell line H9C2, and Annexin V/PI, Western blot analysis, co-immunoprecipitation, RT-PCR, immunohistochemistry, and TUNEL assay were carried out.
Results
We show that Bim was significantly up-regulated by CIH in cardiomyocytes, and the function of Bim in CIH-induced apoptosis was supported by the genetic suppression of Bim with si-RNA. We also observed that CIH-motivated expression of Bim was directly related to fork head box class O1 (FOXO1), which is increased in CIH. Genetic ablation and pharmacological inhibition of FOXO1 in cardiomyocytes attenuated CIH-induced apoptosis, hypertrophy, and features of perivascular fibrosis in cardiomyocytes in vitro and in vivo.
Conclusions
FOXO1 is a key integrator of the apoptosis signal transduction pathway, driving chronic intermittent hypoxia-induced cardiac hypertrophy, and inhibition of FOXO1 provides a potential target for the treatment of OSAS with cardiac hypertrophy.
MeSH Keywords: bcl-2-Associated X Protein; Cell Hypoxia; Forkhead Transcription Factors; Hypertrophy, Left Ventricular; Sleep Apnea, Obstructive
Background
Obstructive sleep apnea syndrome (OSAS) is a whole-body disease characterized by recurrent episodes of apnea and/or hypopnea; it leads to chronic intermittent hypoxia (CIH), increased cardiac hypertrophy and ultimately, heart failure, and death [1,2]. In recent years, although greater progress has been made in the study of genetics, molecular biology, and the treatment of cardiac hypertrophy caused by CIH, the high mortality rate has not improved [3,4], especially in most patients with cardiac hypertrophy [5]. Thus, actively exploring new molecular and signal transmission pathways is of great significance to find effective therapeutic targets.
Currently, cardiac hypertrophy is characterized by cardiomyocyte hypertrophy and apoptosis, myocardial inflammation, and fibrosis. Cardiomyocyte apoptosis is a major decisive factor of cardiac hypertrophy [6,7]. Bim, a BH3-only Bcl-2 protein, initiates apoptosis by promoting the release of mitochondrial cytochrome C, which in turn activates caspase3 and eventually leads to apoptosis [8]. It has been reported that Bim exists in 3 major isoforms (BimS, BimL, and BimEL) that are generated by alternative splicing of a number of transcripts [9]. The FOXO family of forkhead transcription factors, which consists of FOXO1, FOXO3, FOXO4, and FOXO6, plays a vital role in a variety of basic biological processes, such as proliferation and apoptosis [10,11]. These FOXO transcription factors regulate a variety of physiological and pathological processes [12]. The activation of FOXO1 has recently been demonstrated to modulate the expression of genes involved in apoptosis, including Bim [13]. Previous data have suggested that Bim is a downstream target of FOXO transcription factors in a number of cell types. Recently, Bim has been shown to be mediated by FOXO3- and FOXO1-induced apoptosis [14]. However, the exact mechanism by which FOXO regulates the expression of Bim isoforms and their roles in CIH-induced apoptosis in myocardial cells has not been elucidated.
Here, by using the cardiomyocyte cell line H9C2 in a CIH model, we provide evidence that Bim plays a potent role in cardiomyocyte apoptosis under CIH. Furthermore, among the FOXO1and FOXO3 isoforms, FOXO1 is shown to be centrally involved in the pro-apoptosis phenotype of cardiomyocytes. Moreover, we demonstrate that Bim is a direct transcriptional target of FOXO1. In the present study, we identified that FOXO1 inhibition abrogates the induction of Bim and reverses chronic intermittent hypoxia-induced cardiac hypertrophy and apoptosis. We report a hitherto undescribed cellular response of CIH-induced cardiomyocytes apoptosis, and our results reveal that FOXO1/Bim plays an important role in mediating cardiac apoptosis in chronic intermittent hypoxia-induced cardiac hypertrophy.
Material and Methods
Ethics Statement
The Huazhong University of Science and Technology Committee and the Tongji Medical College Ethics Committee, Tongji Hospital approved all experiments.
Reagents
Dimethyl sulfoxide (DMSO) and AS1842856 were purchased from Sigma (Invitrogen, Carlsbad, CA, USA). Polyclonal antibodies against Bax, Bcl-2, caspase-3, BimEL, BimL, BimS, p-FOXO1, FOXO1, and FOXO3 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-GAPDH, goat anti-mouse IgG, and goat anti-rabbit IgG antibodies were purchase from Proteintech Group (Rosemont, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Gibco (Invitrogen, Carlsbad, USA). The protease inhibitor cocktail was purchased from Roche (Switzerland). The BCA protein assay reagent kit and enhanced chemiluminescent (ECL) plus reagent kit were obtained from Pierce (Pierce Biotech, USA).
Cell culture and exposure to intermittent hypoxia (IH) induction
The cardiomyocyte cell line H9C2 was cultured in DMEM containing 2.25 g/L glucose (Gibco, USA), 10% FBS (Gibco, USA), 100 mg/mL streptomycin, and 100 U/mL penicillin. The cells were grown in an incubator in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The cells synchronized for 24 h before exposing to normoxia (N) or IH for 0 h, 2 h, 4 h, or 6 h, with some modifications [15]. For induction of IH, the cells were exposed to alternating cycles of 5% O2 for 5 min followed by 21% O2 for 10 min at 37°C in a ProOx C21 cell incubator (BioSpherix, USA).
Animals and treatments
Sprague-Dawley rats (adult males, weighing 200–250 g) were purchased from the Laboratory Animal Center, Tongji Medical College. The chronic intermittent hypoxic (CIH) rats were placed in a normobaric chamber (Oxycycler modelA84XOV; BioSpherix, Lacona, NY), and then the O2 level was reduced from the normal level to 8–10% within 120 s and maintained at approximately 8–10% for 120 s. Subsequently, the oxygen concentration was quickly increased to 21% within 50 s and maintained at this level for 300 s. This process was repeated 8 h/day for a total of 6 weeks, consistent with the experimental procedure of our previous study [16]. The CIH rats were given a dose of AS1842856 (5 mg/kg) or vehicle on days 28 to 42 at random. Rats injected with AS1842856 (5 mg per kg body weight) or vehicle on days 28 to 42 and exposed subject to normal oxygen condition were used as the control group. AS1842856 was dissolved into mixed liquor (saline, Cremophor, and ethanol), and the mixed liquor was used as the vehicle control [17].
Histology of the left ventricle
Hearts from the rats were harvested for histological analysis of the left ventricle. After removal, the hearts were weighed to calculate the heart weight/body weight (HW/BW) ratio. Then, they were cut into small chunks and fixed in paraformaldehyde for further processing. Standard hematoxylin and eosin (HE) staining was performed for routine histology, and Masson’s trichrome staining was performed to assess the deposition of collagen, which was stained blue.
Immunohistochemistry
We deparaffinized 4-μm-thick paraffin-embedded rats heart tissue sections in xylene twice (15 min each), rehydrated with a gradient of ethanol to PBS. Antigens were repaired by microwave treatment in citrate buffer (pH 6.0) using a Quick kit (Biyuntian, China). The primary antibodies were diluted as follows: anti-FOXO1, 1: 100; anti-VEGF, 1: 50; anti-Bim, 1: 50; and TUNEL, 1: 60. Slides were then incubated with primary antibody at 4°C overnight, following the manufacturer’s instructions.
Flow cytometry
Cells were plated in 10-cm culture dishes at a density of 3×106 cells per plate and incubated for 2 days at 37°C in an atmosphere containing 5% CO2. After incubation, the cells were harvested and washed twice with PBS and then resuspended in Annexin V-PI from a dual-staining assay kit for 10 min at room temperature. The fluorescence signals from Annexin V and PI were measured by flow cytometry with a FACS Calibur™ platform (BD Bioscience), and the data were analyzed using Cell Quest Pro software (BD Bioscience).
Immunoblotting and coimmunoprecipitation
The heart tissue and cell lysates were prepared with 10% polyacrylamide gels, and then transferred to a polyvinylidene difluoride nitrocellulose membrane. For co-immunoprecipitation, cell lysates were incubated with the following antibodies: anti-FOXO1 (1: 1000), anti-FOXO3 (1: 1000), anti-p-FOXO1 (1: 1000), anti-BimEL (1: 500), anti-BimL (1: 1000), anti-BimS (1: 1000), anti-BAX (1: 1000), anti-Bcl2 (1: 800), anti-Caspase3 (1: 500), and anti-GAPDH (as the loading control; 1: 2000). Then, the membranes were subjected to a 1-h incubation with HRP-conjugated secondary antibodies with horseradish peroxidase (HRP). Western blot analyses were performed as described previously [18].
Transfection with siRNA
The pre-designed siRNAs of the cardiomyocyte cell line H9C2 were transfected by targeting Bim (5′-CAAUUGUCUACCUUCUCGG-3′), FOXO1 (5′-GAGCGTGCCCTACTTCAAG-3′), or the nonspecific siRNA (control; 5′-GACCACGAGUAAAAGUAGU-3′) using Lipofectamine RNAi MAX Transfection Reagent (Invitrogen) according to the manufacturer’s instructions.
Lentiviral vector constructs and transduction
Full-length cDNA of the FOXO1 gene was purchased from GeneChem Biotech (Shanghai, China). For viral transduction, the transfection lentiviral vector was added to H9C2 cells at 70–80% confluency and transduced at MOI of 25. After an additional 48-h incubation, the cells were then harvested and used for the indicated assays.
Reverse transcription-PCR (RT-PCR)
Total RNA was transcribed using TRIzol reagent (Takara, Dalian, China). β-actin mRNA was used for the internal control. The primers are as follows: β-actin, forward 5′-GAGGATGGAGATGGCCTTAG-3′, and reverse 5′-CTTAAGGGT CGCCTTCGACC-3′; Bim, forward 5′-AGAGG AAATTGCATGT GAAC-3′; reverse 5′-GACAATGTAACGTAACAGTG-3′; and FOXO1, forward 5′-CTCCCATACCCACCCTG-3′; and reverse 5′-AATAACATGCCATCCAAG-3′.
TUNEL assay
Harvested cells were fixed in 4% paraformaldehyde in PBS for 30 min and then permeabilized with 0.05% Triton X-100 on ice for 5 min. The cells were subjected to TdT-mediated dUTP nick-end labeling while being incubated in a humidified chamber at 37°C for 60 min in the dark. The cells were washed 3 times with PBS; anti-fluorescence quenching solution was then added and allowed to react for 5 min. Finally, the cells were examined using a fluorescence microscope.
Statistical analysis
The data are presented as the means ±SEM. Statistical significance was determined using Student’s t-test or one-way analysis of variance (ANOVA). The results were analyzed using GraphPad Prism 5. Differences were considered statistically significant at P≤0.05.
Results
Bim activation is responsible for chronic intermittent hypoxia-induced cardiomyocyte apoptosis
We examined the apoptotic effects of chronic intermittent hypoxia(CIH) in H9C2 cells using flow cytometry. H9C2 cells were exposed to CIH for 2, 4, and 6 h. As shown in Figure 1A and 1B, CIH exposure resulted in a time-dependent increase in the apoptosis of H9C2 cells compared with the normoxic cells by Annexin V-FITC staining.
Figure 1.

CIH induces apoptosis in human cardiomyocyte cell line H9C2. (A). Representative flow cytometry scatter plots of propidium iodide (PI) (Y axis) vs. annexin V-fluorescein isothiocyanate (FITC) (X axis) and (B). Quantitation data of the average of 3 independent flow cytometry experiments. (C). Effect of CIH on apoptosis protein expression of BimEL, BimL, BimS, BAX, Bcl2, and caspase3 by Western blot analysis. (D) and (E) Quantitation data of (C). * P<0.05, ** P<0.01 compared with control, n=3.
Recent studies have demonstrated that Bcl-2 family members induce cell apoptosis by manifold factors [19]. To further investigate the effect of Bcl-2 family members on CIH-induced cardiomyocyte apoptosis, we used Western blot analysis to evaluate the expression of Bcl-2, Bax, and Bim, which are Bcl-2 family proteins. BimEL, BimL, and BimS are 3 major alternative splicing isoforms of Bim. As shown in Figure 1C and 1D, only the level of BimEL was readily increased in this study in a time-dependent manner. Additionally, the expression ratio of Bax and Bcl2 was noticeably changed in these cells under the same conditions (Figure 1C, 1E). Furthermore, the expression of caspase 3, which is cleaved during apoptosis, was examined. As shown in Figure 1C and 1E, CIH induced caspase 3 cleavage in H9C2 cells in a time-dependent manner. These results confirmed that CIH induced cardiomyocyte apoptosis and expression of Bim proteins. Also, the above results demonstrated that Bim plays a principal role in inducing apoptosis in H9C2 cells in response to CIH.
To further explore these results, H9C2 cells were transfected with siRNA against Bim or control and then exposed to CIH. As shown in Figures 2A–2C, compared with the control cells, the ratio of BAX/Bcl2 and caspase 3 cleavage was reversed after down-regulation of Bim in response to CIH. Interestingly, the number of apoptotic cells assayed by flow cytometry after CIH exposure in H9C2 cells decreased after Bim-specific siRNA transfection (Figure 2D, 2E). These data suggest that the activation of Bim plays a major role CIH-induced cardiomyocyte apoptosis.
Figure 2.

Activation of Bim is responsible for CIH-induced apoptosis. (A). Cells were transiently transfected with Bim siRNA or a control RNAi and then treated with CIH. Western blot analysis of BimEL, BAX, Bcl2, and caspase3 proteins expression. (B, C). Quantitation data of (A). (D) Representative flow cytometry scatter plots of propidium iodide (PI) (Y axis) vs. Annexin V-fluorescein isothiocyanate (FITC) (X axis), and (E) Quantitation data of average of 3 independent flow cytometry experiments. * P<0.05, ** P<0.01 compared with control, n=3.
Chronic intermittent hypoxia increases phosphorylation activity and expression of FOXO1
It is reported that FOXO3 and FOXO1, especially FOXO3, are master upstream regulators of Bim [20,21]. Thus, we investigated whether the activation of FOXO3 and FOXO1 is also regulated by the CIH-induced up-regulation of Bim. Interestingly, the expression of FOXO1 mRNA was clearly up-regulated after CIH exposure (Figures 3A 3B). However, CIH was unable to induce the expression of FOXO3 mRNA. Further, Western blot analysis was used to evaluate the effect of CIH on the expression of FOXO1 and FOXO3 proteins. Consistent with the results of RT-PCR, the Western blot analysis showed (Figure 3C, 3D) that FOXO1 was constitutively phosphorylated in response to CIH. Notably, the level of FOXO1 was also increased after CIH exposure. Conversely, the expression of FOXO3 was not detectable. These results suggested the possibility that FOXO1 is responsible for the induction of Bim in response to CIH to mediate apoptosis in H9C2 cells.
Figure 3.

CIH induces an increase in FOXO1 expression. (A) and (B) RT-PCR to detect FOXO1 and FOXO3 expression. (C). Full-length blots of p-FOXO1, FOXO1, FOXO3, and GAPDH are presented. (D). Quantitation data of (C). * P<0.05, ** P<0.01 compared with control, n=3.
FOXO1 regulates the expression of Bim in response to chronic intermittent hypoxia in H9C2 cells
To further investigate whether there is a cause-and-effect relationship between the increased FOXO1 and Bim expression in response to CIH in H9C2 cells, we explored whether FOXO1 expression is necessary for the subsequent expression of Bim in response to CIH in H9C2 cells. A lentiviral vector over-expressing FOXO1 was devised and transfected into H9C2 cells. As shown in Figure 4A and 4B, the over-expression of FOXO1 augmented Bim expression under normoxia and CIH. To further elucidate these results, H9C2 cells were transfected with FOXO1-specific siRNA or control and then subjected to CIH. The expression of FOXO1 dropped by 60% after siRNA transfection. Bim expression also fell compared with that in the control group in response to CIH (Figure 4C, 4D).
Figure 4.

FOXO1 mediates Bim expression in response to CIH. (A) H9C2 cells were transfected with lentiviral vector and subjected to Western blot analysis to identify FOXO1 or Bim expression. (B) Quantitation data of (A). (C) H9C2 cells were transiently transfected with FOXO1 siRNA or a control RNAi and then treated with CIH. The cells were then harvested and subjected to Western blot analysis to identify the expression of FOXO1 and Bim. (D) Quantitation data of (C). (E) The potential interaction between FOXO1 and Bim was investigated via co-immunoprecipitation and immunoblotting in H9C2 cells exposed to either normoxia or CIH. (F) Full-length blots of FOXO1 and Bim and GAPDH are presented. (G) The expression levels of FOXO1 and Bim were quantified by immunoblots in HPASMCs transfected with siRNA exposed to CIH. (H) Results were quantified by densitometry analysis of the band form (G) and then normalization to GAPDH protein. * P<0.05, ** P<0.01 compared with control, n=3.
Moreover, to confirm the correlation between FOXO1 and Bim in H9C2 cells, a coimmunoprecipitation assay was performed (Figure 4E, 4F). We found that FOXO1 and Bim interact with each other. Furthermore, the results demonstrated that the CIH-treated cells express more proteins than in the normoxic controls. The results of Western blot showed that we used consistent input amounts for the assay. Finally, the levels of FOXO1 and Bim were detected in H9C2 cells when transfected with FOXO1 and Bim gene-targeted siRNAs under CIH conditions. When compared with the normoxic controls, the expression level of Bim was clearly reduced in the silencing of FOXO1 group. However, knockdown of Bim did not affect the expression of FOXO1 (Figure 4G, 4H). These results clearly show that FOXO1 is required for the expression of Bim in response to chronic intermittent hypoxia in H9C2 cells.
Silencing of FOXO1 prevents the induction of apoptosis by chronic intermittent hypoxia in cardiomyocytes
To determine the biological role of FOXO1 in CIH-mediated apoptosis of cardiomyocytes, siRNA targeting FOXO1 was utilized to disrupt the expression of FOXO1. To this end, H9C2 cells were subjected to CIH, siRNA treatment, or the combination of both. Our study showed that CIH-induced apoptotic cell death was decreased in the presence of FOXO1 knockdown, as demonstrated by both TUNEL assay (Figure 5A, 5B) and flow cytometry (Figure 5C, 5D) to detect the apoptosis ratio. Collectively, our studies suggest that the CIH-induced increase in FOXO1 plays a pro-apoptotic role in cardiomyocytes.
Figure 5.

Silencing of FOXO1 abrogates induction of apoptosis by chronic intermittent hypoxia in cardiomyocytes. (A) H9C2 cells were transiently transfected with FOXO1 siRNA or a control RNAi and then treated with CIH. The cells were then harvested. TUNEL assay was performed to measure the ratio of apoptotic cells. TUNEL-positive cells were counted from at least 100 random fields (200×). (B) Bar chart showing Quantitation data of average of 3 independent TUNEL assay experiments. (C) Quantitation data of average of 3 independent flow cytometry experiments. (D) Representative flow cytometry scatter plots of propidium iodide (PI) (Y axis) vs. Annexin V-fluorescein isothiocyanate (FITC) (X axis). * P<0.05, ** P<0.01 compared with control, n=3.
FOXO1 and Bim expression increases in chronic intermittent hypoxia-induced cardiac hypertrophy in vivo
We examined whether the expressions of FOXO1 and Bim are up-regulated in the cardiac tissues in response to CIH. Immunostaining for FOXO1and Bim showed that the immunoreactivity to both proteins was enhanced in the hearts of rats exposed to CIH compared with those of the normoxic controls (Figure 6A–6C). Western blotting results also demonstrated the up-regulation of FOXO1and Bim in the hearts from CIH-exposed rats compared with those of the normoxic controls (Figure 6D, 6E). Our results confirm that FOXO1 and Bim expression increase in chronic intermittent hypoxia-induced cardiac hypertrophy in vivo.
Figure 6.

Increased levels of FOXO1 and Bim in CIH rats. (A). Heart tissue sections were stained immunohistochemically for FOXO1 and Bim (magnification, 400×). (B, C). The percentage of FOXO1 and Bim marker positive (%) was evaluated. (D) Immunoblots of FOXO1 and Bim using hearts isolated from the rats exposed to either normoxia or CIH (magnification, 400×). (E) Results were quantified by densitometry analysis of the band form (D) and then normalization to GAPDH protein. * P<0.05, ** P<0.01 compared with control, n=6–12.
FOXO1 inhibition abrogates the induction of Bim and reverses chronic intermittent hypoxia-induced cardiac hypertrophy and apoptosis
A recent report suggested that treatment with the FOXO1-selective inhibitor AS1842856 reduced phosphorylation of FOXO1 and decreased its expression [15]. To elucidate whether FOXO1inhibition abrogates the induction of Bim and reverses CIH-induced cardiac hypertrophy and apoptosis, we treated the rats exposed to CIH with the FOXO1 antagonist AS1842856. AS1842856 application started 4 weeks after the rats were exposed to CIH at a time point when cardiac hypertrophy was already well established. CIH exposure induced cardiomegaly in the rats and a significant increase of cardiac hypertrophy and the administration of AS1842856 significantly reduced the degeneration of cardiomyocytes, including myofibrillar lysis, disarray of myofibers, and interstitial fibrosis in comparison with the CIH-exposed group (Figure 7A–7D). Moreover, Bim expression was virtually eliminated in rats subjected to FOXO1 inhibition in comparison with the vehicle control-treated animals. Furthermore, we tested whether FOXO1 inhibition alleviates CIH-induced cardiomyocyte apoptosis, using the TUNEL assay. As shown in Figure 8A and 8B, the number of TUNEL-positive cells in the left ventricle of the cardiac myocardium was significantly elevated in the CIH-exposed group, whereas it was significantly reduced in the CIH-exposed group and the AS1842856-treated group.
Figure 7.

FOXO1 inhibition abrogates induction of Bim and reverses chronic intermittent hypoxia-induced cardiac hypertrophy. (A) Representative light micrographs, cardiomyocyte cross-sectional area, and collagen content (%fibrosis) in the LV myocardium of CIH-treated rats. CIH caused cardiac hypertrophy cardiomyocyte degeneration, increased perivascular fibrosis, and increased expression of VEGF, FOXO1, and Bim. These abnormalities were significantly suppressed by injection of FOXO1 inhibitor AS1842856 (magnification, 400×). (B). The heart weight (HW)/body weight (BW) ratio was calculated after the hearts were isolated. (C). CIH significantly increased the cardiomyocyte cross-sectional area and (D) interstitial fibrosis, whereas injection of FOXO1 inhibitor significantly suppressed these changes. Each experiment was performed 3 times. * P<0.05, ** P<0.01 compared with control, n=6. (NC, Normoxia group; T, AS1842856)
Figure 8.

FOXO1 inhibition reverses CIH-induced cardiac apoptosis. (A) TUNEL staining of LV myocardium in CIH and CIH-treated with FOXO1 inhibition AS1842856 (magnification, 400×). (B) TUNEL-positive cells are significantly increased in the rats exposed to CIH, whereas these cells are significantly decreased by injection of FOXO1 inhibitor AS1842856. * P<0.05, ** P<0.01 compared with control, n=6. (NC, Normoxia group; T, AS1842856.)
Discussion
Here, we showed that up-regulation of FOXO1 in cardiomyocytes is central in the pathogenesis of CIH-induced cardiac hypertrophy, and that inhibition of the expression of FOXO1 has the potential future in therapies to reverse the cardiac remodeling underlying CIH (Figure 9). Additionally, the present study provides a novel mechanism by which CIH induces apoptosis in cardiomyocytes through the FOXO1/Bim pathway in vitro and in vivo.
Figure 9.
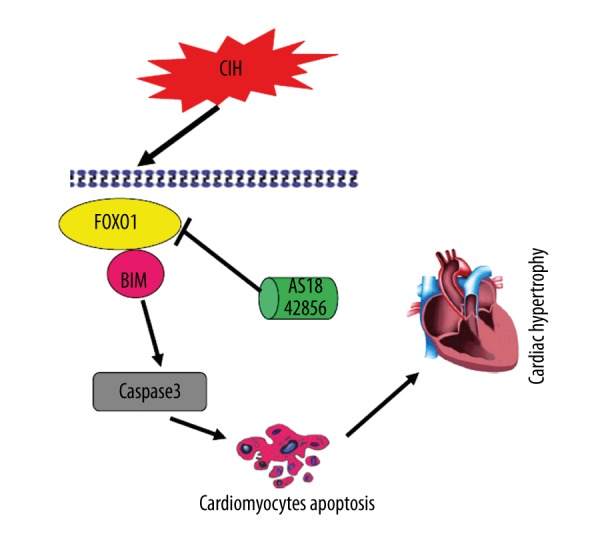
Graphical summary illustrating FOXO1 activates Bim expression to mediate cardiac apoptosis in chronic intermittent hypoxia-induced cardiac hypertrophy.
Several pro-apoptosis pathways have been implicated in the abnormal apoptosis of cardiomyocytes. Two major pro-apoptosis pathways have been reported to date: the extrinsic pathway and the intrinsic pathway [22]. The intrinsic pathway is controlled by the balance between pro-apoptotic (Bax, Bak, Bim, and Bad) and anti-apoptotic (Bcl-2 and Bcl-xL) members of the Bcl-2 family [23,24]. The up-regulation of Bim induces the dissociation of cytochrome C from the mitochondria and promotes the formation of apoptosomes and the activation of its effector caspase3 [25]. Moreover, recent reports have demonstrated increased expression of pro-apoptotic cytokines and their receptors in patients with OSAS-induced cardiac hypertrophy. Our results also show that Bim is a critical pro-apoptotic integrator of cardiomyocytes induced by CIH.
Recent studies have implicated Bim as a downstream target of FOXO1 and FOXO3. The FOXO family of transcription factors, including FOXO1 and FOXO3, is emerging as a key regulator for cell survival and apoptosis [26]. Activation of FOXO induces apoptosis by up-regulating several apoptotic genes, including those encoding the ligand for the death receptor Fas, CD95, and Bim [27]. However, the specific mechanisms of these apoptotic regulators are not well understood. There is no definitive information available on the molecular mechanism by which FOXO proteins mediate Bim expression, and it is not clear if Bim is a direct or indirect target of FOXO. Previous studies have showed that in several tumor cell lines, the FOXO3/Bim pathway plays a potent part in inducing apoptosis in reaction to various stimuli [28]. FOXO1 is even known to activate the promoter of the gene encoding Bim and thus induce FOXO1-dependent apoptosis [29]. In our study, our results clearly demonstrated that FOXO1, but not FOXO3, takes part in Bim-regulated apoptosis in response to CIH exposure. Furthermore, this study indicates that Bim is a direct transcriptional target of FOXO1.
FOXO1 is a member of the FOXO family and functions as a key regulator of cell survival and apoptosis [30]. Although FOXO1 activation induces apoptosis, the mechanism for the induction of FOXO1 itself is largely unknown. In this study, we showed that FOXO1 increased the expression of its pro-apoptotic target, Bim, at the transcriptional level. Moreover, FOXO1, -specifically siRNA, significantly rescued the cells from CIH-induced apoptosis. Our findings indicate that FOXO1 may contribute to the induction of apoptosis in cardiomyocytes in response to CIH. Furthermore, under sustained conditions of CIH stress, FOXO1 induced expression of the pro-apoptosis gene, Bim, to trigger programmed death of cardiomyocytes [31]. The ability of FOXO1 to induce apoptosis at multiple steps has generated considerable interest, and efforts have been made to assess its roles in CIH-induced cardiac hypertrophy. Several lines of evidence have shown that Bim is the major mediator of apoptosis in various diseases [32]. Therefore, the induction of Bim by FOXO1 may have a key role in maintaining the level of Bim in cardiomyocytes and dictating their response to CIH. Thus, manipulation of the level of FOXO1 in patients may become important in the therapy of OSAS with cardiac hypertrophy.
The assumed core function of FOXO1 in the mechanisms of CIH-induced cardiac hypertrophy requires further evidence by in vivo functional omission studies. In animal models with FOXO1inhibition, we demonstrated the down-regulated expression of FOXO1, a descended of its expression and the corresponding inhibition of Bim, with its downstream target gene, FOXO1. We observed that CIH accelerated the degeneration of cardiomyocytes and aggravated systolic dysfunction in rats, whereas these deleterious effects of CIH were reversed in the FOXO1-inhibited groups. The group exposed to CIH displayed cardiomyocyte hypertrophy and increased expression of VEGF. On the other hand, injection of a low concentration of FOXO1 inhibitor attenuated cardiomyocyte hypertrophy and perivascular fibrosis in the LV myocardium, resulting in the preservation of cardiac function. However, this study did not use constitutive gene knockout in rats, and we did not further access its role in CIH-induced cardiac hypertrophy. Our data show that weakening of FOXO1 in cardiomyocytes is enough to attenuate CIH-induced cardiac hypertrophy.
In the last decade, great advances in the therapy of OSAS with cardiac hypertrophy have been achieved, but there is no cure for this devastating sickness to date, primarily because there are various pro-apoptotic pathways that induce the cardiac remodeling that underlies OSAS. Our study reveals that FOXO1 has a central effect on pro-apoptotic cardiomyocytes, and offers new avenues for therapeutic intervention.
Conclusions
FOXO1 is a key integrator of the apoptosis signal transduction pathway, driving chronic intermittent hypoxia-induced cardiac hypertrophy. Inhibition of FOXO1 provides a potential target for the treatment of OSAS with cardiac hypertrophy.
Footnotes
Conflicts of interest
None.
Source of support: Departmental sources
References
- 1.Basner RC. Cardiovascular morbidity and obstructive sleep apnea. N Engl J Med. 2014;370(24):2339–41. doi: 10.1056/NEJMe1404501. [DOI] [PubMed] [Google Scholar]
- 2.Sarioglu N, Demirpolat G, Erel F, Kose M. Which is the ideal marker for early atherosclerosis in obstructive sleep apnea (OSA) – carotid intima-media thickness or mean platelet volume? Med Sci Monit. 2017;23:1674–81. doi: 10.12659/MSM.900959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mokhlesi B, Ayas NT. Cardiovascular events in obstructive sleep apnea – can CPAP therapy SAVE lives? N Engl J Med. 2016;375(10):994–96. doi: 10.1056/NEJMe1609704. [DOI] [PubMed] [Google Scholar]
- 4.Saraç S, Afşar GÇ, Oruç Ö, et al. Impact of patient education on compliance with positive airway pressure treatment in obstructive sleep apnea. Med Sci Monit. 2017;23:1792–99. doi: 10.12659/MSM.902075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.West SD, McBeath HA, Stradling JR. Obstructive sleep apnoea in adults. BMJ. 2009;338:b1165. doi: 10.1136/bmj.b1165. [DOI] [PubMed] [Google Scholar]
- 6.Baguet JP, Barone-Rochette G, Tamisier R, et al. Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol. 2012;12:679–88. doi: 10.1038/nrcardio.2012.141. [DOI] [PubMed] [Google Scholar]
- 7.Niroumand M, Kuperstein R, Sasson Z, Hanly PJ. Impact of obstructive sleep apnea on left ventricular mass and diastolic function. Am J RespirCrit Care Med. 2001;163(7):1632–36. doi: 10.1164/ajrccm.163.7.2007014. [DOI] [PubMed] [Google Scholar]
- 8.Carter MJ, Cox KL, Blakemore SJ, et al. PI3Kδ inhibition elicits anti-leukemic effects through Bim-dependent apoptosis. Leukemia. 2017;31(6):1423–33. doi: 10.1038/leu.2016.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sionov RV, Vlahopoulos SA, Granot Z. Regulation of Bim in health and disease. Oncotarget. 2015;6(27):23058–134. doi: 10.18632/oncotarget.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xin Z, Ma Z, Jiang S, et al. FOXOs in the impaired heart: New therapeutic targets for cardiac diseases. Biochim Biophys Acta. 2017;1863(2):486–98. doi: 10.1016/j.bbadis.2016.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Webb AE, Brunet A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem Sci. 2014;39(4):159–69. doi: 10.1016/j.tibs.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martins R, Lithgow GJ, Link W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 2016;15(2):196–207. doi: 10.1111/acel.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.La Colla A, Vasconsuelo A, Milanesi L, Pronsato L. 17β-estradiol protects skeletal myoblasts from apoptosis through p53, Bcl-2, and FoxO families. J Cell Biochem. 2017;118(1):104–15. doi: 10.1002/jcb.25616. [DOI] [PubMed] [Google Scholar]
- 14.Shukla S, Rizvi F, Raisuddin S, Kakkar P. FoxO proteins’ nuclear retention and BH3-only protein Bim induction evoke mitochondrial dysfunction-mediated apoptosis in berberine-treated HepG2 cells. Free Radic Biol Med. 2014;76:185–99. doi: 10.1016/j.freeradbiomed.2014.07.039. [DOI] [PubMed] [Google Scholar]
- 15.Xie S, Deng Y, Pan YY, et al. Chronic intermittent hypoxia induces cardiac hypertrophy by impairing autophagy through the adenosine 5′-monophosphate-activated protein kinase pathway. Arch Biochem Biophys. 2016;606:41–52. doi: 10.1016/j.abb.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Pan YY, Deng Y, Xie S, et al. Altered Wnt signaling pathway in cognitive impairment caused by chronic intermittent hypoxia: Focus on glycogen synthase kinase-3β and β-catenin. Chin Med J (Engl) 2016;129(7):838–45. doi: 10.4103/0366-6999.178969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savai R, Al-Tamari HM, Sedding D, et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20(11):1289–300. doi: 10.1038/nm.3695. [DOI] [PubMed] [Google Scholar]
- 18.Li G, He Y, Yao J, et al. Angelicin inhibits human lung carcinoma A549 cell growth and migration through regulating JNK and ERK pathways. Oncol Rep. 2016;36(6):3504–12. doi: 10.3892/or.2016.5166. [DOI] [PubMed] [Google Scholar]
- 19.D’Orsi B, Mateyka J, Prehn JH. Control of mitochondrial physiology and cell death by the Bcl-2 family proteins Bax and Bok. Neurochem Int. 2017 doi: 10.1016/j.neuint.2017.03.010. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20.Chi HC, Chen SL, Cheng YH, et al. Chemotherapy resistance and metastasis-promoting effects of thyroid hormone in hepatocarcinoma cells are mediated by suppression of FoxO1 and Bim pathway. Cell Death Dis. 2016;7(8):e2324. doi: 10.1038/cddis.2016.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng H, Du B, Jiang H, Gao J. Over-expression of CHAF1A promotes cell proliferation and apoptosis resistance in glioblastoma cells via AKT/FOXO3a/Bim pathway. BiochemBiophys Res Commun. 2016;469(4):1111–16. doi: 10.1016/j.bbrc.2015.12.111. [DOI] [PubMed] [Google Scholar]
- 22.Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10(3):161–67. doi: 10.1038/sj.cr.7290045. [DOI] [PubMed] [Google Scholar]
- 23.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–84. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 24.Diao SL, Xu HP, Zhang B, et al. Associations of MMP-2, BAX, and Bcl-2 mRNA and protein expressions with development of atrial fibrillation. Med Sci Monit. 2016;22:1497–507. doi: 10.12659/MSM.895715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faber AC, Ebi H, Costa C, Engelman JA. Apoptosis in targeted therapy responses: The role of BIM. Adv Pharmacol. 2012;65:519–42. doi: 10.1016/B978-0-12-397927-8.00016-6. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Gan B, Liu D, Paik JH. FoxO family members in cancer. Cancer Biol Ther. 2011;12(4):253–59. doi: 10.4161/cbt.12.4.15954. [DOI] [PubMed] [Google Scholar]
- 27.Roy SK, Chen Q, Fu J, et al. Resveratrol inhibits growth of orthotopic pancreatic tumors through activation of FOXO transcription factors. PLoS One. 2011;6(9):e25166. doi: 10.1371/journal.pone.0025166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilk A, Urbanska K, Grabacka M, et al. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell Cycle. 2012;11(14):2660–71. doi: 10.4161/cc.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song HM, Song JL, Li DF, et al. Inhibition of FOXO1 by small interfering RNA enhances proliferation and inhibits apoptosis of papillary thyroid carcinoma cells via Akt/FOXO1/Bim pathway. Onco Targets Ther. 2015;8:3565–73. doi: 10.2147/OTT.S95395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tikhanovich I, Cox J, Weinman SA. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol. 2013;28(Suppl 1):125–31. doi: 10.1111/jgh.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang C, Li J, Hong K, et al. BH3-only protein Bim is upregulated and mediates the apoptosis of cardiomyocytes under glucose and oxygen-deprivation conditions. Cell Biol Int. 2015;39(3):318–25. doi: 10.1002/cbin.10392. [DOI] [PubMed] [Google Scholar]
- 32.Gillings AS, Balmanno K, Wiggins CM, et al. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 2009;276(21):6050–62. doi: 10.1111/j.1742-4658.2009.07329.x. [DOI] [PubMed] [Google Scholar]