

ABSTRACT
Monoclonal antibodies developed for therapeutic or diagnostic purposes need to demonstrate highly defined binding specificity profiles. Engineering of an antibody to enhance or reduce binding to related antigens is often needed to achieve the desired biologic activity without safety concern. Here, we describe a deep sequencing-aided engineering strategy to fine-tune the specificity of an angiopoietin-2 (Ang2)/vascular endothelial growth factor (VEGF) dual action Fab, 5A12.1 for the treatment of age-related macular degeneration. This antibody utilizes overlapping complementarity-determining region (CDR) sites for dual Ang2/VEGF interaction with KD in the sub-nanomolar range. However, it also exhibits significant (KD of 4 nM) binding to angiopoietin-1, which has high sequence identity with Ang2. We generated a large phage-displayed library of 5A12.1 Fab variants with all possible single mutations in the 6 CDRs. By tracking the change of prevalence of each mutation during various selection conditions, we identified 35 mutations predicted to decrease the affinity for Ang1 while maintaining the affinity for Ang2 and VEGF. We confirmed the specificity profiles for 25 of these single mutations as Fab protein. Structural analysis showed that some of the Fab mutations cluster near a potential Ang1/2 epitope residue that differs in the 2 proteins, while others are up to 15 Å away from the antigen-binding site and likely influence the binding interaction remotely. The approach presented here provides a robust and efficient method for specificity engineering that does not require prior knowledge of the antigen antibody interaction and can be broadly applied to antibody specificity engineering projects.
KEYWORDS: Angiopoietin-1, Angiopoietin-2, deep mutational scanning, deep sequencing, dual action Fab, dual specificity antibody, drug discovery, phage, specificity engineering, vascular endothelial growth factor
Abbreviations
- Ang1
Angiopoietin-1
- Ang2
Angiopoietin-2
- VEGF
vascular endothelial growth factor
- ER
enrichment ratio
- DAF
dual action Fab
- HC
heavy chain
- LC
light chain
- CDR
complementary-determining region
- PDB
protein databank
Introduction
A classical paradigm of immunology states that monoclonal antibodies are monospecific and typically recognize a single antigen exclusively (reviewed by Eisen and Chakraborty1). However, as recognized over 40 years ago, certain antibodies do exhibit binding activity to more than one antigen (multi-specificity).2 One reason for multi-specificity is that the same or a very similar epitope is present on more than one antigen. Typical examples include species cross-reactive antibodies that recognize orthologous proteins in different species3,4 or antibodies that interact with different members of a conserved protein family.5-7 Another underlying mechanism in multi-specificity lies in the plasticity of the antigen binding site of some antibodies, which allows for the recognition of structurally unrelated epitopes by the same antigen binding site.8
Specificity engineering is often required during the development of monoclonal antibodies for diagnostic or therapeutic use, and has been applied to add on or improve the recognition of related epitopes, for example, by increasing cross- species specificity,9 targeting multiple toxin serotypes using one antibody,10,11 or extending the binding against related haptens12,13 or various members of a protein family.14,15 Specificity engineering has also been used to shave off binding function, for example, to abolish or reduce binding to a closely related antigen,16-18 and can be critically important for reducing the potential of off-target toxicity.19 As a more extreme example of specificity engineering, we previously reported a step-wise engineering strategy for generating dual-specific antibodies de novo, called Two-in-One Antibody with dual action Fab (DAF), which are capable of recognizing two structurally unrelated antigens using a highly overlapping antigen binding site. The first proof-of-concept DAF binds vascular endothelial growth factor (VEGF) and human epithelial growth factor receptor (HER)2.20 One of the Two-in-One antibodies subsequently generated, duligotuzumab (MEHD7945A), which targets EGFR and HER3, has advanced into clinical Phase 2 studies, demonstrating the therapeutic utility of this engineering strategy.20-22
To tune the specificity of antibodies, various engineering approaches have been used, and they commonly involve the use of phage or yeast display together with various types of combinatorial library design strategies, such as computational based design,9 structural guided design,17 random mutagenesis (e.g., error-prone PCR)11-14,18 or a combination thereof.15,19 Recently, a new approach called deep mutational scanning, which adds deep sequencing (or NextGen Sequencing) to the combination of single- or multiple-site saturated mutagenesis libraries and a selection by a display system, has enabled large-scale assessment of the effects of mutations on the functional fitness of a protein, e.g., ligand binding function.23 Mutations that are enriched may have positive effects on the protein fitness, while mutations that are depleted likely have negative effects on the fitness. Deep mutational scanning has primarily been used to identify mutations that improve the antigen binding affinity of antibodies.24-27 However, one can argue that in typical engineering where finding affinity optimizing mutation is the aim, deep sequencing is not necessarily required because highly enriched mutations can be found without sequencing a large number of clones. Here, we describe an application of the deep mutational scanning in antibody specificity engineering where accurate depletion information only possible with deep sequencing is a necessity.
Using deep mutational scanning, we previously reported the affinity maturation of an angiopoietin-2 (Ang2)/VEGF DAF, 5A12, resulting in DAF variants with sub-nanomolar affinity against the two angiogenic growth factors,28 which represent the highest dual affinity DAFs reported to date. One of the affinity-matured DAFs, 5A12.1, unintentionally obtained nano-molar affinity against Ang1 during the affinity maturation process. Ang1, which is highly homologous to Ang2, belongs to the angiopoietin family (Ang1, Ang2, Ang3 and Ang4), and all four proteins are ligands of the tyrosine kinase receptor Tie-2.29 While Ang2 is believed to be an antagonist against Ang1 in activating Tie-2 signaling for vascular stabilization, both Ang1 and Ang2 are thought to be agonist for Tie-2 signaling important for lymphatic stability. Blocking Ang2 together with VEGF is aimed for a condition called wet age-related macular degeneration, where uncontrolled vascular growth and leakage play a role in the pathogenesis.30 Ang1 cross reactivity therefore may increase the safety risk of an Ang2/VEGF blocking antibody like 5A12.1.31 To remove Ang1 binding of 5A12.1, without affecting the Ang2 and VEGF binding function, we turned again to deep mutational scanning. From the mutational libraries panned against all three targets, Ang1, Ang2 and VEGF in parallel, we identified the mutations that are depleted from Ang1 panning but enriched or not depleted from VEGF and Ang2 panning. We then confirmed some of the top mutations by binding studies of the Fab variants and examined the mechanism of the mutation by mapping these mutations on the structure and structural model of Fab-Ang2/1 complex.
Results
5A12.1Ang1 binding and phage panning
5A12.1 has a KD for VEGF and Ang2 of 0.2 and 0.7 nM, respectively, which is greatly improved over the parental DAF 5A12 (KD for VEGF: 5 nM, KD for Ang2: 5 nM) (Table 2).28 However, further characterization of the antibody revealed that while 5A12 had no detectable binding to the angiopoietin family member Ang1 (Biacore data not shown), the affinity maturation process led inadvertently to recruiting significant Ang1 binding. The overall degree of sequence identity between Ang1 and Ang2 is 60% (comparing 495 amino acids). However, the epitope of 5A12 on Ang2 resides in a domain of greater homology between Ang1 and Ang2, and there are only five substitutions in the 16 residues that make up the structural epitope (69% identical residues between Ang1 and Ang2) (Fig. 1A).
Table 2.
Kinetic binding data of 5A12.1 and 5A12.1 Fab variants to the three antigens. The clones are numbered the same way as in the scatter plot in Fig. 3. In addition, for each position it is listed “C” if the position is in contact with Ang2 (< 5Å away from Ang2) in the 5A12:Ang2 crystal structure (PDB 4ZFG). Further, we list if a position is surface exposed, “S” or buried, “B” as listed in 28.
| VEGF |
Ang2 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Clone # | Mutated chain | mutation | Ang2 contact | Solvent accessibility | ka (1/Ms) | kd (1/s) | Kd (M) | ka (1/Ms) | kd (1/s) | Kd (M) | Ang1 Kd (M) |
| 1 | wt | wt | wt | wt | 9.18E+05 | 5.67E-04 | 6.18E-10 | 3.99E+05 | 3.12E-04 | 7.83E-10 | 4.42E-009 |
| 2 | HC | I29T | N | B | 4.64E+05 | 7.13E-04 | 1.54E-09 | 3.96E+05 | 1.87E-03 | 4.71E-09 | none |
| 3 | HC | I29Q | N | B | 4.47E+05 | 7.25E-04 | 1.62E-09 | 6.29E+05 | 2.07E-03 | 3.29E-09 | none |
| 4 | HC | I29K | N | B | 3.47E+05 | 7.20E-04 | 2.07E-09 | 6.30E+05 | 3.14E-03 | 5.00E-09 | none |
| 5 | HC | S30G | N | S | 4.71E+05 | 7.88E-04 | 1.67E-09 | 7.25E+05 | 1.01E-03 | 1.40E-09 | none |
| 6 | HC | Y32A | N | S | 2.03E+05 | 8.51E-04 | 4.18E-09 | 7.23E+05 | 1.58E-03 | 2.19E-09 | some |
| 7 | HC | I34Q | N | B | 5.19E+05 | 8.76E-04 | 1.69E-09 | 3.93E+05 | 3.67E-04 | 9.33E-10 | some |
| 8 | HC | H35D | C | B | 2.68E+05 | 5.81E-04 | 2.17E-09 | 7.72E+05 | 4.25E-04 | 5.50E-10 | none |
| 9 | HC | P52aE | N | S | 1.63E+05 | 2.15E-03 | 1.32E-08 | 3.54E+05 | 7.28E-04 | 2.06E-09 | some |
| 10 | HC | Y56A | N | S | 4.52E+05 | 9.17E-04 | 2.00E-09 | 5.44E+05 | 6.00E-04 | 1.10E-09 | some |
| 11 | HC | T57H | N | S | 4.43E+05 | 5.87E-04 | 1.32E-09 | 3.54E+05 | 3.91E-03 | 1.10E-08 | none |
| 12 | HC | T57D | N | S | 4.13E+05 | 6.12E-04 | 1.48E-09 | 2.77E+05 | 3.19E-03 | 1.15E-08 | none |
| 13 | HC | Y58I | C | S | 3.56E+05 | 5.65E-04 | 1.59E-09 | 1.55E+06 | 2.05E-03 | 1.32E-09 | none |
| 14 | HC | Y58W | C | S | 4.17E+05 | 4.51E-04 | 1.08E-09 | 3.69E+05 | 4.65E-04 | 1.26E-09 | some |
| 15 | HC | Y58L | C | S | 3.45E+05 | 5.31E-04 | 1.54E-09 | 1.09E+06 | 2.24E-03 | 2.06E-09 | none |
| 16 | HC | A93S | N | B | 2.72E+05 | 6.50E-04 | 2.40E-09 | 4.26E+05 | 7.75E-04 | 1.82E-09 | some |
| 17 | HC | F95M | N | B | 3.84E+05 | 1.35E-03 | 3.51E-09 | 4.25E+05 | 2.32E-03 | 5.45E-09 | some |
| 18 | HC | V96T | N | B | 3.03E+05 | 6.79E-04 | 2.24E-09 | 6.17E+05 | 1.23E-03 | 2.00E-09 | some |
| 19 | HC | L99A | C | S | 4.25E+05 | 1.71E-03 | 4.00E-09 | 2.69E+05 | 1.19E-03 | 4.43E-09 | some |
| 20 | LC | L28M | C | S | 5.11E+05 | 6.27E-04 | 1.23E-09 | 3.33E+05 | 1.22E-03 | 3.66E-09 | none |
| 21 | LC | L28A | C | S | 4.98E+05 | 7.22E-04 | 1.45E-09 | 3.10E+05 | 3.42E-04 | 1.10E-09 | some |
| 22 | LC | Y55A | N | S | 5.19E+05 | 7.22E-04 | 1.39E-09 | 1.95E+05 | 6.91E-04 | 3.55E-09 | none |
| 23 | LC | Y55E | N | S | 5.44E+05 | 1.14E-03 | 2.09E-09 | 2.69E+05 | 7.03E-04 | 2.61E-09 | some |
| 24 | LC | S56Q | N | S | 4.95E+05 | 6.61E-04 | 1.33E-09 | 3.45E+05 | 4.15E-04 | 1.20E-09 | some |
| 25 | LC | H89M | N | B | 5.74E+05 | 8.66E-04 | 1.51E-09 | 2.47E+05 | 9.31E-04 | 3.85E-09 | some |
| 26 | LC | S94P | N | S | 2.83E+05 | 2.64E-03 | 9.32E-09 | 6.73E+05 | 8.33E-04 | 1.24E-09 | some |
Figure 1.
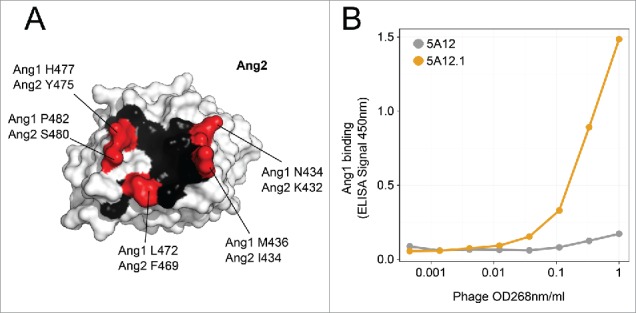
(A) Structural epitope of 5A12 on Ang2 as observed in 5A12:Ang2 crystal structure (PDB 4ZFG) is mapped onto the surface of Ang2. Epitope positions (< 5Å from 5A12) that are conserved between Ang1 and Ang2 are colored in black, while positions that differ between the two proteins are labeled red. (B) Phage titration ELISA to measure the binding of phage displaying either the parental Ang2/VEGF DAF 5A12 (gray) or the affinity maturated version 5A12.1 (orange) to Ang1.
We first tested the feasibility of 5A12.1 panning against Ang1. 5A12 and 5A12.1 Fab were displayed on phage as described.32 A phage binding titration ELISA was performed against Ang1 (Fig. 1B), which demonstrated Ang1 binding of the displayed 5A12.1 Fab should be sufficient to perform phage panning. To identify the effects of all possible single CDR mutations on all three antigens, we decided to utilize a deep mutational approach. In our approach, we used our previously published 3NNK library design28 in which each clone contains three mutations encoded by the NNK codons, one in each of the three CDRs. The 3NNK library design not only allows us to calculate enrichment ratios (ERs) as a measure of the effect of mutations on the antibody fitness, but also allows mutations to a wider range of CDR positions, including those that are not in direct contact with the antigens, increasing the chance to identify specificity modulating mutations that act in an allosteric manner. Two libraries were generated, one containing mutations in the heavy chain CDRs and one in the light chain CDRs. Both libraries were panned against all three antigens, i.e., Ang1, Ang2 and VEGF (Table 1). Ang2 and VEGF pannings were performed using their respective biotinylated antigen in solution. Ang1 pannings were performed using both biotinylated antigen in solution and plate-bound antigen. Plate bound panning can potentially yield more information because it is less stringent than solution panning and may reveal mutations to further reduce the weak interaction between Ang1 and 5A12.1.
Table 1.
Sequencing statistics for calculating enrichment ratios of the different panning samples. For each data set the total number of sequencing reads, the number of reads that passed quality control and further processing, as well as the number of observed versus the expected number of single NNK mutations in all three CDRs of given sample are listed.
| Library name | Panning Antigen | Panning type | Final panning condition | Library | No. of raw reads | Reads after filtering and quality control |
|---|---|---|---|---|---|---|
| T28P_HCNNK_presort | — | — | — | HC | 3105123 | 890518 |
| T28P_HCNNK_V0.2 | VEGF | Solution | 0.2nM VEGF @ RT | HC | 2909108 | 1848223 |
| T28P_HCNNK_V0.2comp37 | VEGF | Solution | 0.2nM VEGF +1000x competition @ 37°C | HC | 2821201 | 1835315 |
| T28P_HCNNK_A20.2 | Ang2 | Solution | 0.2nM Ang2 @ RT | HC | 1250730 | 794404 |
| T28P_HCNNK_A20.2comp | Ang2 | Solution | 0.2nM Ang2 + 1000x competition @ RT | HC | 1980579 | 1213739 |
| T28P_HCNNK_A1 | Ang1 | Plate | 5ug/ml Ang1 coated | HC | 2230805 | 869890 |
| T28P_HCNNK_A120 | Ang1 | Solution | 10nM Ang1 @ RT | HC | 2091304 | 1265479 |
| T28P_LCNNK_presort | — | — | — | LC | 3085954 | 361233 |
| T28P_LCNNK_V0.2 | VEGF | Solution | 0.2nM VEGF @ RT | LC | 3192859 | 1930882 |
| T28P_LCNNK_V0.2comp37 | VEGF | Solution | 0.2nM VEGF +1000x competition @ 37°C | LC | 2068266 | 1241521 |
| T28P_LCNNK_A20.2 | Ang2 | Solution | 0.2nM Ang2 @ RT | LC | 2799196 | 1350784 |
| T28P_LCNNK_A20.2comp | Ang2 | Solution | 0.2nM Ang2 + 1000x competition @ RT | LC | 1244163 | 123207 |
| T28P_LCNNK_A1 | Ang1 | Plate | 5ug/ml Ang1 coated | LC | 1096430 | 59751 |
| T28P_LCNNK_A120 | Ang1 | Solution | 10nM Ang1 @ RT | LC | 1828262 | 914787 |
The phage pools from the final two most stringent panning conditions for each antigen together with the original library (pre-sorted) were sent for sequencing (Table 1). The Ang2 panning conditions for both the heavy and light chain library were 0.2 nM biotinylated Ang2 (A20.2) and 0.2 nM biotinylated Ang2 in the presence of 200 nM non-biotinylated Ang2 as competitor (A20.2comp). For VEGF, panning with 0.2 nM biotinylated VEGF (V0.2) and panning at 37°C with 0.2 nM biotinylated VEGF in the presence of 200 nM non-biotinylated VEGF (V0.2comp37) were chosen for sequencing. For Ang1, we used the final round of panning using 20 nM biotinylated Ang1 (A120). In addition, a less stringent panning procedure using plate immobilized Ang1 (A1p) was sent for deep sequencing. By using two panning conditions for each antigen, we aimed to increase the odds for identifying the best mutations that strongly reduce Ang1 binding while maintaining high VEGF and Ang2 binding.
Enrichment ratio determination and sample cross correlation
In total, DNA from 14 samples (two pre-sorted libraries and 12 sorted libraries) was deep sequenced using MiSeq (Table 1). The ERs for all CDR mutations were determined by dividing the frequency of a given mutation at a given position in the sorted samples by the frequency in the pre-sorted sample. In total, the ER of 483 LC mutations and 609 HC mutations under six panning conditions (2898 and 3654 ER, respectively) were obtained. Fig. 2 shows a comparison of all ER (as Log2 ER) between the six heavy chain data sets and the six light chain data sets. As expected the samples from panning against the same antigen using similar panning protocol showed the highest Pearson correlation coefficient, e.g., V0.2 in comparison with V0.2comp37, while samples from panning against different antigens showed very low correlation e.g., Ang20.2 in comparison to V0.2. In addition, the panning protocol used at times has a strong influence on the ER, e.g., the two Ang1 pannings (A120 and A1p), which differed in their antigen immobilization methods showed low correlation.
Figure 3.
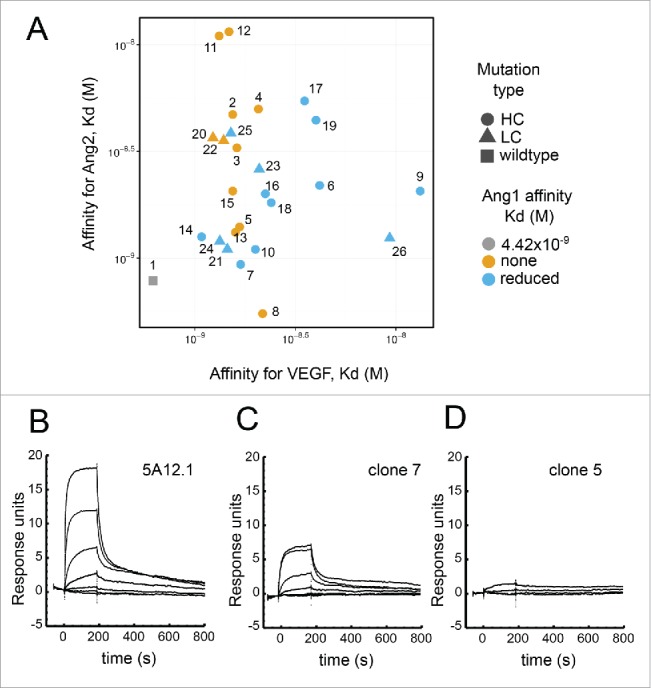
Binding of selected Fab variants. (A) The scatter plot visualizes the KD of Fab with selected mutations for VEGF and Ang2 as measured by Biacore. The shape of each data point indicates the location of the mutation with circle indicating mutation in HC, triangle in the LC and the gray square indicating 5A12.1wt with an affinity for Ang1 of KD 4 nM, with (B) showing the BIAcore sensorgram trace. The color of each data point encodes for Ang1 binding. Blue is for mutations that show much reduced yet still residual Ang1 binding with (C) showing the BIAcore sensorgram trace of such an example; while orange is for mutation showing no detectable binding with (D) showing the BIAcore sensorgram trace of an example.
Figure 2.
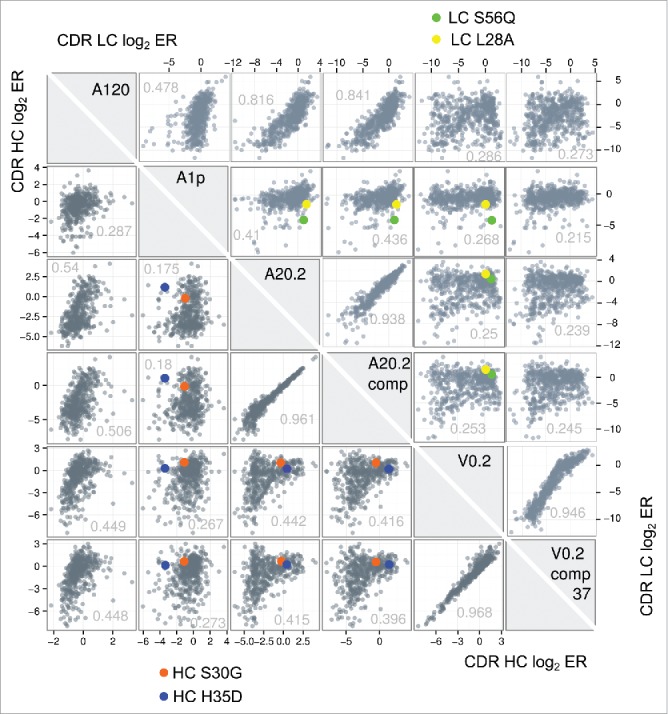
A cross-correlation plot of the enrichment ratios (ER) of each single mutation from 6 NNK phage library pannings is shown to visualize the distribution of ERs obtained from the different panning conditions and how they relate to each other. Each box shows a correlation between two ER data sets obtained from different phage panning tracks as scatter plot of all HC CDR single mutations (lower left triangle) or LC CDR single mutations (upper right triangle). The respective X- and Y-axes represent the respective log2ER for the sample labels in the diagonal gray boxes. To visualize the behavior of selected mutations that passed our selection filter (see main text for details) for candidates that reduce Ang1 binding while maintaining Ang2/VEGF binding the enrichment ratios of those mutants are highlighted as circles in blue and orange. The Pearson correlation coefficient for each panning pair is listed in the scatter plots.
Identification of specificity improving residues and affinity determination
To promote the desired specificity of 5A12.1, we filtered for mutations that were strongly depleted from Ang1 panning, while showing enrichment or only minimal depletion in the Ang2 and VEGF panned tracks. The filter was applied to all iterations of Ang1, Ang2 and VEGF data sets. 26 heavy chain mutations (number of mutations H1: 10, H2: 9, H3: 7) and 9 light chain mutations (number of mutations L1: 3, L2: 4, L3: 2) satisfied the filter condition. Interestingly, diversifying our Ang1 panning approach was helpful, as the only mutations identified in the light chain were from the Ang1 plate panning data set, while the heavy chain mutations from both Ang1 plate and solution panning passed the filter. Most selected mutations are located in CDR-H1. The finding that fewer light chain mutations are identified is consistent with the fact that Ang2, and thus presumably Ang1 binding, is facilitated mainly through the light chain.28 CDR-L3 especially has been shown to contain hotspot residues for the 5A12:Ang2 interaction that do not tolerate mutation.28
Notably, 18 heavy and seven light chain mutations were identified at least twice in the different iterations of the different panning data sets, and were selected for further screening. The 25 5A12.1 Fab variants were expressed and purified as described previously.28 The affinity of these variants toward all three antigens was measured by Biacore and, indeed, all variants showed significantly reduced Ang1 binding. 11 variants showed essentially no Ang1 binding by Biacore under the conditions tested (single cycle kinetics using 1 uM of Ang1 as analyte). The other 14 variants retained some Ang1 binding, but the weak binding data could not to be fitted to determine KD. In addition, most of mutations retained sub-nanomolar to single digit nanomolar affinity toward Ang2 and VEGF. For example, the mutation HC-S30G recognizes VEGF with a KD of 1.7 nM and Ang2 with a KD of 1.4 nM, while showing no detectable Ang1 binding. Similarly, another mutation HC-Y58I has a KD of 1.6 nM for VEGF and 1.32 nM for Ang2 while also showing no Ang1 binding activity.
Non-specific binding properties and stability of the identified 5A12.1 variants
Extensive engineering of antibodies carries the risk of introducing mutations that could unintentionally change biophysical properties. We therefore measured some of the identified mutants' biophysical properties, which are linked to the “developability” of antibodies.33 A high melting temperature of the Fab portion correlates with an increase in antibody expression titers.33,34 We thus used differential scanning fluorimetry (DSF) to determine the melting temperature (Tm) of 5 Fab variants that showed no or strongly reduced affinity for Ang1 (HC.S30G, HC.Y58I, HC.Y58W, LC.S56Q and LC.H89M) and compared it to wildtype 5A12.1 (Table 3). All variants have a very similar or even slightly higher melting temperature than 5A12.1 (ΔTM between -0.4 and +2.4)
Table 3.
Developability profiles of 5A12.1 Fab and 5 5A12.1 Fab variants. The melting temperature (Tm) of the 5A12.1 and five selected 5A12 Fab variants as determined by DSF is listed. In addition, non-specific binding properties of the IgGs were determined by baculovirus ELISA. The normalized baculovirus ELISA score is listed. The low binding and high binding antibody controls had normalized baculovirus ELISA score as 0.15 and 3.23, respectively.
| Clone | Tm (°C) | Baculovirus ELISA |
|---|---|---|
| T28P | 81.8 | 0.31 |
| HC.S30G | 80.8 | 0.4 |
| HC.Y58I | 82.2 | 0.78 |
| HC.Y58W | 82 | 0.51 |
| LC.S56Q | 82 | 0.35 |
| LC.H89M | 84.2 | 0.42 |
We next tested whether the specificity engineered mutants exhibit non-specific binding. Non-specific binding can lead to fast clearance and shortened half-life of antibodies in vivo. We used the previously described baculovirus ELISA35 to measure the non-specific binding properties of 5A12.1 and the 5 specificity engineered variants (Table 3). All the variants had normalized baculovirus ELISA score <1, indicating lower risk for fast clearance. In summary, both assays, DSF and baculovirus ELISA, indicate all variants identified have favorable developability profiles.
Structural mapping of mutations generates a specificity landscape of 5A12.1
We next mapped the 25 mutations on to the previously published structure of Ang2:5A12 complex to obtain a structural map of specificity modulating residues (Fig. 4). To compare the spatial arrangement of the CDR mutations of the antibody to the location of non-conserved residues on the Ang2 epitope (Fig. 1A), we structurally aligned Ang1 with Ang2 in the Ang2:5A12 structure, thereby constructing an Ang1:5A12 model (Fig. 4). When mapping the specificity changing mutations onto the model, one hotspot of specificity modifying mutations consisting of three adjacent CDR-H2 positions, HC-56, HC-57 and HC-58, and two adjacent CDR-H1 positions, CDR-H34 and HC-H35 becomes visible. Of these five structurally adjacent residues, we noted that HC-35 and HC-58 are in direct contact of Ang2 at a non-conserved position, Ang2-I434, which is Ang1-M436. This suggests that the improved discrimination between Ang2 and Ang1 of these variants is based on a differential interaction with the Ang2-I434/Ang1-M436 site.
Figure 4.
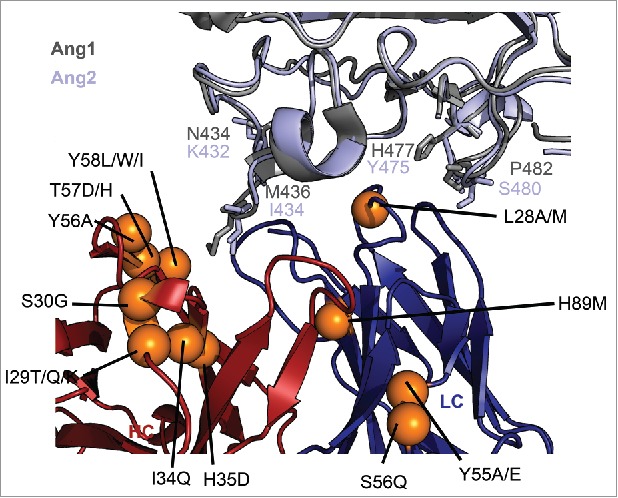
Structural mapping of the specificity determining mutations. The crystal structure of the 5A12:Ang2 complex (PDB 4ZFG) is shown in cartoon representation. The heavy chain is shown in blue, the light chain in red and Ang2 in light blue. The crystal structure of Ang1 (PDB 4JYO) has been structurally aligned onto Ang2 and colored in gray. The orange spheres represent location of mutations that greatly reduce Ang1 binding while maintaining VEGF and Ang2 binding affinity. The side chains of residues different in Ang1 and Ang2 that are located in the structural epitope of Ang2 are shown in stick representation.
The other mutations are fairly equally distributed over the other CDRs and do not cluster spatially. Interestingly, many of these specificity-modifying CDR mutations are not in direct contact with Ang2 (Table 2), and thus very likely also not in contact with Ang1, hence affecting the binding specificity allosterically. A striking example is LC-S56Q, which greatly reduces Ang1 binding while being more than 15 Å away from Ang2 or Ang1 (based on the model). Another example is a structurally buried position at the stem of CDR-L3, LC-H89M, which also reduces Ang1 binding of 5A12.1. While the importance of antigen-distal residues in affinity and specificity engineering has been noted in previous reports,11,13,26 it is difficult to predict this kind of mutations based on structural information. Identification of these remote mutations is an advantage of our full-mapping approach.
Discussion
Previous applications of deep scanning mutations (for affinity engineering) mainly focused on finding strongly enriched mutations to improve the affinity of a given binder.24-26 However, the identification of strongly enriched mutations do not necessarily require deep sequencing, as in many cases limited sequencing by Sanger method can identify strongly enriched mutation.36-38 The advantage of deep mutational scanning for protein engineering is realized in applications that require a large data set to accurately determine the depletion of a mutation. For example, tracking the change from 1 in 100 to 1 in over 300 in occurrences for identifying the mutations that have negative effects on the selected binding function can only be accomplished with thousands of sequences.28 Here, we demonstrate the use of depletion data for specificity engineering, which allowed us to greatly reduce the binding of a DAF against Ang1 while maintaining high affinity binding against Ang2 and VEGF.
Although the method introduced here represents an extreme case of Two-in-One DAF engineering for binding three distinct antigens, it is applicable not only to DAFs, but also generally to fine-tune the affinity and specificity of any antibody or protein binder. When we re-analyzed our data focusing only on Ang2 and Ang1 binding with a very strict filter (ER for Ang1 < -2 and ER for Ang2 > 1), we identified seven more mutations that can selectively bias Ang2 over Ang1 binding (data not shown). This observation suggests that the engineering of the specificity of Ang2 over Ang1 without considering VEGF binding can be achieved very easily using deep mutational scanning. Thus, the approach demonstrated here is a robust and efficient way to map specificity-determining positions in antibody-antigen and protein-protein interactions.
Material and methods
Antigens
Phage ELISA, phage panning and subsequent Biacore was performed using the receptor binding domain (RBD) of human Ang2 with a C-terminal His-tag (hAng2his), human Fc c-terminally fused to RBD of human Ang1 protein (Fc.hAng1) and human VEGF 8–109 (hVEGF109).
NNK library generation and phage panning
To test if selection of 5A12.1 could be performed on Fc.hAng1, 2 ug/ml of Fc.hAng1 was coated on an ELISA plate (Nunc Maxisorp). After blocking using 0.5% bovine serum albumin (BSA), 0.1% Tween-20 in phosphate-buffered saline (PBS), serially diluted phage was added. Bound phage were detected using a horseradish peroxidase-conjugate anti-M13 antibody (GE healthcare, catalog number 27–9420–01). NNK CDR walking phage libraries of 5A12.1 for deep mutational scanning were constructed from 5A12 Fab format with either LC or HC CDR residues mutated using a mixture of degenerate oligonucleotides for each CDR loop and a Kunkel mutagenesis protocol as described previously.28 Each oligonucleotide randomizes one of the CDR sites with an NNK codon which encodes for all 20 amino acids using all 32 codons including a stop codon (N = G, A, T and C and K = G and T in equal portions). Libraries were designed to allow one NNK mutation in each of the three LC or HC CDRs called 3NNK libraries. Library DNA was electroporated into E. coli XL1 cells yielding ∼5–109 transformants. Libraries were sorted against biotinylated hVEGF109, hAng2his or hAng1-FC using a described previously solution panning protocol,39 which increased the stringency of successive rounds by incubating phage with decreasing concentration of antigens. Antigen concentrations ranged from 5 nM - 0.2 nM for hVEGF109 panning, from 100 nM - 0.2 nM for hAng2his panning and from 100 nM - 20 nM for Fc.hAng1. In addition, to solution panning, panning with on plate immobilized Fc.hAng1 was performed using a described previously protocol.40
Illumina sequencing and data analysis
For deep sequencing, phagemid double-stranded DNA was isolated from selected rounds. The VH and the VL segment from each sample were amplified by an 18-cycle PCR amplification using Phusion DNA polymerase (New England Biolabs). The amplicon was purified on a 2% agarose gel. Amplicons were prepared using the TruSeq Nano DNA library preparation kit from Illumina. Multiplexed adaptor-ligated libraries with unique barcodes were sequenced on the Illumina MiSeq, for 2 × 300 cycle, paired-end sequencing. Sequencing data were analyzed using the statistical programming language R41 and ShortRead.42 Quality control was performed on identified CDR sequences, where each CDR sequence was checked for the correct length and was allowed to carry only up to one NNK mutation and no non-NNK mutation. Calculating the frequency of all mutations, of every randomized position, generated position weight matrices. ERs for all mutations were calculated by dividing the frequency of a given mutation at a given position in the sorted sample by the frequency of the very same mutation in the unsorted sample, as described previously.23 To identify specificity improving mutations, we applied the following filter: ERx Ang1 < -1 & ERx Ang2 > -0.5 & ERx VEGF > 0, where ERx is the log2 enrichment ratio of a given mutation X. Mutations which passed this filter in various iterations of the data sets obtained from panning different antigens were chosen for further characterization. Data was plotted using ggplot2.43
Antibody characterization
The VL and VH of selected phage clones were cloned into vectors previously designed for transient human Fab expression in mammalian cells.44 Fabs were purified by affinity chromatography. For KD determination, Fab was used as analyte in Biacore surface plasmon resonance measurements using a CM5 sensor chip immobilized with low density (RU) of hVEGF109, hAng2his or Fc.Ang1 at 25°C to determine monovalent affinities. For thermal melt temperature (Tm), we used DSF, which monitors thermal unfolding of proteins in the presence of a fluorescent dye SYPRO orange dye (Invitrogen). The diluted dye (1:20) 1 µl was added into 24 µl Fab protein (∼100 µg/ml). The fluorescence intensity during temperature increase from 20°C to 100°C was plotted and Tm, the inflection point of the transition curve was calculated using the Boltzmann equation.45 For baculovirus ELISA VH and VL sequences of selected variants were cloned into a mammalian IgG vector for expression and purification by affinity chromatography. 1% baculovirus particle suspension was prepared in coating buffer (0.05 M sodium carbonate pH 9.6) and 25 μl was added per well in 384-well plate (Nunc-Immuno Plate ThermoFisher) at 4°C overnight. The wells were blocked with 50μl blocking buffer (PBS containing 0.5% BSA and 10PPM Proclin) for 1 h at room temperature. After rinsing the plate three times with washing buffer (PBS), 25 ul of purified selected IgGs in blocking buffer were loaded in duplicate wells for 1 h at room temperature. The plate was washed six times with washing buffer and 25 μl at 10 ng/ml goat anti-human Fc fragment specific conjugated to horseradish peroxidase (Jackson ImmunoResearch) was added per well for 1 h at room temperature. Then the plate was washed six times with washing buffer and 25 ul of TMB substrate (Moss, Inc.) was added per well for 15 min at room temperature. The reaction was stopped by adding 25 ul of 1 M phosphoric acid per well. Absorbance was measured at 450 nm using a plate reader. A high binding antibody with fast pharmacokinetic clearance, a mid-level binding antibody with borderline pharmacokinetic clearance, and a low binding antibody with normal pharmacokinetic clearance are run along with the samples. The OD values of samples are normalized to our mid binding antibody control value at 100 nM as normalized baculovirus ELISA score. Samples with normalized baculovirus ELISA score >1 have a higher risk for fast clearance and the score <1 indicates a lower risk for fast clearance.
Structural analysis
The previously published 5A12:Ang2 complex structure (PDB 4ZFG) was used as a template to generate a simple model of a 5A12:Ang1 complex. The 5A12:Ang1 model was generated by structurally aligning the crystal structure of Ang146 (PDB 4JYO) with Ang2 of 5A12:Ang2 complex (PDB 4ZFG) using Pymol (The PyMOL Molecular Graphics System, Schrödinger, LLC.). The structural epitope of 5A12 on Ang2 was identified using the program ‘Contact’ in the CCP4 suite and a cutoff value of 5Å.47 Crystal structures were visualized using Pymol.
Disclosure of potential conflicts of interest
All authors are or were paid employees of Genentech Inc., a member of the Roche Group, and are inventors for a patent application based on the work described herein.
Acknowledgments
We like to thank Vasant Janakiraman and Jeremy Stinson for performing sequence library preparation and Illumina Miseq sequencing runs.
References
- 1.Eisen HN, Chakraborty AK. Evolving concepts of specificity in immune reactions. Proc Natl Acad Sci 2010; 107:22373-80; PMID:21173256; https://doi.org/ 10.1073/pnas.1012051108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michaelides MC, Eisen HN. The strange cross-reaction of menadione (vitamin K3) and 2,4-dinitrophenyl ligands with a myeloma protein and some conventional antibodies. J Exp Med 1974; 140:687-702; PMID:4138007; https://doi.org/ 10.1084/jem.140.3.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CV, Hymowitz SG, Wallweber HJ, Gordon NC, Billeci KL, Tsai S-P, Compaan DM, Yin J, Gong Q, Kelley RF, et al.. Synthetic anti-BR3 antibodies that mimic BAFF binding and target both human and murine B cells. Blood 2006; 108:3103-11; PMID:16840730; https://doi.org/ 10.1182/blood-2006-03-011031 [DOI] [PubMed] [Google Scholar]
- 4.Fuh G, Wu P, Liang W-C, Ultsch M, Lee CV, Moffat B, Wiesmann C. Structure-function studies of two synthetic anti-vascular endothelial growth factor fabs and comparison with the avastin™ Fab. J Biol Chem 2006; 281:6625-31; PMID:16373345; https://doi.org/ 10.1074/jbc.M507783200 [DOI] [PubMed] [Google Scholar]
- 5.Hamdani N, van der Velden J. Lack of specificity of antibodies directed against human beta-adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol 2009; 379:403-7; PMID:19156400; https://doi.org/ 10.1007/s00210-009-0392-1 [DOI] [PubMed] [Google Scholar]
- 6.Pradidarcheep W, Stallen J, Labruyère WT, Dabhoiwala NF, Michel MC, Lamers WH. Lack of specificity of commercially available antisera against muscarinergic and adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol 2009; 379:397-402; PMID:19198807; https://doi.org/ 10.1007/s00210-009-0393-0 [DOI] [PubMed] [Google Scholar]
- 7.Sanders BM, Martin LS, Nakagawa PA, Hunter DA, Miller S, Ullrich SJ. Specific cross-reactivity of antibodies raised against two major stress proteins, stress 70 and chaperonin 60, in diverse species. Environ Toxicol Chem 1994; 13:1241-9; https://doi.org/ 10.1002/etc.5620130805 [DOI] [Google Scholar]
- 8.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science 2003; 299:1362-7; PMID:12610298; https://doi.org/ 10.1126/science.1079731 [DOI] [PubMed] [Google Scholar]
- 9.Farady CJ, Sellers BD, Jacobson MP, Craik CS. Improving the species cross-reactivity of an antibody using computational design. Bioorg Med Chem Lett 2009; 19:3744-7; PMID:19477127; https://doi.org/ 10.1016/j.bmcl.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riaño-Umbarila L, Contreras-Ferrat G, Olamendi-Portugal T, Morelos-Juárez C, Corzo G, Possani LD, Becerril B. Exploiting cross-reactivity to neutralize two different scorpion venoms with one single chain antibody fragment. J Biol Chem 2011; 286:6143-51; PMID:21156801; https://doi.org/ 10.1074/jbc.M110.189175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, Geren I, Stevens RC, Marks JD. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat Biotechnol 2007; 25:107-16; PMID:17173035; https://doi.org/ 10.1038/nbt1269 [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki C, Iba Y, Yamada Y, Takahashi H, Sawada J, Kurosawa Y. Changes in the specificity of antibodies by site-specific mutagenesis followed by random mutagenesis. Protein Eng 1999; 12:407-15; PMID:10360981; https://doi.org/ 10.1093/protein/12.5.407 [DOI] [PubMed] [Google Scholar]
- 13.Korpimäki T, Rosenberg J, Virtanen P, Lamminmäki U, Tuomola M, Saviranta P. Further improvement of broad specificity hapten recognition with protein engineering. Protein Eng 2003; 16:37-46; PMID:12646691; https://doi.org/ 10.1093/proeng/gzg010 [DOI] [PubMed] [Google Scholar]
- 14.Fagète S, Ravn U, Gueneau F, Magistrelli G, Kosco-Vilbois MH, Fischer N. Specificity tuning of antibody fragments to neutralize two human chemokines with a single agent. mAbs 2009; 1:288-96; PMID:20069756; https://doi.org/ 10.4161/mabs.1.3.8527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin Y, Djakovic S, Marsters S, Tien J, Peng J, Tremayne J, Lee G, Neve RM, Wu Y, Merchant M, et al.. Redesigning a monospecific anti-FGFR3 antibody to add selectivity for FGFR2 and expand antitumor activity. Mol Cancer Ther 2015; 14:2270-8; PMID:26269606; https://doi.org/ 10.1158/1535-7163.MCT-14-1050 [DOI] [PubMed] [Google Scholar]
- 16.Dubreuil O, Bossus M, Graille M, Bilous M, Savatier A, Jolivet M, Ménez A, Stura E, Ducancel F. Fine tuning of the specificity of an anti-progesterone antibody by first and second sphere residue engineering. J Biol Chem 2005; 280:24880-7; PMID:15878862; https://doi.org/ 10.1074/jbc.M500048200 [DOI] [PubMed] [Google Scholar]
- 17.Chames P, Coulon S, Baty D. Improving the affinity and the fine specificity of an anti-cortisol antibody by parsimonious mutagenesis and phage display. J Immunol 1998; 161:5421-9; PMID:9820517 [PubMed] [Google Scholar]
- 18.Lamminmäki U, Westerlund-Karlsson A, Toivola M, Saviranta P. Modulating the binding properties of an anti-17β-estradiol antibody by systematic mutation combinations. Protein Sci Publ Protein Soc 2003; 12:2549-58; PMID:14573866; https://doi.org/ 10.1110/ps.0353903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bumbaca D, Wong A, Drake E, Reyes AE, Lin BC, Stephan J-P, Desnoyers L, Shen B-Q, Dennis MS. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. mAbs 2011; 3:376-86; PMID:21540647; https://doi.org/ 10.4161/mabs.3.4.15786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bostrom J, Yu S-F, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, et al.. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 2009; 323:1610-4; PMID:19299620; https://doi.org/ 10.1126/science.1165480 [DOI] [PubMed] [Google Scholar]
- 21.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, Totpal K, Wong A, Lee CV, Stawicki S, et al.. A Two-in-One antibody against HER3 and EGFR Has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011; 20:472-86; PMID:22014573; https://doi.org/ 10.1016/j.ccr.2011.09.003 [DOI] [PubMed] [Google Scholar]
- 22.Lee CV, Koenig P, Fuh G. A Two-in-One antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. mAbs 2014; 6:622-7; PMID:24618680; https://doi.org/ 10.4161/mabs.28483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S. High resolution mapping of protein sequence–function relationships. Nat Methods 2010; 7:741-6; PMID:20711194; https://doi.org/ 10.1038/nmeth.1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, et al.. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol 2012; 30:543-8; PMID:22634563; https://doi.org/ 10.1038/nbt.2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forsyth CM, Juan V, Akamatsu Y, DuBridge RB, Doan M, Ivanov AV, Ma Z, Polakoff D, Razo J, Wilson K, et al.. Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. mAbs 2013; 5:523-32; PMID:23765106; https://doi.org/ 10.4161/mabs.24979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujino Y, Fujita R, Wada K, Fujishige K, Kanamori T, Hunt L, Shimizu Y, Ueda T. Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem Biophys Res Commun 2012; 428:395-400; PMID:23103372; https://doi.org/ 10.1016/j.bbrc.2012.10.066 [DOI] [PubMed] [Google Scholar]
- 27.Koenig P, Lee CV, Walters BT, Janakiraman V, Stinson J, Patapoff TW, Fuh G. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc Natl Acad Sci 2017; 114(4):E486-95:201613231; PMID:28057863; https://doi.org/ 10.1073/pnas.1613231114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koenig P, Lee CV, Sanowar S, Wu P, Stinson J, Harris SF, Fuh G. Deep Sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular degeneration. J Biol Chem 2015; 290:21773-86; PMID:26088137; https://doi.org/ 10.1074/jbc.M115.662783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett 2013; 328:18-26; PMID:22922303; https://doi.org/ 10.1016/j.canlet.2012.08.018 [DOI] [PubMed] [Google Scholar]
- 30.van Lookeren Campagne M, LeCouter J, Yaspan BL, Ye W. Mechanisms of age-related macular degeneration and therapeutic opportunities: Pathology, genetics, animal models, and therapeutic rationale of AMD. J Pathol 2014; 232:151-64; PMID:24105633; https://doi.org/ 10.1002/path.4266 [DOI] [PubMed] [Google Scholar]
- 31.Thomson BR, Heinen S, Jeansson M, Ghosh AK, Fatima A, Sung H-K, Onay T, Chen H, Yamaguchi S, Economides AN, et al.. A lymphatic defect causes ocular hypertension and glaucoma in mice. J Clin Invest 2014; 124:4320-4; PMID:25202984; https://doi.org/ 10.1172/JCI77162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowman HB, Bass SH, Simpson N, Wells JA. Selecting high-affinity binding proteins by monovalent phage display. Biochemistry (Mosc) 1991; 30:10832-8; PMID:1932005; https://doi.org/ 10.1021/bi00109a004 [DOI] [PubMed] [Google Scholar]
- 33.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al.. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci 2017; 114:944-9; PMID:28096333; https://doi.org/ 10.1073/pnas.1616408114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang F, Sen S, Zhang Y, Ahmad I, Zhu X, Wilson IA, Smider VV, Magliery TJ, Schultz PG. Somatic hypermutation maintains antibody thermodynamic stability during affinity maturation. Proc Natl Acad Sci 2013; 110:4261-6; PMID:23440204; https://doi.org/ 10.1073/pnas.1301810110 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012; 4:8-7; PMID:23778268; https://doi.org/ 10.4161/mabs.22189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Fouts AE, Stengel K, Luan P, Dillon M, Liang W-C, Feierbach B, Kelley RF, Hötzel I. In vitro affinity maturation of a natural human antibody overcomes a barrier to in vivo affinity maturation. mAbs 2014; 6:437-45; PMID:24492299; https://doi.org/ 10.4161/mabs.27875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, Takeuchi T, Lerner RA, Crea R. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci U S A 2005; 102:8466-71; PMID:15939870; https://doi.org/ 10.1073/pnas.0503543102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang W-P, Green K, Pinz-Sweeney S, Briones AT, Burton DR, Barbas CF III. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J Mol Biol 1995; 254:392-403; PMID:7490758; https://doi.org/ 10.1006/jmbi.1995.0626 [DOI] [PubMed] [Google Scholar]
- 39.Bostrom J, Lee CV, Haber L, Fuh G. Improving antibody binding affinity and specificity for therapeutic development [Internet] In: Dimitrov AS, (Ed.), Therapeutic antibodies. Totowa, NJ: Humana Press; 2009, p 353-76 [DOI] [PubMed] [Google Scholar]
- 40.Koenig P, Fuh G. Selection and screening using antibody phage display libraries In: Ossipow V, Fischer N, (Eds.), Monoclonal antibodies. Totowa, NJ: Humana Press; 2014. page 133-49 [DOI] [PubMed] [Google Scholar]
- 41.R Development Core Team R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. Available from: http://www.R-project.org. [Google Scholar]
- 42.Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, Gentleman R. ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 2009; 25:2607-8; PMID:19654119; https://doi.org/ 10.1093/bioinformatics/btp450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer; 2009. [Google Scholar]
- 44.Lee CV, Liang W-C, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol 2004; 340:1073-93; PMID:15236968; https://doi.org/ 10.1016/j.jmb.2004.05.051 [DOI] [PubMed] [Google Scholar]
- 45.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2007; 2:2212-21; PMID:17853878; https://doi.org/ 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 46.Yu X, Seegar TCM, Dalton AC, Tzvetkova-Robev D, Goldgur Y, Rajashankar KR, Nikolov DB, Barton WA. Structural basis for angiopoietin-1–mediated signaling initiation. Proc Natl Acad Sci 2013; 110:7205-10; PMID:23592718; https://doi.org/ 10.1073/pnas.1216890110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collaborative Computational Project N 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50:760-3; PMID:15299374; https://doi.org/ 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]