

ABSTRACT
Phage display antibody libraries are a rich resource for discovery of potential therapeutic antibodies. Single-chain variable fragment (scFv) libraries are the most common format due to the efficient display of scFv by phage particles and the ease by which soluble scFv antibodies can be expressed for high-throughput screening. Typically, a cascade of screening and triaging activities are performed, beginning with the assessment of large numbers of E. coli-expressed scFv, and progressing through additional assays with individual reformatting of the most promising scFv to full-length IgG. However, use of high-throughput screening of scFv for the discovery of full-length IgG is not ideal because of the differences between these molecules. Furthermore, the reformatting step represents a bottle neck in the process because each antibody has to be handled individually to preserve the unique VH and VL pairing. These problems could be resolved if populations of scFv could be reformatted to full-length IgG before screening without disrupting the variable region pairing. Here, we describe a novel strategy that allows the reformatting of diverse populations of scFv from phage selections to full-length IgG in a batch format. The reformatting process maintains the diversity and variable region pairing with high fidelity, and the resulted IgG pool enables high-throughput expression of IgG in mammalian cells and cell-based functional screening. The improved process led to the discovery of potent candidates that are comparable or better than those obtained by traditional methods. This strategy should also be readily applicable to Fab-based phage libraries. Our approach, Screening in Product Format (SiPF), represents a substantial improvement in the field of antibody discovery using phage display.
KEYWORDS: Antibody, phage display, scFv, IgG, Screen in Product Format (SiPF)
Introduction
Phage display antibody libraries are an important source of therapeutic and reagent antibodies.1,2 The small size and good solubility of phage particles and the combinatorial nature of most antibody libraries allow as many as 1011–1012 unique antibodies to be displayed and selected. In addition, the ability to tailor in vitro selection parameters for defined antibody properties and the high-throughput nature of phage display system make it a very powerful platform.3 Phage display antibody libraries, and other in vitro antibody display methods such as ribosome and mRNA display, usually make use of antibody fragments such as antigen-binding fragments (Fabs) or single-chain variable fragments (scFvs) due to difficulties in bacterial expression, folding and display of full-length IgG molecules. scFv phage libraries in particular are common because of the simplicity of the display vector and higher expression levels of scFv in Escherichia coli (E. coli) compared with Fab.4,5 Following in vitro selection, soluble scFv from single colonies of E. coli can rapidly be expressed in sufficient amounts for high-throughput screening (HTS),6-9 which helps to reduce the number of scFv antibodies for further characterization. However, since the predominant antibody drug format is full-length IgG, this screening is surrogate in nature and has disadvantages, including the lack of consistency between the activity of different formats, inability to assay for properties that are dependent on bi-valent binding or Fc-mediated function of an antibody, and the tendency of scFv antibodies to aggregate, which leads to false positive or negative results.9 In addition, endotoxin-sensitive, cell-based functional assays are not compatible with scFv expressed in bacteria. This is a major drawback since functional assays are typically the most valuable in determining the most relevant antibodies. Moreover, these assays also often require purification of scFv samples, which reduces the throughput of the screens.
Through HTS, scFvs are triaged and then converted to whole IgG on an individual basis to preserve pairing of the variable heavy (VH) and variable light (VL) chains. This is time-consuming, labor intensive and low throughput. Therefore, despite great progress made in HTS technologies, the functional mining of large and diverse scFv phage display libraries remains sub-optimal because the number of antibodies ultimately assessed as full-length IgG is only a small fraction of the selected repertoire. A recent trend in the field has been to screen phage library outputs as scFv.Fc fusions, which resemble IgG and are easier to make.10-13 These approaches add great value to the screening and triaging of scFv antibodies for IgG conversion, but do not overcome the known pitfalls associated with converting scFv to IgG. For example, we and others have repeatedly found that, during reformatting of scFv to IgG, there is not only significant attrition, but also changes in properties of the molecules such as affinity and activity14-16 (unpublished results). We and others17-22 therefore suggest that screening of selected phage display libraries directly as IgG would be a preferred approach for antibody discovery compared with surrogate approaches using scFv or scFv.Fc fusion proteins.
Several solutions to aid rapid scFv to IgG reformatting process have been described. For example, Sanmark et al21 used Type IIS restriction enzymes to perform high throughput conversion of single framework-based scFvs to IgG. This approach, however, cannot be applied to naïve libraries consisting of many different frameworks of VH and VL. Others have used rapid and efficient ligation-independent methods for cloning scFv variable regions or Fab chains into IgG expression vectors.17,18 Both techniques rely on ligation of multiple DNA fragments at once, but require conversion on an individual basis and are limited in the number of antibodies that can be tested as full-length IgG. This limitation can be removed if scFv phage display selection outputs, which are typically 105–107 in size, can be converted to IgG in a batch format.
There are 2 reports on batch conversion of scFvs to IgGs. Renaut et al19 inserted restriction enzyme sites flanking the linker sequence of a single scFv construct from which they made complementarity-determining region (CDR) variants for affinity maturation. Using restriction enzyme digestion and ligation, they selected scFvs and batch converted them to IgG by substituting the linker with IgG expression elements and constant domain sequences in a 2-step process. This approach is appropriate for limited germlines because restriction sites cannot be added to the V-domain-Linker boundaries without introducing mutations in the V-genes. Using a model scFv, Batonick et al22 reported batch reformatting of scFv to IgG via the phiC31 phage integrase system in E.coli and expression of IgG through the mammalian trans-splicing machinery. It is not known if this technology can be applied in a library setting because the (Gly4Ser)3 scFv linker23 used for making scFv must be substituted by the 13 amino acid long attP site. This is further complicated by the insertion of 2 artificial 13 amino acid long attL and attR peptides at the N-terminus of VL and between the VH and CH1 domains, respectively. We developed a novel platform that would: (1) enable batch conversion of diverse scFv populations into IgG (in the size range of 105–107) without introducing any mutations in the V-domains, (2) minimize molecular cloning steps, (3) preserve the original pairs of VH and VL chains from phage libraries, and (4) maintain the enrichment and diversity of the different sequences in the phage library output. We named this platform SiPF (Screening in Product Format), and provide here multiple examples to show that SiPF leads to the discovery of diverse and functional antibodies. The strategy is easily adaptable in any molecular biology laboratory using either scFv or Fab libraries to enable more effective mining of phage display libraries for the discovery of better antibodies.
Results
Batch conversion of scFv to full-length human IgG for SiPF
We used a 2-step strategy for the scFv batch conversion. In step 1, the (G4S)3 scFv linker was replaced by a DNA segment coding for the hinge and constant domains for (HC) and the promoter and signal sequence for (LC). This was achieved by PCR of the phagemid library using 3′ primers annealing to the framework (FW)4 of the VH and forward or 5′ primers annealing to the FW1 of the VL regions (the orientation of V-domains in our scFv are VH-linker-VL). This is a critical step because it ensures the linkage between VH-VL pairings throughout the conversion process (Fig. 1A). The linearized phagemids are complementary to the 5′ and 3′ ends, of a fragment that is PCR amplified from a donor vector, pmDV (Fig. 1B). The donor fragment encodes the following segments: (1) hinge and CH1-CH3 of human IgG1; (2) translation stop site; (3) poly adenylation site; and (4) a cytomegalovirus (CMV) promoter and signal sequence for the LC. The linearized plasmid and the donor fragment are ligated in frame by In-Fusion technology. This results in an intermediate pool of phagemids that contains all components necessary for IgG expression in mammalian cells except the promoter and signal sequence for the heavy chain and the constant domain of the LC (Fig. 1C).
Figure 1.
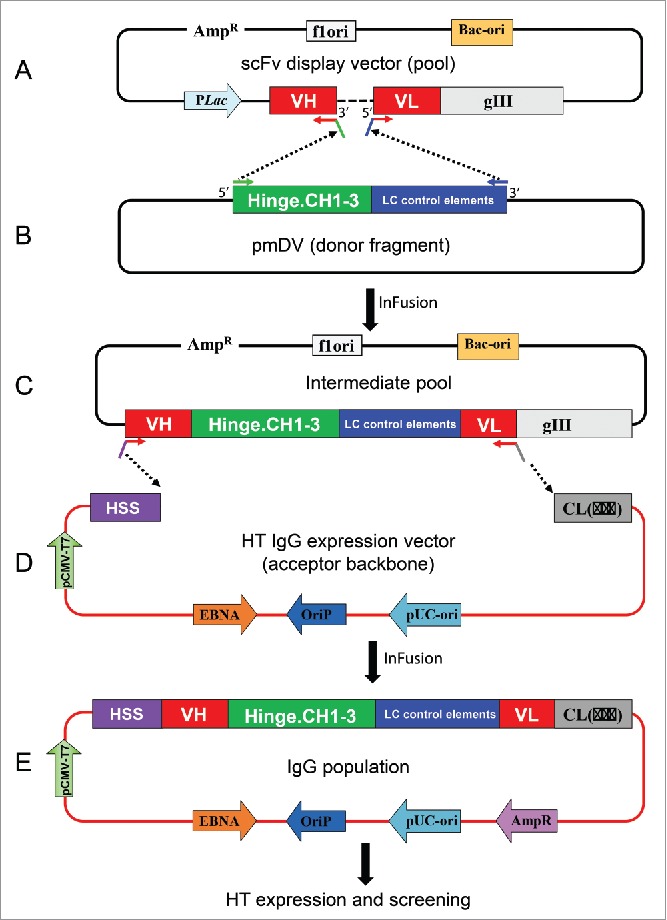
Schematic representation of batch reformatting of scFvs to IgGs. (A) Upper panel: Amplification of the phage display vector with 5′ primers annealing to VL FW1 and 3′ primers annealing to VH FW4. This results in the amplification of the whole plasmid except the linker between the VH and VL (dotted line). The primers carry complementary overhangs corresponding to the 5′ and 3′ ends of a donor fragment (B). This donor fragment represents the hinge and IgG1 constant domains followed by translation stop and polyA sites (in green) and the light chain control elements consisting of the LC promoter and signal sequence (in blue). This intermediate fragment is then fused in frame with VH and VL by means of In-Fusion technology. The resulting pool of plasmids is shown in the bottom panel (C, IgG intermediate pool). The IgG cassette from the intermediate vector pool is PCR amplified using 5′ primers that anneal to the FW1 of VH and 3′ primers that anneal to the FW4 of VL. The primers have overhangs to anneal to the signal sequence of HC and 5′ end of the Cκ or Cλ. The PCR products are then cloned into the IgG vector (D). The result is a pool of plasmid that codes for the entire HC and LC and can express IgG in mammalian cells. HSS, heavy chain signal sequence.
In step 2, the incomplete IgG cassette from step 1 is PCR amplified using 5′ primers that anneal to the FW1 of VH and add an overhang that anneals to the HC signal sequence in the IgG vector (Fig. 1D), and 3′ primers that anneal to the FW4 of Vκ and Vλ and add an overhang complementary to the light chain (LC) constant domains in the IgG vector. The purified PCR products were cloned into the IgG vector using In-fusion cloning to give rise to IgG expression vectors that contain all the elements for high level expression of the HC and LC. The final expression vector is bicistronic with both HC and LC under the control of a CMV promoter (Fig. 1E).
For any cloning step performed between the selection and functional screening of an antibody population, it is essential that the process maintains the original characteristics of the selected population as much as possible, i.e., the population should retain its sequence diversity, clonal representation and the VH-VL pairings that have been enriched by the selection process. However, inherent differences in PCR amplification or expression bias of different scFv in the pool, as well as the potential for mispairing of VH and VL regions by incomplete extension in the PCR stages, are all potential factors that can influence the composition of the IgG population. The optimized SiPF population cloning process described here maintains the selected scFv population well, as described below.
VH-VL pairing is maintained during batch reformatting of scFv to IgG
Precise identification of selected VH-VL pairs following population sub-cloning of very large (e.g., 106) and diverse antibody repertoires is challenging due to limitations on sampling size and the inability to discriminate between newly created VH-VL pairings or previously non-sampled VH-VL pairings. While next generation sequencing (NGS) technologies allow large sequence data sets to be generated, e.g., for 106 scFv antibody fragments, the sub-cloned populations in IgG format used here are too large in terms of template length for current high-fidelity NGS capabilities. Therefore, a ‘mini-library’ comprising 31 individual scFv of known sequences was used to evaluate the maintenance of correct VH-VL pairs. Batch conversion of the mini-library to full-length IgG resulted in 55% of the IgG sequences having the correct VH-VL pairing, with the remaining IgG representing antibodies with newly created VH and VL pairings (Fig. 2A). We also observe an ∼50% pairing rate when this experiment is performed using 2 or 10 scFv, and we suggest the pairing rate in diverse selection outputs may also be maintained in the order of 50%, as indicated by the tracking of common sequences that present in the SiPF population, with half of the antibodies having VH regions paired with the same VL region that was seen in the phagemid sample of sequences (data not shown). This result is not surprising given the length of the PCR products generated (∼5.5 kb at Step 1 and ∼3 kb at Step 2) and the fact that the sequences of the amplicons are highly similar, for example in the vector backbone regions. It is therefore highly likely that VH and VL swapping occurs during PCR due to incomplete extension and heterologous priming in subsequent PCR cycles.24,25 In our initial work carrying out batch reformatting for SiPF using standard PCR, including the studies presented here, we were able to identify functional antibodies from selections, indicating that a pairing rate of ∼55% can be sufficient for success. However, a higher degree of original VH-VL pairing is desired and may be essential in some cases. This can be achieved by performing the antibody amplification steps using emulsion PCR, which separates the individual templates into micelles and limits recombination between different antibody sequences. When batch reformatting of the mini-library is performed using emulsion PCR, the accuracy of the VH-VL pairing increases to 90% (Fig. 2B).
Figure 2.
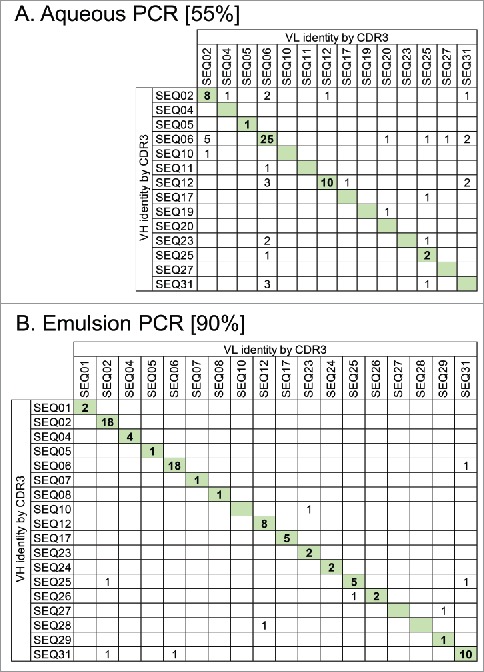
Maintenance of VH and VL pairing during batch reformatting of a mini-library of 31 scFvs with known sequences to full-length IgG. Antibody amplification steps of the reformatting process were done using either standard aqueous PCR conditions (A) or using emulsion PCR where DNA templates are separated within water-in-oil droplets (B). scFv DNA sequences from each pool were identified by their VH and VL CDR3 sequences. Numbers in the table show IgG sequences observed with the certain VH and VL combinations indicated. IgG having the original VH-VL pairing are indicated by shading. Values in square brackets indicate the % correct pairing rate in each population from 86 or 87 scFv sequences analyzed.
The enrichment of antibody sequences achieved during phage display selection is maintained
An important requirement of any batch reformatting process is the preservation of the diversity and relative abundance of the various antibody sequences that have been selected from the library. To evaluate these features, we compared the distribution and diversity of the VH CDR3 sequences as scFv in a selected phagemid library compared with IgG in the corresponding SiPF population after batch reformatting. Sequencing of 129 and 176 individual colonies from the 2 pools, respectively, indicated that about 84% of the scFv population, comprising 25 unique VH CDR3 sequences, is represented in the IgG population. The 3 most prevalent VH CDR3 sequences comprising 43% of the scFv population (19% of SEQ1, 16% of SEQ2, 8% of SEQ3) remain the most common in the IgG population, comprising 49% of the population (19% of SEQ1, 13% of SEQ2, 17% of SEQ3) (Fig. 3). The 16% of scFv sequences that were not seen in the IgG population are the least frequent members present at only 1 instance in the scFv population. These results, which are representative of many populations subjected to the batch reformatting process, indicate that the clonal enrichment obtained during phage display selection is broadly preserved when batch-converted to IgG for SiPF.
Figure 3.
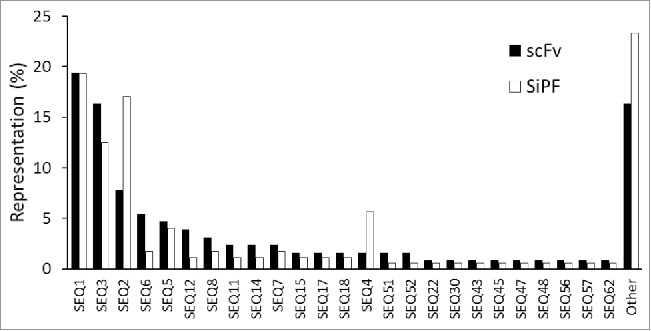
Representation of VH CDR3 sequences in scFv populations before and after batch reformatting for SiPF. Randomly chosen individual colonies were given an arbitrary identity based on their VH CDR3 sequence. Sequence representation is shown as a percentage frequency in scFv (129 sequences) or SiPF (176 sequences) populations. The 3 most frequent VH CDR3 sequences, and the representation of other sequences, is broadly the same for both populations. 16% (scFv) and 23% (SiPF) of the sequences in each population do not overlap, most likely due to sample size and the low probability of re-sampling relatively rare sequences.
Enrichment for full-length antibody sequences during population cloning
One added advantage of the batch reformatting process is that it can effectively enrich for antibodies having full-length variable domains and reduce the number of truncated sequences in the population. It is known that phage display naïve antibody libraries tend to contain a small percentage of members having in-frame deletions in their variable domains,,26,27 and these may be enriched during selection to various extents and more commonly in selections of naïve libraries on complex antigens such as whole cells (unpublished observations). The SiPF cloning process we describe here relies on the presence of FW4 of VH and FW1 of VL, respectively, for amplification by PCR. Furthermore, SiPF conversion involves steps of PCR product purification from agarose gels where only products of correct sizes are recovered, while incomplete scFv sequences are removed in the cloning process. This is illustrated in Table 1. Phage display selection performed on an ion channel protein resulted in a scFv population with 66% of the population being full-length at round 2. After batch reformatting for SiPF, antibodies with full-length variable domains comprise 92% of the final pool.
Table 1.
Comparison of variable domain sequences of phagemid scFv from Round 2 of selection and as VH and VL in IgG after batch reformatting to IgG.
| |
PRE SiPF (phagemid) |
SiPF Vector |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Library 1 |
Library 2 |
Library 1 |
Library 2 |
|||||||||||
| λ | λ | κ | ||||||||||||
| λ | 352 | κ | λ | 264 | κ | VH | VL | VH | κ | VH | VL | VH | VL | |
| Total reads | 229 | 5 | 136 | 27 | 176 | 176 | 176 | 176 | 176 | 176 | 176 | 176 | ||
| Full length, in-frame scFv | 66% | 62% | 157 | 166 | 148 | 167 | 161 | 165 | 158 | 164 | ||||
| 89% | 94% | 84% | 95% | 91% | 94% | 90% | 93% | |||||||
SiPF allows mining of the Vκ population in phage scFv library selection output
After selection of scFv phage libraries, the resulting antibody populations often show a strong bias for Vλ scFv, as shown in the example presented in Table 1. Such a bias for Vλ scFv has been reported previously,15,17 and maybe due to the better expression and secretion in E.coli of Vλ scFv, resulting in their dominance during display and subsequent selection. One way to avoid this Vλ dominance from compromising the ability to sample the Vκ population thoroughly is to perform separate selection of Vλ and Vκ only libraries. However, for existing libraries in which Vλ and Vκ are mixed, the use of separate primer pools for amplifying Vλ and Vκ containing scFvs without cross-amplifying the other isotype allows the Vκ population to be amplified separately from Vλ and screened as IgG.
Expression levels of IgG in SiPF platform are sufficient for screening
A good expression level is an important attribute for a robust high throughput IgG screening platform. Our previous experience with IgG expression in 293F cells in 96-well plate formats yielded expressions in the range of 0.4–40 μg/ml in about 90% of the wells, with the remaining 10% showing no or little expression (results not shown). While this is sufficient for simple binding assays, functional screens often require higher antibody concentrations. In addition, some assays are inhibited by components of the conditioned media, and dilution to remove the inhibition further reduces the antibody concentration. To increase the final antibody concentration, we investigated extending the expression time. Using a high-expressing antibody across 2 96-well plates, the mean titer rose from 44 ± 5 μg/ml at day 7 to 76 ± 10 μg/ml at day 12. However, at day 12 the viability of the cells had dropped to below 50%, and we were concerned about an increase in host cell proteins from lysed cells remaining in the conditioned media. We then investigated the Expi293F system using the same high-expressing antibody. After 6 d the titer was very high (457 ± 71 μg/ml) across 2 96-well plates and cell viability remained above 90%. This represents a 10-fold increase in expression level compared with the 293F cells. The high titer of the Expi293F system was confirmed in a panel of 1496 individual SiPF samples. We obtained expression levels ranging from less than 1 μg/ml (6% of samples) to over 200 μg/ml (5% of samples), with a mean titer of 70 μg/ml (Fig. 4A). These values are higher than those reported for 96-well plate expression of IgG from stable cell lines selected by fluorescence-activated cell sorting for high expression of the IgG chains.28
Figure 4.
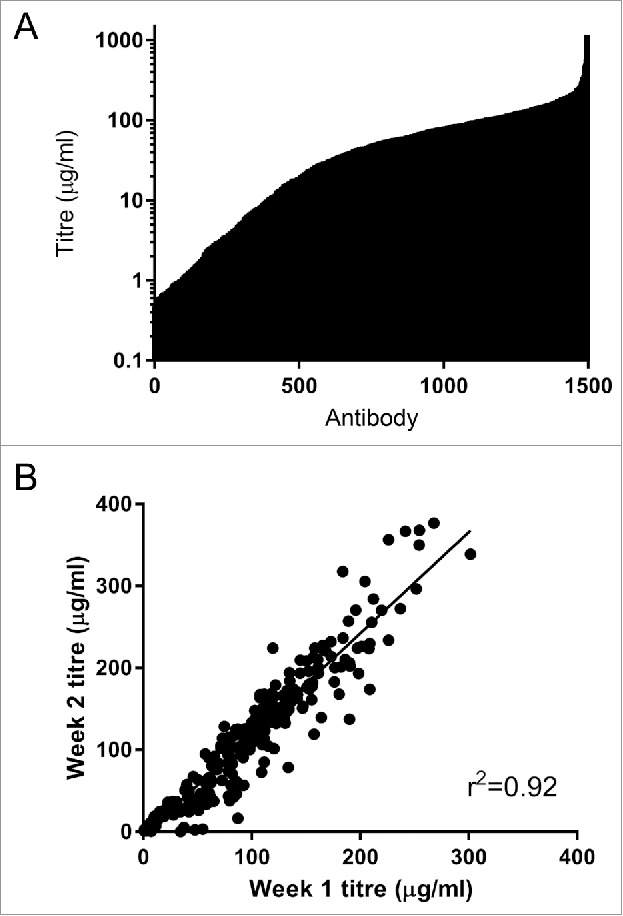
(A) Expression of 1496 IgG in Expi293F cells. Cells were transfected with mini-prep DNA, harvested after 6 d and IgG expression levels was determined using octet protein A. The mean titer for this set of antibodies was 70 µg/ml. (B) Reproducibility of the high-throughput mammalian expression system. A set of 352 antibodies were expressed in 4 96-well blocks and the titer determined after 6-days (week 1). Two weeks later the same antibodies were re-expressed under the same conditions (week 2). The expression of each antibody in each of the separate instance is highly comparable.
To determine whether the results was reproducible, we expressed 352 SiPF samples in 2 different experiments. Although the titers in the second experiment were higher than those seen in the first experiment (p < 0.0001 Wilcoxon matched pairs test), there was an excellent correlation between the 2 (r2 = 0.924). The variation between the 2 experiments is likely a property of the host cell passage number (unpublished observations). The variations in individual titers are most likely a property of the antibodies themselves rather than well-to-well variation within the process (Fig. 4B). The higher expression level possible with Expi293F cells opens up a much wider range of functional assays to the SiPF platform. It also enables increased dilution of conditioned media to remove inhibitory factors for sensitive assays.
SiPF identifies potent, functional antibodies
We have compared traditional methods of scFv or scFv.Fc-based screening to those obtained by SiPF in 2 separate antibody generation campaigns. For comparison to scFv.Fc screening, we used the identification of antibodies targeting glucocorticoid-induced tumor necrosis factor receptor ligand (GITRL) as an example. The modulation of the ligand's interaction with its receptor GITR by monoclonal antibodies is considered one strategy to modulate the human immune responses in treating autoimmune diseases and cancer.29 For the identification of neutralizing antibodies against GITRL, phage display selections were either screened as scFv.Fc13 or batch-converted to IgG format, as described herein. From the scFv.Fc screening platform, 5 neutralizing antibodies were obtained from a total of 6,000 scFv.Rc screened. These were then converted to IgG and ranked by their inhibitory potencies. For SiPF, 192 SiPF colonies were used for plasmid preparation and transfection in 96-well format. The IgG-containing supernatants were tested for both GITRL binding and Jurkat-hGR-NFkB-Luc cell-based anti-hGITRL functional analysis. Among the 192 colonies picked, 39 (20%) showed hGITRL binding activity, and among these 8 IgG displayed comparable or better GITRL inhibitory activity than the top antibodies identified through scFv.Fc. Therefore, both platforms were able to identify similar panels of functional antibodies, with some overlap including the most potent IgG. This was achieved, however, much more efficiently by the use of SiPF (4% of individual colonies evaluated) compared with the pSplice platform (0.8% of individual colonies evaluated).
For comparison to scFv screening, we used the identification of antagonistic antibodies against a cytokine as an example. Phage display selection of 4 naïve scFv libraries30-32 was performed. Antibodies from the second round of selection of all 4 libraries were subjected to high-throughput screening using a traditional scFv approach where single-point screening of bacterially expressed periplasmic scFv samples (3348 scFv tested) was followed by IC50 profiling of a smaller number of purified scFv (61 scFv tested). Finally, IC50 profiling of the most potent of these antibodies was performed using full-length IgG format, which required individual subcloning of VH and VL genes into IgG expression plasmids (10 IgG tested). Single-point screening and potency assessment of the most active IgG molecules were performed using a cell-based receptor inhibition reporter assay. This strategy successfully identified a panel of neutralizing antagonistic antibodies, the most potent being Ab048, which had an IC50 in the reporter assay of 49 nM (Fig. 5). In parallel to this work, 2 of the 4 selection outputs were used to validate the SiPF process, where the selected scFv phagemid populations were converted to full-length IgG using the population cloning process described here. Single-point screening of conditioned media from cells expressing IgG was performed using the aforementioned reporter assay. In total, 528 SiPF IgG samples were used for single-point screening and 22 unique IgG were chosen for re-expression and purification to determine EC50 values in the same cell-based assay. The most potent antibody identified from this approach was Ab009, having an IC50 in the reporter assay of 2.3 nM, which is 10 times more potent than the best clone from the conventional approach (Ab048; Fig. 5).
Figure 5.
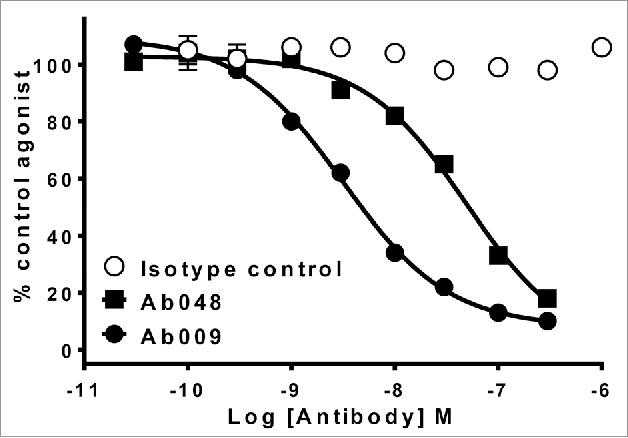
More potent anti-cytokine antibodies are identified by SiPF than by scFv screening. Comparison of potency of the 2 leading antibodies identified from scFv screening (Ab048) or SiPF screening (Ab049) of phage display selections that were performed to identify an antagonistic antibody for a soluble cytokine. Neutralization of 1nM cytokine-induced receptor activation by Ab048 and Ab009 are shown in a cell-based receptor reporter enzyme complementation assay (PathHunter, DiscoverRx).
In both of these examples, SiPF enabled the more efficient discovery of the most potent antibodies while avoiding surrogate screening steps. A typical comparison of SiPF and scFv format based screening is shown in Fig. 6.
Figure 6.
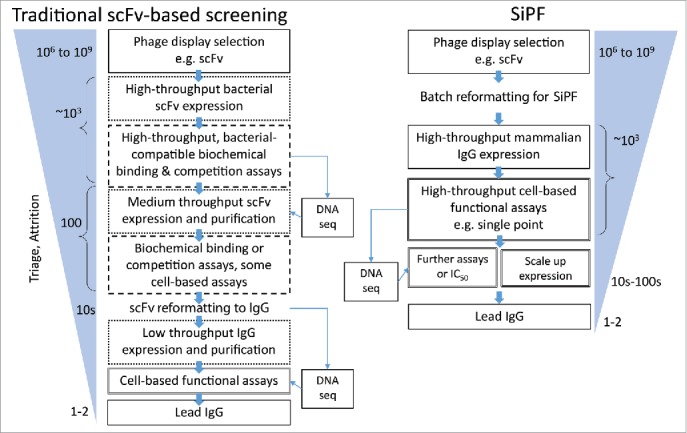
Flow diagrams comparing typical routes from phage display selection to the identification of functional antibodies, highlighting the utility of the SiPF platform in reducing surrogate steps (e.g., scFv format, binding based / biochemical) assays. Rapid conversion of phage scFv to IgG allows earlier analysis of larger numbers of IgG in more relevant, functional cell-based assays.
SiPF facilitates antibody screening for binding to complex membrane proteins
Complex membrane proteins such as G-protein-coupled receptors (GPCRs) are valuable but challenging targets for antibody discovery. Formyl peptide receptor-1 (FPR1) is a class A GPCR with important roles in inflammation and represents an example of a therapeutically relevant, complex membrane target for antibody discovery. In several experiments, we have used scFv libraries as a source of antibodies to FPR1, whereby scFv libraries were selected by phage display on FPR1 over-expressing cells, followed by high-throughput screening of scFv for binding to the same cells using fluorescence micro volume assay technology (FMAT). We typically see high-level, non-specific binding to both positive and negative cells in these experiments (data not shown); therefore, it is our experience that bacterially expressed scFv are not suitable for this type of screening cascade. Using the SiPF platform for FMAT screening on FPR1 over-expressing cells, several novel antibodies with specific binding to FPR1 were identified (Fig. 7). This highlights the utility of SiPF to facilitate the identification of antibodies binding to complex membrane protein such as GPCRs through improved cell-based assay.
Figure 7.
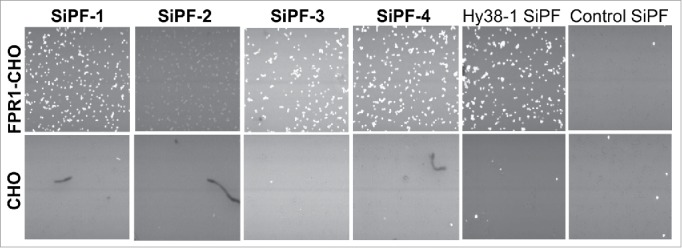
FMAT analysis of cell-binding of anti FPR1 antibodies as IgG supernatant generated using the SiPF platform. Four different FPR1 binding antibodies identified from cell-based phage display selections are shown. Hy38-1 SiPF is a positive control antibody known to recognize FPR133 that has been expressed using the SiPF platform.
Discussion
The small particle size, phenotype-genotype linkage and use of E.coli expression for filamentous phage display of antibody libraries confers unique properties not achieved by any other system and provides a trove of valuable antibody molecules.1,2 However, antibody phage display libraries also face several challenges that arise due to the E.coli-based expression and screening of scFvs, given that the molecular nature and bio-physical properties of scFv differ significantly from IgG, which is typically the format in which the antibodies are ultimately required. In fact, it has long been appreciated that direct screening of phage display libraries as IgG expressed in mammalian cells is a more desirable path.17,20 This has not been possible to date because reformatting of antibody fragments from scFv to IgG requires the maintenance of the VH and VL pairing by cloning each antibody individually and rendering the conversion of large numbers of scFv to IgG, extremely labor intensive.
Here, we described a novel strategy for conversion of scFv populations to IgG that does not require any specific library design. This strategy uses a 2-step batch reformatting process in which the first step is linearization of the phagemid scFv population through PCR amplification of the VH and VL regions linked by the phagemid vector backbone. This is fundamentally different from all the previous cloning strategies which amplify VH and VL of individual antibodies through separate PCR reactions, thereby creating the pairing dilemma. The steps that follow ensure the resulting IgG pools maintain the clonal diversity, as well as the enrichment of the parental phage display selection pools. An essential and unique aspect to this process is the use of water-in-oil emulsions for PCR amplification steps to ensure correct VH-VL pairing is maintained with high fidelity.
We coupled this novel cloning strategy with an extremely high titer mammalian expression system. Generating mean antibody concentrations of ∼70μg/ml in only 6 d has enabled the SiPF platform to be used in a wide variety of functional assays. In addition, the conditioned media can be diluted while retaining a high antibody concentration to remove any inhibitory components for sensitive screens. The robustness of the SiPF platform has been demonstrated through multiple quality control experiments and sample parallel campaigns.
Interestingly, our examples of the utility of the SiPF platform have yielded antibodies that meet or exceed the potency of those obtained through the screening of larger numbers of scFv or scFv.Fc. Overall, the SiPF platform does not merely serve to maintain the VH-VL pairings during the conversion, but also provides other benefits such as: the separate amplification of κ and λ clones ensures that the severely underrepresented Vκ clones would get an increased likelihood of being triaged; the stringent PCR quality control in each of the amplification step determine that only full-length antibody sequences are represented in the final IgG pools; and the mammalian cell-expressed IgG in the protein-free medium enables more relevant cell-based functional screening. However, given the small number of campaigns performed so far this represents only a promising first step in a new direction, and more data comparing SiPF with other screening strategies should be accumulated in the future.
It should be noted that our current SiPF strategy is generally applicable to essentially all existing scFv- or Fab-based phage display antibody libraries through appropriate design of suitable primers. An alternative IgG screening platform after Fab-based phage library selection was reported previously, but this depends on a specially designed phagemid vector for constructing new libraries.34 Furthermore, the same principle adopted by the SiPF platform can be used in any setting where pairwise cloning of large number of samples in a library is desired.
Finally, the flexibility in the SiPF's design easily allows the introduction of various mutations to the Fc of the IgG vector, thus accommodating special high throughput screening, such as is needed for antibody-drug conjugate discovery programs. We predict that the SiPF platform will provide an enabling shift in the field of phage display and functional screening for antibody discovery.
Material and methods
Phage display selection
scFv phage display libraries30-32 were used for selections to identify antibodies against various target proteins. For selection on purified protein in solution, phage libraries were first blocked with MPBS [1X phosphate-buffered saline (PBS), 3% (w/v) non-fat dried milk powder] and deselected on Dynal M-280 streptavidin-coated beads (Invitrogen) before incubating with the biotin-labeled antigen (25–100 nM) at room temperature for one hour. Phage-antigen complexes were then captured on streptavidin-coated magnetic beads and washed 3 times with PBST (1XPBS, 0.1% (v/v) Tween 20), PBST-L (1XPBS, 0.01% (v/v) Tween 20) or PBS alone. Following washing, phage particles were eluted by incubation with 10 µg/ml trypsin in 1XPBS at 37°C for 30 min. Eluted phage populations were recovered and amplified for use in a further round of selection by infection of logarithmic phase TG1 E. coli cells. Typically, 3 rounds of selection were performed. For cell-surface panning on GITRL, cells expressing the target antigen were biotinylated by Sulfo-NHS-LC-LC-Biotin (Thermo Scientific) according to the manufacturer's instructions and selections were performed as described above except that phage libraries were incubated with ∼2 × 106 target-expressing cells and a 10-fold excess of non-labeled parental cells not expressing the target, and selections were washed with cold 1X PBST and 1X PBS. Cell-surface selection on FPR1 was performed essentially as described above for soluble protein, except that both cells and phage particles were pre-blocked with growth media containing 10% (v/v) FCS, 6 to 8 washes to remove non-bound phage were performed after selection by brief centrifugation to pellet the cells and gentle suspension of cells in PBS, and finally phage were eluted using 250 µg/ml trypsin.
Vector construction
Three plasmid vectors, a donor plasmid and 2 acceptor plasmids, were constructed to allow batch reformatting of scFvs from phagemid vectors into full-length mammalian IgG in a high-throughput manner. The donor plasmid (pmDV) was constructed on a pUC vector backbone with a complete IgG1 HC constant region (hinge, CH1, CH2 and CH3), translation stop and transcription termination signals, followed by a CMV promoter and signal peptide for LC expression (Fig. 1B). Two different acceptor plasmids were constructed using an in-house pOE vector backbone with OriP and EBNA for high mammalian protein expression.13 These plasmids contain a CMV promoter, a signal peptide for HC expression, a cloning junction and finally C-kappa (pmIgGκ or C-lambda (pmIgGλ) for generating kappa and lambda IgG pools, respectively (Fig. 1D).
Mini-library creation
To evaluate the maintenance of VH-VL pairing during batch reformatting of scFv populations to full-length IgG, a mini-library was prepared that contained 31 scFv of known sequence. The scFv antibodies comprising the mini-library were derived from various pre-existing phage display selection outputs, and were chosen essentially at random to assemble a population broadly representing a ‘typical’ phage display antibody selection output in terms of antibody sequence diversity. The scFv were derived from phage display selections performed against different target antigens (8 in total that included a mix of soluble cytokines and membrane proteins), all VH and VL regions were unique and CDR3 regions ranged in length from 4 to 25 amino acids. Overall, the 31 scFv contained a wide range of human antibody germline families as would be expecting in a typical phage display selection output. (VL germlines comprised Vk1_A3, n = 1; Vk2_DPK15_(A19,A3), n = 2; Vk4_DPK24_(B3), n = 1; Vlambda1_DPL1_(1a), n = 1; Vlambda1_DPL2_(1c), n = 3; Vlambda1_DPL3_(1 g), n = 5; Vlambda1_DPL8_(1e), n = 8; Vlambda2_2c, n = 1; Vlambda2_DPL11_(2a2), n = 4; Vlambda3_DPL16_(3l), n = 2; Vlambda3_DPL23_(3r), n = 1; Vlambda6_6a, n = 1; Vlambda7_DPL19_(7b), n = 1. VH germlines comprised Vh1_DP-10_(1–69), n = 4; Vh1_DP-14_(1–18), n = 3; Vh1_DP-25_(1–03), n = 1; Vh1_DP-7_(1–46), n = 4; Vh1_DP-8,75_(1–02), n = 2; Vh1_DP-88_(1-e), n = 2; Vh3_DP-38_(3–15), n = 1; Vh3_DP-46_(3–30.3), n = 1; Vh3_DP-47_(3–23), n = 8; Vh3_DP-49_(3–30.5), n = 1; Vh4_DP-65_(4–30.1), n = 1; Vh5_5-a, n = 1; Vh5_DP-73_(5–51), n = 1; Vh6_DP-74_(6–1), n = 1).
Batch reformatting of scFv populations to full-length IgG
The process of converting phage display scFv populations to full-length IgG populations includes 2 major steps. Firstly, the scFv population is linearized by PCR and the donor fragment is introduced to generate an intermediate pool. Secondly, the partial IgG expression cassette from the intermediate pool is amplified by PCR and inserted into kappa or lambda acceptor vectors to generate the final pool for full-length IgG expression (Fig. 1). The 2 steps of PCR can be performed using standard aqueous conditions as described below, or ideally can be performed using emulsion PCR to maintain a higher fidelity of VH-VL pairing during the batch reformatting process.
Phagemid DNA was purified from phage display selection outputs and amplified by PCR using pools of 5′ VL FW1 primers and 3′ VH FW4 primers, resulting in linearization of the phagemid pool. This linearization step results in a DNA fragment containing all the elements of the display vector except the linker between the VH and VL regions. A total of 6 V-kappa FW1, 11 V-lambda FW1 and 3 VH FW4 primers were used for maximal coverage of antibody germline genes (Table 2). These 5′ and 3′primers carry overhangs complementary to overhangs on the 3′ and 5′ primers used to PCR amplify HC constant regions and the LC control elements from the donor plasmid pmDV (Fig. 1A and B). Kappa and lambda reactions were performed separately to minimize cross-priming. PCR was performed using KOD Xtreme Hot Start DNA Polymerase (Novagen) or CloneAmp HiFi PCR premix (Takara) and reactions were typically performed for 17 cycles with denaturation at 98°C for 10 sec, annealing at 60°C for 30 sec and extension at 68°C for 5 min. The donor fragment containing the IgG1 HC regions and control elements described above was prepared by amplification using the primers 5′TCGACCAAGGGCCCATCCGTCTTCC and 5′ ACAGCGCGCCCCGGGGAGCCAGAGC (Fig. 1B). The donor fragment was then cloned into the linearized phagemid vector by In-Fusion cloning using the In-Fusion HD Cloning Kit (Takara). The resulting intermediate pool consists of antibody cassettes with complete IgG1 HC and a partial LC region, but lacks the HC control elements and the LC constant domain. Plasmid DNA was then purified from the intermediate pool and the antibody cassette was amplified by PCR using pools of 5′ VH FW1 and 3′ VL FW4 primers. A total of 9 VH FW1, 3 V-kappa FW4 and 3 V-lambda FW4 primers were used to amplify the antibody cassettes in separate kappa and lambda reactions (Table 3). PCR was performed for 18 cycles with denaturation at 98°C for 10 sec, annealing at 62°C for 30 sec and extension at 68°C for 3 min. The amplified antibody cassettes were In-Fusion cloned into the acceptor vectors pmIgGκ and pmIgGλ to generate the final kappa and lambda full-length IgG pools from the single starting scFv population. Throughout the process, all DNA fragments were gel purified to remove non-specific or truncated products. Individual bacterial colonies from phage display selection outputs as well as intermediate and final cloned pools were grown overnight and used for Sanger DNA sequencing to assess diversity and representation of the antibody populations.
Table 2.
Primers for intermediate pool generation. Sequences in red are complementary to the 5′ end of VL FW1 (5′ primers) or the 3′ end of VH FW4 (3′ primers). Sequences in blue are overhangs in 5′ primers that are complementary to the LC control elements in the donor fragment. Sequences in green are overhangs in 3′ primers that are complementary to the hinge region of IgG1 in the donor fragment.
| 5′ kappa_1 | GCTCCCCGGGGCGCGCTGT GAC ATC CAG WTG ACC CAG TCT |
|---|---|
| 5′ Kappa_2 | GCTCCCCGGGGCGCGCTGT GAT ATT GTG ATG ACC CAS ACT |
| 5′ Kappa_3 | GCTCCCCGGGGCGCGCTGT GAA ATT GTG WTG ACR CAG TCT |
| 5′ Kappa_4 | GCTCCCCGGGGCGCGCTGT GAT RTT GTG ATG ACW CAG TCT |
| 5′ Kappa_5 | GCTCCCCGGGGCGCGCTGT GAA ACG ACA CTC ACG CAG TCT |
| 5′ Kappa_6 | GCTCCCCGGGGCGCGCTGT GAA ATT GTG CTG ACT CAG TCT |
| 5′ lambda_1 | GCTCCCCGGGGCGCGCTGT CAG TCT GTG YTG ACK CAG CCR |
| 5′ lambda_2 | GCTCCCCGGGGCGCGCTGT CAG TCT GCC CTG ACT CAG CCT |
| 5′ lambda_3 | GCTCCCCGGGGCGCGCTGT TCC TAT GWG CTG ACT CAG CYA |
| 5′ lambda_4 | GCTCCCCGGGGCGCGCTGT CWG CCT GTG CTG ACT CAG CCM |
| 5′ lambda_5 | GCTCCCCGGGGCGCGCTGT TCT TCT GAG CTG ACT CAG GAC |
| 5′ lambda_6 | GCTCCCCGGGGCGCGCTGT AAT TTT ATG CTG ACT CAG CCC |
| 5′ lambda_7 | GCTCCCCGGGGCGCGCTGT CAG YCT GTA CTG ACT CAA CCG |
| 5′ lambda_8 | GCTCCCCGGGGCGCGCTGT CAG RCT GTG GTG ACY CAG GAG |
| 5′ lambda_9 | GCTCCCCGGGGCGCGCTGT CAG CCT GTG CTG ACT CAA TC |
| 5′ lambda_10 | GCTCCCCGGGGCGCGCTGT CAG GCA GGG CTG ACT CAG CCA |
| 3′ heavy_1 | TGGGCCCTTGGTCGACGC TGA GGA GAC RGT GAC CAG GGT KCC |
| 3′ heavy_2 | TGGGCCCTTGGTCGACGC TGA AGA GAC GGT GAC CAT TGT CCC |
| 3′ heavy_3 | TGGGCCCTTGGTCGACGC TGA GGA GAC GGT GAC CGT GGT CCC |
Table 3.
Primers for final IgG pool generation. Sequences in red are complementary to 5′ end of VH FW1 (5′ primers) or the 3′ end of VL FW4 (3′ primers). Sequences in blue are overhangs in 5′ primers that are complementary to the HC signal sequence region of the IgG acceptor vector. Sequences in black are overhangs in 3′ primers that are complementary to the 5′ end of either the kappa or lambda CL region of the IgG acceptor vector.
| 5′ heavy_1 | TCTCCACAGGTGTACACTCC CAG RTG CAG CTG GTG CAR T |
|---|---|
| 5′ heavy_2 | TCTCCACAGGTGTACACTCC SAG GTC CAG CTG GTR CAG T |
| 5′ heavy_3 | TCTCCACAGGTGTACACTCC CAG RTC ACC TTG AAG GAG T |
| 5′ heavy_4 | TCTCCACAGGTGTACACTCC SAG GTGCAG CTG KTG GAG |
| 5′ heavy_5 | TCTCCACAGGTGTACACTCC CAG GTG CAG CTA CAG CAG T |
| 5′ heavy_6 | TCTCCACAGGTGTACACTCC CAG GTA CAG CTG CAG CAG TC |
| 5′ heavy_7 | TCTCCACAGGTGTACACTCC GAR GTG CAG CTG GTG CAG |
| 5′ heavy_8 | TCTCCACAGGTGTACACTCC CAG STG CAG CTG CAG GAG TC |
| 5′ heavy_9 | TCTCCACAGGTGTACACTCC GAG GTG CAG CTG TTG GAG TCT |
| 3′ kappa_1 | GTGCAGCCACCGTACGTTT GAT HTC CAC YTT GGT CCC |
| 3′ kappa_2 | GTGCAGCCACCGTACGTTT GAT CTC CAG CTT GGT CCC |
| 3′ kappa_3 | GTGCAGCCACCGTACGTTT AAT CTC CAG TCG TGT CCC |
| 3′ lambda_1 | AGTGACCGAGGGCGCCGCCTTGGGCTGACC TAG GAC GGT GAC CTT GGT CCC |
| 3′ lambda_2 | AGTGACCGAGGGCGCCGCCTTGGGCTGACC TAG GAC GGT CAG CTT GGT CCC |
| 3′ lambda_3 | AGTGACCGAGGGCGCCGCCTTGGGCTGACC GAG GAC GGT CAG CTG GGT |
Emulsion PCR
The PCR amplification of antibody populations of Step 1 and Step 2 can be performed using water in oil emulsions to limit cross-hybridization of incomplete extension products. Emulsions were prepared for PCR using the Micellula DNA Emulsion & Purification Kit (Roboklon, E3600), according to manufacturer's recommendations. Emulsion PCR was optimized for template amount, i.e., the number of DNA molecules per micelle, the ideal amount being a balance between fidelity and yield (lower DNA amounts give more micelles that contain only one template, but yields are also lower). We have found the use of 100 ng of DNA template in a 200 µl aqueous PCR reaction emulsified with 1.2 ml micelle components gives an optimal yield and pairing fidelity. An emulsion component premix was prepared by combining 880 μl Component 1, 80 μl Component 2 and 240 μl Component 3 per reaction. This was mixed vigorously using a high speed shaker, e.g., a Qiagen TissueLyser at 25 Hz for 5 min. 200 µl standard aqueous PCR reaction mix containing 100 ng DNA template were prepared using the reagents and primers described previously and added to the 1.2 ml emulsion premix. Water-in-oil droplets ready for PCR were created by high-speed shaking of the aqueous phase and emulsion mix at 15 Hz for 5 min. The emulsion was aliquoted into tubes suitable for use in a thermal cycler and 25 cycles of amplification were performed using the thermal cycling conditions as described previously for standard PCR. Following amplification, aliquots of the same reaction were pooled into a single tube and the emulsion was opened by the addition of 1.5 ml isobutanol. DNA was then extracted from the opened emulsion PCR reactions using the column purification reagents provided.
Detection of anti-GITRL antibodies using a Jurkat-hGR-NFkB-Luc based assay
This method is based on the finding that GITRL binds to its receptor on GITR-expressing cells and triggers NF-kB activation. A Jurkat cell line (Jurkat-hGR-NFkB-Luc) stably expressing human GITRL receptor (hGR) and carrying a NF-kB-controlled luciferase gene was generated in-house. Recombinant trimeric hGITRL was also generated in-house. For screening of hGITRL inhibitory antibodies, 50 µl cell culture supernatant from high throughput IgG transfection serially diluted in RPMI (Life Technology, #11875–093) was mixed with 10 µl of 1 µg /ml hGITRL and 40 µl of Jurkat-hGR-NFkB-Luc at a density of 1.25 × 106/ml. The resulting reaction mixture contained 0.1 µg /ml hGITRL, 50K Jurkat-hGR-NFkB-Luc cells, and various concentrations of anti-hGITRL IgGs in a volume of 100 µl. Antibodies s7F5 and s1F9, derived from the conventional scFv.Fc screening platform,13 were used as positive controls, and CAT2 IgG was used as negative control. Purified IgGs were spiked into spent culture medium to a final concentration of 140 µg/ml, put through the same serial dilution process performed for all other supernatants, and used in the screening assay. The mixture was incubated in a white, 96-well plate with a flat, clear bottom (Corning Costar #3903) at 37°C for 4 hours. The plate was then centrifuged at 500 g for 4 min at room temperature. Supernatant was then discarded, followed by the addition of 20 µl/well 2x CCLR (Cell Culture Lysis Reagent, 5x, Promega # E1531). The plate was shaken vigorously on a plate shaker at 500 rpm for 5 min at ambient temperature. 100 µl/well luciferase substrate, prepared as suggested by the manufacturer (Luciferase Assay System, Promega, #E1501), was added to each well and luminescence intensity was measured immediately on an Envision Multi-label plate reader (Perkin Elmer). The inhibitory effect of the IgGs was calculated as (1-test IgGCPS /control IgGCPS) × 100 where “test IgGCPS” represents the luciferase activity from a well containing an IgG with unknown activity and “control IgGcps” represents the luciferase activity from a well containing a control IgG without any inhibitory activity.
Detection of anti-cytokine neutralizing antibodies using an enzyme fragment complementation assay
Inhibition of cytokine-induced signaling was measured using a commercially available cell line from the DiscoverX PathHunter® range. This cell line is engineered to express the cognate human cytokine receptor and 2 inactive fragments of β-galactosidase, such that receptor activation leads to enzyme fragment complementation and detection of activity in the form of chemiluminescence. For high-throughput screening, conditioned media from cells expressing IgG (final concentration of 10–25% in the final assay volume) were incubated with cytokine (final concentration of 1 nM) for 30 mins to 1 hour at room temperature. 10 µl of this premix was then added to 20 µl of PathHunter® cells in quadruplicate and incubated for 3 hours at room temperature. 12 µl of a detection reagent and substrate mix was added to each well for a further hour at room temperature and luminescence was measured using an Envision plate reader. For IC50 measurement, serial dilutions of purified test IgG samples were assayed as described above. Appropriate scFv and IgG negative controls were included in all assays. Data were calculated as mean % of control agonist ± SD (wells with cytokine alone) and plotted in GraphPad Prism 5.00 for Windows (GraphPad Software Inc.) using sigmoidal 4 parameter nonlinear regression to determine IC50 values.
High throughput screening for the identification of antibodies binding to FPR1 using FMAT®
FMAT® technology was used to measure the ability of full-length IgG antibodies to bind to FPR1-expressing cells as an example of the utility of SiPF for screening for binding to complex membrane proteins. Conditioned media from HEK293-expressing IgG were combined with anti-human AlexaFluor 647 labeled antibody (Life Technologies) and either cynomolgus FPR1-expressing Chinese hamster ovary (CHO) cells or non-transfected CHO cells. Assay plates were incubated at room temperature for 3 hours before fluorescence was measured using an FMAT 8200 cellular detection system (Applied Biosystems). Appropriate IgG controls were included in all assays. Data was analyzed using the FMAT analysis software and events were gated based on fluorescence (FL1). A minimum count of 20 events was set as a threshold before data was reported for each well.
Expression and quantitation of full-length IgG
Individual colonies from full-length IgG populations were grown overnight in 96-well microtiter plates for high-throughput plasmid DNA purification using the Agencourt CosMCPrep kit (Beckman Coulter) on a Biomek FX (Beckman Coulter) liquid handling robot with a Cytomat Microplate hotel (Beckman Coulter). Using this set up 16 microtiter plates can be processed by one person in a day delivering 90 µL of DNA at 100 ng/µL for each plasmid. Purified DNA was then transfected into Expi293F cells (Thermo Fisher, UK) using the ExpiFectamine 293 transfection kit (Thermo Fisher, UK) in a volume of 300 µL in 96 deep-well blocks. Transfection was performed using a MiniTrak liquid handling robot (Perkin Elmer, UK) installed in a Robotic Bio Enclosure (CAS, UK) to reduce contamination levels. Transfected plates were grown at 37°C with 5% CO2 shaking at 350 rpm in a Khuner ISF1-X shaking incubator. Culture supernatant containing secreted IgG was harvested after 6 d by pelleting cells with centrifugation at 2000 g for 15 min and transferring 250 µL cell-free supernatant to a 96-well plate. IgG concentrations of culture supernatants were quantitated by Bio-Layer Inferometry using Protein A biosensors in conjunction with the OctetRED384 system (ForteBio). The rate of binding to the Protein A sensors was measured and sample IgG concentrations determined by comparison to a standard curve of known IgG concentrations. The IgG containing supernatant was then used in high-throughput biochemical or cell-based assays as described.
For further characterization of individual IgG, expression was scaled up using a proprietary high-yielding CHO transient system.24 Expressed IgG was purified from the culture medium using MabSelect SuRe (GE Healthcare, UK) and buffer exchanged into PBS using a PD10 column (GE Healthcare, UK). The concentration of IgG was determined spectrophotometrically using an extinction coefficient based on the amino acid sequence of the IgG. Purified IgG were assessed for purity and integrity using SDS-PAGE and HPLC-SEC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Thie H, Meyer T, Schirrmann T, Hust M, Dubel S. Phage display derived therapeutic antibodies. Curr Pharm Biotechnol 2008; 9:439-46; PMID: 19075684; https://doi.org/ 10.2174/138920108786786349 [DOI] [PubMed] [Google Scholar]
- 2.Chan CE, Lim AP, MacAry PA, Hanson BJ. The role of phage display in therapeutic antibody discovery. Int Immunol 2014; 26:649-57; PMID: 25135889; https://doi.org/ 10.1093/intimm/dxu082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nixon AE, Sexton DJ, Ladner RC. Drugs derived from phage display: From candidate identification to clinical practice. mAbs 2014; 6:73-85; PMID: 24262785; https://doi.org/ 10.4161/mabs.27240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbabi-Ghahroudi M, Tanha J, MacKenzie R. Prokaryotic expression of antibodies. Cancer Metastasis Rev 2005; 24:501-19; PMID: 16408159; https://doi.org/ 10.1007/s10555-005-6193-1 [DOI] [PubMed] [Google Scholar]
- 5.Steinwand M, Droste P, Frenzel A, Hust M, Dubel S, Schirrmann T. The influence of antibody fragment format on phage display based affinity maturation of IgG. mAbs 2014; 6:204-18; PMID: 24262918; https://doi.org/ 10.4161/mabs.27227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallborn J, Carlsson R. Automated screening procedure for high-throughput generation of antibody fragments. BioTechniques 2002; Suppl:30-7; PMID: 12514927 [PubMed] [Google Scholar]
- 7.Schofield DJ, Pope AR, Clementel V, Buckell J, Chapple S, Clarke KF, Conquer JS, Crofts AM, Crowther SR, Dyson MR, et al.. Application of phage display to high throughput antibody generation and characterization. Genome Biol 2007; 8:R254; PMID: 18047641; https://doi.org/ 10.1186/gb-2007-8-11-r254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turunen L, Takkinen K, Soderlund H, Pulli T. Automated panning and screening procedure on microplates for antibody generation from phage display libraries. J Biomol Screen 2009; 14:282-93; PMID: 19224869; https://doi.org/ 10.1177/1087057108330113 [DOI] [PubMed] [Google Scholar]
- 9.Hayhurst A, Happe S, Mabry R, Koch Z, Iverson BL, Georgiou G. Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J Immunol Methods 2003; 276:185-96; PMID: 12738372; https://doi.org/ 10.1016/S0022-1759(03)00100-5 [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Katayama A, Wang Y, Yu L, Favoino E, Sakakura K, Favole A, Tsuchikawa T, Silver S, Watkins SC, et al.. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res 2011; 71:7410-22; PMID: 22021902; https://doi.org/ 10.1158/0008-5472.CAN-10-1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon H, Song JM, Ryu CJ, Kim YG, Lee EK, Kang S, Kim SJ. An efficient strategy for cell-based antibody library selection using an integrated vector system. BMC Biotech 2012; 12:62; PMID: 22989299; https://doi.org/ 10.1186/1472-6750-12-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jager V, Bussow K, Wagner A, Weber S, Hust M, Frenzel A, Schirrmann T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotech 2013; 13:52; PMID: 23802841; https://doi.org/ 10.1186/1472-6750-13-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao X, Chen Y, Mugabe S, Gao C, Tkaczyk C, Mazor Y, Pavlik P, Wu H, Dall'Acqua W, Chowdhury PS. A novel dual expression platform for high throughput functional screening of phage libraries in product like format. PLoS One 2015; 10:e0140691; PMID: 26468955; https://doi.org/ 10.1371/journal.pone.0140691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazor Y, Van Blarcom T, Carroll S, Georgiou G. Selection of full-length IgGs by tandem display on filamentous phage particles and Escherichia coli fluorescence-activated cell sorting screening. FEBS J 2010; 277:2291-303; PMID: 20423457; https://doi.org/ 10.1111/j.1742-4658.2010.07645.x [DOI] [PubMed] [Google Scholar]
- 15.Chan CE, Chan AH, Lim AP, Hanson BJ. Comparison of the efficiency of antibody selection from semi-synthetic scFv and non-immune Fab phage display libraries against protein targets for rapid development of diagnostic immunoassays. J Immunol Methods 2011; 373:79-88; PMID: 21856306; https://doi.org/ 10.1016/j.jim.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schirrmann T, Meyer T, Schutte M, Frenzel A, Hust M. Phage display for the generation of antibodies for proteome research, diagnostics and therapy. Molecules 2011; 16:412-26; PMID: 21221060; https://doi.org/ 10.3390/molecules16010412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jostock T, Vanhove M, Brepoels E, Van Gool R, Daukandt M, Wehnert A, Van Hegelsom R, Dransfield D, Sexton D, Devlin M, et al.. Rapid generation of functional human IgG antibodies derived from Fab-on-phage display libraries. J Immunol Methods 2004; 289:65-80; PMID: 15251413; https://doi.org/ 10.1016/j.jim.2004.03.014 [DOI] [PubMed] [Google Scholar]
- 18.Jones ML, Seldon T, Smede M, Linville A, Chin DY, Barnard R, Mahler SM, Munster D, Hart D, Gray PP, et al.. A method for rapid, ligation-independent reformatting of recombinant monoclonal antibodies. J Immunol Methods 2010; 354:85-90; PMID: 20153332; https://doi.org/ 10.1016/j.jim.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 19.Renaut L, Monnet C, Dubreuil O, Zaki O, Crozet F, Bouayadi K, Kharrat H, Mondon P. Affinity maturation of antibodies: Optimized methods to generate high-quality ScFv libraries and isolate IgG candidates by high-throughput screening. Methods Mol Biol 2012; 907:451-61; PMID: 22907368; https://doi.org/ 10.1007/978-1-61779-974-7_26 [DOI] [PubMed] [Google Scholar]
- 20.Chen CG, Fabri LJ, Wilson MJ, Panousis C. One-step zero-background IgG reformatting of phage-displayed antibody fragments enabling rapid and high-throughput lead identification. Nucleic Acids Res 2014; 42:e26; PMID: 24253301; https://doi.org/ 10.1093/nar/gkt1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanmark H, Huovinen T, Matikka T, Pettersson T, Lahti M, Lamminmaki U. Fast conversion of scFv to Fab antibodies using type IIs restriction enzymes. J Immunol Methods 2015; 426:134-9; PMID: 26271437; https://doi.org/ 10.1016/j.jim.2015.08.005 [DOI] [PubMed] [Google Scholar]
- 22.Batonick M, Kiss MM, Fuller EP, Magadan CM, Holland EG, Zhao Q, Wang D, Kay BK, Weiner MP. pMINERVA: A donor-acceptor system for the in vivo recombineering of scFv into IgG molecules. J Immunol Methods 2016; 431:22-30; PMID: 26851519; https://doi.org/ 10.1016/j.jim.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science 1988; 242:423-6; PMID: 3140379; https://doi.org/ 10.1126/science.3140379 [DOI] [PubMed] [Google Scholar]
- 24.Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, Griffiths AD. Amplification of complex gene libraries by emulsion PCR. Nat Methods 2006; 3:545-50; PMID: 16791213; https://doi.org/ 10.1038/nmeth896 [DOI] [PubMed] [Google Scholar]
- 25.Shao K, Ding W, Wang F, Li H, Ma D, Wang H. Emulsion PCR: A high efficient way of PCR amplification of random DNA libraries in aptamer selection. PLoS One 2011; 6:e24910; PMID: 21949784; https://doi.org/ 10.1371/journal.pone.0024910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol 2008; 26:1135-45; PMID: 18846087; https://doi.org/ 10.1038/nbt1486 [DOI] [PubMed] [Google Scholar]
- 27.Harrison JL, Williams SC, Winter G, Nissim A. Screening of phage antibody libraries. Methods Enzymol 1996; 267:83-109; PMID: 8743311; https://doi.org/ 10.1016/S0076-6879(96)67007-4 [DOI] [PubMed] [Google Scholar]
- 28.Shi S, Condon RG, Deng L, Saunders J, Hung F, Tsao YS, Liu Z. A high-throughput automated platform for the development of manufacturing cell lines for protein therapeutics. Journal of visualized experiments: JoVE 2011; PMID: 21968840; https://doi.org/ 10.3791/3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Placke T, Kopp HG, Salih HR. Glucocorticoid-induced TNFR-related (GITR) protein and its ligand in antitumor immunity: Functional role and therapeutic modulation. Clin Dev Immunol 2010; 2010:239083; PMID: 20936139; https://doi.org/ 10.1155/2010/239083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol 1996; 14:309-14; PMID: 9630891; https://doi.org/ 10.1038/nbt0396-309 [DOI] [PubMed] [Google Scholar]
- 31.Lloyd C, Lowe D, Edwards B, Welsh F, Dilks T, Hardman C, Vaughan T. Modelling the human immune response: Performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel 2009; 22:159-68; PMID: 16730741; https://doi.org/ 10.1093/protein/gzn058 [DOI] [PubMed] [Google Scholar]
- 32.Groves M, Lane S, Douthwaite J, Lowne D, Rees DG, Edwards B, Jackson RH. Affinity maturation of phage display antibody populations using ribosome display. J Immunol Methods 2006; 313:129-39; PMID: 16730741; https://doi.org/ 10.1016/j.jim.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 33.Douthwaite JA, Sridharan S, Huntington C, Hammersley J, Marwood R, Hakulinen JK, Ek M, Sjogren T, Rider D, Privezentzev C, et al.. Affinity maturation of a novel antagonistic human monoclonal antibody with a long VH CDR3 targeting the Class A GPCR formyl-peptide receptor 1. mAbs 2015; 7:152-66; PMID: 25484051; https://doi.org/ 10.4161/19420862.2014.985158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tesar D, Hotzel I. A dual host vector for Fab phage display and expression of native IgG in mammalian cells. Protein Eng Des Sel 2013; 26:655-62; PMID: 24065833; https://doi.org/ 10.1093/protein/gzt050 [DOI] [PubMed] [Google Scholar]