

Abstract
The ClusPro server (https://cluspro.org) is a widely used tool for protein-protein docking. The server provides a simple home page for basic use, requiring only two files in Protein Data Bank format. However, ClusPro also offers a number of advanced options to modify the search that include the removal of unstructured protein regions, applying attraction or repulsion, accounting for pairwise distance restraints, constructing homo-multimers, considering small angle X-ray scattering (SAXS) data, and finding heparin binding sites. Six different energy functions can be used depending on the type of proteins. Docking with each energy parameter set results in ten models defined by centers of highly populated clusters of low energy docked structures. This protocol describes the use of the various options, the construction of auxiliary restraints files, the selection of the energy parameters, and the analysis of the results. Although the server is heavily used, runs are generally completed in < 4 hours.
INTRODUCTION
Protein-protein interactions are important for understanding cellular function and organization. Substantial progress has been made toward generating potential protein-protein interaction networks using high-throughput proteomics studies, primarily yeast two-hybrid assays1,2 and mass spectrometry3,4. Mechanistic interpretation of the interactions frequently requires atom-level details, ideally obtained by X-ray crystallography. However, some of the biologically important interactions occur in transient complexes, and hence experimental structure determination may be very difficult, even when the structures of the component proteins are known. Therefore, computational docking methods have been developed that, starting from the structures of component proteins, attempt to determine the structure of their complexes targeting an accuracy close to that provided by X-ray crystallography5–7. Docking usually generates a number of detailed models that define the positions of all atoms, but the current scoring functions are usually not accurate enough for reliable model discrimination, and in most cases the model closest to the native structure cannot be identified solely by computational tools. However, model selection can be based on additional information obtained by lower resolution methods such as site-directed mutagenesis or chemical cross-linking, and the selected models generated by the docking provide atom-level details.
Docking methods can be classified as direct or template-based. Based on thermodynamics, direct methods attempt to find the structure of the target complex located at the minimum of Gibbs free energy in the conformational space, and thus require a computationally feasible free energy evaluation model and an effective minimization algorithm8. As will be discussed, direct docking methods may give good results if the conformational changes upon protein-protein association are moderate. Template-based docking is based on the observation that interacting pairs sharing above 30% sequence identity often interact in the same way, and hence the structure of the target complex can be obtained by homology modeling tools if an appropriate template complex of known structure is available9. Although the applicability of template-based docking has been extended based on the observation that partial structures representing the interface region can provide templates10, the coverage of the template space at present is still limited and hence direct methods are generally more useful in many applications.
This protocol describes ClusPro, a web based server for the direct docking of two interacting proteins. ClusPro was introduced in 200411,12 but since then has been substantially modified and expanded13–15. The server performs three computational steps as follows: (1) rigid body docking by sampling billions of conformations, (2) root-mean-square deviation (RMSD) based clustering of the 1000 lowest energy structures generated to find the largest clusters that will represent the most likely models of the complex, and (3) refinement of selected structures using energy minimization (Figure 1). The rigid body docking step uses PIPER16, a docking program based on the Fast Fourier Transform (FFT) correlation approach. The FFT approach, introduced by Katchalski-Katzir and co-workers17 in 1992, led to major progress in rigid body protein-protein docking. In this method, one of the proteins (which we will call the receptor) is placed at the origin of the coordinate system on a fixed grid, the second protein (which we will call the ligand) is placed on a movable grid, and the interaction energy is written in the form of a correlation function (or as a sum of a few correlation functions). The numerical efficiency of the methods stems from the fact that such energy functions can be efficiently calculated using Fast Fourier Transforms, and results in the ability of exhaustively sampling billions of the conformations of the two interacting protein, evaluating the energies at each grid point. Thus, the FFT based algorithm enables docking of proteins without any a priori information on the structure of the complex. Katchalski-Katzir et al.17 used a simple scoring function that accounted only for shape complementarity. However, subsequent methods based on the FFT correlation approach to docking introduced more complex and more accurate scoring functions that also included terms representing electrostatic interactions18,19, or both electrostatic and desolvation contributions20. A key to the success of rigid body methods is that the shape complementarity term allows for some overlaps, and hence the methods are able to tolerate moderate differences between bound and unbound (separately crystallized) structures. As will be discussed, one of the distinguishing characteristics of PIPER, the docking program used in the current version of ClusPro, is that this implementation of the FFT correlation method employs a scoring function that includes a structure-based pairwise interaction term, and the combination with the other terms in the energy function substantially increases the accuracy of docking, resulting in more near-native structures16.
FIGURE 1.
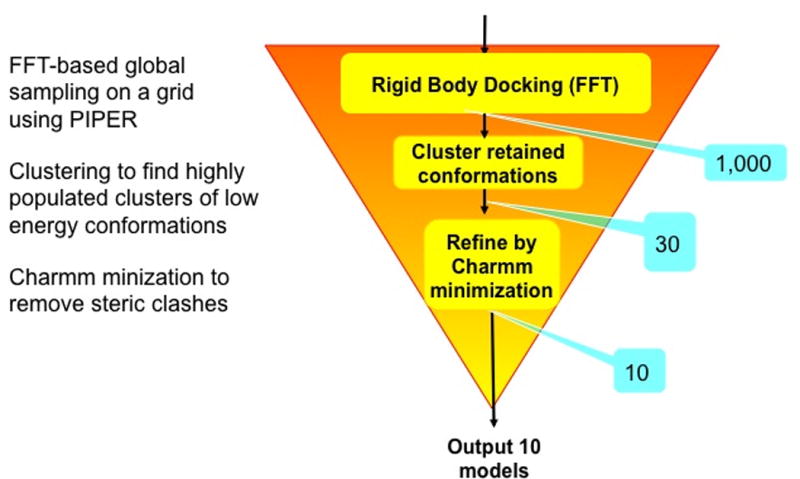
Outline of the ClusPro algorithm. After each step, the number of structures retained is shown in a blue box.
Rigid body methods including PIPER perform exhaustive sampling of the conformational space on a dense grid, and hence certainly sample some near-native structures. However, the need for tolerating some steric clashes due to docking unbound protein structures requires the use of approximate scoring functions, and reducing sensitivity to conformational differences also reduces specificity. Therefore, the docked conformations that are close to the native structure do not necessarily have the lowest energies, whereas low energy conformations may occur far from the X-ray structures. In PIPER we retain the 1000 lowest energy docked structures for further processing and hope that this set includes at least some that are close to the native structure of the complex. Another unique feature of ClusPro is that we select the centers of highly populated clusters of the low energy structures rather than simply the lowest energy conformations as predictions of the complex21. As will be further discussed, the size of each cluster represents the width of the corresponding energy well, and hence provides some information on entropic contributions to the free energy. While the largest clusters do not necessarily contain the most near-native structures, we have shown12 that the 30 largest clusters include near-native structures for 92% of complexes in a protein-protein benchmark set22–25. However, the success rates are higher for certain classes of complexes such as enzyme-inhibitor pairs26,27, and hence it is generally sufficient to retain only ten or fewer highly populated clusters. Supplementary Table 1 lists some performance characteristics of ClusPro 2.0 for the different classes of complexes in benchmark 4.0. The characteristics shown are the number of complexes with at least one cluster of docked structures within 10 Å interface root mean square deviation (IRMSD) from the native complex, the average number of docked structures with less than 10 Å IRMSD within the 1000 lowest energy structures, and the average value of the lowest IRMSD achieved. Note that to define the IRMSD of a docked structure from the native complex we first select the interface residues as the ligand residues that have any atom within 10 Å of any receptor atom. Then we superimpose the receptors in the two structures, and calculate the Cα RMSD for the selected interface residues. According to Supplementary Table 1, a large fraction of enzyme-inhibitor pairs are in the “easy” category, and in almost all cases we have a cluster with its center within 10 Å IRMSD, with the average IRMSD of 2.94 Å. In contrast, for the “other” type complexes such clusters are found only in about 50% of cases, and the average IRMSD is 7.54 Å even in the easy category. The success rate is similar for antibody-antigen pairs if both proteins are independently crystallized. However, as will be discussed, the performance is improved if the non-CDR regions of the antibody are masked and hence cannot be in the interface.
The current version, ClusPro 2.0, is heavily used; by June 2016 it had over 17,000 users (among them over 7,000 registered, which is not required), and completed over 172,000 docking calculations, currently adding about 3,000 per month. Models built by the server have been reported in over 400 publications. Later in this protocol we describe the types of the applications that frequently occur in these papers.
The PIPER docking algorithm
As stated, the ClusPro server is based on PIPER, which performs the sampling. The center of mass of the receptor is fixed at the origin of the coordinate system, and the possible rotational and translational positions of the ligand are evaluated at the given level of discretization. The rotational space is sampled on a sphere-based grid that defines a subdivision of a spherical surface in which each pixel covers the same surface area as every other pixel28. The 70,000 rotations we consider correspond to about 5 degrees in terms of the Euler angles. The step size of the translational grid is 1 Å. It is easy to see that for an average size protein this amounts to sampling 109–1010 conformations.
PIPER represents the interaction energy between two proteins using an expression of the form E = w1Erep + w2Eattr + w3Eelec + w4EDARS, where Erep and Eattr denote the repulsive and attractive contributions to the van der Waals interaction energy, and Eelec is an electrostatic energy term. EDARS is a pairwise structure-based potential constructed by the Decoys as the Reference State (DARS)29 approach, and it primarily represents desolvation contributions, i.e., the free energy change due to the removal of the water molecules from the interface16. The coefficients w1, w2, w3, and w4 define the weights of the corresponding terms, and are optimally selected for different types of docking problems. Unless specified otherwise in Advanced Options, the server generates four sets of models using the scoring schemes called (1) balanced, (2) electrostatic-favored, (3) hydrophobic-favored, and (4) van der Waals + electrostatics (see Table 1). As will be described, for complexes that do not fit into these categories and are classified as “others” in the protein-protein docking benchmarks22–25, ClusPro generates structures using three different coefficient sets (Table 1). To understand the magnitude of these coefficients we note that Erep and Eattr, while defined on the grid, are scaled to the van der Waals energy16, and hence w1 < 1.0 and w2 <1.0 yield “softening” of both repulsive and attractive van der Waals terms. Such softening is necessary, since the bound and unbound conformations of the proteins to be docked generally differ, in some cases substantially. The DARS potential has been parameterized on a set of complexes that included a large number of enzyme-inhibitor pairs and multi-subunit proteins, and hence the resulting potential assumes good shape and electrostatic complementarity29. Since EDARS is scaled to the magnitudes of protein-protein binding free energies, w4 =1.0 is the “neutral” choice. Eelec is represented by a truncated and smoothed Coulomb expression, also defined on the grid16. Since the charges are given as multiples of the electronic charge and the interatomic distance is measured in Å, obtaining the electrostatic interaction energy in kcal/mol using the Coulomb expression and these units30 would require the coefficient w3 =332. However, the truncation and smoothing reduce the magnitude of Eelec relative to the value based on the Coulomb expression, and hence we use w3=600 in the balanced option of the parameter set. This set was shown to generally provide very good results for enzyme-inhibitor complexes. If it is known or assumed that the association of two proteins is mainly driven by their electrostatic interactions, then we select results obtained by the electrostatic-favored weights, in which the weight of the electrostatics is doubled relative to the balanced energy expression. In contrast, for complexes primarily stabilized by hydrophobic interactions we use the hydrophobic-favored potential, which thus doubles the weight of the desolvation term. In the fourth option, van der Waals + electrostatics, the pairwise potential Epair is not used. The need for this option occurs for proteins that are very different from the ones used for the parameterization of EDARS. The selection of the parameters in the Others Mode will be discussed further in this protocol. We recognize that the weights in the PIPER energy function are somewhat arbitrary, and were selected to optimize the success rates for different classes of protein complexes. However, as described below, the most likely models of the complex are selected on the basis of cluster population rather than energy value. In fact, PIPER does not aim to estimate the true interaction energy, and the score provided by ClusPro should not be considered as a measure of binding affinity.
TABLE 1.
Weighting coefficients of PIPER energy terms in various docking modes
| Coefficient set | Energy term weight coefficients
|
|||
|---|---|---|---|---|
| Erep | Eattr | Eelec | EDARS | |
| Balanced | 0.40 | −0.40 | 600.0 | 1.0 |
| Electrostatic-favored | 0.40 | −0.40 | 1200.0 | 1.0 |
| Hydrophobic-favored | 0.40 | −0.40 | 600.0 | 2.0 |
| Van der Waals + electrostatics | 0.40 | −0.40 | 600.0 | 0.0 |
| Others Mode, Set 1 | 0.30 | −0.30 | 300.0 | 1.25 |
| Others Mode, Set 2 | 0.50 | −0.20 | 300.0 | 0.50 |
| Others Mode, Set 3 | 0.50 | −0.20 | 300.0 | 0.0 |
Model selection by cluster population
The second step of ClusPro is clustering the lowest energy 1000 docked structures using pairwise IRMSD as the distance measure.12,21 We calculate IRMSD values for each pair among the 1000 structures, and find the structure that has the highest number of neighbors within 9Å IRMSD radius. The selected structure will be defined as the center of the first cluster, and the structures within the 9Å IRMSD neighborhood of the center will constitute the first cluster. The members of this cluster are then removed, and we select the structure with the highest number of neighbors within the 9Å IRMSD radius among the remaining structures as the next cluster center. These neighbors will form the next cluster. Up to 30 clusters are generated in this manner. After clustering we minimize the energy of the retained structures for 300 step with fixed backbone using only the van der Waals term of the Charmm potential31. The minimization removes the steric overlaps but generally yields only very small conformational changes. In its basic operation ClusPro outputs the structures at the centers of the 10 most populated clusters.
Considering the centers of the large clusters of low energy structures rather than simply low energy structures appears to be unique to ClusPro,32 and may implicitly account for some entropic effects.33 We have recently shown that under fairly natural assumptions the cluster populations are proportional to cluster probability. This argument is based on considering the approximate partition function Q = Σj exp(-Ej /RT), where Ej is the energy of the jth pose, and the summation includes all structures generated by the docking. Similarly, for the kth low energy cluster the partition function can be approximated by Qk = Σj exp(Ej /RT), where now we consider only the structures in the cluster. In terms of these approximate partition functions the probability of the kth cluster is given by Pk = Qk/Q. Furthermore, within the low energy cluster we introduce the approximation Ej=E, i.e., we assume that that the energy values are the same for all structures in the cluster, because the energies are from a narrow range that is comparable to the uncertainty of the energy function. Due to this assumption the probability of the cluster is given by Pk=exp(-E/RT)×Nk/Q, and thus it is proportional to the number Nk of the structures in the kth cluster. Therefore we suggest finding the most populated clusters rather than the lowest energy structures and using the centers of the clusters as putative models. Although this approach to model selection is somewhat unusual and has not been widely adopted, we believe that the consistent success of ClusPro in the CAPRI experiment34–38 supports its use (see the section Comparison to other methods for details on the performance of ClusPro). Since it was requested by a number of users, ClusPro also outputs the PIPER energies of cluster centers, as well as the lowest PIPER energy within each cluster. However, since these values do not include entropic contributions and the weights of the energy components are selected to yield near-native structures rather than correct thermodynamics of binding, the PIPER energy does not provide valid information on binding free energy, and should not be used for ranking the models. We repeatedly emphasize that the latter is based on cluster population rather than cluster energy score.
Applications of the method
The usage of the ClusPro server has been growing beyond our expectations. By June 2016 the server had over 17,000 users (among them over 7,000 registered, which is not required), and completed more than 172,000 docking calculations, currently adding about 3,000 per month. Models built by the server have been reported in over 400 publications. These statistics demonstrate that there is substantial need for protein-protein docking, and that implementing our algorithms as a server expanded the availability of the method to parts of the research community without experience in computational structural biology. The large number of publication also implies that we can follow how the server is employed. We describe here some of the typical applications, each illustrated by examples.
Docking X-ray or NMR structures of proteins
This is the most basic and straightforward application of ClusPro. As an early example, Chance and co-workers constructed the three-dimensional structure of cofilin, an important actin binding protein, bound to monomeric actin39,40. The binding model was validated by hydroxyl radical-mediated oxidative protein footprinting, and identified key ionic and hydrophobic interactions at the binding interface39. Models can be frequently used for mechanistic interpretation. For example, Luxán et al.41 docked JAG1, a cell surface protein that interacts with receptors in the mammalian Notch signaling pathway, to the dimers of the E3 ubiquitin-protein ligase MIB1, and used the results to show that certain mutations of MIB1 arrest chamber myocardium development, preventing trabecular maturation and compaction, and thereby cause left ventricular noncompaction cardiomyopathy.
Modeling antibody-antigen interactions
The analysis of antibody-antigen interactions is a particularly important modeling and docking problem, required in biotechnology and vaccine design applications. For example, Tran et al.42 crystallized the Fab fragments of two vaccine-elicited monoclonal antibodies (mAbs) binding to HIV-1 gp120. Alanine scanning of their complementarity-determining regions, coupled with epitope scanning of their epitopes on gp120, revealed putative contact residues. Using this information they docked the Fabs to gp120. Coupled with EM reconstructions of the mAb-gp120 complexes, the docking results suggested that the antibodies use a distinct approach to the HIV-1 primary receptor binding site, and this information was used for vaccine redesign.
Constructing the structure of large multi-domain proteins
We reviewed the work43 by Kuriyan and co-workers44, who combined computational docking, small-angle x-ray scattering, mutagenesis, and calorimetry to study the histone domain of the Ras-specific nucleotide exchange factor son of sevenless (SOS). They have shown that the domain folds into the rest of SOS and interacts with a helical linker that connects the pleckstrin-homology (PH) and Dbl-homology (DH) domains to the catalytic domain of SOS. Results suggested that the histone domain is a potential mediator of membrane-dependent activation signals44.
Building homo-oligomers of proteins
Examples are the construction of the human p53-controlled ribonucleic reductase (p53R2) homodimer, which was used to explain mutations that cause mitochondrial DNA depletion45, and the modeling of an L-type Ca2+ channel used for the characterization of interactions with 1,4-dihydropyridines46. Determination of homo-oligomeric proteins by docking from monomeric structures solved by NMR spectroscopy is frequently required, because NMR determination of multimers by NMR is far from simple due to the lack of chemical shift perturbation data and the difficulty of obtaining a sufficient number of intermonomer distance restraints. For example, Zweckstetter and co-workers47 determined the solution structure of the 15.4 kDa homodimer CylR2, the regulator of cytolysin production from Enterococcus faecalis, by combining the available experimental information with docking.
Protein-peptide docking
Although in the present form ClusPro is not an appropriate tool for docking very flexible peptides to proteins, the server can be used if some information on the protein-bound structure of the peptide is available. For example, to study activating mutations of STAT5B in lymphomas, Chan and co-workers48 docked a phosphorylated self-peptide to the homology model of the SH2 domain of a STAT5B mutant. In spite of docking a flexible peptide to a homology model, which may have some structural deviations from the X-ray structure, this analysis was feasible, because highly homologous templates of the SH2 domain with bound peptides were available, and provided both a peptide structure and the key binding residues of STAT5B.
Docking of homology models
The ClusPro server is frequently used for docking homology models. For example, Williams and co-workers49 studied the specificity of binding of KirCII, a trans-acting acyltransferase, to a panel of acyl carrier proteins, by docking a homology model of KirCII. In addition to ClusPro, the author used the PatchDock server50, and considered the convergence of the two methods on very similar models as part of the validation. Steeland et al.51 studied the binding of small single domain antibodies to human tumor necrosis factor receptor (TNFR) using homology models of both proteins. Results were used for the design of selective inhibitors of the TNF/TNFR interaction51. However, while we explored relatively well the performance of ClusPro for docking X-ray structures16, we have somewhat limited experience with homology models, and our incomplete analysis suggests that even moderate errors in key side chains or loops may substantially reduce the accuracy of docking results. Therefore we have recently collected a benchmark set of proteins to facilitate the development of methods that integrate homology modeling and docking52.
Experimental Design
We describe here a number of advanced options for use in ClusPro as follows.
Structure Modification
Structure modification is suggested if any of the proteins has an unstructured or uncertain terminal region. Such regions may occur as the result of chemical tagging in the purification process, or may be created by homology modeling programs due to the lack of templates for the given region. Removal of such regions is frequently advantageous, because they can interfere with rigid body docking. When using this option, the server removes terminal residues until a regular secondary structure (alpha helix, extended strand, 3-helix, π-helix, or a hydrogen bonded turn) would be reached within 2 amino acid residues along the sequence. The modification can be requested for the receptor, the ligand, or both, and applies to both ends of the chain.
Attraction and Repulsion
If experimental information is available that indicates certain residues are involved in binding, whereas other residues remain surface accessible upon complex formation, this can be used to influence the results of the docking by setting attraction and repulsion, respectively, on these residues. To specify attraction, the user must enter the residues in one or both sides of the interface that are believed to participate in the binding. In the docking calculations an extra attractive force is applied to the selected residues. Repulsion can be specified similarly, by selecting a number of residues that are not expected to be in the interface, and hence are the origins of repulsive interactions in the docking. Alternatively one can upload a protein data bank (PDB) file containing the residues that are not supposed to be in the interface and hence are “masked” during the docking calculations by adding a repulsive force component. The masking files specify the amino acids that do not participate in intermolecular interaction, and hence substantially restrict the conformational space that can be occupied by the complex.
Restraints
We have recently added to ClusPro the option to define pairwise distance restraints. Such restraints can be derived, e.g., from NMR Nuclear Overhauser Effect (NOE) experiments or by chemical crosslinking. A pairwise distance restraint can be defined by two sets of atoms, S1 and S2 and a distance range, dmin to dmax. The restraint is considered satisfied if there are at least one atom in S1 and at least one atom in S2 such that the distance between them falls in this range. While the implementation allows for arbitrary sets of atoms to be used to define a restraint, most frequently these involve a single atom or residue on each side of the interface. Given multiple restraints, users may want to require a certain number of restraints out of a group to be satisfied. In addition, restraints may be based on sources with varying reliability, requiring different cutoff values. Our implementation allows for grouping restraints into restraint groups, and restraint groups into restraint sets. Restraint groups are considered satisfied when more than a user specified number of restraints in the group are satisfied, and a restraint set is satisfied when more than a user specified number of its groups are satisfied. When a restraint set is provided, ClusPro will only report solutions that satisfy the restraints. To do this efficiently, for each rotation we first generate the set of translations that satisfy each individual restraint, called the feasible translation set for the particular restraint. We then consider the intersection of feasible translation sets for the restraints in each restraint group, and select the translations that appear more often than the cutoff for the restraint group. The selected feasible translation sets for each restraint group are merged in a similar way to generate the feasible translation set for an entire restraint set. Providing restraints can actually decrease the running times, since only the van der Waals interaction energy is computed for each feasible translation, and translations that result in unacceptable clashes are skipped. After selecting the solutions that satisfy the restraints, 1000 structures with the lowest PIPER energies are clustered and minimized as customary in ClusPro.
The Others Mode
The Others Mode uses a special scoring scheme to target the complexes that are classified as “others” in the established protein-protein docking benchmark22–25,53. The motivation for developing a special scoring function is their diverse nature and their generally limited shape and electrostatic complementarity. To overcome these challenges we use three different sets of weighting coefficients, generate 500 structures with each, and cluster the resulting 1500 conformations (Table 1). This is the only case when we consider 1500 rather than 1000 docked structures. In view of the assumed weaker shape complementarity and higher structural uncertainty we reduce the coefficients of Eattr and Eelec, and use three different values for the coefficient of EDARS. The 1500 structures are mixed and clustered together, and the centers of well-populated clusters are considered as models of the complex. Our docking studies15 confirm that the strategy substantially improves the overall success rates for the “other” type of complexes, in agreement with the assumption that the interaction energy of these complexes cannot be well described by a single set of coefficients.
The Antibody Mode
In the “Antibody Mode” PIPER uses a potential developed for docking antibody and antigen pairs.54 Analysis of antibody–protein antigen complexes has revealed inherent asymmetry between the two sides of the interface. Specifically, phenylalanine, tryptophan and tyrosine residues highly populate the paratope of the antibody but not the epitope of the antigen. Since this asymmetry cannot be adequately modeled using the symmetric pairwise potential generally used in PIPER, we have removed the usual assumption of symmetry. Interaction statistics were extracted from antibody–protein complexes under the assumption that the interaction preferences of an atom of a particular type on the antibody differ from the preferences of an atom of the same type on the antigen. The use of the new potential significantly improved the performance of docking for antibody–protein antigen complexes54. The method allows for the masking of non-paratope residues, either by using automated selection or by providing a masking file as described for the Attraction and Repulsion option.
Multimer Docking
A subclass of interactions between proteins is the case where two or more identical proteins interact to form a homomultimer. The construction of such multimers is frequently required, because a number of proteins have been solved as monomers but exist in a homomultimeric state in vivo. We have developed a special mode in our docking where we limit the rotations sampled by the docking algorithm to rotations that satisfy either C2 or C3 symmetry, respectively, for dimers and trimers55. The updated method we currently use is similar to the one developed by the Weng lab and implemented in the M-ZDOCK program56, with some differences that result in a slightly simpler algorithm. The difference is that Pierce et al.56 rotate the receptor protein in the process of generating symmetric structures, whereas ClusPro rotates the coordinate system. Since the new method has not been published, we provide the steps of the algorithm here. (1) Center the receptor (the input monomer) at the origin. (2) Select values for the Euler angles ψ and θ to define an axis of rotation. (3) Copy the receptor, and rotate it by 360°/N around the axis of rotation to create the ligand, where N=2 for a dimer and N=3 for a trimer. (4) Discretize both the ligand and receptor, with a grid spacing of 1 Å. (5) Perform FFT search in the plane perpendicular to the axis of rotation for the best scoring multimer position. Since the search space is now restricted to S2 rather than the rotational group S3, a set of points uniformly distributed on the sphere is generated using the S2 sequence code from https://mitchell-lab.biochem.wisc.edu/SOI/index.php. This set of points is used as the basis for symmetric rotations of 180 degrees (dimer) and 120 degrees (trimer). Symmetry is enforced during docking by considering only translations within 2Å of the plane defined by the axis of rotation and passing through the center of rotation. (6) Repeat Steps 2 through 5 for all ψ and θ values on a grid with 5 degrees step size. The 1000 lowest energy structures are retained and clustered as in the general ClusPro docking.
SAXS Profile
Small Angle X-ray Scattering (SAXS) experiments are based on observing the X-ray scattering of a macromolecule in solution at different scattering angles, resulting in a one-dimensional scattering profile. While the SAXS profile provides information about the shape and size of the molecule57, the amount of such information is much lower than the one that can be obtained by X-ray crystallography, and on its own does not provide atom-level resolution. This makes docking a natural complement to SAXS for the determination of complex structures. ClusPro can account for SAXS experimental data by retaining a number of docked structures that best agree with the provided SAXS profile58. These structures are then ranked by the PIPER scoring function and clustered as usual in ClusPro, thus the information from SAXS is used only to filter the structures generated by PIPER and the search is not biased by modifying the scoring function as done in a number of other methods that account for SAXS results59,60. The advantage of this approach is that the docked structures are restrained rather than determined by the SAXS data, and hence the method can be used even with moderately informative SAXS profiles58.
Heparin Ligand
Many proteins bind the polysaccharides heparan sulfate and heparin. Heparan sulfate (HS) consists of alternating hexuronic acid and D-glucosamine disaccharide units. Variations in sulfation and hexuronic acid structure result in various HS molecules61–64. Heparin, a member of the HS family, consists of highly-sulfated disaccharides, and is frequently used as a model compound in studies of protein-HS interactions65. Since crystallizing protein-heparin complexes for structure determination is generally difficult, docking can be a useful approach for understanding specific interactions, and hence we have extended PIPER and ClusPro to heparin docking66. The method generates and evaluates close to a billion poses of a heparin tetrasaccharide. The docked structures are clustered using pairwise RMSD as the distance measure. However, since we use a generic heparin structure as a probe, and since there are not enough protein-heparin complex structures to improve the interaction potential, clustering the docked heparin structures and selecting the clusters with the highest protein-ligand contacts predict only the heparin binding sites rather than bound heparin poses66. Nevertheless, the cluster centers can provide starting points for further refinement of heparin positions using methods that account for flexibility, e.g., molecular dynamics.
Comparison to existing methods
Table 2 shows a classification of direct protein-protein docking methods based on the amount of the information that is required in addition to the structures of the component proteins8. Each class of methods has its own strengths and limitations. Global methods are the most useful when no a priori information on the complex is available, and hence the entire 6D conformational space must be sampled. Since such methods use rigid body approximation, they allow only for moderate conformational change upon binding, which is a major limitation. The medium-range methods such as RosettaDock67 and ATTRACT68 can sample only selected regions of the conformational space and hence require some information on the putative complex, but the Monte Carlo or similar search algorithms can account for some level of flexibility, primarily in side chains. Therefore these methods are particularly useful when side chain conformations are very uncertain, for example when one of the component protein structures is a homology model, but some information is available on the structure of the complex to identify the region of interest in the conformational space. Finally, restraint based docking, exemplified by the program HADDOCK (High Ambiguity Driven biomolecular DOCKing)69,70, incorporates interaction restraints into the scoring function to guide the search toward regions of the conformational space in which the restraints are satisfied. The method can work very well if a sufficient number of correct restraints are available, but performance may deteriorate without them.
TABLE 2.
Important classes of direct docking algorithms
| Method class | Properties
|
Examples | |
|---|---|---|---|
| Search Method | Protein Flexibility | ||
| Global systematic rigid body docking | Fast Fourier Transform; Geometric matching | Minimal; smooth potential allows for some overlaps | ZDOCK53, GRAMM78, PIPER16, DOCK/PIERR120 |
| Medium-range methods: Localized searches over selected regions | Monte Carlo minimization; Multi-start quasi-Newton minimizer with side chain search | Moderate, mostly side chains, some loops; motion along normal modes | RosettaDock67 ICM-DISCO121 ATTRACT68 SWARMDOCK74 |
| Restraint-based docking | Supported by a priori information in the scoring function | Can be more substantial if restraints are available | HADDOCK69 |
The quality of docking methods is continuously monitored by CAPRI (Critical Assessment of Predicted Interactions), the ongoing communitywide experiment devoted to protein docking71. In the CAPRI challenge, initiated in 2003, participating research groups and automated servers are given prediction targets, each being an unpublished experimentally determined structure of a protein-protein complex. Given the atomic coordinates of the component proteins or of their homologues, the participants have to model the complexes. Servers must submit results within two days, whereas human predictor groups have several weeks and can use any available information. Each group can submit ten predictions for each target. The submitted models are evaluated by independent assessors. For each group or server, the assessors report the number of targets with acceptable or better quality predictions, and also note the number of targets for which highly accurate (***) or medium accuracy (**) models were submitted. These categories have been defined on the basis of the fraction of native contacts, the backbone root mean square deviation of the ligand (LRMSD) from the reference ligand structure after superimposing the receptor structures, and the backbone RMSD of the interface residues (IRMSD). The calculation of these measures and the exact definitions of categories were given in the first CAPRI evaluation paper72. Although the CAPRI quality score is based on a number of characteristics including several RMSD measures and the predicted fraction of native contacts, for simplicity we focus on LRMSD, and note that for the highly accurate, medium accuracy, acceptable, and incorrect models the ligand LRMSD is given by LRMSD < 1Å, 1Å < LRMSD < 5Å, 5Å < LRMSD < 10Å, and LRMSD > 10Å, respectively.
Since its release in 2004, ClusPro has consistently been the best automated server in the CAPRI challenge. Table 3 shows the performance of the four best servers based on the results of the last three CAPRI evaluation meetings in 200936, 201337, and 2016. These results confirm that the global systematic sampling of the entire conformational space as performed by ClusPro is useful when essentially no a priori information on the structure of the target complex can be used, which is generally the case for servers that must submit results within two days, directly produced by the server. The only freedom we had using ClusPro was the choice of the energy coefficient set. The comparison of different methods is more complex if the performance of both “human” predictor groups and servers is considered (Table 4). Because predictors can use additional information, medium-range methods such as ATTRACT by Zaccharias68,73, SWARMDOCK by Bates74, and FRODOCK by the Chacon group75, recently combined with a new scoring scheme by Guerois76, performed well. The restraint based HADDOCK method by Bonvin and co-workers also provided good predictions for many targets69,70. However, good results were also obtained by several methods that, similarly to ClusPro, perform global search assuming rigid proteins, including ZDOCK53,77, GRAMM78, and PatchDock50. Our own group’s “human” predictions are based on running ClusPro, followed by refinement and selection of most likely clusters”79, in some cases involving Monte Carlo minimization runs either using RosettaDock67 or our own implementation of the Monte Carlo algorithm80. For a few targets the refinement improved medium accuracy structures into highly accurate ones, but generally the impact was moderate, and in 2016 we actually lost an acceptable prediction. According to Table 4, the relative performance of ClusPro has been improving over the years, and based on the 2016 results the server appears to be competitive with the best “human” predictor groups. We note that the PIPER scoring function has been recently used in an algorithm that expands FFT-based sampling to five rotational coordinates81. Working in a space with spherical coordinates defined as a manifold, the new 5D algorithm retains the accuracy of PIPER for globular proteins while providing at least 10-fold speedup. However, moderate loss of accuracy may occur for proteins that are very elongated along some direction. An additional advantage of the method is that increasing the number of correlation function terms is computationally inexpensive, which enables using even more complex energy functions as well as accounting for any number of pairwise distance constraints81.
TABLE 3.
Server performance based on the last three CAPRI evaluation meetingsa
| CAPRI evaluation meeting year and number of targets
|
||||||
|---|---|---|---|---|---|---|
| 2009, 12 Targets | 2013, 14 Targets | 2016, 14 Targets | ||||
|
| ||||||
| Rank | Server | Success | Server | Success | Server | Success |
| 1 | ClusPro14 (Vajda/Kozakov) |
5/1***/3** | ClusPro15 (Vajda/Kozakov) |
6/4** | ClusPro15 (Vajda/Kozakov) |
9/3** |
| 2 | HADDOCK69 (Bonvin) |
4/1***/1** | HADDOCK69 (Bonvin) |
4/1***/1** | DOCK/PIERR120 (Elber) |
6/2** |
| 3 | GRAMM-X78, (Vakser) |
2/2** | SWARMDOCK74 (Bates) |
4/1** | LzerD122 (Kihara) |
4/1***/3** |
| 4 | SKE-DOCK123 (Umeyama) |
2/1** | DOCK/PIERR120 (Elber) |
3/1** | HADDOCK69 (Bonvin) |
4/2** |
Number of targets with acceptable or better quality predictions / number of targets with highly accurate (***) predictions / number of target with medium accuracy (**) predictions.
TABLE 4.
Predictor group performance based on the last three CAPRI evaluation meetingsa
| CAPRI evaluation meeting, year and targets
|
||||||
|---|---|---|---|---|---|---|
| 2009, 12 Targets | 2013, 14 Targets | 2016, 14 Targets | ||||
|
| ||||||
| Rank | Group | Success | Group | Success | Group | Success |
| 1 | Vajda/Kozakov16 | 6/4***/2** | Bonvin69 | 9/1***/3** | Guerois76 | 10/1***/8** |
| 2 | Zacharias124 | 6/4***/1** | Bates74 | 8/2** | Zacharias124 | 10/3***/2** |
| 3 | Zou125 | 6/3***/2** | Vakser78 | 7/1*** | ClusPro15 | 9/3** |
| 4 | Eisenstein126 | 6/3***/1** | Kozakov/ Vajda16 | 6/2***/3** | Kozakov/ Vajda16 | 8/3***/2** |
| 5 | Wolfson127 | 6/3***/1** | Shen128 | 6/1***/3** | Seok129 | 8/3***/2** |
| 6 | Weng53 | 6/2***/2** | Fernandez-Recio130 | 6/1***/3** | Fernandez-Recio130 | 7/1***/3** |
| 7 | Zhou131 | 6/2***/2** | ClusPro15 | 6/4** | Zou125 | 7/1***/2** |
| 8 | Bonvin69 | 6/1***/4** | Zou125 | 6/1***/2** | Weng53 | 6/1***/4** |
| 9 | ClusPro14 | 5/1***/3** | Zacharias124 | 6/1*** | Vakser78 | 6/2***/2** |
| 10 | Fernandez-Recio130 | 5/2** | Eisenstein126 | 5/1***/2** | Bates74 | 6/3** |
Number of targets with acceptable or better quality predictions / number of targets with highly accurate (***) predictions / number of target with medium accuracy (**) predictions.
Limitations
PIPER performs rigid body docking in the 6D space of rotations and translations. The rigid body assumption is a good approximation for several classes of protein complexes, e.g., for most enzyme-inhibitor complexes (Supplementary Table 1). In fact, PIPER uses a “soft” potential that allows for certain steric overlaps. However, the protein-protein docking benchmark set22–25 also includes a number of “difficult” targets, i.e., complexes in which at least one of the proteins substantially changes backbone conformation upon binding. Neither PIPER nor any other rigid body method can produce acceptable docked structures for such complexes. However, we note that extending ClusPro to docking short flexible peptides to proteins is in preparation.
As discussed, unless requesting the “other” mode, ClusPro yields four sets of docked structures using the scoring schemes called (1) balanced, (2) electrostatic-favored, (3) hydrophobic-favored, and (4) van der Waals + electrostatics. In the “other” mode ClusPro generates structures using three different scoring schemes. The differences in the weighting of energy coefficients represent real differences in the biophysical forces that dominate interactions between two proteins, as the association in some complexes is driven primarily by electrostatic interactions, while in others desolvation of hydrophobic interfaces may be the main driving force. Unfortunately at present we are unable to perform automated selection of the best scoring scheme. Thus, it is left to the user to make such selection based on the assumed properties of the particular complex. If no such information is available, we suggest using the balanced option, and the “other” mode for signal transduction complexes. It is generally a good sign if the selected parameters lead to a cluster of low energy structures that is substantially more populated than the others. It is also useful if different parameter sets yield the same model. Once a model is selected, the user can also explore whether the assumption used was correct by analyzing the properties of the interface. Nevertheless, we recognize that the problem of parameter selection is not resolved, and further research is needed.
Even assuming that a parameter set is selected, ClusPro returns ten clusters of low energy structures, the center of each cluster representing a putative model of the complex. Again, the existence of a large cluster provides some level of certainty, but generally additional information is needed for reliable model selection. As mentioned, we found over 400 publications that reported models constructed by ClusPro. In many applications the models were validated by experimental techniques, including site-directed mutagenesis with NMR, calorimetry, FRET, or surface plasmon resonance44,82–101, cross-linking102–106, various spectroscopic methods107–113, X-ray scattering114, electron self-exchange reaction115, radiolytic protein footprinting with mass spectrometry39,40, hydrogen/deuterium exchange116, or intermolecular Nuclear Overhauser Effect restraints47. ClusPro results can also be used to design low resolution and hence cost effective validation experiments. We believe that such combination of computational and experimental tools is the most meaningful use of docking.
In addition to the above fundamental limitations we have several technical shortcomings that we hope remove in the future. Some of these shortcomings are as follows: (1) At this point the molecules to be docked can include only standard amino acids, nucleic acids to define an RNA molecule as the receptor, and heparin as the ligand. Thus, the docking cannot account for the presence of co-factors or other small ligands that can be important in the modulation of protein-protein interactions. (2) Similarly, we are unable to account for non-standard or modified amino acids, e.g. phosphorylation, another important factor that can affect the interactions. (3) Currently ClusPro can create only dimers and trimers in the Multimer Docking mode, and does not provide an option for constructing more complex multimers. (4) There are two limitations on size of the proteins to be docked. First, uploaded files cannot be more than 10MB each. Second, the total grid size cannot be more than roughly 40×106 Å3, which corresponds to a cube of ~350 Å in each dimension. This grid has to contain both the receptor and ligand in each potential relative orientation of the two molecules.
Availability
The server can be used without registration, but in this case the results will be publicly accessible. The advantage of registering is that the job does not show up on the website, but this options is available only to users with educational or governmental email addresses. The server provides options to view the results online, but protein visualization tools allow for more convenient analyses. We use and recommend PyMOL, which was used to demonstrate the analysis of docking results in this Protocol.
MATERIALS
EQUIPMENT
A computer with internet access and a web browser. The program is also available for download by contacting https://structure.bu.edu/contact.
Atomic resolution structures of biomolecules under investigation, in PDB format. The PDB ID can be used to directly fetch the structure or the structure may be uploaded from the computer.
Access to PyMOL or similar structure viewing software is recommended but not required. PyMOL can be downloaded www.pymol.org. Alternatively, you can use any molecular viewer that supports the visualization of multiple structures in one PDB file.
PROCEDURE
CRITICAL The current version of ClusPro provides a very simple home page for the basic use of the server, and a number advanced options that modify the docking process. Some options can be requested by simply clicking on a label, while others require providing additional information.
Input data for docking TIMING ~1 to 2 min
-
1| Locate the server at https://cluspro.org. ClusPro can be used without a user account or with a user account if you have an educational or governmental email address. If you decide to use the server without a user account, click ‘Use the server without the benefits of your own account’. To create an account, register on the ClusPro server website. A password will be sent to the email address and can be later changed by clicking on the label Preferences. You can also request an e-mail to be sent to you when your job is completed. If you already have a username and password, fill in the boxes and click Login. Either with or without a user account you will get to the server home screen (Fig. 2). From this page you will be able to submit a new docking job and select a number of Advanced Options.
CRITICAL STEP Users who run ClusPro without an account will have their results publicly accessible.
2| (Optional) Provide a job name for this submission. If you choose to leave this blank, a unique ID will be created for this field.
3| Select the type of the computer the job will use. Selecting cpu will lead it to use computer clusters at the Massachusetts Green High Performance Computing Center (MGHPC). Selecting gpu will lead it to use the graphic computing units, also located at MGHPC. Since we generally cannot predict the actual usage on these systems, selecting cpu versus gpu has no predictable consequence for the user. Currently we use the cpu version, because ample computer time is available and usually the jobs start immediately after submission.
4| Input the coordinates of the receptor structure using PDB format. Only atoms of 20 standard amino acid residues and nucleotides will be retained. All HETATM (hetero atom) records, including waters, ligands, and cofactors, will be automatically removed. There are two options for inputting a structure: use Option A to import coordinates from the PDB, or option B to upload a structure.
(A) Import coordinates from PDB
i) Import coordinates directly from the PDB by typing the four digit PDB ID into the field PDB ID.
(B) Upload structure
-
i) Upload a structure from your computer by clicking on the Upload PDB option under the PDB ID field. Select Browse to upload a file containing a structure in PDB format.
CRITICAL STEP At this point only structures containing standard amino acid residues and nucleotides will be docked. ATOM records of nonstandard amino or nucleic acids will cause an error.
CRITICAL STEP Since the protein considered the receptor is placed on a fixed grid whereas the protein considered the ligand is placed on a rotating and translating grid, it is computationally advantageous to consider the larger protein as the receptor.
CRITICAL STEP If the target is a protein-RNA complex, the RNA must be defined as the receptor rather than the ligand.
(Optional) To see the supported RNA residue and atom names click on the label RNA.
? TROUBLESHOOTING
5| In the Chains field, enter the protein chains of the receptor that you wish to include in the docking. List chains using their chain id and separate multiple chains with a whitespace. If no chains are specified, then all chains in the PDB file will be used for docking.
-
6| Input the coordinates of the ligand structure using PDB format. Only atoms of 20 standard amino acid residues or a standard heparin structure can be used. If the ligand is a protein, you can use Option A to import coordinates from the PDB, or option B to upload a structure directly as in Step 4.
CRITICAL STEP If you will use the Advanced Option Multimer Docking, no ligand structure needs to be specified.
CRITICAL STEP If you will use the Advanced Option Heparin Docking, no ligand structure needs to be specified.
? TROUBLESHOOTING
7| In the Chains field, enter the chains of the ligand protein that you wish to include in the docking as in Step 5.
FIGURE 2.
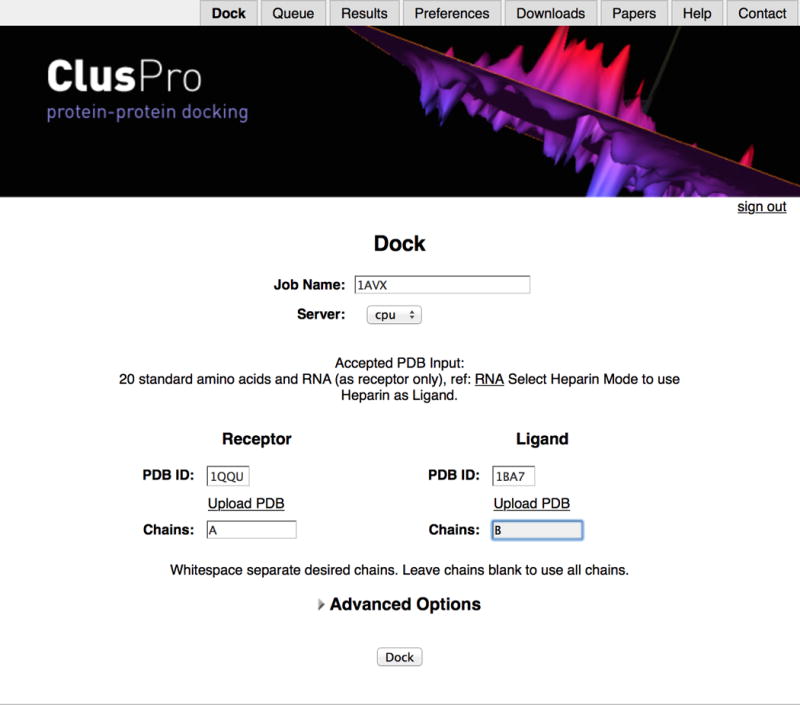
Home screen of the ClusPro server. The example shows submission of the job to dock soybean trypsin inhibitor (ligand, PDB ID 1BA7) to the X-ray structure of porcine trypsin (receptor, PDB ID 1QQU).
Selection of Advanced Options TIMING ~5 to 45 min (see below)
- 8| (Optional) At this point, it is possible to define additional advanced options depending on your requirements. See the following table for a summary of these options. These options are accessible by clicking the “Advanced Options” label.
Step Option Description
8A Structure Modification Removal of unstructured terminal residues
8B Attraction and Repulsion Setting attraction or repulsion on selected residues
8C Restraints Selection of pairwise distance restraints
8D Others Mode Selecting a special scoring scheme for “other” type complexes
8E Antibody Mode Selecting a special scoring scheme for docking antibody-antigen pairs
8F Multimer Docking Constructing homodimers or homotrimers
8G SAXS Profile Accounting for experimental SAXS data
8H Heparin Ligand Global docking of a generic heparin molecule
- Selecting Structure Modification TIMING ~5 min
-
Structure modification is suggested if any of the proteins has an unstructured terminal region. Click on Advanced Options and select Structure Modification. You can then request removal of unstructured terminal residues from the receptor, the ligand, or both.CRITICAL STEP This mode can be combined with other advanced options. However, editing the PDB files manually provides greater flexibility, e.g., the potential for removing flexible loops with uncertain structure that would interfere with docking.
-
- Selecting Attraction and Repulsion TIMING ~30 min
- If a priori experimental information indicates that certain residues are in the binding interface, you can influence the results of the docking by setting attraction on these residues. Alternatively, if some information indicates that certain residues remain surface accessible upon binding, you can influence the results of the docking by setting repulsion on these residues. Click on Advanced Options and select Attraction and Repulsion. You will then see the entry fields for attraction and repulsion.
- Attracting and repulsing residues can be selected by typing whitespace separated “chain-residue” entries, e.g., a-23 a-25 a-26 a-27, in the appropriate boxes.
-
(Optional) Repulsing residues can also be selected by uploading a masking file that includes residues you do not want to be in the interface. Click on the Use PDB Masking File option, and select Browse to upload a pre-constructed masking file. To generate a PDB masking file, we recommend using PyMOL. Open in PyMOL the PDB file of the protein to be docked, and click on the button marked with “S” in the lower right corner to see the sequence of the protein. You can then select the residues to avoid in the interface by clicking on their one-letter amino acid code in the sequence. This will create a selection object called “sele” on the right side of your screen that holds these residues. You can also see the selection by what residues have dots placed on them in the viewer. You can then choose File->Save Molecule from the PyMOL menu. This will give you a window where you choose to save “sele”. You can then upload this PDB file as your masking file.CRITICAL STEP This mode can be combined with other advanced options.
- Selecting Restraints TIMING ~30 to 45 min
- Pairwise distance restraints may be available, e.g., from NMR Nuclear Overhauser Effect (NOE) experiments or from chemical crosslinking. To account for such restraints in docking, click on Advanced Options and select Restraints.
-
To use this option you need to prepare a restraint file. There are two options for restraint file format: either the AIR (Ambiguous Interaction Restraints) format, which is also used by the HADDOCK program, or our JSON format. To create a restraint file in AIR format, you can download a sample AIR type restraint file from https://cluspro.org/examples/ja026939x_s2.txt. To provide restraints as a JSON file, you can download a sample JSON type restraint file from https://cluspro.org/examples/ja026939x_s2.json. We have developed a web based restraint set generator to help the preparation of the restraint file in JSON format (see Fig. 3), available at https://cluspro.org/generate_restraints.html.CRITICAL STEP Restraints can either be a JSON file with a .json suffix, or an AIR restraint file from HADDOCK with any other suffix.
- Choose the restraint file.
- Selecting Others Mode TIMING ~5 min
- The use of Others Mode is suggested for complexes that may have less perfect shape and electrostatic complementarity than the enzyme-inhibitor complexes. Examples are the “other” type complexes in the protein-protein docking benchmark set22–25.Click on Advanced Options and select Others Mode. As described, this mode uses three different sets of energy weight coefficients and selects 500 low energy structures from each. The 1500 conformations are clustered together to select the most populated clusters.
- Selecting Antibody Mode TIMING ~10 to 45 min
- Using the Antibody Mode is suggested for antibody-antigen docking. Click on Advanced Options, and select Antibody Mode. Click on the label Use Antibody Mode. As described, this mode will use a scoring function specifically developed for antibody-antigen complexes.
- (Optional) Avoiding non-CDR (non-complementarity determining region) residues in the interface may improve the docking result. There are two options for specifying CDR-residues: either automatic or manual selection. For automatic masking of non-CDR residues, click on the label “Automatically Mask Non-CDR Regions”. For manual masking of non-CDR residues using the Attraction and Repulsion option, you need to click on the label Attraction and Repulsion. You will then see the entry fields for attraction and repulsion. Repulsing residues can be selected by typing them as whitespace separated “chain-residue” entries as shown in Step 8B(ii). Alternatively, repulsing residues can be selected by uploading a masking file generated as described in Step 8B(iii). In both cases it is necessary to identify the CDRs of the specific antibody for selecting non-CDR residues as repulsive or listed in a masking file. To find CDRs, you can consult the website http://www.bioinf.org.uk/abs/#cdrid, or use the annotation tools in the abYsis server at http://www.abysis.org. On the home page of the abYsis server, select Sequence Input Key Annotation, enter the sequence of either the heavy or the light chain of the antibody in FASTA format, and click Submit. Click on the label Numbering and Regions. The resulting page provides three different annotations of CDR regions (Chothia, ABM, and Kabat). Select the amino acid residues that are reliably not part of any CDR, and generate the masking file as described in Step 8B(ii).
- Selecting Multimer Docking TIMING ~5 min
-
Multimer docking is special in that the receptor and ligand are the same molecule. Therefore you only need to fill in the information for your molecule in the receptor field. Similarly, if you are specifying information such as attraction and repulsion, you only need to specify this information for the receptor. Click on Advanced Options and select Multimer Docking. You can then enter either 2 or 3 subunits for either a dimer or a trimer.CRITICAL STEP The current multimer mode in ClusPro supports only dimers and trimers.
-
- Selecting SAXS Profile TIMING ~10 to 30 min
- If you have SAXS data, you can submit it to ClusPro for use during the docking process. The server will fit the theoretical profiles generated from the docked structures against the uploaded SAXS profile using the chi scores to filter the structures before clustering. To use SAXS data click on Advanced Options and select SAXS Profile.
- To use this option you need to prepare a file containing the SAXS profile. The file should contain the SAXS results in a 3-column format. The first column should be the angle (q) in units of 1/Å. The second column should be the scattering intensity (I). The third column is optional, and should be the experimental error if it is available. You can download a zip file with sample inputs from https://cluspro.org/examples/saxs.zip. The zip file contains two pdb files, rec.pdb and lig.pdb, as well as a sample SAXS profile file, SalGEc_new_autoRg.dat.
- Choose the file containing the SAXS profile.
- Selecting Heparin Ligand TIMING ~5 min
- Click on the labels Advanced Option and Heparin Ligand, and then select Use Heparin as Ligand.
FIGURE 3.
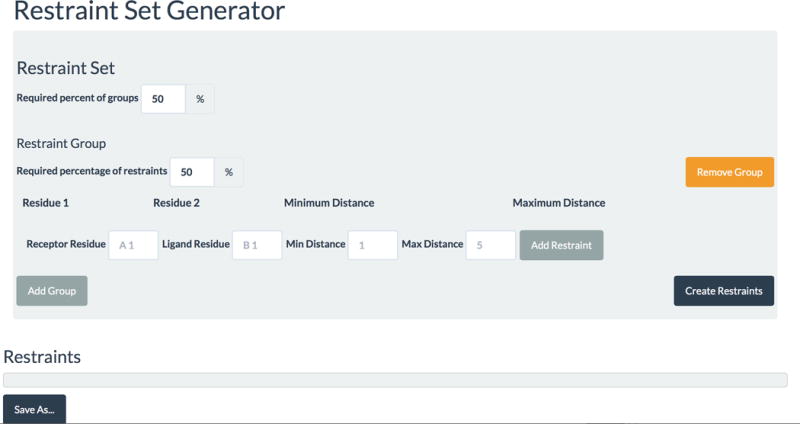
Screen image of the restraint file generator web tool to prepare a JSON file. The generator is available at the URL https://cluspro.org/generate_restraints.html.
Running ClusPro TIMING ~ 1 to 8 hours
9| Click on the DOCK button to begin the docking calculation. The status of the job can be immediately checked on the Queue page, which also shows the jobs that are ahead of yours. The jobs will run in order of their submission. Each job is listed with ID number, job name, user name, and a status update. To see a detailed Status page, click on the ID of your job. The Status page shows the job ID number, job name, job status, and submission time. See Box 1 for the list of status abbreviations. The Status page also shows small pictorial representations of the uploaded and processed input structures (Fig. 4).
-
10| If requested, an email will be sent when the job has completed or if an error occurred (see Box 2 for a listing of possible errors and their meanings). The email provides a link to the Results page or to an error message. Click the link to get to the Results page. Alternatively, locate the results under the Results tab on the server. All user results will be listed in order of ID number.
CRITICAL STEP We will store the result files on the server for at least 2 months. After this time, the results may be deleted.
? TROUBLESHOOTING
BOX 1. STATUS UPDATES FOR CLUSPRO RUNS.
The progress of the ClusPro run can be monitored in the “Queue” tab. Note that since the timings for each step are highly dependent on the input structure, they are not provided here.
Processing pdb files
Downloading PDB file from the www.pdb.org web site, processing chain information, extracting the chains the user specified
Pre-docking minimization
Running CHARMM to add missing atoms and polar hydrogens, minimizing the added atoms in the presence of the protein
Copying to supercomputer
Copying the PDB files to the cluster where ClusPro will run
Held on supercomputer
Files are on cluster, but job is not yet submitted
In queue on supercomputer
Job has been submitted on the cluster, but has not started running
Running on supercomputer
Job has begun running on the cluster
Clustering and minimization
Clustering top structures, selecting representative from top ranked clusters for minimization. Cluster are ranked by cluster size
Finalizing job
Compresses files on server for transfer
Copying to local computer
Results are being copied from the cluster back to the ClusPro server
Finished
Everything is complete
Computing Saxs profiles
Preliminary to docking (Saxs Profile mode only)
Error on local system
Processing PDB files fails. Check error messages
Error on supercomputer
Job fails to run on the cluster. Check error messages
FIGURE 4.
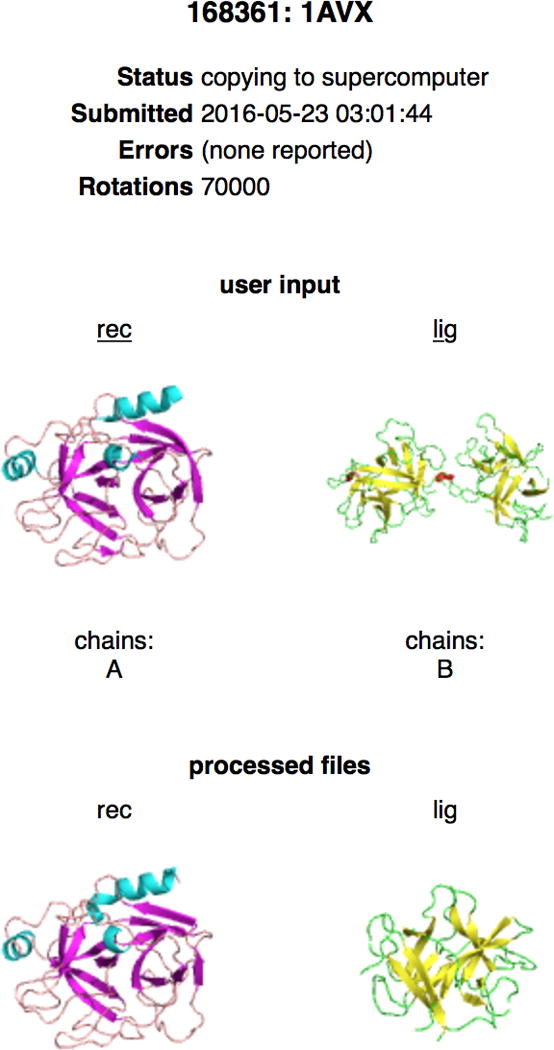
Screen image of the ClusPro Status page. The screen was obtained by clicking on the job Id on the queue page, for docking of soybean trypsin inhibitor (ligand) to the porcine trypsin (receptor). The Status page shows the job ID number, job name, job status, and submission time stamp. The page also shows pictorial representations of the uploaded and processed input structures The PDB file for the ligand includes two chains, but only chain B is used for docking. Clicking on the labels rec and lig will download the submitted coordinate files as read by the server.
BOX 2. ERROR MESSAGES AND THEIR MEANINGS.
When the calculation encounters an error, the job will be terminated and the user will receive an email with an error reason. The error will also be listed next to the corresponding job id in the “Results” tab. The error codes and explanations are listed below.
xxxx not found in PDB
xxxx is the four letter PDB ID. This error occurs when the computer is unable to download the entered PDB ID from the website, www.pdb.org. Usually this error occurs when the PDB ID does not exist, but it can also occur when the PDB website is down.
Unknown residue xxx in receptor. Please remove
xxx is the three letter amino acid code. This error occurs when a residue in an atom record is not recognizable by ClusPro and thus ClusPro does not have parameters for it. Check to make sure that the proper three letter code is used and the amino acid is one of the 20 naturally occurring amino acids.
Unknown residue xxx in ligand. Please remove
xxx is the three letter amino acid code. This error occurs when a residue in an atom record is not recognizable by ClusPro and thus ClusPro does not have parameters for it. Check to make sure that the proper three letter code is used and the amino acid is one of the 20 naturally occurring amino acids.
Receptor chains must be fewer than 20 characters
Chain specification is incorrect.
Receptor chains must be white space separated alphanumeric characters
Incorrect or missing receptor chain specification.
Receptor PDB id must be 4 alphanumeric characters
Invalid PDB id.
Receptor file too large
PDB file exceeds maximum allowed size. May happen if the protein has multiple domains. Some domains can be docked separately.
Receptor file only partially uploaded
PDB file is either too large or a network error has occurred during upload.
Ligand chains must be fewer than 20 characters
Chain specification is incorrect.
Ligand chains must be white space separated alphanumeric characters
Incorrect or missing ligand chain specification.
Ligand PDB id must be 4 alphanumeric characters
Invalid PDB id.
Ligand file too large
PDB file exceeds maximum allowed size. May happen if the protein has multiple domains. Some domains can be docked separately.
Ligand file only partially uploaded
PDB file is either too large or a network error has occurred during upload.
Copy of receptor failed
File did not transfer to computing cluster. Check input for consistency.
Copy of ligand failed
File did not transfer to computing cluster. Check input for consistency.
Processing failed on receptor
This error occurs during the initial steps when Charmm is used to add missing hydrogen atoms and minimize the structure. Usually, this occurs when the protein structure has sterically clashing atoms or the structure generally does not make physical sense in terms of bonds.
Processing failed on ligand
This error occurs during the initial steps when Charmm is used to add missing hydrogen atoms and minimize the structure. Usually, this occurs when the protein structure has sterically clashing atoms or the structure generally does not make physical sense in terms of bonds.
Not enough lines in output for xxx
Restraints are wrong or cannot be satisfied.
Job ran out of memory on server
Receptor and/or ligand are too large.
Ligand repulsion file too large
If the protein is large, the repulsion file is even larger (Attraction and Repulsion mode only).
Ligand attraction/repulsion file only partially uploaded. Please try again
A network error may have occurred during upload. (Attraction and Repulsion mode only).
Ligand attraction/repulsion must be whitespace separated chain-residue
Incorrect format (Attraction and Repulsion mode only).
Receptor repulsion file too large
(Attraction and Repulsion mode only).
Receptor attraction/repulsion file only partially uploaded. Please try again
A network error may have occurred during upload. (Attraction and Repulsion mode only).
Receptor attraction/repulsion must be whitespace separated chain-residue
Incorrect format (Attraction and Repulsion mode only).
Number of multimers must be expressed as a number
Format error (Multimer mode only).
Number of multimers must be an integer
Format error (Multimer mode only).
Number of multimers must be 2 or greater
Format error (Multimer mode only).
Copy of heparin failed
(Heparin Ligand mode only). Restart job.
‘SAXS’ profile only partially uploaded. Please try again
File is either too large or a network error has occurred during upload (Saxs Profile mode only).
Restraints file too large
If the protein is large, the restraints file is even larger (Restraints mode only).
Restraints file only partially uploaded. Please try again
File is either too large or a network error has occurred during upload (Restraints mode only).
Error converting AIR restraints to JSON. Please consult the help page
Submitted restraints may not be formatted properly. A sample AIR type restraint file can be downloaded from the help page (Restraints mode only).
Restraints file has incorrect format. Please consult the help page
Restraints files must be in either AIR format or in JSON format. See the help page to use tool for generating restraints in JSON format and for sample AIR and JSON formatting (Restraints mode only).
Restraints file refers to unknown residues
Check residue names (Restraints mode only).
Analyzing results from ClusPro TIMING ~ 30–45 min
-
11| View the results by clicking on the ID number of the job under the Results tab. On the Result page you can view and download the results of your docking. The page (see Fig. 5) starts with the job name. We recall that a single job actually performs 4 docking calculations with 4 different sets of energy parameters. By default the page shows results for the Balanced set (Table 1). Results for electrostatic-favored, hydrophobic-favored, or van der Waals + electrostatics sets can be viewed by clicking on the corresponding label. For each parameter set the Result page shows small pictures representing the top 10 models, but the user can request more models (up to 30 or less if fewer than 30 clusters have been created). Clicking on the number above a picture will download that model as a PDB file for your viewing. You can also download all displayed models or all models for all coefficients (see Step 13). Optionally, you can click on the label Job details at the top of the Results page opens the Status page that was available from the Queue page while the job was running. The Status page now shows the job ID number, job name, job status, submission time stamp, and the error message if there was an error. The page also shows pictorial representations of the uploaded and processed input structures.
CRITICAL STEP In the Heparin Ligand Mode, the Results page also shows a small picture of the target protein in surface representation with the putative heparin binding site indicated in atomic charge colors (blue for N, red for O, and white for C), whereas the non-contact region is dark. In addition, the page has the label Atom Contacts. Clicking on the label downloads the file atom_contacts.csv, which lists the contact atom pairs on the two sides of the heparin-protein interface.
-
12| View model scores by clicking on the corresponding label on the results page. Depending on the selected set of energy parameters, the page shows the actual weighting coefficients of the PIPER energy terms, and a table that lists the requested number of clusters of docked structures in the order of cluster size (Fig. 6). For each cluster, the table shows the size (i.e., the number of docked structures), the PIPER energy of the cluster center (i.e., the structure that has the highest number of neighbor structures in the cluster), and the energy of the lowest energy structure in the cluster.
CRITICAL STEP Although we show energy values, it is emphasized that model selection is based on cluster size rather than on energy. In fact, the energy calculated by PIPER does not directly relate to binding affinity. However, low energy regions tend to generate large clusters of docked structures, and the size of a cluster is approximately proportional to its probability, and thus the energy landscape indirectly determines the most likely conformation of the complex.
CRITICAL STEP In the Other Mode, the table shows the size for each cluster (i.e., the number of docked structures). No energy values are shown, as the clusters may include structures obtained using different energy functions.
-
13| (optional) From the Results page, download PDB files of the displayed models, or also the models for all energy coefficients. This latter option downloads a large .tar file that includes 4 times 30 PDB files (or fewer if for some of the energy coefficients ClusPro generates fewer than 30 clusters). Each PDB file includes a structure at the center of a cluster. The naming conventions are as follows: files model.000.00.pdb through model.000.29.pdb include the structures generated using the balanced parameter set; files model.002.00.pdb through model.002.29.pdb are from the electrostatics favored calculations; files model.004.00.pdb through model.004.29 are hydrophobicity-favored; and finally, files model.006.00.pdb through model.006.29 have the structures obtained using the parameters we have defined as van der Waals + electrostatics. Since the models generated by the four different energy parameters have different names, structures selected from the four sets can also be opened simultaneously in PyMol. To do this, click on the firs selected model to open it in PyMOL, and use the PyMOL command “load filename” to add further structures.
CRITICAL STEP In the Others Mode, the PDB files are named model.003.00.pdb through model.003.29.pdb.
CRITICAL STEP In the Antibody Mode, the PDB files are named model.000.00.pdb through model.000.29.pdb.
-
14| Use the appropriate software to visualize the protein structure files. PyMOL provides a convenient tool for the visualization of the structures generated by ClusPro. When you open a downloaded structure, e.g., model.000.00.pdb in PyMOL, it actually creates two structures named rec.pdb for the receptor and lig.000.00.pdb for the ligand. You can add further models using the PyMOL command load fname, where fname is the name of the file to be loaded. In these cases only a new ligand is opened, e.g., load model.000.01.pdb will create the structure lig.000.01.pdb, as the receptor is named rec.pdb in all result files. In test cases you can also load the X-ray structure of the complex (Fig. 7). Further examples of visualization by PyMOL are shown in the Anticipated Results section.
? TROUBLESHOOTING
FIGURE 5.

Screen image of the ClusPro Results page for docking of soybean trypsin inhibitor (ligand) to the porcine trypsin (receptor). The page shows the job name and, by default, results for the balanced set of scoring function coefficients. Results for electrostatic-favored, hydrophobic-favored, or van der Waals + electrostatics sets can be viewed by clicking the corresponding labels. For each parameter set the result page shows small pictures representing the top 10 models, but the user can display more models (up to 30 or less if fewer than 30 clusters have been created). Clicking on the number above a picture will download that model as a PDB file for your viewing. The page also provides the label to download models for all coefficients.
FIGURE 6.
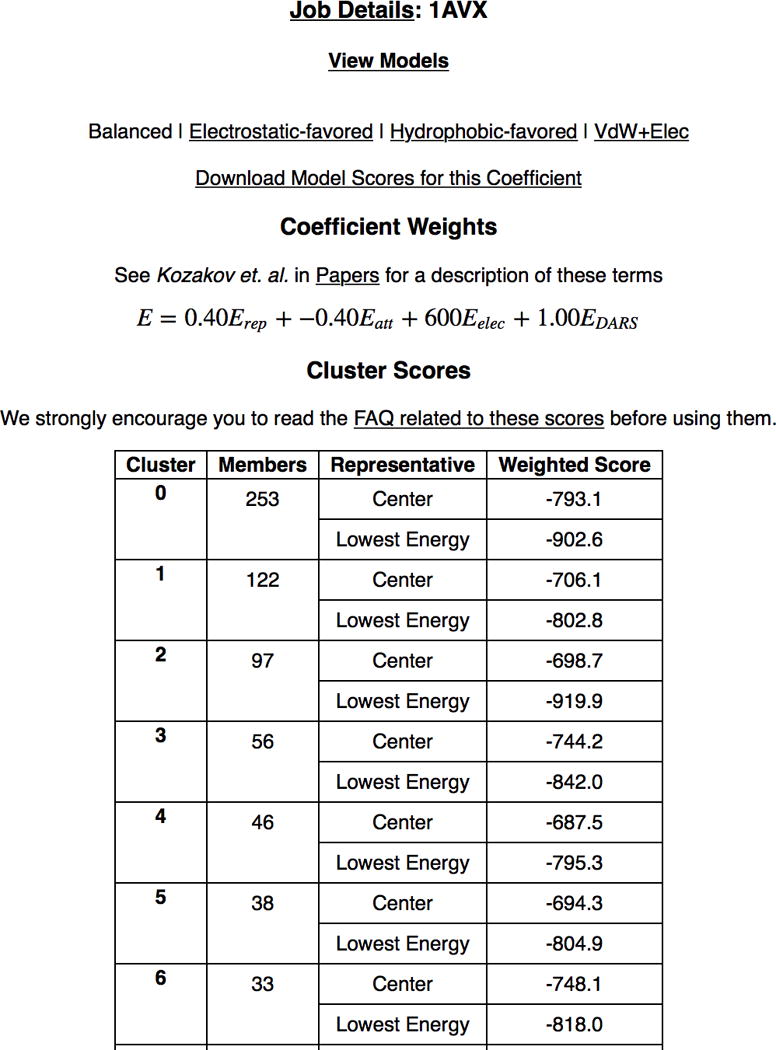
Screen image of the ClusPro Results page showing model scores for the balanced coefficient set when docking soybean trypsin inhibitor to porcine trypsin. The page shows the actual weighting coefficients of the energy terms, and a table that lists the clusters of docked structures in the order of cluster size. For each cluster, the table shows the size (i.e., the number of docked structures), the energy of the cluster center (i.e., the structure that has the highest number of neighbor structures in the cluster), and the energy of the lowest energy structure in the cluster.
FIGURE 7.
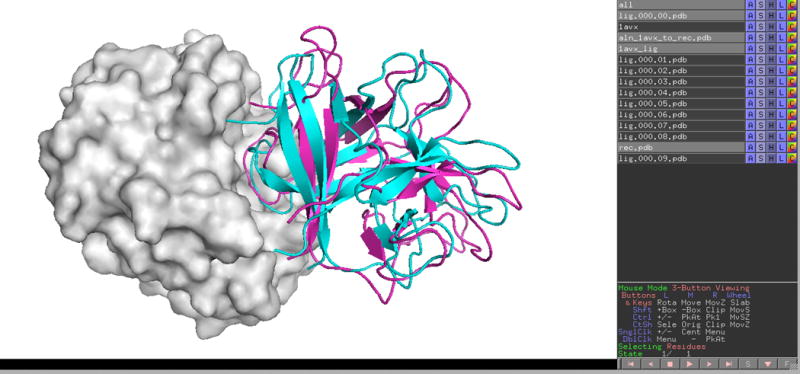
Visualization by PyMol of the structure at the center of the most populated cluster in docking soybean trypsin inhibitor (ligand) to porcine trypsin (receptor). The docked ligand structure (lig.000.00.pdb) is shown as cyan cartoon, whereas the receptor is shown as grey surface. For comparison we also loaded the X-ray structure of the enzyme-inhibitor complex (PDB ID 1AVX) into PyMol, aligned it with the structure of the receptor, and show the native pose of the inhibitor as magenta cartoon.
TIMING
Steps 1–7, inputting the coordinates of the component proteins: ~ 1 to 2 min
Step 8 (A), selecting Structure Modification: ~ 5 min
Step 8 (B), selecting Attraction and Repulsion: ~ 30 min
Step 8 (C), selecting Restraints: ~30 to 45 min
Step 8 (D), selecting Others Mode: ~5 min
Step 8 (E), selecting Antibody Mode: ~10 to 45 min
Step 8 (F), selecting Multimer Docking: ~ 5 min
Step 8 (G), selecting SAXS Profile: ~ 10 to 30 min
Step 8 (H), selecting Heparin Ligand: ~ 5 min
Steps 9–10, running ClusPro: ~ 1 to 8 hours; depends on the sizes of the proteins and the number of jobs in the queue. On average, results are returned in less than 4 hours.
Steps 11–14, analysis of ClusPro results for selecting putative models of the complex, considering result obtained by four different scoring functions: ~ 30–45 min
TROUBLESHOOTING
Troubleshooting advice can be found in Table 5.
TABLE 5.
Troubleshooting table.
| Step | Problem | Possible reason | Possible Solution |
|---|---|---|---|
| 4 | I uploaded a structure from my computer, but as soon as I submitted the job failed with the message: “Processing failed on receptor.” What is wrong? | Inconsistency with the PDB format. Note that homology modeling tools frequently fail to produce correct PDB format, e.g., by not placing a chain identifier. | Make sure that your file is in PDB format and has the required spacing between columns. Documentation with respect to the PDB file format can be found at: http://www.wwpdb.org/docs.html. Also, make sure that all ATOM records the PDB file contain only standard amino or nucleic acids. |
| I entered a HETATM record to include in the calculation, but it was not recognized. What do I do? | At this point there is no option for considering heteroatoms (HETATM entries). | Remove the HETATM records. | |
| 6 | I uploaded a structure from my computer, but as soon as I submitted the job failed with the message: “Processing failed on ligand.” What is wrong? | Inconsistency with the PDB format. Note that homology modeling tools frequently fail to produce correct PDB format, e.g., by not placing a chain identifier. | See TROUBLESHOOTING for Step 4. |
| 10 | My job keeps crashing even though my input file looks fine. | FFT grids are too large. This is typically caused by uploading a file output by homology modeling that has extremely large regions without a template, resulting in a tail that basically floats off into space. | Use the Structure Modification Option to remove the questionable parts of the model. |
| 14 | I cannot load the docking results into my molecular viewer. | Your viewer likely does not support multiple structures in one PDB file. | Try using PyMol, available at www.pymol.org |
ANTICIPATED RESULTS
Docking an enzyme-inhibitor pair
The first enzyme-inhibitor target in the protein-protein docking benchmark25 is docking the x-ray structure of soybean trypsin inhibitor (PDB ID 1BA7) to the X-ray structure of porcine trypsin (PDB ID 1QQU). As shown in Fig. 2, we select the enzyme as the receptor. Since the only chain in the file is A, the Chain ID can be provided or omitted. The PDB file 1BA7 has two copies of the inhibitor structure, of which chain B is slightly more complete (169 rather than 165 residues) and hence it is used as the ligand, providing B as the Chain ID. For both proteins we import the coordinates from the PDB. As soon as the job shows up on the Queue page, we can check its status. The Status page shows the job ID number, job name, job status, submission time stamp, and PDB ID. The page also shows that both files have been processed by the server, and only one of the chains was used for the ligand (Fig. 3). Once the job is completed, by default the Results page (Fig. 4) shows pictures of the 10 models (i. e., the centers of the 10 most populated clusters). Clicking on the label View Model Scores opens a page that shows the weighting coefficients of the PIPER energy terms and a table of cluster scores (Fig. 5). For each cluster, this table shows the size (i.e., the number of members), the weighted energy score of the cluster center (i.e., the structure that has the highest number of neighbor structures in the cluster), and the energy score of the lowest energy structure in the cluster. Note that the top cluster, with 253 members, is substantially larger than the second largest cluster (122 members), indicating a well-defined set of encounter complexes. Such outcome generally provides some level of assurance that the clustering of docked structures occurs in the neighborhood of the native state. We suggest downloading selected models for visualization in PyMOL. Fig. 6 shows the receptor, porcine trypsin, as grey surface. The ligand at the center of the largest cluster (lig.000.00.pdb) is shown as cyan cartoon. We also loaded the X-ray structure of the enzyme-inhibitor complex (PDB ID 1AVX) into PyMol, aligned it with the structure of the receptor, and show the native pose of the inhibitor as magenta cartoon in Fig. 6. The agreement is good, resulting in the Cα IRMSD of 3.3 Å. This is a typical outcome for many enzyme-inhibitor complexes, because both proteins are fairly rigid, the conformational change upon association is small, and the two molecules have excellent shape complementarity. Although the results are shown for the balanced set of energy coefficients, the top model remains the same for the electrostatic favored energy expression, with an even larger difference in the populations of the largest and second largest clusters, with 268 and 100 members, respectively. The stability of the model upon variation in the energy parameter further increases confidence in its correctness.
Predicting an “others” type complex
Docking the component proteins of “other” type of complexes is the same as the basic ClusPro run, apart from selecting Others Mode in Advanced Options. As an example, we dock the X-ray structure of the cytoplasmic domain of the unphosphorylated type 1 TGF-beta receptor (PDB ID 1IAS, Chain A, 342 residues), which actually was defined here as the ligand, to the X-ray structure of the FK506 binding protein (FKBP, PDB ID 1D6O, Chain A, 107 residues). Fig. 8 shows the PyMOL visualization of the results. As in the previous example, the receptor (FKBP in this case) is shown as grey surface, the ligand lig.003.00.pdb at center of the largest cluster represented by the cyan cartoon, and the ligand extracted and superimposed from the complex (PDB ID 1B6C) is shown in magenta. We note that the IRMSD between free and bound structures of the TGF-beta receptor is 1.9 Å, and the structural differences between free and bound states are clearly seen in Fig. 8. Nevertheless, docking of the unbound structures yields a large cluster with 267 members, whereas the second largest cluster has only 100 members, again suggesting a stable native complex, occupying a well-defined energy well on the energy landscape. Indeed, the interface is predicted well, resulting in the IRMSD value of 2.96 Å, in spite of the overall conformational change in the ligand.
FIGURE 8.
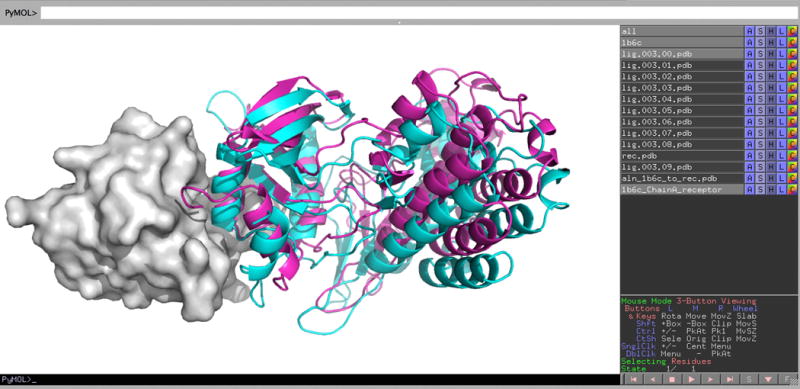
Screen image of exploring the results of running ClusPro in Others Mode. Results are shown using PyMOL from docking the X-ray structure of the ligand, to the X-ray structure of the FK506 binding protein (FKBP). The receptor, FKBP, is shown as grey surface, and the ligand (lig.003.00.pdb) at center of the largest cluster is shown as cyan cartoon. For comparison we superimposed the native complex (PDB ID 1B6C) on the receptor in the docked structure, and the corresponding ligand pose is shown as magenta cartoon.
Antibody-antigen docking
We docked the X-ray structure of the extracellular domain of the human tissue factor (PDB ID 1TFH) to the unbound X-ray structure of the FAB domain of the inhibitory antibody 5G9 (PDB ID 1FGN). Both the heavy and light chains were used to represent the receptor. We first used the Antibody Mode with automatic masking of non-CDR regions, selected in Advanced Options. As shown in Fig. 9, we have an acceptable model, lig.000.05.pdb, with IRMSD of 4.7 Å from the ligand position in the native complex (PDB ID 1AHW). However, this model is the center of the 6th largest cluster with only 48 members, whereas the largest cluster has 118 members, but much larger IRMSD from the native. Thus, while docking produces a good model, without a priori information we would not have been able to identify it. We also prepared a sequence-specific masking file to place repulsion on the non-CDR residues as suggested in Step 8E(ii). The abYsis server provides three different annotations of CDR regions (Chothia, ABM, and Kabat). We put repulsion only on residues that were not considered to be in a CDR by any of these annotations. The manual masking substantially improved the results, and a model (lig.000.01.pdb) with the IRMSD of 4.8 Å moved to the 2nd most populated cluster with 73 members. The largest cluster, with 190 members, was still a false positive, emphasizing the uncertainty of docking results for antibody-antigen complexes. Nevertheless, the example also shows that manual preparation of a sequence-specific masking file can be useful.
FIGURE 9.
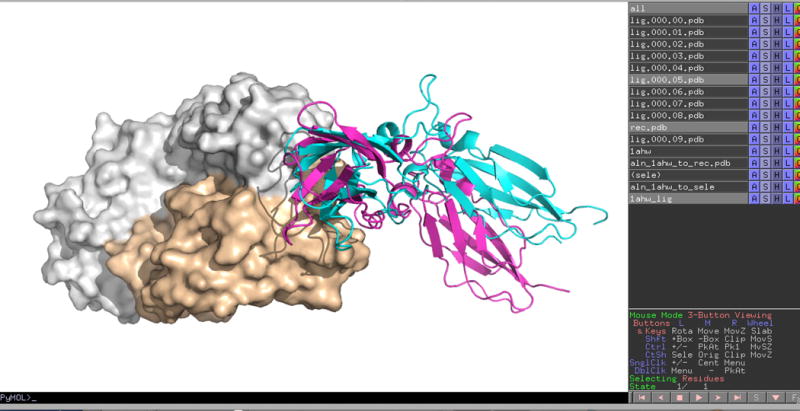
Screen image of the PyMOL visualization of the results of running ClusPro in Antibody Mode. We docked the X-ray structure of the extracellular domain of the human tissue factor (PDB ID 1TFH) to the unbound X-ray structure of the FAB domain of the inhibitory antibody 5G9 (PDB ID 1FGN). Both the heavy and light chains were used to represent the receptor. The center of the 6th most populated cluster, lig.000.05.pdb, is shown as cyan cartoon, whereas the antibody is shown in surface representation. The antigen in the native complex (PDB ID 1AHW) is shown as magenta cartoon. The IRMSD between native and predicted ligand poses is 4.7 Å.
Accounting for pairwise distance restraint
As an example we present constructing the complex from the X-ray structures of the Escherichia coli glucose-specific phosphocarrier protein IIAglc (E2A, PDB ID 1F3G, considered the receptor) and the signal transducing protein HPr (PDB ID 1POH, considered the ligand)117. ClusPro works very well for this problem even without any restraint, as the center of the second ranked cluster, has the IRMSD of 3.78 Å from the native state of the complex (PDB ID 1GGR). As shown in in Fig. 10A, this model (cyan cartoon) is oriented similarly to the native ligand (magenta cartoon), but its position is slightly shifted. However, this second cluster has only 221 members, whereas the top cluster has 424, but the ligand structures in this cluster are rotated relative to the native structure, resulting in the IRMSD of more than 20 Å. NOE (Nuclear Overhauser Effect) measurement were available for this complex117, resulting in 20 intermolecular distance restraints. Docking with these restraints was also performed by HADDOCK using a set of AIRs69 (https://cluspro.org/examples/ja026939x_s2.txt). We have prepared a JSON file using https://cluspro.org/generate_restraints.html, the on-line restrain set generator tool (see Fig. 3). The JSON file is shown at https://cluspro.org/examples/ja026939x_s2.json. Accounting for the restraints further improves the result, and the center of the largest cluster, with population 268, is shifted to 2.88 Å IRMSD. Indeed, as shown in Fig. 10B, the model (shown as blue cartoon) is now turned by a few degrees around an axis perpendicular to the middle of the receptor binding site, and this yields only small IRMSD. While accounting for the restraints improved the result, the identification of the best structure without additional information still would be difficult, as the second largest cluster has 235 members, thus it is almost as large as the top cluster.
FIGURE 10.
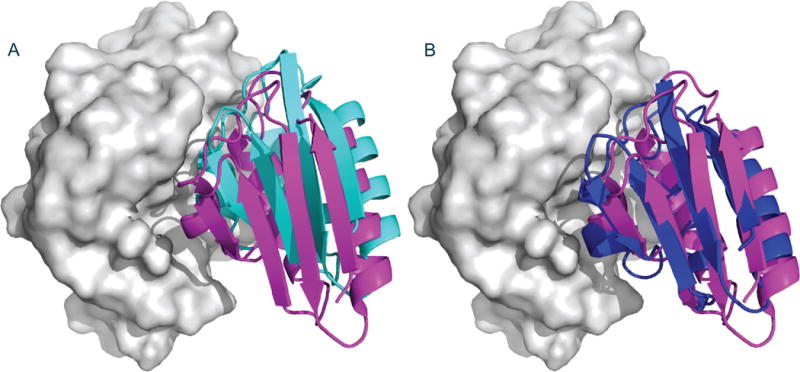
Best models from docking the signal transducing protein HPr (PDB ID 1POH) to the glucose-specific phosphocarrier protein E2A (PDB ID 1F3G) without and with restraints. The receptor protein, E2A, is shown as grey surface. A. Ligand position at the center of the second largest cluster (shown as cyan cartoon) from docking without restraints. The position is slightly shifted relative to the native ligand binding position, shown as magenta cartoon. B. Ligand position at the center of the largest cluster (shown as blue cartoon) from docking with restraints. The ligand is now turned a few degrees around an axis perpendicular to the center of the receptor.
Accounting for experimental SAXS data
Target 58 of the CAPRI docking challenge was determining the complex between the salmon cold-active goose-type lysozyme and the e. coli PliG lysozyme inhibitor using SAXS data as restraints118. A model of salmon lysozyme was built using the MODELLER v9.0119 program using the template of black swan goose lysozyme (PDB ID 1GBS, 57.8% sequence identity). Aromatic residues (Tyr, Phe, and Trp) that were not present in the template were placed in the most probable non-clashing rotamer positions. Other side chains that were not present in the template were not modeled since they have uncertain localization. The input files for this target are given at https://cluspro.org/examples/saxs.zip. The zip file contains two PDB files, rec.pdb, which provides coordinates of the model for the receptor, the salmon goose-type lysozyme, and lig.pdb, the e. coli PliG lysozyme inhibitor (PDB ID 4DY3). The zip file also includes the relevant SAXS profile file, SalGEc_new_autoRg.dat. Details of the SAXS profile were described in our report on extending ClusPro to accounting for SAXS data58.
ClusPro provided a near-native model even without the use of the SAXS restraints, but it was the center of the 6th largest cluster. Essentially the same near-native model was obtained when accounting for the SAXS data (see cyan cartoon in Fig. 11), but now as the center of the 3rd largest cluster. The conformation of the ligand, the e. coli PliG lysozyme inhibitor, is shown as magenta cartoon for reference (Fig. 11). The Others Mode can be combined with the SAXS Profile Mode, and this was used for the calculation.
FIGURE 11.
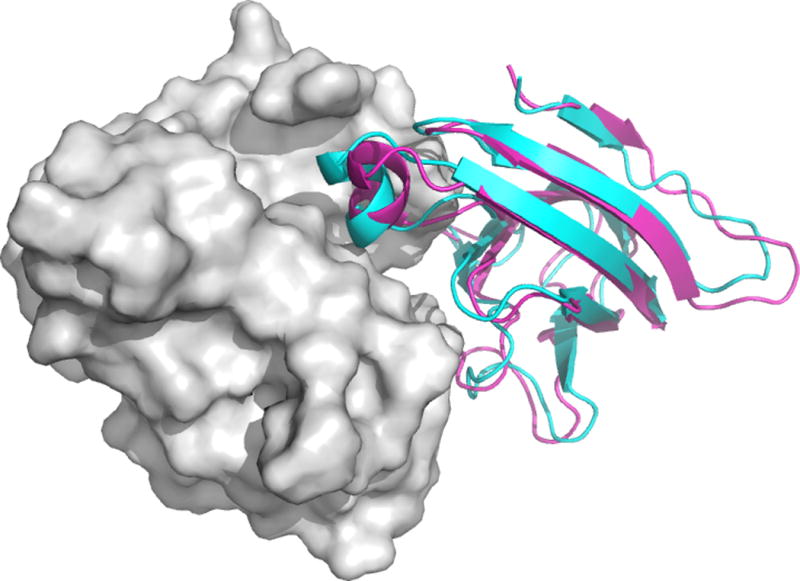
Docking the E. coli PliG lysozyme inhibitor to the salmon goose-type lysozyme using SAXS data as restraints. A near-native model (shown cyan cartoon) was obtained as the center of the 3rd largest cluster. The X-ray conformation of the inhibitor is shown as magenta cartoon.
Constructing a dimer in Multimer Mode
As an example of multimer modeling, we constructed the dimer of the sugar aminotransferase AtmS13 from Actinomadura melliaura. Since no monomeric structure of the protein was available, we have first constructed a homology model. The sequence of AtmS13 from Actinomadura melliaura (Uniprot entry Q0H2X1) was downloaded from the Uniprot website at http://www.uniprot.org in FASTA format. To build a homology model using the HHpred server http://toolkit.tuebingen.mpg.de/hhpred, the amino acid sequence was pasted into the box provided on the HHpred home page, and submitted for template selection and alignment using the pdb70 database for hidden Markov model (HMM) construction. All other parameters were left at their default values. HHPred returned a large number of potential template sequences aligned to the AtmS13 sequence, with the sugar aminotransferase CalS13 from Micromonospora echinospora (PDB ID 4ZAS) at the top of the list with the highest similarity score (63% sequence identity). The next step was clicking on the label Create model, which provided the options of either creating a model from the manually selected templates or automatically selecting the best templates. We have used the first option. Notice that building a homology model by HHpred requires the user to have a license to the MODELLER program119. The license is freely available for academic users and easily obtainable at http://salilab.org/modeller/registration.shtml. Entering the coordinates of the homology model constructed as the receptor, we used the Multimer Docking option of ClusPro with the number of subunits set to 2 and left all the other options at their default values. Once the docking was completed, the resulting models were compared to the available crystal structure of AtmS13 homodimer (PDB ID 4XAU, chains A and B). In agreement with our prior observation for dimers, the balanced coefficient provided the best ranking of near-native models. In particular, a near native docking model with IRMSD of 2.62 Å was ranked second (Fig. 12).
FIGURE 12.
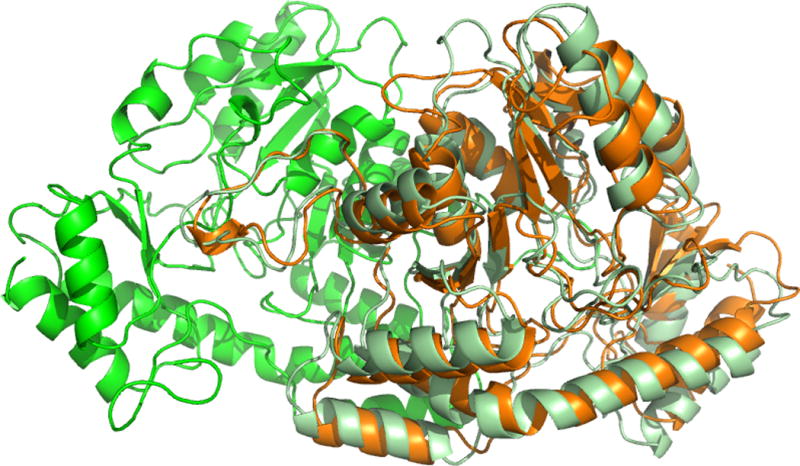
Constructing the dimer of the sugar aminotransferase AtmS13 from Actinomadura melliaura by homology modeling and multimer docking. Chains A and B of the target dimer are shown in green and light green, respectively. Chain B, predicted from the homology model of chain A with the IRMSD of 2.62 Å, is shown as orange cartoon.
Finding heparin binding sites
As an example, we docked the heparin tetramer probe to the ligand-free structure of the basic fibroblast growth factor (PDB ID 1BFG) that has also been co-crystallized with a heparin hexamer (PDB ID 1BFC). The docking and clustering yield eight clusters, with the two largest clusters having very similar populations, 387 and 378, respectively. Fig. 13 shows the center of the second largest cluster (cyan sticks), since some of the sulfate groups interacting with the protein have better overlap with the bound heparin (magenta sticks) than the sulfate groups in the top model, which is reoriented tail-to-head relative to the models shown. However, all eight models interact with the relatively deep pocket in the heparin binding site. Since we dock a generic heparin structure rather than the specific ligand, it is no surprise that the results have somewhat limited accuracy, but generally identify the most likely region of heparin binding.
FIGURE 13.
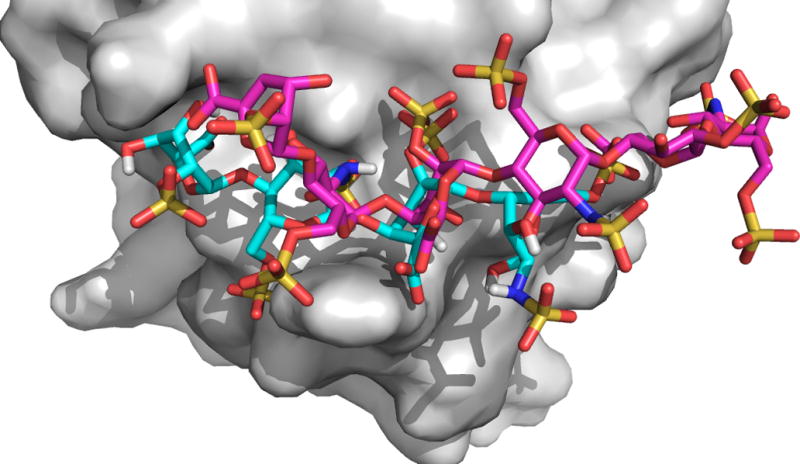
Docking of the heparin tetramer probe to the ligand-free structure of the basic fibroblast growth factor. The center of the second largest cluster is shown as cyan sticks. The X-ray structure of the bound hexamer shown in magenta.
Supplementary Material
TABLE S1 | Performance of ClusPro on Docking Benchmark 4.025
EDITORIAL SUMMARY.
ClusPro is a web server that performs rigid body docking of two proteins by sampling billions of conformations. Low energy docked structures are clustered, and centers of the largest clusters are used as likely models of the complex.
Acknowledgments
This investigation was supported by grants R35 GM118078 and R01 GM061867 from the National Institute of General Medical Sciences, and grants DBI 1147082, DBI 1458509, and AF 1527292 from the National Science Foundation.
Footnotes
Author contributions statements
D.K., D.R.H., B.X., and D.B. developed the server; D.K., D.R.H., D.P., B.X., and K.P. performed experiments; S.V., K.P, D.R.H., and C.Y. prepared the manuscript.
Competing financial interests
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/nprot/index.html.
TWEET: The ClusPro web server predicts models of the complex based on the structures of two interacting proteins.
References
- 1.Ito T, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 3.Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 4.Ewing RM, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith GR, Sternberg MJ. Prediction of protein-protein interactions by docking methods. Curr Opin Struct Biol. 2002;12:28–35. doi: 10.1016/s0959-440x(02)00285-3. [DOI] [PubMed] [Google Scholar]
- 6.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie DW. Recent progress and future directions in protein-protein docking. Curr Protein Pept Sci. 2008;9:1–15. doi: 10.2174/138920308783565741. [DOI] [PubMed] [Google Scholar]
- 8.Vajda S, Kozakov D. Convergence and combination of methods in protein-protein docking. Curr Opin Struct Biol. 2009;19:164–170. doi: 10.1016/j.sbi.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aloy P, Ceulemans H, Stark A, Russell RB. The relationship between sequence and interaction divergence in proteins. J Mol Biol. 2003;332:989–998. doi: 10.1016/j.jmb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Sinha R, Kundrotas PJ, Vakser IA. Protein docking by the interface structure similarity: how much structure is needed? PLoS One. 2012;7:e31349. doi: 10.1371/journal.pone.0031349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 13.Comeau SR, et al. ClusPro: performance in CAPRI rounds 6–11 and the new server. Proteins. 2007;69:781–785. doi: 10.1002/prot.21795. [DOI] [PubMed] [Google Scholar]
- 14.Kozakov D, et al. Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins. 2010;78:3124–3130. doi: 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozakov D, et al. How good is automated protein docking? Proteins. 2013;81:2159–2166. doi: 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 17.Katchalski-Katzir E, et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci U S A. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabb HA, Jackson RM, Sternberg MJE. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 19.Mandell JG, et al. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001;14:105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 20.Chen R, Weng ZP. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins. 2002;47:281–294. doi: 10.1002/prot.10092. [DOI] [PubMed] [Google Scholar]
- 21.Kozakov D, Clodfelter KH, Vajda S, Camacho CJ. Optimal clustering for detecting near-native conformations in protein docking. Biophys J. 2005;89:867–875. doi: 10.1529/biophysj.104.058768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen R, Mintseris J, Janin J, Weng Z. A protein-protein docking benchmark. Proteins. 2003;52:88–91. doi: 10.1002/prot.10390. [DOI] [PubMed] [Google Scholar]
- 23.Mintseris J, et al. Protein-Protein Docking Benchmark 2.0: an update. Proteins. 2005;60:214–216. doi: 10.1002/prot.20560. [DOI] [PubMed] [Google Scholar]
- 24.Hwang H, Pierce B, Mintseris J, Janin J, Weng Z. Protein-protein docking benchmark version 3.0. Proteins. 2008;73:705–709. doi: 10.1002/prot.22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang H, Vreven T, Janin J, Weng Z. Protein-protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vajda S, Camacho CJ. Protein-protein docking: is the glass half-full or half-empty? Trends Biotechnol. 2004;22:110–116. doi: 10.1016/j.tibtech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Vajda S. Classification of protein complexes based on docking difficulty. Proteins. 2005;60:176–180. doi: 10.1002/prot.20554. [DOI] [PubMed] [Google Scholar]
- 28.Yershova A, Jain S, LaValle SM, Mitchell JC. Generating Uniform Incremental Grids on SO(3) Using the Hopf Fibration. Int J Robot Res. 2010;29:801–812. doi: 10.1177/0278364909352700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chuang GY, Kozakov D, Brenke R, Comeau SR, Vajda S. DARS (Decoys As the Reference State) potentials for protein-protein docking. Biophys J. 2008;95:4217–4227. doi: 10.1529/biophysj.108.135814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilson MK, Honig B. Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins. 1988;4:7–18. doi: 10.1002/prot.340040104. [DOI] [PubMed] [Google Scholar]
- 31.Brooks BR, et al. Charmm - a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 32.Lorenzen S, Zhang Y. Identification of near-native structures by clustering protein docking conformations. Proteins. 2007;68:187–194. doi: 10.1002/prot.21442. [DOI] [PubMed] [Google Scholar]
- 33.Shortle D, Simons KT, Baker D. Clustering of low-energy conformations near the native structures of small proteins. Proc Natl Acad Sci U S A. 1998;95:11158–11162. doi: 10.1073/pnas.95.19.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendez R, Leplae R, Lensink MF, Wodak SJ. Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures. Proteins. 2005;60:150–169. doi: 10.1002/prot.20551. [DOI] [PubMed] [Google Scholar]
- 35.Lensink MF, Mendez R, Wodak SJ. Docking and scoring protein complexes: CAPRI 3rd Edition. Proteins. 2007;69:704–718. doi: 10.1002/prot.21804. [DOI] [PubMed] [Google Scholar]
- 36.Lensink MF, Wodak SJ. Docking and scoring protein interactions: CAPRI 2009. Proteins. 2010;78:3073–3084. doi: 10.1002/prot.22818. [DOI] [PubMed] [Google Scholar]
- 37.Lensink MF, Wodak SJ. Docking, scoring, and affinity prediction in CAPRI. Proteins. 2013;81:2082–2095. doi: 10.1002/prot.24428. [DOI] [PubMed] [Google Scholar]
- 38.Lensink MF, et al. Prediction of homo- and hetero-protein complexes by protein docking and template-based modeling: a CASP-CAPRI experiment. Proteins. 2016 doi: 10.1002/prot.25007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamal JK, Benchaar SA, Takamoto K, Reisler E, Chance MR. Three-dimensional structure of cofilin bound to monomeric actin derived by structural mass spectrometry data. Proc Natl Acad Sci U S A. 2007;104:7910–7915. doi: 10.1073/pnas.0611283104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamal JK, Chance MR. Modeling of protein binary complexes using structural mass spectrometry data. Protein Sci. 2008;17:79–94. doi: 10.1110/ps.073071808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luxan G, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19:193–201. doi: 10.1038/nm.3046. [DOI] [PubMed] [Google Scholar]
- 42.Tran K, et al. Vaccine-elicited primate antibodies use a distinct approach to the HIV-1 primary receptor binding site informing vaccine redesign. Proc Natl Acad Sci U S A. 2014;111:E738–E747. doi: 10.1073/pnas.1319512111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwede T, et al. Outcome of a workshop on applications of protein models in biomedical research. Structure. 2009;17:151–159. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sondermann H, Nagar B, Bar-Sagi D, Kuriyan J. Computational docking and solution x-ray scattering predict a membrane-interacting role for the histone domain of the Ras activator son of sevenless. Proc Natl Acad Sci U S A. 2005;102:16632–16637. doi: 10.1073/pnas.0508315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourdon A, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 46.Cosconati S, Marinelli L, Lavecchia A, Novellino E. Characterizing the 1,4-dihydropyridines binding interactions in the L-type Ca2+ channel: Model construction and docking calculations. J Med Chem. 2007;50:1504–1513. doi: 10.1021/jm061245a. [DOI] [PubMed] [Google Scholar]
- 47.Rumpel S, Becker S, Zweckstetter M. High-resolution structure determination of the CylR2 homodimer using paramagnetic relaxation enhancement and structure-based prediction of molecular alignment. J Biomol NMR. 2008;40:1–13. doi: 10.1007/s10858-007-9204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kucuk C, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Comm. 2015;6:6025. doi: 10.1038/ncomms7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye Z, Musiol EM, Weber T, Williams GJ. Reprogramming acyl carrier protein interactions of an Acyl-CoA promiscuous trans-acyltransferase. Chem Biol. 2014;21:636–646. doi: 10.1016/j.chembiol.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steeland S, et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J Biol Chem. 2015;290:4022–4037. doi: 10.1074/jbc.M114.617787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bohnuud T, et al. A benchmark testing ground for integrating homology modeling and protein docking. Proteins. 2016 doi: 10.1002/prot.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 54.Brenke R, et al. Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics. 2012;28:2608–2614. doi: 10.1093/bioinformatics/bts493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Comeau SR, Camacho CJ. Predicting oligomeric assemblies: N-mers a primer. J Struct Biol. 2005;150:233–244. doi: 10.1016/j.jsb.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Pierce B, Tong W, Weng Z. M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics. 2005;21:1472–1478. doi: 10.1093/bioinformatics/bti229. [DOI] [PubMed] [Google Scholar]
- 57.Yang S. Methods for SAXS-based structure determination of biomolecular complexes. Adv Materials. 2014;26:7902–7910. doi: 10.1002/adma.201304475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia B, et al. Accounting for observed small angle X-ray scattering profile in the protein-protein docking server cluspro. J Comput Chem. 2015;36:1568–1572. doi: 10.1002/jcc.23952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pons C, et al. Structural characterization of protein-protein complexes by integrating computational docking with small-angle scattering data. J Mol Biol. 2010;403:217–230. doi: 10.1016/j.jmb.2010.08.029. [DOI] [PubMed] [Google Scholar]
- 60.Schneidman-Duhovny D, Hammel M, Sali A. Macromolecular docking restrained by a small angle X-ray scattering profile. J Struct Biol. 2011;173:461–471. doi: 10.1016/j.jsb.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernfield M, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 62.Lindahl U. Heparan sulfate-protein interactions - A concept for drug design? Thromb Haemostasis. 2007;98:109–115. [PubMed] [Google Scholar]
- 63.Fugedi P. The potential of the molecular diversity of heparin and heparan sulfate for drug development. Mini Rev Med Chem. 2003;3:659–667. doi: 10.2174/1389557033487755. [DOI] [PubMed] [Google Scholar]
- 64.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forster M, Mulloy B. Computational approaches to the identification of heparin-binding sites on the surfaces of proteins. Biochem Soc Trans. 2006;34:431–434. doi: 10.1042/BST0340431. [DOI] [PubMed] [Google Scholar]
- 66.Mottarella SE, et al. Docking server for the identification of heparin binding sites on proteins. J Chem Inf Model. 2014;54:2068–2078. doi: 10.1021/ci500115j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gray JJ, et al. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 68.Zacharias M. ATTRACT: protein-protein docking in CAPRI using a reduced protein model. Proteins. 2005;60:252–256. doi: 10.1002/prot.20566. [DOI] [PubMed] [Google Scholar]
- 69.Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 70.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 71.Janin J, et al. CAPRI: a Critical Assessment of PRedicted Interactions. Proteins. 2003;52:2–9. doi: 10.1002/prot.10381. [DOI] [PubMed] [Google Scholar]
- 72.Mendez R, Leplae R, De Maria L, Wodak SJ. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins. 2003;52:51–67. doi: 10.1002/prot.10393. [DOI] [PubMed] [Google Scholar]
- 73.de Vries SJ, Schindler CE, Chauvot de Beauchene I, Zacharias M. A web interface for easy flexible protein-protein docking with ATTRACT. Biophys J. 2015;108:462–465. doi: 10.1016/j.bpj.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moal IH, Bates PA. SwarmDock and the use of normal modes in protein-protein docking. Int J Mol Sci. 2010;11:3623–3648. doi: 10.3390/ijms11103623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramirez-Aportela E, Lopez-Blanco JR, Chacon P. FRODOCK 2.0: fast protein-protein docking server. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btw141. [DOI] [PubMed] [Google Scholar]
- 76.Yu J, et al. InterEvDock: a docking server to predict the structure of protein-protein interactions using evolutionary information. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS One. 2011;6:e24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tovchigrechko A, Vakser IA. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006;34:W310–W314. doi: 10.1093/nar/gkl206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kozakov D, Schueler-Furman O, Vajda S. Discrimination of near-native structures in protein-protein docking by testing the stability of local minima. Proteins. 2008;72:993–1004. doi: 10.1002/prot.21997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moghadasi M, et al. The impact of side-chain packing on protein docking refinement. J Chem Inf Model. 2015;55:872–881. doi: 10.1021/ci500380a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Padhorny D, et al. Protein-protein docking by fast generalized Fourier transforms on 5D rotational manifolds. Proc Natl Acad Sci U S A. 2016;113:E4286–E4293. doi: 10.1073/pnas.1603929113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gruschus JM, Greene LE, Eisenberg E, Ferretti JA. Experimentally biased model structure of the Hsc70/auxilin complex: substrate transfer and interdomain structural change. Protein Sci. 2004;13:2029–2044. doi: 10.1110/ps.03390504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. 2006;26:5827–5837. doi: 10.1128/MCB.00441-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee DY, et al. Mutagenesis and modeling of the peroxiredoxin (Prx) complex with the NMR structure of ATP-bound human sulfiredoxin implicate aspartate 187 of Prx I as the catalytic residue in ATP hydrolysis. Biochemistry. 2006;45:15301–15309. doi: 10.1021/bi061824h. [DOI] [PubMed] [Google Scholar]
- 85.Watson AA, et al. The crystal structure and mutational binding analysis of the extracellular domain of the platelet-activating receptor CLEC-2. J Biol Chem. 2007;282:3165–3172. doi: 10.1074/jbc.M610383200. [DOI] [PubMed] [Google Scholar]
- 86.Martin MC, Allan LA, Mancini EJ, Clarke PR. The docking interaction of caspase-9 with ERK2 provides a mechanism for the selective inhibitory phosphorylation of caspase-9 at threonine 125. J Biol Chem. 2008;283:3854–3865. doi: 10.1074/jbc.M705647200. [DOI] [PubMed] [Google Scholar]
- 87.Liu C, et al. The SH3-like domain switches its interaction partners to modulate the repression activity of mycobacterial iron-dependent transcription regulator in response to metal ion fluctuations. J Biol Chem. 2008;283:2439–2453. doi: 10.1074/jbc.M706580200. [DOI] [PubMed] [Google Scholar]
- 88.Pilpa RM, et al. Functionally distinct NEAT (NEAr Transporter) domains within the Staphylococcus aureus IsdH/HarA protein extract heme from methemoglobin. J Biol Chem. 2009;284:1166–1176. doi: 10.1074/jbc.M806007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liang S, et al. Mapping of a microbial protein domain involved in binding and activation of the TLR2/TLR1 heterodimer. J Immunol. 2009;182:2978–2985. doi: 10.4049/jimmunol.0803737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cohavi O, Tobi D, Schreiber G. Docking of antizyme to ornithine decarboxylase and antizyme inhibitor using experimental mutant and double-mutant cycle data. J Mol Biol. 2009;390:503–515. doi: 10.1016/j.jmb.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 91.Hofmann WA, et al. SUMOylation of nuclear actin. J Cell Biol. 2009;186:193–200. doi: 10.1083/jcb.200905016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arthur CJ, et al. Structure and malonyl CoA-ACP transacylase binding of streptomyces coelicolor fatty acid synthase acyl carrier protein. ACS Chem Biol. 2009;4:625–636. doi: 10.1021/cb900099e. [DOI] [PubMed] [Google Scholar]
- 93.Nuth M, Cowan JA. Iron-sulfur cluster biosynthesis: characterization of IscU-IscS complex formation and a structural model for sulfide delivery to the [2Fe-2S] assembly site. J Biol Inorg Chem. 2009;14:829–839. doi: 10.1007/s00775-009-0495-7. [DOI] [PubMed] [Google Scholar]
- 94.Stauch B, et al. Model structure of APOBEC3C reveals a binding pocket modulating ribonucleic acid interaction required for encapsidation. Proc Natl Acad Sci U S A. 2009;106:12079–12084. doi: 10.1073/pnas.0900979106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Venkatachari NJ, et al. Human immunodeficiency virus type 1 Vpr: oligomerization is an essential feature for its incorporation into virus particles. Virol J. 2010;7:119. doi: 10.1186/1743-422X-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fredericks WJ, et al. The bladder tumor suppressor protein TERE1 (UBIAD1) modulates cell cholesterol: Implications for tumor progression. DNA Cell Biol. 2011;30:851–864. doi: 10.1089/dna.2011.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lira-Navarrete E, et al. Structural insights into the mechanism of protein o-fucosylation. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li D, et al. A comprehensive model of the spectrin divalent tetramer binding region deduced using homology modeling and chemical cross-linking of a mini-spectrin. J Biol Chem. 2010;285:29535–29545. doi: 10.1074/jbc.M110.145573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang GF, et al. Ligand-independent antiapoptotic function of estrogen receptor-beta in lung cancer cells. Mol Endocrinol. 2010;24:1737–1747. doi: 10.1210/me.2010-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Naudin C, et al. The occluding loop of cathepsin B prevents its effective inhibition by human kininogens. J Mol Biol. 2010;400:1022–1035. doi: 10.1016/j.jmb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 101.Guzman L, et al. Blockade of ethanol-induced potentiation of glycine receptors by a peptide that interferes with Gbetagamma binding. J Pharmacol Exp Ther. 2009;331:933–939. doi: 10.1124/jpet.109.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xi J, et al. Interaction between the T4 helicase-loading protein (gp59) and the DNA polymerase (gp43): A locking mechanism to delay replication during replisome assembly. Biochemistry. 2005;44:4600–4600. doi: 10.1021/bi0479508. [DOI] [PubMed] [Google Scholar]
- 103.Nelson SW, Yang JS, Benkovic SJ. Site-directed mutations of T4 helicase loading protein (gp59) reveal multiple modes of DNA polymerase inhibition and the mechanism of unlocking by gp41 helicase. J Biol Chem. 2006;281:8697–8706. doi: 10.1074/jbc.M512185200. [DOI] [PubMed] [Google Scholar]
- 104.Sobrado P, et al. Identification of the binding region of the [2Fe-2S] ferredoxin in stearoyl-acyl carrier protein desaturase: insight into the catalytic complex and mechanism of action. Biochemistry. 2006;45:4848–4858. doi: 10.1021/bi0600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kedlaya RH, Bhat KM, Mitchell J, Darnell SJ, Setaluri V. TRP1 interacting PDZ-domain protein GIPC forms oligomers and is localized to intracellular vesicles in human melanocytes. Arch Biochem Biophys. 2006;454:160–169. doi: 10.1016/j.abb.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li D, Tang HY, Speicher DW. A structural model of the erythrocyte spectrin heterodimer initiation site determined using homology modeling and chemical cross-linking. J Biol Chem. 2008;283:1553–1562. doi: 10.1074/jbc.M706981200. [DOI] [PubMed] [Google Scholar]
- 107.Krauss U, Losi A, Gartner W, Jaeger KE, Eggert T. Initial characterization of a blue-light sensing, phototropin-related protein from Pseudomonas putida: a paradigm for an extended LOV construct. Phys Chem Chem Phys. 2005;7:2804–2811. doi: 10.1039/b504554a. [DOI] [PubMed] [Google Scholar]
- 108.Surolia I, Reddy GB, Sinha S. Hierarchy and the mechanism of fibril formation in ADan peptides. J Neurochem. 2006;99:537–548. doi: 10.1111/j.1471-4159.2006.04072.x. [DOI] [PubMed] [Google Scholar]
- 109.Alminaite A, et al. Oligomerization of hantavirus nucleocapsid protein: analysis of the N-terminal coiled-coil domain. J Virol. 2006;80:9073–9081. doi: 10.1128/JVI.00515-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alminaite A, Backstrom V, Vaheri A, Plyusnin A. Oligomerization of hantaviral nucleocapsid protein: charged residues in the N-terminal coiled-coil domain contribute to intermolecular interactions. J Gen Virol. 2008;89:2167–2174. doi: 10.1099/vir.0.2008/004044-0. [DOI] [PubMed] [Google Scholar]
- 111.Juszczyk P, et al. Binding epitopes and interaction structure of the neuroprotective protease inhibitor cystatin c with beta-amyloid revealed by proteolytic excision mass spectrometry and molecular docking simulation. J Med Chem. 2009;52:2420–2428. doi: 10.1021/jm801115e. [DOI] [PubMed] [Google Scholar]
- 112.Brown KA, Dayal S, Ai X, Rumbles G, King PW. Controlled assembly of hydrogenase-CdTe nanocrystal hybrids for solar hydrogen production. J Am Chem Soc. 2010;132:9672–9680. doi: 10.1021/ja101031r. [DOI] [PubMed] [Google Scholar]
- 113.Raab M, Parthasarathi L, Treumann A, Moran N, Daxecker H. Differential binding of ICln in platelets to integrin-derived activating and inhibitory peptides. Biochem Biophys Res Commun. 2010;392:258–263. doi: 10.1016/j.bbrc.2009.12.088. [DOI] [PubMed] [Google Scholar]
- 114.Nan R, et al. Zinc binding to the Tyr402 and His402 allotypes of complement factor H: possible implications for age-related macular degeneration. J Mol Biol. 2011;408:714–735. doi: 10.1016/j.jmb.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sato K, Crowley PB, Dennison C. Transient homodimer interactions studied using the electron self-exchange reaction. J Biol Chem. 2005;280:19281–19288. doi: 10.1074/jbc.M500842200. [DOI] [PubMed] [Google Scholar]
- 116.Man P, et al. Accessibility changes within diphtheria toxin T domain when in the functional molten globule state, as determined using hydrogen/deuterium exchange measurements. FEBS J. 2010;277:653–662. doi: 10.1111/j.1742-4658.2009.07511.x. [DOI] [PubMed] [Google Scholar]
- 117.Wang GS, et al. Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIA(Glucose) of the Escherichia coli phosphoenolpyruvate : sugar phosphotransferase system. EMBO J. 2000;19:5635–5649. doi: 10.1093/emboj/19.21.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leysen S, Vanderkelen L, Weeks SD, Michiels CW, Strelkov SV. Structural basis of bacterial defense against g-type lysozyme-based innate immunity. Cell Mol Life Sci. 2013;70:1113–1122. doi: 10.1007/s00018-012-1184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 120.Viswanath S, Ravikant DV, Elber R. DOCK/PIERR: web server for structure prediction of protein-protein complexes. Methods Mol Biol. 2014;1137:199–207. doi: 10.1007/978-1-4939-0366-5_14. [DOI] [PubMed] [Google Scholar]
- 121.Fernandez-Recio J, Totrov M, Abagyan R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins. 2003;52:113–117. doi: 10.1002/prot.10383. [DOI] [PubMed] [Google Scholar]
- 122.Esquivel-Rodriguez J, Filos-Gonzalez V, Li B, Kihara D. Pairwise and multimeric protein-protein docking using the LZerD program suite. Methods Mol Biol. 2014;1137:209–234. doi: 10.1007/978-1-4939-0366-5_15. [DOI] [PubMed] [Google Scholar]
- 123.Terashi G, et al. The SKE-DOCK server and human teams based on a combined method of shape complementarity and free energy estimation. Proteins. 2007;69:866–872. doi: 10.1002/prot.21772. [DOI] [PubMed] [Google Scholar]
- 124.May A, Zacharias M. Protein-protein docking in CAPRI using ATTRACT to account for global and local flexibility. Proteins. 2007;69:774–780. doi: 10.1002/prot.21735. [DOI] [PubMed] [Google Scholar]
- 125.Huang SY, Zou X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins. 2008;72:557–579. doi: 10.1002/prot.21949. [DOI] [PubMed] [Google Scholar]
- 126.Eisenstein M, Ben-Shimon A, Frankenstein Z, Kowalsman N. CAPRI targets T29–T42: proving ground for new docking procedures. Proteins. 2010;78:3174–3181. doi: 10.1002/prot.22793. [DOI] [PubMed] [Google Scholar]
- 127.Andrusier N, Mashiach E, Nussinov R, Wolfson HJ. Principles of flexible protein-protein docking. Proteins. 2008;73:271–289. doi: 10.1002/prot.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shen Y. Improved flexible refinement of protein docking in CAPRI rounds 22–27. Proteins. 2013;81:2129–2136. doi: 10.1002/prot.24404. [DOI] [PubMed] [Google Scholar]
- 129.Heo L, Lee H, Seok C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci Rep. 2016;6:32153. doi: 10.1038/srep32153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cheng TM, Blundell TL, Fernandez-Recio J. pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins. 2007;68:503–515. doi: 10.1002/prot.21419. [DOI] [PubMed] [Google Scholar]
- 131.Zhou HX, Qin S. Interaction-site prediction for protein complexes: a critical assessment. Bioinformatics. 2007;23:2203–2209. doi: 10.1093/bioinformatics/btm323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 | Performance of ClusPro on Docking Benchmark 4.025