

A scorpion toxin uses protons in the venom as coactivators to target nociceptive ion channel TRPV1 and cause pain.
Abstract
Venomous animals use peptide toxins for hunting and self-defense. To achieve these goals, toxins need to bind to their targets with high affinity due to the small amount that a single bite or sting can deliver. The scorpion toxin BmP01 is linked to sting-induced excruciating pain; however, the reported minimum concentrations for activating TRPV1 channel or inhibiting voltage-gated potassium (Kv) channels (both in the micromolar range) appear too high to be biologically relevant. We show that the effective concentration of BmP01 is highly pH-dependent—it increases by about 10-fold in inhibiting Kv channels upon a 1-U drop in pH but decreases more than 100-fold in activating TRPV1. Mechanistic investigation revealed that BmP01 binds to one of the two proton-binding sites on TRPV1 and, together with a proton, uses a one-two punch approach to strongly activate the nociceptive channel. Because most animal venoms are acidic, proton-facilitated synergistic action may represent a general strategy for maximizing toxin potency.
INTRODUCTION
Venom has evolved to arm many otherwise defenseless animals with a lethal weapon (1). This effective strategy for survival is beautifully demonstrated by scorpions, snakes, spiders, cone snails, and sea anemones (the 5S’s). Most animal venoms contain multiple peptide toxins highly selective for their perspective targets, many of which are ion channels (2, 3). For example, charybdotoxin blocks voltage-gated Kv channels with exquisite potency [Ki (inhibitor constant) in the subnanomolar range] (4), causing uncontrollable hyperexcitability of the nervous system (5, 6). Hanatoxins achieve a similar feat by inhibiting voltage sensor movements (7). With each constituent being optimized through evolution to agitate or nullify its target, a tiny drop of venom can paralyze or even kill a large mammal (8). Achieving these feats requires toxins to be effective at extremely low concentrations (picomolar to nanomolar). Mechanistic investigation of animal toxins has revealed many fundamental biological processes, greatly advanced our understanding of toxin target molecules (3), and facilitated drug development (9–11).
Many scorpions can deliver lethal stings. Although legendary for their toxicity, scorpion stings are also known to be extremely painful, apparently to deter larger predators that cannot be overpowered by venom toxicity alone. Like other venomous species, scorpions use a cocktail of toxins to simultaneously manipulate multiple targets. BmP01, a member of the inhibitor cystine knot motif toxins, is a small scorpion peptide toxin known to target the nociceptive TRPV1 ion channel to produce pain (12). Here, we report a mechanistic study of BmP01, which has revealed a novel cooperative action between the toxin and protons, both from the scorpion venom, that yields high potency in activating the toxin’s target.
RESULTS
The Buthidae scorpions, living in East Asian countries, produce more than a hundred peptide toxins, most of which remain poorly understood (13). BmP01, originally identified from Mesobuthus martensii (Fig. 1A) (4, 14), is one of these less-studied toxins. BmP01 is a short 29–amino acid peptide (Fig. 1B). Like many peptide toxins, BmP01 forms a compact structure using three pairs of disulfide bonds to cross-link a short α helix and two β-strands (Fig. 1C) (15). The polymodal nociceptive TRPV1 ion channel is a likely target of BmP01 to produce the excruciating pain associated with scorpion sting (12). Similar to the TRPV1-selective agonist capsaicin, BmP01 induced strong calcium influx in TRPV1-expressing human embryonic kidney (HEK) 293 cells but not in untransfected cells (Fig. 1, D and E). Single-channel recording further confirmed direct activation of TRPV1 by BmP01 (Fig. 1F and fig. S1). BmP01-induced TRPV1 activation exhibited two properties characteristic of classic ligand-host interaction. First, BmP01 is highly selective for TRPV1 over its close analogs, such as TRPV3 (Fig. 1G). Second, BmP01-induced TRPV1 activity is strongly concentration-dependent (Fig. 1H). However, it was noticed that TRPV1 activities were observed only when micromolar concentrations of BmP01 were used [median effective concentration (EC50) at 40.4 ± 12.3 μM, n = 10]. Similar properties were previously reported in BmP01-mediated inhibition of voltage-gated Kv channels (14, 16). Therefore, given the fact that high concentrations inside the prey are apparently unachievable by a single sting, it appears that BmP01 has evolved to target TRPV1 to produce pain and/or Kv to produce hyperactivity.
Fig. 1. BmP01 directly activates TRPV1 channel.
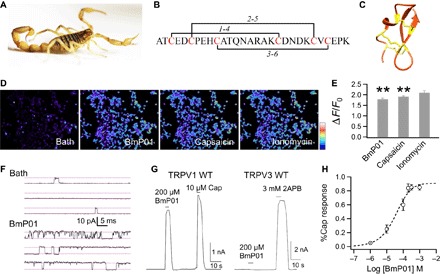
(A) Image of the scorpion M. martensii. (B) Peptide sequence and disulfide bonds of BmP01. (C) Solution structure of BmP01 [Protein Data Bank (PDB) ID: 1WM7]. Three disulfide bonds are colored in yellow. (D) Calcium imaging of hTRPV1-expressing HEK293 cells challenged sequentially with BmP01 (200 μM), capsaicin (10 μM), and ionomycin (1 mM). The scale bar represents 126 to 314 arbitrary units. (E) Fluorescence signals of hTRPV1-expressing HEK293T cells in response to BmP01, capsaicin, and ionomycin. **P < 0.001 (n = 3 to 5). (F) Representative single-channel current traces recorded in the absence or presence of 100 μM BmP01 (pH 7.0). (G) Representative whole-cell current response to BmP01 and agonists at saturating concentration [for hTRPV1, 10 μM capsaicin; for mTRPV3, 3 mM 2-aminoethoxydiphenyl borate (2APB)]. (H) Concentration-response relationship of BmP01 and hTRPV1 (n = 10) fitted to a Hill equation with the following parameters: EC50, 40.4 ± 12.3 μM; slope factor, 0.80 ± 0.05.
In search of an explanation for the apparent paradox regarding the biological role of BmP01, we noticed that scorpions produce venom that is acidic (Fig. 2, A and B). We therefore examined the possibility that under acidic conditions, BmP01 may exhibit higher potencies. This turned out to be the case. Although it took at least 300 μM BmP01 to induce detectable TRPV1 activity under basic conditions (pH 8.0 to 8.5), we observed that the effective concentration dropped substantially at lower pH levels (Fig. 2C). Although there was barely detectable direct activation of TRPV1 by protons alone at pH 6.5, BmP01 induced robust channel currents at 0.1 to 1 μM concentrations. In support of a synergistic action between protons and BmP01, we found that BmP01’s concentration dependence curve exhibited a leftward shift and became shallower at lower pH levels (Fig. 2D). Observations from TRPV1-transfected cells could be reliably reproduced in animal tests. We found that injection of BmP01 in an acidic solution was much more potent in inducing pain behavior in mice, whereas the same increase in acidity did not appreciably potentiate capsaicin-evoked pain behavior (Fig. 2E). Lowering pH antagonized the BmP01-mediated inhibition of Kv channels, making the toxin less potent (fig. S2). Our findings thus suggest that TRPV1, but not the Kv channel, is the biological target for BmP01, which exploits the acidity of the venom milieu to boost toxin potency.
Fig. 2. Extracellular acidification strongly potentiates BmP01 in activating TRPV1 and inducing pain.
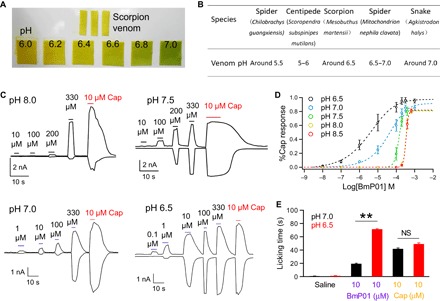
(A) Determination of the acidity value of venom from M. martensii. (B) Comparison of the acidity value of different venoms. (C) Representative whole-cell currents from hTRPV1 at various BmP01 concentrations and pH levels. (D) Concentration-response relationships of BmP01 at various pH levels fitted to a Hill equation; n = 5 to 10. (E) Paw-licking behavior of mice following injection of either saline, BmP01 (pH 7.0 or 6.5), or capsaicin (pH 7.0 or 6.5). Ten microliters of each reagent was injected at the plantar surface of the left hind paw. NS, not significant. **P < 0.01.
To better understand the underlying molecular mechanism for synergistic action between BmP01 and protons, we searched for BmP01-binding site on TRPV1 by taking advantage of the toxin’s high selectivity for TRPV1 over its homolog TRPV3. A series of TRPV1-TRPV3 chimera was generated (Fig. 3A), and subjected to BmP01 challenge. The results indicate that the outer pore is a major determinant for BmP01 binding; however, it is unlikely that BmP01 binds to the pore turret, because chimera containing TRPV3’s turret (V1/3S) remained responsive to the toxin (Fig. 3, B and C, and fig. S3). This finding, confirmed by direct mutagenesis tests (Fig. 3, A and B), is noteworthy in that two previously characterized TRPV1 toxins, RhTx from centipede (17) and DkTx from spider (18–20), also bind to the outer pore but differ in their binding sites from each other and from BmP01. It is therefore possible that the three toxins may activate TRPV1 through distinct mechanisms. These observations support the suggestion that the outer pore of TRPV1 is an important and complex gating structure (21).
Fig. 3. BmP01 targets the pre-S6 region of TRPV1.
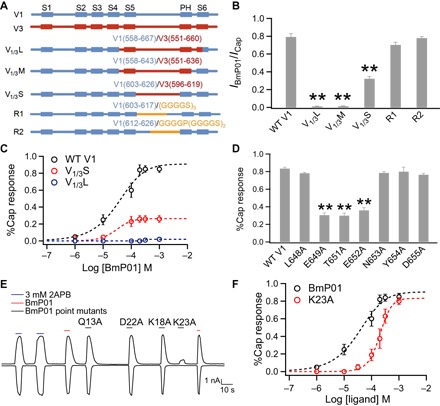
(A) Schematic representation of the chimeras between mTRPV1 (blue) and mTRPV3 (red), and mTRPV1 mutants containing a sequence replacement in the turret (yellow). (B) Comparison of the BmP01 response (shown as percentage of the capsaicin response). mTRPV1 was used as wild type (WT). **P < 0.001 (n = 3 to 5). (C) Concentration-response relationships of WT mTRPV1, V1/3S, and V1/3L fitted to a Hill equation. (D) Comparison of the BmP01 response of WT hTRPV1 and point mutants. **P < 0.001 (n = 3 to 5). (E) Representative whole-cell current recording from hTRPV1 treated with BmP01 and its point mutants (200 μM each) and 3 mM 2APB. (F) Concentration-response relationships for WT BmP01 and K23A fitted to a Hill equation (n = 5 to 10).
Guided by results from chimeric tests, our follow-up site-directed mutagenesis screening identified three charged or polar residues in the pre-S6 region that likely participate in mediating BmP01 binding. An alanine mutation to each of these residues substantially reduced the toxin’s potency (Fig. 3D and fig. S4). These results revealed that the pre-S6 loop, an important TRPV1 gating element (22), is a key channel structure targeted by BmP01.
We next searched for the channel-interacting site on BmP01. Given that mutations to charged or polar residues in TRPV1 had a strong impact on toxin potency, we screened the charged/polar residues in BmP01. The charged fractions of residue side chains in BmP01 are little affected by lowering pH from 7.0 to 6.5 (table S1). We found that, although most mutant toxins containing an alanine substitution remained potent, K23A mutant toxin exhibited a diminished potency in eliciting a channel response: 10 μM of the mutant toxin was not able to elicit pain response in mice (Fig. 3E and fig. S5). Because K23A caused a substantial right shift of the concentration-response relationship without a significant change in the maximal response (Fig. 3F), the mutant toxin’s low potency is most likely due to weakened binding affinity. Therefore, it appeared that electrostatic interactions may contribute to toxin-channel interaction.
To test the hypothesis that BmP01 and TRPV1 form direct electrostatic interactions via charged/polar residue(s), we carried out a thermodynamic mutant cycle analysis (23, 24) between K23 of BmP01 and each of the three key pre-S6 residues in TRPV1 (Fig. 4A and fig. S6). We found that the coupling energy between K23 and E649 (but not the other two residues) is substantially higher than 1.5 kT, indicating the possibility that a direct salt bridge may exist between these two residues (25).
Fig. 4. Bimodal activation of TRPV1 by BmP01 and proton.
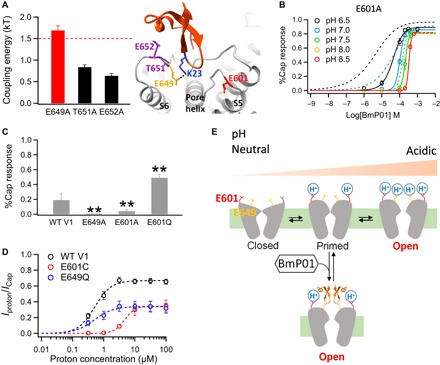
(A) A salt bridge mediates BmP01-TRPV1 interaction. Left: Comparison of coupling energy between K23 of BmP01 and the pre-S6 channel residues (n = 4 to 7) with the 1.5-kT threshold for direct interaction indicated with a dashed line. Right: Structural model of the BmP01-TRPV1 complex, with the two proton-binding sites (E601 and E649) highlighted in red and orange, respectively. (B) Concentration-response relationships of E601A at varying pH levels fitted to a Hill equation (n = 5 to 10). Concentration-response relationships of WT hTRPV1 at the same pH levels (Fig. 2D) are superimposed as dashed lines for comparison. (C) Comparison of responses by WT hTRPV1 and its point mutants to 100 μM BmP01 at neutral acidity (pH 7.5) (n = 3 to 5). **P < 0.01. (D) Proton concentration-response relationships of WT mTRPV1, E601Q, and E649Q mutants fitted to a Hill equation (n = 4 to 7). (E) Cartoon summarizing the bimodal activation of TRPV1 by proton and BmP01. At pH 6.5, proton binding to the high-affinity site E601 potentiates TRPV1; under this situation, BmP01 binding to E649 leads to strong activation. At even lower pH levels, protons bind to both E601 and E649 to open the channel without BmP01.
Using this crucial structural constraint, we conducted Rosetta-based molecular docking (24, 26) of BmP01 onto the TRPV1 outer pore. Our predicted toxin-channel complex model suggests that BmP01 binds to the outer pore via direct contact to the pre-S6 loop and its surrounding structures (Fig. 4A). Although many details of the toxin-channel interaction remain to be elucidated, the location of BmP01 binding has important indications. For example, cryo–electron microscopy (cryo-EM) studies suggest that TRPV1 has two activation gates: a lower gate at the S6 bundle crossing and an upper gate at the selectivity filter (27). By interacting with the outer pore, BmP01 may directly affect the closed-open equilibrium of the upper gate.
How does this structural insight help to explain BmP01’s biological function as a potent pain inducer? TRPV1 is a polymodal nociceptor for a wide array of physical and chemical stimuli, including heat, capsaicin, animal toxins, and protons (17, 28–33). Two protonation sites on TRPV1 are thought to mediate proton activation but differ in their functional roles (34). Protonation of E601 at the extracellular end of S5 (Fig. 4A), which is the higher-affinity site for protons, potentiates the channel, whereas protonation of E649—the lower-affinity site for protons and the residue that K23 of BmP01 interacts with—leads to channel activation (34). Strong activation of TRPV1 by protons requires binding of both sites; synergistic proton binding to multiple sites gives rise to the observed steep pH-dependent TRPV1 activation that ensures high sensitivities in detecting tissue damages, such as inflammation (29). Our finding that BmP01 binds to the lower-affinity but high-potency site E649 suggests that BmP01 may act like a proton to promote channel activation. This would allow strong activation of TRPV1 under less acidic conditions that would only yield occupation of E601. Therefore, there is a synergy between protons and BmP01 in activating TRPV1.
The predicted synergistic action by BmP01 and protons was fully supported by functional tests. We found that when protonation of E601 was prevented by the E601A mutation, the potency and pH dependence of BmP01 was substantially reduced (Fig. 4B). At neutral or high pH levels, both the E601A mutant channel and the WT channel responded similarly to BmP01, confirming the mutation’s selective effect on protonation. On the other hand, mimicking protonation by an E601Q mutation substantially enhanced BmP01’s potency at neutral pH (Fig. 4C). Furthermore, consistent with their different functional roles, proton binding to E601 and E649 in sequential order was determined by their pKa (where Ka is the acid dissociation constant) values. A point mutation to E601 caused a significant rightward shift in pH-dependent activation (EC50 of 4.72 ± 0.08 μM, n = 4), but a point mutation to E649 did not (EC50 of 0.43 ± 0.14 μM, n = 7; for WT, EC50 of 0.48 ± 0.06 μM, n = 7) (Fig. 4D), indicating that the pKa value for E601 is likely much higher than the pKa value for E649.
In summary, our results show that BmP01 is not a potent activator at neutral pH because it occupies only the E649 site. Under acidic conditions, however, when the E601 sites (four per channel) are protonated, occupation of E649 by BmP01 produces strong channel activation (Fig. 4E). The cooperative action of BmP01 and proton causes strong pain locally at the sting site.
DISCUSSION
Bimodal activation of TRPV1 by BmP01 and proton reveals an ingenious strategy that scorpions use for self-defense. Taking advantage of the intrinsic acidity of its venom, scorpions produce a peptide toxin that targets TRPV1 with a one-two punch approach, mimicking what protons do to TRPV1 at high concentrations. However, the presence of toxin makes it unnecessary for scorpions to further acidify their venom, which may be incompatible with other aspects of venom biology. Nevertheless, delivering BmP01 in an acidic package can achieve potent TRPV1 activation that is comparable to activation by capsaicin, a pungent irritant in chili peppers (24, 28, 35). Strong boost in potency comes from the cooperative interaction of BmP01 and proton with TRPV1.
BmP01 belongs to the α-KTX8 toxin subfamily (36), whose members share similar topology (16) and sequence features (fig. S5A). For instance, K23, which is critical to BmP01-TRPV1 interaction, is highly conserved. We have observed that other α-KTX8 members, such as Tx203 and OdK1, can also activate TRPV1 at neutral pH (fig. S5B). Increasing the acidity to pH 6.5 largely potentiated TRPV1 activation by either Tx203 or OdK1 just like BmP01 (fig. S5B). These observations indicate that the bimodal activation mechanism may be applicable to other toxins within the α-KTX8 subfamily. However, because protein-protein interactions are affected by many factors, such as size, shape, and hydrophobicity in the microenvironment, case-by-case investigations are needed to determine the exact structural mechanism underlying bimodal activation of TRPV1 by other members of the α-KTX8 subfamily.
Toxins within the α-KTX8 subfamily have long been known as blockers of potassium channels (36). Our observations that some of them can strongly activate TRPV1 channels under acidic conditions are intriguing. Evolutionarily, both Kv and TRP channels belong to the same ion channel superfamily (37), and share a similar topology in the transmembrane domains. The opposite effects of α-KTX8 toxins on these channels reflect important structural differences in the pore regions. Cryo-EM examination of TRPV1 structure reveals that the TRPV1 selectivity filter adopts a “flatter” conformation compared to the similar section of voltage-gated potassium channels, despite almost identical amino acid sequences (19, 27, 38, 39). Structural differences in TRPV1 and potassium channels may cause toxins, such as BmP01, to use different strategies target these channels. Our study here demonstrates that BmP01 uses K23 to bind to the pore helix—a well-known hotspot for polymodal gating (22)—to open TRPV1 (Fig. 4A), whereas this toxin may use K18 as a “plug” to block the selectivity filter of a potassium channel (16). In light of these findings, it would be attractive to test in vivo whether both ion channels are effectively targeted by BmP01 and other members of the α-KTX8 subfamily, because activating TRPV1 and inhibiting Kv channels would both contribute to excitation of nociceptive neurons. Being able to simultaneously target both ion channels would give a clear metabolic and biochemical advantage to scorpions, a group of very old animals that have exhibited little anatomic change through evolution.
Virtually all known ion channels are allosteric proteins with a multimeric structural design that incorporates cooperativity in sensing a wide range of physical and chemical stimuli (40, 41). Although BmP01-induced activation of TRPV1 involves highly specific molecular interactions, it is unlikely that this bimodal activation is a unique phenomenon to the α-KTX8 subfamily of toxins. Animal venoms are generally acidic (42). Scorpion venom is only mildly acidic, whereas those of spider and centipede can reach pH 5.0 to 5.5 (Fig. 2B). In this aspect, venoms are similar to the synaptic vesicle milieu (43–45), Golgi complex, and secretory granules. The acidic environment in membrane-bound cellular compartments is recognized to be crucial for biosynthesis, sorting, and trafficking of proteins (46). Animal toxins delivered in this acidic package must have undergone optimization through evolution to better perform their biological functions. We suggest that, in addition to using a cocktail of toxins, bimodal activation may represent another general survival strategy used by venomous animals.
MATERIALS AND METHODS
Cell transfection
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 mg/ml) at 37°C with 5% CO2. Cells were plated on glass coverslips 24 hours before transfection. Transient transfection was conducted by adding 4 μl of Lipofectamine 2000 (Invitrogen) and 4 μg of plasmid DNA into Opti-MEM and then stored for 20 min. The mixer was added in a 35-mm cell culture dish. Electrophysiological experiments and fluorescence imaging recordings were performed between 24 and 48 hours after transfection.
Toxin peptides synthesis and purification
Syntheses of linear BmP01 and point mutations were carried out on an automatic peptide synthesizer (PerSeptive Biosystems) using a 9-fluorenyl methoxycarbonyl/tert-butyl strategy and HOBt/TBTU/NMM coupling method. Crude reduced peptides were purified by reversed-phase high-performance liquid chromatography (HPLC). The purity of peptides was checked to be higher than 95% by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry and HPLC techniques. Linear peptide was refolded under the solution with 10 mM glutathione and 100 mM oxidized glutathione, adjusted to pH 7.0, and placed at room temperature for 24 hours. The successfully refolded peptide was eluted by nearly 30% acetonitrile.
Electrophysiological recordings
Macroscopic and single-channel currents from TRPV1-expressing cells were recorded in whole-cell or outside-out patches using a HEKA EPC10 amplifier controlled with PATCHMASTER software (HEKA). All recordings were performed at room temperature. Patch pipettes were pulled from thin-wall borosilicate glass (A-M Systems) and fire-polished to a resistance of ~2 megohms. Both bath and pipette solutions contained 130 mM NaCl, 0.2 mM EDTA, and 3 mM Hepes (pH 7.2). There was no Ca2+ in the solution so as to reduce desensitization to capsaicin. For whole-cell recordings, the capacity current was minimized by amplifier circuitry, and the series resistance was compensated by 30 to 65%. The membrane potential was held on 0 mV, and currents were elicited by a protocol consisting of a 300-ms step to +80 mV followed by a 300-ms step to −80 mV at 1-s intervals. Capsaicin at 10 μM concentration was used to maximally activate TRPV1 WT and mutant channels.
Calcium fluorescence imaging
HEK293T cells stably expressing hTRPV1 channels were loaded with 2 μM Fluo-4 AM (Invitrogen Life Technologies) in Ringer’s solution [140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 10 mM d-glucose, 10 mM Hepes, and 2 mM CaCl2, (pH 7.2)] without Ca2+ for 20 min at room temperature, after washing twice with the same solution. Normal Ringer’s solution (with 2 mM Ca2+) was used during imaging. These cells were challenged sequentially by 200 μM BmP01, 10 μM capsaicin, and 1 μM ionomycin. Fluorescence images of HEK293T cells were acquired with an Olympus IX-81 microscope with a Hamamatsu R2 charge-coupled device camera controlled by MetaFluor software. Fluo-4 was excited by a mercury vapor light source with a 494 ± 20–nm excitation filter, whereas fluorescence emission was detected with a 516 ± 30–nm emission filter. Change in fluorescence intensity, F, was normalized by the baseline fluorescence, F0, in each cell and was expressed as (F − F0/F0) × 100%.
Channel mutagenesis and point mutations
All chimeras used in this study (V1/3L, V1/3M, and V1/3S) were based on the mouse TRPV1 and mouse TRPV3. These chimeras were generated by the overlapping extension method and confirmed by DNA sequencing, as described previously (17). For mutations R1 and R2, the segments of WT mouse TRPV1 (603GKNNSLPVESPPHKCRG619 and 613PPHKCRGSACRPGN626) were replaced by GGGGSGGGGSGGGGS and GGGGPGGGGSGGGGS, respectively (47). All human TRPV1 point mutations were constructed using the QuikChange II XL site-directed mutagenesis kit following the manufacturer’s instruction.
Activity-related restraint-docking of BmP01 to the TRPV1 channel
To prepare structures of BmP01 (PDB ID: 1WM7) and rTRPV1 channel (toxin-bound state; PDB ID: 3J5Q) for molecular docking, they were first relaxed in Rosetta 3.5 (48). For each structure, 10,000 models were generated. The top 10 lowest-energy models converged well, and the lowest-energy model was chosen for docking. To dock the toxin, membrane environment was first set up on the channel model using RosettaScripts (26, 49). On the basis of results of a thermodynamic mutant cycle analysis (Fig. 4A), three distance restraints were set up: because E649 on TRPV1 and K23 on BmP01 exhibited specific interaction, the distance between their Cβ atoms was set to be less than 4.0 Å. On the other hand, the distances between Cβ atoms of the T651/K23 pair and the E652/K23 pair were set to be larger than 6.0 Å because coupling energies between these residue pairs indicated no specific interaction between them. A total of 20,000 docking models were generated, from which the top 1000 total energy score models were identified. From this pool, the one model that was in agreement with distance restraints was chosen as the final docking model. All numbering in protein sequence was based on mouse TRPV1.
Animal assay
All mice (C57BL/6J) used in this study were purchased from the Model Animal Research Center of Nanjing University, China. For the paw-licking assay, pain was induced in mice by intraplantar injection of capsaicin or BmP01. Each of the testing materials was dissolved in 100-μl saline. Mice were injected 10 μl of each drug with an appropriate concentration at the plantar surface of the left hind paw. Control mice were injected with the same volume of saline (at pH 7.0 or 6.5). The time of paw licking was recorded immediately after injection by taking videos for 30 min. All efforts were made to reduce the number of animals used and to minimize the suffering of animals.
Data analysis
Experimental data of electrophysiological recordings were acquired and analyzed using the PATCHMASTER program and Igor Pro. Data obtained from experiments involving animals were analyzed by GraphPad Prism 5 (GraphPad Software Inc.). All statistical values are shown as means ± SEM of n repeats of the experiments. Statistical analysis was carried out using Student’s t test; double asterisks were used to indicate the presence of a significant difference at the P < 0.001 level. NS indicates no significance.
For double-mutant cycle analysis, EC50 values of four channel-toxin combinations (WT channel, WT BmP01: EC50–1; mutant channel, WT BmP01: EC50–2; WT channel, mutant BmP01: EC50–3; and mutant channel, mutant BmP01: EC50–4) were determined separately. The strength of coupling was determined by the coupling energy (kT multiplied by lnΩ, where k is the Boltzmann constant and T is temperature in Kelvin) (24, 25). lnΩ was calculated using the following equation: lnΩ = ln[(EC50–1)(EC50–4)/(EC50–2)(EC50–3)]. All values of EC50 [or IC50 (median inhibitory concentration)] and Hill coefficient of concentration-response curves were summarized in tables S2 to S4.
Acknowledgments
We are grateful to our laboratory members for assistance and insightful discussion. Funding: This work was supported by funding from the Ministry of Science and Technology of China (2013CB911304), National Science Foundation of China (331372208 and U1302221), Chinese Academy of Sciences (XDA12040209 and QYZDJ-SSW-SMC012), and Yunnan Province (2015HA023) to R.L. and from the National Science Foundation of China (31640071) and Chinese Academy of Sciences (XDA12020334 and Youth Innovation Promotion Association) to S.Y. This work was also supported by funding from NIH (R01NS072377) to J.Z. and American Heart Association (14POST19820027 and 16PRE26960016) to F.Y. and B.H.L., respectively. Author contributions: S.Y., F.Y., and B.Z. conducted most of the experiments, including toxin peptide synthesis and purification, patch-clamp recording, mutagenesis, calcium imaging, molecular docking, animal behavior tests, and data analysis; B.H.L. also conducted patch-clamp recordings. B.L. and L.L. refolded BmP01. J.Z., F.Y., and S.Y. prepared the manuscript. S.Y., J.Z., and R.L. conceived and supervised the project, as well as participated in data analysis and manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700810/DC1
fig. S1. BmP01 activates TRPV1 by increasing the open probability.
fig. S2. BmP01 inhibits Kv1.3 channel.
fig. S3. Effects of BmP01 on TRPV1, TRPV3, and their chimeras.
fig. S4. Effects of BmP01 on TRPV1 outer pore mutants.
fig. S5. Low pH potentiates TRPV1 current response to toxins in the α-KTX8 toxin subfamily.
fig. S6. Concentration-response curves used for the determination of coupling energy between BmP01 and TRPV1.
table S1. Changes in charged fraction of the side chain of titratable residues in BmP01.
table S2. BmP01 concentration-response parameters for TRPV1 WT and mutants.
table S3. Proton concentration-response parameters for TRPV1 WT and mutants.
table S4. BmP01 concentration-response parameters for Kv1.3 under different pH values.
REFERENCES AND NOTES
- 1.Fry B. G., Roelants K., Champagne D. E., Scheib H., Tyndall J. D. A., King G. F., Nevalainen T. J., Norman J. A., Lewis R. J., Norton R. S., Renjifo C., de la Vega R. C. R., The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 10, 483–511 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Catterall W. A., Cestèle S., Yarov-Yarovoy V., Yu F. H., Konoki K., Scheuer T., Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Kalia J., Milescu M., Salvatierra J., Wagner J., Klint J. K., King G. F., Olivera B. M., Bosmans F., From foe to friend: Using animal toxins to investigate ion channel function. J. Mol. Biol. 427, 158–175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romi-Lebrun R., Lebrun B., Martin-Eauclaire M. F., Ishiguro M., Escoubas P., Wu F. Q., Hisada M., Pongs O., Nakajima T., Purification, characterization, and synthesis of three novel toxins from the Chinese scorpion Buthus martensi, which act on K+ channels. Biochemistry 36, 13473–13482 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Miller C., The charybdotoxin family of K+ channel-blocking peptides. Neuron 15, 5–10 (1995). [DOI] [PubMed] [Google Scholar]
- 6.J. Sack, K. S. Eum, Ion channel inhibitors, in Handbook of Ion Channels, J. Zheng, M. C. Trudeau, Eds. (CRC Press, 2015), chap. 14. [Google Scholar]
- 7.Swartz K. J., MacKinnon R., An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 15, 941–949 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Ismail M., The scorpion envenoming syndrome. Toxicon 33, 825–858 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Teichert R. W., Schmidt E. W., Olivera B. M., Constellation pharmacology: A new paradigm for drug discovery. Annu. Rev. Pharmacol. Toxicol. 55, 573–589 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King G. F., Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 11, 1469–1484 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Vriens J., Appendino G., Nilius B., Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 75, 1262–1279 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Hakim M. A., Jiang W., Luo L., Li B., Yang S., Song Y., Lai R., Scorpion toxin, BmP01, induces pain by targeting TRPV1 channel. Toxins 7, 3671–3687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao Z., Yu Y., Wu Y., Hao P., Di Z., He Y., Chen Z., Yang W., Shen Z., He X., Sheng J., Xu X., Pan B., Feng J., Yang X., Hong W., Zhao W., Li Z., Huang K., Li T., Kong Y., Liu H., Jiang D., Zhang B., Hu J., Hu Y., Wang B., Dai J., Yuan B., Feng Y., Huang W., Xing X., Zhao G., Li X., Li Y., Li W., The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nat. Commun. 4, 2602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J.-J., Dai L., Lan Z.-D., Chi C.-W., Genomic organization of three neurotoxins active on small conductance Ca2+−activated potassium channels from the scorpion Buthus martensi Karsch. FEBS Lett. 452, 360–364 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Wu G., Li Y., Wei D., He F., Jiang S., Hu G., Wu H., Solution structure of BmP01 from the venom of scorpion Buthus martensii Karsch. Biochem. Biophys. Res. Commun. 276, 1148–1154 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Zhu S., Peigneur S., Gao B., Luo L., Jin D., Zhao Y., Tytgat J., Molecular diversity and functional evolution of scorpion potassium channel toxins. Mol. Cell. Proteomics 10, M110.002832 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S., Yang F., Wei N., Hong J., Li B., Luo L., Rong M., Yarov-Yarovoy V., Zheng J., Wang K., Lai R., A pain-inducing centipede toxin targets the heat activation machinery of nociceptor TRPV1. Nat. Commun. 6, 8297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohlen C. J., Priel A., Zhou S., King D., Siemens J., Julius D., A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 141, 834–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Y., Cao E., Julius D., Cheng Y., TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae C., Anselmi C., Kalia J., Jara-Oseguera A., Schwieters C. D., Krepkiy D., Won Lee C., Kim E.-H., Kim J. I., Faraldo-Gómez J. D., Swartz K. J., Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin. eLife 5, e11273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang F., Cui Y., Wang K., Zheng J., Thermosensitive TRP channel pore turret is part of the temperature activation pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 7083–7088 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng J., Ma L., Structure and function of the thermoTRP channel pore. Curr. Top. Membr. 74, 233–257 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Horovitz A., Fersht A. R., Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 214, 613–617 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Yang F., Xiao X., Cheng W., Yang W., Yu P., Song Z., Yarov-Yarovoy V., Zheng J., Structural mechanism underlying capsaicin binding and activation of the TRPV1 ion channel. Nat. Chem. Biol. 11, 518–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiber G., Fersht A. R., Energetics of protein-protein interactions: Analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248, 478–486 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Yarov-Yarovoy V., Schonbrun J., Baker D., Multipass membrane protein structure prediction using Rosetta. Proteins 62, 1010–1025 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao E., Liao M., Cheng Y., Julius D., TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., Julius D., The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Tominaga M., Caterina M. J., Malmberg A. B., Rosen T. A., Gilbert H., Skinner K., Raumann B. E., Basbaum A. I., Julius D., The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Zheng J., Molecular mechanism of TRP channels. Compr. Physiol. 3, 221–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee B. H., Zheng J., Proton block of proton-activated TRPV1 current. J. Gen. Physiol. 146, 147–159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aneiros E., Cao L., Papakosta M., Stevens E. B., Phillips S., Grimm C., The biophysical and molecular basis of TRPV1 proton gating. EMBO J. 30, 994–1002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuypers E., Yanagihara A., Karlsson E., Tytgat J., Jellyfish and other cnidarian envenomations cause pain by affecting TRPV1 channels. FEBS Lett. 580, 5728–5732 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jordt S.-E., Tominaga M., Julius D., Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. U.S.A. 97, 8134–8139 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang F., Zheng J., Understand spiciness: Mechanism of TRPV1 channel activation by capsaicin. Protein Cell 8, 169–177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tytgat J., Chandy K. G., Garcia M. L., Gutman G. A., Martin-Eauclaire M.-F., van der Walt J. J., Possani L. D., A unified nomenclature for short-chain peptides isolated from scorpion venoms: α-KTx molecular subfamilies. Trends Pharmacol. Sci. 20, 444–447 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Yu F. H., Catterall W. A., The VGL-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Science 2004, re15 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Liao M., Cao E., Julius D., Cheng Y., Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Y., Lee A., Chen J., Ruta V., Cadene M., Chait B. T., MacKinnon R., X-ray structure of a voltage-dependent K+ channel. Nature 423, 33–41 (2003). [DOI] [PubMed] [Google Scholar]
- 40.B. Hille, Ion Channels of Excitable Membranes (Sinauer, ed. 3, 2001). [Google Scholar]
- 41.J. Zheng, M. C. Trudeau, Handbook of Ion Channels (CRC Press, Taylor & Francis Group, 2015). [Google Scholar]
- 42.Mackessy S. P., Sixberry N. M., Heyborne W. H., Fritts T., Venom of the brown treesnake, Boiga irregularis: Ontogenetic shifts and taxa-specific toxicity. Toxicon 47, 537–548 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Füldner H. H., Stadler H., 31P-NMR analysis of synaptic vesicles. Status of ATP and internal pH. Eur. J. Biochem. 121, 519–524 (1982). [DOI] [PubMed] [Google Scholar]
- 44.Michaelson D. M., Angel I., Determination of ΔpH in cholinergic synaptic vesicles: Its effect on storage and release of acetylcholine. Life Sci. 27, 39–44 (1980). [DOI] [PubMed] [Google Scholar]
- 45.Miesenböck G., De Angelis D. A., Rothman J. E., Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Weisz O. A., Acidification and protein traffic. Int. Rev. Cytol. 226, 259–319 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Cui Y., Yang F., Cao X., Yarov-Yarovoy V., Wang K., Zheng J., Selective disruption of high sensitivity heat activation but not capsaicin activation of TRPV1 channels by pore turret mutations. J. Gen. Physiol. 139, 273–283 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leaver-Fay A., Tyka M., Lewis S. M., O. F. Lange, Thompson J., Jacak R., Kaufman K. W., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y.-E. A., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P., ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fleishman S. J., Leaver-Fay A., Corn J. E., Strauch E.-M., Khare S. D., Koga N., Ashworth J., Murphy P., Richter F., Lemmon G., Meiler J., Baker D., RosettaScripts: A scripting language interface to the Rosetta macromolecular modeling suite. PLOS ONE 6, e20161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1700810/DC1
fig. S1. BmP01 activates TRPV1 by increasing the open probability.
fig. S2. BmP01 inhibits Kv1.3 channel.
fig. S3. Effects of BmP01 on TRPV1, TRPV3, and their chimeras.
fig. S4. Effects of BmP01 on TRPV1 outer pore mutants.
fig. S5. Low pH potentiates TRPV1 current response to toxins in the α-KTX8 toxin subfamily.
fig. S6. Concentration-response curves used for the determination of coupling energy between BmP01 and TRPV1.
table S1. Changes in charged fraction of the side chain of titratable residues in BmP01.
table S2. BmP01 concentration-response parameters for TRPV1 WT and mutants.
table S3. Proton concentration-response parameters for TRPV1 WT and mutants.
table S4. BmP01 concentration-response parameters for Kv1.3 under different pH values.