

Abstract
Super-resolution microscopy allows optical imaging below the classical diffraction limit of light with currently up to 20 × higher spatial resolution. However, the detection of multiple targets (multiplexing) is still hard to implement and time-consuming to conduct. Here, we report a straightforward sequential multiplexing approach based on the fast exchange of DNA probes which enables efficient and rapid multiplexed target detection with common super-resolution techniques such as (d)STORM, STED, and SIM. We assay our approach using DNA origami nanostructures to quantitatively assess labeling, imaging, and washing efficiency. We furthermore demonstrate the applicability of our approach by imaging multiple protein targets in fixed cells.
Keywords: DNA nanotechnology, dSTORM, multiplexing, SIM, STED
Graphical abstract
Many happy returns: A straightforward sequential multiplexing approach based on the fast exchange of DNA probes has been developed that enables efficient and rapid multiplexed target detection with common super-resolution techniques such as (d)STORM, STED, and SIM.
Super-resolution microscopy allows researchers to obtain images with currently up to 20 × higher spatial resolution than the classical diffraction limit.[1] Although current techniques are already starting to transform research in the life sciences,[2] most implementations are still limited to the observation of only a few molecular species in the same sample, so-called multiplexing. Exchange-PAINT,[3] a recent implementation of the PAINT[4] concept (points accumulation in nanoscale topography) and extension of DNA-PAINT,[5] enables multiplexed super-resolution imaging by using transient, programmable binding between dye-labeled “imager” strands and target-bound complementary “docking” strands during sequential imaging rounds. Although Exchange-PAINT allows spectrally unlimited multiplexing independent of different dye spectra (i.e. by using the same dye for each exchange round), imager strands are not fluorogenic, which firstly limits its applicability beyond total internal reflection (TIR) or oblique illumination away from the coverslip and secondly sets an upper limit for the achievable image speed. Recently, sequential labeling and imaging approaches have been devised for (d)STORM[6] ((direct) stochastic optical reconstruction microscopy), where a target is immunolabeled and imaged, followed by a fluorophore inactivation or quenching step.[7] This procedure is repeated sequentially for the acquisition of all remaining targets. Although these implementations allow spectrally unlimited multiplexing, the fluorophore quenching step followed by immunolabeling of the next target is time-intensive, which overall limits experimental throughput. Furthermore, relabeling and reimaging of targets from previous rounds is difficult to achieve. Recently, Exchange-PAINT was applied to STED[8] (stimulated emission depletion) microscopy.[9] To achieve this, the concentration of imager strands in Exchange-PAINT was increased to render most target strands “occupied” during image acquisition. While this allows for rapid probe exchange between sequential imaging rounds, it comes at the cost of potentially unoccupied target strands (as a result of the stochastic binding and unbinding of strands) and increased background fluorescence because of elevated concentrations of imager strands in solution, both ultimately limiting the achievable image resolution and quality.
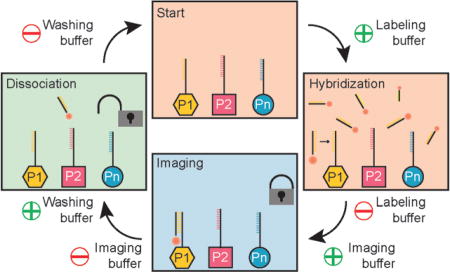
To overcome limitations of current sequential multiplexing approaches and translate DNA-based multiplexing to super-resolution techniques such as (d)STORM, STED, or SIM, we here describe a universal implementation using exchangeable DNA probes. We devised a procedure (Figure 1) that allows us to efficiently attach, image, and detach dye-modified DNA strands (“labeling” strands) to and from corresponding complementary handles coupled to different targets. To achieve this, we designed labeling strands that are optimized for stable binding during image acquisition but can still be efficiently removed from their targets using low-salinity washing buffer containing denaturing agents such as formamide. First, all target species (e.g. proteins P1 to Pn) are labeled with orthogonal DNA strands (e.g. using DNA-conjugated antibodies for proteins) in a one-pot reaction (Figure 1a). Then, buffer containing complementary labeling strands to targets P1 is introduced and DNA hybridization can occur (Figure 1b). Next, the labeling buffer is exchanged by imaging buffer (optimized for dSTORM, STED, or SIM), which does not contain any unbound labeling strands, and image acquisition is performed (Figure 1c). Subsequently, the imaging buffer is exchanged by low-salinity washing buffer containing 30% of the denaturing agent formamide (for more details see the Supporting information for experimental details), thereby facilitating the dissociation of the labeling strands from their targets by virtually “decreasing” the DNA melting temperature.[10] This washing procedure is usually performed for about 10 min, until all the labeling strands have dissociated. Finally, the washing buffer is replaced by hybridization buffer and the whole procedure is repeated for all the remaining target species. In the resulting multiplexed super-resolution micrograph, a unique pseudocolor is assigned to each imaging round (and, thus, each target). Most importantly, multiplexing is not limited by distinct spectral colors anymore, as the labeling strands for each exchange round carry the same spectral dye. The only limitation is the number of orthogonal DNA sequences (as in Exchange-PAINT), which could easily reach hundreds.
Figure 1.

a) Targets 1 – n are labeled with orthogonal approximately 12 nucleotide long DNA sequences P1–Pn. b) Dye-modified “labeling” strands stably hybridize to complementary target strands P1. c) Acquisition is carried out in imaging buffer without free labeling strands in solution. d) Imaging buffer is replaced by denaturing washing buffer to facilitate the dissociation of labeling strands from targets P1. The labeling and washing procedure is repeated for all subsequent targets. Note that the labeling strands are coupled to the same spectral dye (e.g. Alexa 647) in each round, thus enabling spectrally unlimited multiplexing.
To demonstrate the feasibility of our approach, we used self-assembled DNA origami[11] (Figure 2). We designed so-called six-helix-bundle (6HB) structures[12] carrying four orthogonal single-stranded extensions on staple strands (which allows for four labeling and imaging rounds) at specific positions[13] (Figure 2a). For Exchange-STED and Exchange-dSTORM imaging, we arranged the sequences in four spots, approximately 113 nm apart (Figure 2 a). For Exchange-SIM, we opted for a structure displaying three spots spaced about 168 nm apart (Figure S2). Each spot consists of six strands available for hybridization. Representative images of the respective imaging rounds are shown in Figure 2b,c for Exchange-STED and Exchange-dSTORM, respectively (see Figures S3 and S4 for expanded views). To assay the efficiency of our multiplexing approach, we interactively analyzed approximately 100 structures in the Exchange-STED and Exchange-dSTORM experiment. For quantification of correct versus incorrect spots in each labeling and imaging round, it is important to note that false negatives as well as false positives will lead to an “error”; however, these two “failure modes” have different root causes, and are thus important to distinguish. False positives occur when washing is inefficient, that is, labeling strands have not dissociated from their respective targets. False negatives occur when labeling or imaging is inefficient, that is, labeling strands have not hybridized to target strands or dyes are already bleached. The origami platform allows us to uncover both failure modes independently and thus reveals any potential bias of our approach. We analyzed each spot in each round separately to additionally assay for potential biases of different locations on the DNA origami structures. The results from our analysis show that on average about 91%of spots are correct in the Exchange-STED experiments (Figure 2b) and 92% in the Exchange-dSTORM experiments (Figure 2c).
Figure 2.

a) Illustration of the 6HB DNA origami “barcode”. Four spots (with 6 binding sites each), spaced approximately 113 nm apart, can be “decorated” with up to four orthogonal target sequences each (colored in red, green, cyan, and magenta). b) Resulting super-resolution images of four rounds of Exchange-STED (top) with corresponding statistical analysis (bottom). Histograms for each round depict the percentage of correctly identified spots. (i) Statistical analysis showing the number of correct spots per structure in Exchange-STED (14.6 ± 0.7, mean ± standard deviation). c) Corresponding results for Exchange-dSTORM. (ii) Correct spots per structure in Exchange-dSTORM: 14.7 ± 0.4 (mean ± standard deviation). Scale bars: 200 nm.
To demonstrate that the order of Exchange rounds does not affect the experimental outcome, we varied the order for the dSTORM experiments. We found that, indeed, the outcome of the experiment is not affected by the order. We note, that in round 2 of the dSTORM experiment (Figure 2c), we do see a higher than expected number of false positives for spots 3 and 4 (70% and 77% correct, respectively). This potentially suggests insufficient washing between rounds 1 and 2. However, we also note that the expected number of correct spots was restored in round 3. To assay the influence of different washing and hybridization times, we performed additional experiments (Figure S5), where we first decreased the incubation time with the labeling strands from 10 min to 1 min (keeping the washing times constant). In a following experiment, we increased the washing time from 2 × 3 min to 3 × 10 min (keeping the incubation time of 10 min constant). For the shorter probe incubation time, we detect a lower percentage of correctly labeled spots (true positives, see Figure S5). With longer washing times, we observe a similar performance as with our standard conditions. In conclusion, we note that our standard labeling and washing conditions (i.e. 10 min labeling, 2 × 3 min washing) should allow optimal results in exchange experiments. The statistical analysis of both Exchange-STED and -dSTORM experiments further shows that no positional dependency on the DNA origami structure was observed. There was also no bias towards false positives or negatives. Most importantly, there is also no bias towards later washing or labeling rounds, thus indicating that our approach is viable for more extensive multiplexing experiments (i.e beyond four rounds). Over four labeling and imaging rounds, we detected 14.6 ± 0.7 (mean ± standard deviation) correct spots in Exchange-STED (Figure 2b, (i)) and 14.7 ± 0.4 (mean ± standard deviation) correct spots in Exchange-dSTORM (Figure 2c, (ii)) from a total of 16 spots. Detailed experimental conditions and image processing specifics can be found in the Supporting Information.
Next, to translate our multiplexing concept from in vitro DNA origami structures to in situ labeling and imaging of protein targets in cells, we used primary and DNA-conjugated secondary antibodies against alpha-tubulin, LaminB, and TOM20. The respective secondary antibodies were coupled to three of our orthogonal target sequences. Hybridization, imaging, and washing steps were performed similarly to the in vitro studies on DNA origami. To demonstrate in situ imaging, we opted for dSTORM and STED as super-resolution methods (Figure 3), but the same procedure can be performed for SIM as well. The results for the respective three imaging rounds demonstrate the applicability of our labeling, washing, and imaging scheme to in situ cell samples. Relabeling and reimaging of targets from earlier imaging rounds is also possible with similar performance, thus high-lighting the fact that labeling strands indeed dissociate, rather than being bleached and staying bound to their target strands (Figure S6).
Figure 3.

Three-round Exchange-dSTORM and Exchange-STED in situ. a) Alpha-tubulin is imaged in round 1. b) LaminB is imaged in round 2. c) TOM20 is imaged in round 3. d) Overlay of three-round Exchange-dSTORM. e) Zoom-in of the highlighted area from (d) with the corresponding diffraction-limited representation (bottom) demonstrating the increased spatial resolution in dSTORM. f–j) Corresponding Exchange-STED results for the same protein targets. Scale bars: 5 µm (a–d and f–i), 1 µm (e, j).
In conclusion, we have devised a “universal” DNA-based multiplexed labeling and imaging technique that brings the advantages of DNA-PAINT and Exchange-PAINT imaging to super-resolution techniques such as dSTORM, STED, and SIM, while simultaneously overcoming some of the limitations of DNA-PAINT, that is, nonfluorogenic imager strands in solution and slower image acquisition. However, we also note that our presented multiplexing approach—as is the case for all sequential imaging techniques—is limited to fixed cell applications and is not compatible with the imaging of live cells. Our concept has several advantages over previously reported sequential labeling and imaging approaches for multiplexed target detection: 1) Our approach is considerably faster than sequential immunolabeling[7, 14] or DNA strand exchange cascades,[15] as immunolabeling of all target species is performed simultaneously and washing and labeling only takes about 20 min per round. Furthermore, the sample can remain on the microscope, thus no new registration is necessary. 2) Compared to Exchange-PAINT approaches,[9] no free “imager” strands are present in the imaging buffer, as labeling strands stably hybridize to their targets, which furthermore ensures that these are constantly “labeled”. This allows for optimized image-acquisition conditions for the respective super-resolution technique. 3) Targets can be relabeled and reimaged in subsequent rounds, which can provide “resistance” to bleaching and increase image efficiency. Finally, by using DNA origami structures, we were able to assay the efficiency in labeling, imaging, and washing steps in a quantitative fashion.
Supplementary Material
Acknowledgments
We thank J. B. Woehrstein and S. S. Agasti for helpful discussions and Y. Niyaz from Carl Zeiss Microscopy GmbH for the use of their SIM microscope and data acquisition support. This work was supported by the DFG through an Emmy Noether Fellowship (DFG JU 2957/1-1) and the SFB 1032 (Nanoagents for the spatiotemporal control of molecular and cellular reactions), the ERC through an ERC Starting Grant (MolMap, Grant agreement number 680241), the Max Planck Society, the Max Planck Foundation, the Center for Nanoscience (CeNS), and the Nanoinitiative Munich (NIM). M.T.S. acknowledges support from the International Max Planck Research School for Molecular and Cellular Life Sciences (IMPRS-LS). T.S. acknowledges support from the DFG through the Graduate School of Quantitative Biosciences Munich (QBM). P.Y. acknowledges support for from National Institute of Health (1-U01-MH106011-01 and 1R01EB018659-01).
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.201611729.
Conflict of interest
Competing financial interest: A patent application has been filed. P.Y. and R.J. are cofounders of Ultivue, Inc., a startup company with interest in commercializing DNA-PAINT super-resolution technology.
Contributor Information
Florian Schueder, Faculty of Physics and Center for Nanoscience, LMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich (Germany) and Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried (Germany).
Maximilian T. Strauss, Faculty of Physics and Center for Nanoscience, LMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich (Germany) and Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried (Germany)
David Hoerl, Department of Biology II and Center for Nanoscience, LMU Munich, Grosshaderner Strasse 2, 82152 Martinsried (Germany).
Dr. Joerg Schnitzbauer, Faculty of Physics and Center for Nanoscience, LMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich (Germany) and Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried (Germany)
Thomas Schlichthaerle, Faculty of Physics and Center for Nanoscience, LMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich (Germany) and Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried (Germany).
Sebastian Strauss, Faculty of Physics and Center for Nanoscience, LMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich (Germany) and Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried (Germany).
Prof. Peng Yin, Wyss Institute for Biologically Inspired Engineering and Department of Systems Biology, Harvard University, 3 Blackfan Circle, Boston, MA 02115 (USA)
Dr. Hartmann Harz, Department of Biology II and Center for Nanoscience, LMU Munich, Grosshaderner Strasse 2, 82152 Martinsried (Germany)
Prof. Heinrich Leonhardt, Department of Biology II and Center for Nanoscience, LMU Munich, Grosshaderner Strasse 2, 82152 Martinsried (Germany)
Prof. Ralf Jungmann, Faculty of Physics and Center for Nanoscience, LMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich (Germany) and Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried (Germany)
References
- 1.Hell SW, Sahl SJ, Bates M, Zhuang XW, Heintzmann R, Booth MJ, Bewersdorf J, Shtengel G, Hess H, Tinnefeld P, Honigmann A, Jakobs S, Testa I, Cognet L, Lounis B, Ewers H, Davis SJ, Eggeling C, Klenerman D, Willig KI, Vicidomini G, Castello M, Diaspro A, Cordes T. J. Phys. D. 2015;48:443001. [Google Scholar]
- 2.a) Szymborska A, de Marco A, Daigle N, Cordes VC, Briggs JA, Ellenberg J. Science. 2013;341:655–658. doi: 10.1126/science.1240672. [DOI] [PubMed] [Google Scholar]; b) Xu K, Zhong G, Zhuang X. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, Zhuang X. Nature. 2016;529:418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jungmann R, Avendano MS, Woehrstein JB, Dai M, Shih WM, Yin P. Nat. Methods. 2014;11:313–318. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharonov A, Hochstrasser RM. Proc. Natl. Acad. Sci. USA. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jungmann R, Steinhauer C, Scheible M, Kuzyk A, Tinnefeld P, Simmel FC. Nano Lett. 2010;10:4756–4761. doi: 10.1021/nl103427w. [DOI] [PubMed] [Google Scholar]
- 6.a) Rust MJ, Bates M, Zhuang X. Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Angew. Chem. Int. Ed. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008;120:6266–6271. [Google Scholar]
- 7.a) Tam J, Cordier GA, Borbely JS, Sandoval Alvarez A, Lakadamyali M. PLoS One. 2014;9:e101772. doi: 10.1371/journal.pone.0101772. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Valley CC, Liu S, Lidke DS, Lidke KA. PLoS One. 2015;10:e0123941. doi: 10.1371/journal.pone.0123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hell SW, Wichmann J. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 9.Beater S, Holzmeister P, Lalkens B, Tinnefeld P. Opt. Express. 2015;23:8630–8638. doi: 10.1364/OE.23.008630. [DOI] [PubMed] [Google Scholar]
- 10.a) McConaughy BL, Laird CD, McCarthy BJ. Biochemistry. 1969;8:3289–3295. doi: 10.1021/bi00836a024. [DOI] [PubMed] [Google Scholar]; b) Blake RD, Delcourt SG. Nucleic Acids Res. 1996;24:2095–2103. doi: 10.1093/nar/24.11.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jungmann R, Liedl T, Sobey TL, Shih W, Simmel FC. J. Am. Chem. Soc. 2008;130:10062–10063. doi: 10.1021/ja8030196. [DOI] [PubMed] [Google Scholar]
- 11.Rothemund PW. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 12.Douglas SM, Chou JJ, Shih WM. Proc. Natl. Acad. Sci. USA. 2007;104:6644–6648. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin C, Jungmann R, Leifer AM, Li C, Levner D, Church GM, Shih WM, Yin P. Nat. Chem. 2012;4:832–839. doi: 10.1038/nchem.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F. Proc. Natl. Acad. Sci. USA. 2013;110:11982–11987. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Duose DY, Schweller RM, Hittelman WN, Diehl MR. Bioconjugate Chem. 2010;21:2327–2331. doi: 10.1021/bc100348q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schweller RM, Zimak J, Duose DY, Qutub AA, Hittelman WN, Diehl MR. Angew. Chem. Int. Ed. 2012;51:9292–9296. doi: 10.1002/anie.201204304. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012;124:9426–9430. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.