

Abstract
Background
DiGeorge syndrome affects more than 3.5 million persons worldwide. Partial DiGeorge syndrome (pDGS), which is characterized by a number of gene deletions in chromosome 22, including the chicken tumor virus number 10 regulator of kinase (Crk)–like (CrkL) gene, is one of the most common genetic disorders in human subjects. To date, the role of natural killer (NK) cells in patients with pDGS remains unclear.
Objective
We sought to define the effect of pDGS-related Crk haploinsufficiency on NK cell activation and cytotoxic immunological synapse (IS) structure and function.
Methods
Inducible CrkL-silenced NK cells were used to recapitulate the pDGS, CrkL-haploinsufficient phenotype. Findings were validated by using NK cells from patients with actual pDGS. Ultimately, deficits in the function of NK cells from patients with pDGS were restored by lentiviral transduction of CrkL.
Results
Silencing of CrkL expression inhibits NK cell function. Specifically, pDGS haploinsufficiency of CrkL inhibits accumulation of activating receptors, polarization of cytolytic machinery and key signaling molecules, and activation of β2-integrin at the IS. Reintroduction of CrkL protein restores NK cell cytotoxicity.
Conclusion
CrkL haploinsufficiency causes functional NK deficits in patients with pDGS by disrupting both β2-integrin activation and activating receptor accumulation at the IS. Our results suggest that NK cell IS quality can directly affect immune status, providing a potential target for diagnosis and therapeutic manipulation in patients with pDGS and in other patients with functional NK cell deficiencies.
Keywords: CrkL, β2-integrin, partial DiGeorge syndrome, natural killer cells, functional natural killer deficiency, immunological synapse
Human DiGeorge syndrome (DGS) results from a common heterozygous deletion within chromosome 22q11.1–3 Three major genes (chicken tumor virus number 10 regulator of kinase–like [CrkL], T-box protein 1 [Tbx1], and extracellular signal-regulated kinase 2 [Erk2]) appear to be responsible for DGS phenotypes, which range from athymia with no circulating T cells (complete DGS, less than 1% of all cases) to thymic hypoplasia, decreased numbers of T cells, and less severe forms of immunodeficiency (partial DiGeorge syndrome [pDGS]).4–7 To date, a definitive DGS genotype-phenotype correlation cannot explain the clinical or immunologic differences found in patients with DGS. Intriguingly, however, deletion of CrkL alone (Crkl−/−) results in mice with a phenotype consistent with human DGS,5 highlighting the importance of this particular gene.
Crk family proteins, including Crk8 and CrkL,9 are known to play a pivotal role in controlling human natural killer (NK) cell activation and inhibition.10,11 Specifically, Crk family proteins are required for the movement of human IgG1 Fc portion (a ligand for activating receptor CD16) microclusters and phosphorylated Vav-1 (pVav-1, a central regulator of immune responses) in human NK cells. Degranulation by Crk-silenced NK cells stimulated with an antibody to CD16 is significantly reduced.10 Both target cell lysis and microtubule organization center (MTOC) polarization are impaired in NKG2D receptor (a critical natural cytotoxicity receptor in NK cells)–stimulated, CrkL-silenced NK cells, confirming that Crk family proteins also play an important role in NKG2D-mediated NK cytotoxicity.12 Although patients with pDGS appear to harbor normal numbers of NK cells,4 NK cell function has not been fully explored in these patients. Here we document impaired function in Crk-silenced and CrkL-haploinsufficient (pDGS) NK cells through modulated integrin and activating receptor signaling, with a particular focus on immunological synapse (IS) structure and function.
METHODS
The methods used in this study are described in the Methods section in this article’s Online Repository at www.jacionline.org.
RESULTS
CrkL silencing inhibits NK cell function by disrupting both lytic granule polarization and pVav-1 accumulation to the IS
NK cells recognize and kill target cells through 2 distinct mechanisms: natural cytotoxicity through receptors, such as NKG2D, and antibody-dependent cellular cytotoxicity (ADCC) through CD16.13 Optimal NK cytotoxicity occurs in a precise stepwise fashion, proceeding from conjugation with target cells to degranulation and ultimately to cytotoxicity.14 As a first step toward understanding the relationship between CrkL and NK cell function in patients with pDGS, we developed an inducible short hairpin RNA knockdown system15 to specifically silence CrkL protein (see Fig E1 in this article’s Online Repository at www.jacionline.org) in CD16-KHYG-1 cells, a human NK cell line capable of both ADCC and natural cytotoxicity.16 Effective CrkL silencing in CD16-KHYG-1 cells was verified by means of Western blotting (see Fig E2 in this article’s Online Repository at www.jacionline.org). First, we examined conjugation between NK cells and MHC class I–deficient K562 target cells, an event mediated mainly by activating receptors, such as NKG2D.17 Conjugation of CrkL-silenced NK cells was significantly less than that of wild-type (WT) and scrambled short hairpin RNA (control) NK cells (see Fig E3, A, in this article’s Online Repository at www.jacionline.org). Conjugation was also reduced in the setting of ADCC (see Fig E3, B). CrkL-silenced CD16-KHYG-1 cells conjugated less with anti–Schneider Drosophila Line 2 (S2) serum-coated S2 cells18 transfected with human intercellular adhesion molecule 1 (ICAM-1), a ligand for integrin leukocyte function–associated antigen 1 (LFA-1), than did WT and control CD16-KHYG-1 cells.
Next, we examined degranulation using surface expression of CD107a as a marker.18,19 CD107a+ staining of CrkL-silenced NK cells was decreased after both K562 (see Fig E3, C) and S2–ICAM-1 (see Fig E3, D) stimulation, showing that CrkL silencing inhibits both natural cytotoxicity– and ADCC-induced degranulation. Finally, we tested CrkL’s effect on cytotoxicity using standard chromium51 (51Cr) release (killing) assays. Both natural cytotoxicity (see Fig E3, E) and ADCC (see Fig E3, F) were impaired in CrkL-silenced CD16-KHYG-1 cells. Together, we conclude that CrkL is required for effective NK target conjugation, degranulation, and killing in both NKG2D-mediated natural cytotoxicity and CD16-mediated ADCC.
Because the IS plays a critical role in effective target cell killing in both antigen-specific cytotoxic T lymphocytes and NK cells,20–22 we hypothesized that CrkL silencing would negatively affect the structure, function, and signaling cascades of the NK cytotoxic IS. To test this, we mixed inducible CrkL knockdown CD16-KHYG-1 cells with K562 target cells and then imaged perforin (a cargo molecule in the lytic granules), tubulin (a marker of MTOC), F-actin (a critical structural molecule required for IS reorganization), and pVav-1 (Fig 1, A). CrkL silencing inhibited lytic granule polarization as measured by MTOC to IS distance in both natural cytotoxicity (K562 targets; Fig 1, B), and CD16-mediated ADCC (S2-ICAM targets, see Fig E4 in this article’s Online Repository at www.jacionline.org). Both F-actin and pVav-1 failed to accumulate at the IS in CrkL-silenced NK cells (Fig 1, C and D), indicating that CrkL silencing in CD16-KHYG-1 cells inhibits signaling cascades at the NK cytotoxic IS.
FIG 1.
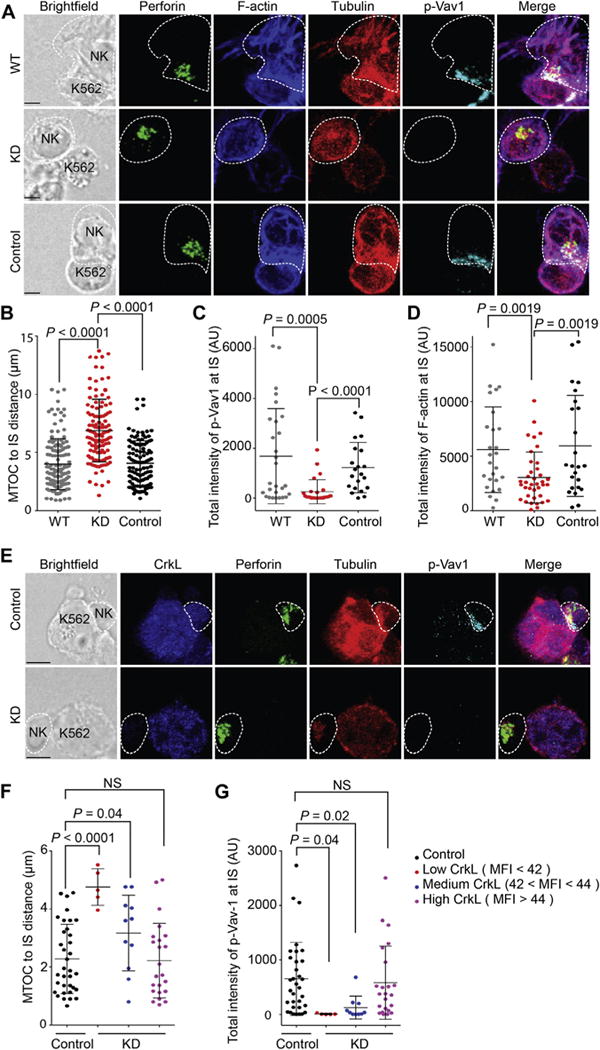
CrkL silencing disrupts both lytic granule polarization to the IS and pVav-1 accumulation. A, Confocal microscopy of WT, CrkL short hairpin RNA (shRNA) knockdown (KD), and scrambled shRNA (Control) lentivirus-transduced CD16-KHYG-1 cells in conjugate with K562 cells. Cells were stained with antibodies to visualize perforin (green), F-actin (blue), β-tubulin (red), and pVav-1 (cyan). B–D, Quantification of the distance between the MTOC and the IS (Fig 1, B) and total intensity of pVav-1 (Fig 1, C) and F-actin (Fig 1, D) at the IS. E, Confocal images of IL-2–activated primary NK cells transfected with either CrkL small interfering RNA (KD) or control small interfering RNA (Control) in conjugate with K562 cells. Cells were stained with antibodies to visualize CrkL (blue), perforin (green), β-tubulin (red), and pVav-1 (cyan). F and G, Quantitative analysis of the distance between the MTOC and the IS (Fig 1, F) and total intensity of pVav-1 at the IS (Fig 1, G) in small interfering RNA–treated NK cells stratified by CrkL MFI values. Bright-field images are shown on the left. Error bars represent SDs. Scale bars −3.0 μm. Data are from 3 independent experiments. NS, Not significant.
To validate our model system, we investigated the role of CrkL in human primary NK cells. IL-2–activated NK cells were first transfected with small interfering RNA (CrkL or scrambled) and then mixed with K562 target cells (Fig 1, E). To overcome incomplete CrkL silencing, we selected only those cells with the highest degree of silencing (by means of quantitative CrkL fluorescence10) for further analysis. Degree of CrkL silencing correlated positively with MTOC to IS distance (Fig 1, F) and inversely with pVav-1 intensity (Fig 1, G), confirming that CrkL silencing in primary NK cells inhibits lytic granule polarization and IS signaling (through pVav-1).
CrkL silencing inhibits activating receptor accumulation and integrin clustering
F-actin regulates the accumulation of activating receptors.23,24 We hypothesized that the inhibition of IS F-actin accumulation seen with CrkL silencing would, in turn, result in decreased activating receptor and signaling molecule accumulation at the IS. To test this hypothesis, we imaged NK cells on glass-supported ICAM-1– and anti-CD16–containing planar lipid bilayers using 3-dimensional confocal resolution and super-resolution stimulated emission depletion microscopy. In experiments with WT and control NK cells, ICAM-1 and anti-CD16 clustered normally (Fig 2, A). As a control, neither CD16-KHYG-1 nor human primary NK cells responded to bilayers containing an isotype control anti-mouse IgG1 (see Fig E5 in this article’s Online Repository at www.jacionline.org). In contrast, after CrkL silencing, dispersed anti-CD16 clusters were observed (Fig 2, A), which is consistent with observations from the previous Crk knockdown Fc of human IgG1 (a ligand for CD16)–containing bilayer studies.10 This defect in central accumulation of anti-CD16 and ICAM-1 likely results in some of the functional IS defects (eg, impaired lytic granule polarization) we observed. Perforin intensity at the focal bilayer plane (a marker of lytic granule polarization to the NK cell–bilayer synapse) was significantly less in CrkL-silenced NK cells compared with that seen in WT and control cells (Fig 2, B), supporting a defect in activating receptor signaling at the NK cytotoxic IS.
FIG 2.
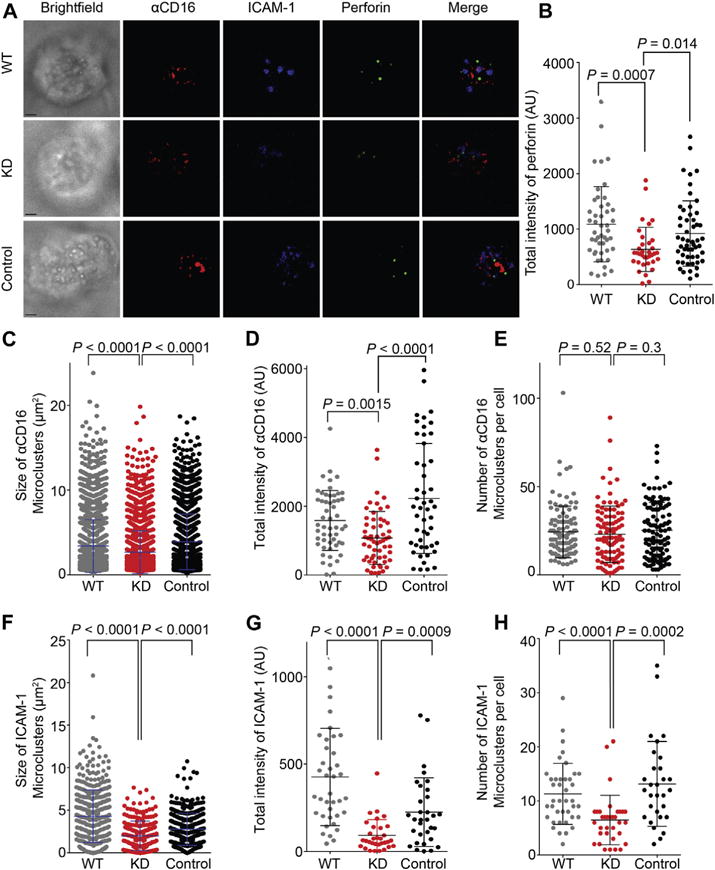
CrkL silencing inhibits activating receptor CD16 and integrin clustering. A, Confocal images of WT, CrkL short hairpin (shRNA; KD), and scrambled shRNA (Control) CD16-KHYG-1 cells activated on lipid bilayers carrying anti-CD16–Alexa Fluor 568 (red) and ICAM-1–Alexa Fluor 647 (blue). Fixed and permeabilized NK cells were stained for perforin, followed by Alexa Fluor 488–conjugated secondary antibody (green) to visualize lytic granules. B, Quantification of the total intensity of polarized perforin at the lipid bilayer focal plane. C–E, Quantification of the size (Fig 2, C), total intensity (Fig 2, D), and number (Fig 2, E) of anti-CD16 microclusters on lipid bilayers carrying ICAM-1 and anti-CD16. F–H, Quantification of the size (Fig 2, F), total intensity (Fig 2, G), and number (Fig 2, H) of ICAM-1 microclusters on lipid bilayers. Error bars represent SDs. Scale bars −3.0 μm. Data represent 2 to 6 independent experiments.
Given the importance of activating receptor microclusters in immune responses,20,25 we further quantified IS accumulation of CD16, NKG2D, and 2B4, as well as the integrin LFA-1, which is required for activating receptor movement.20,26 The size and total intensity of anti-CD16 on CrkL-silenced NK cells were decreased (Fig 2, C and D), although the number of anti-CD16 microclusters was unaffected (Fig 2, E). We suspect that the latter might simply be related to overexpression of CD16 on the CD16-KHYG-1 cell line.16 Size, total intensity, and number of ICAM-1 (the ligand for LFA-1) microclusters were also significantly reduced on CrkL-silenced compared with WT and control NK cells (Fig 2, F to H). To test whether CrkL similarly affects natural cytotoxicity receptors, we imaged NK cells on bilayers containing anti-NKG2D and anti-2B4 in the presence of ICAM-1. This time, in addition to decreased total intensity of polarized perforin at the focal bilayer plane, we observed decreased size, total intensity, and number of anti-NKG2D microclusters with CrkL-silenced NK cells (see Fig E6 in this article’s Online Repository at www.jacionline.org). To rule out the possibility of decreased overall expression of activating receptors on CrkL-silenced NK cells, we measured the mean fluorescence intensity (MFI) of CD16, NKG2D, CD11a/CD18 (2 components of LFA-1), ICAM-1, and F-actin using flow cytometry and found comparable overall expression levels on WT and CrkL-silenced NK cells (see Fig E7 in this article’s Online Repository at www.jacionline.org). Thus CrkL silencing reduced the clustering of activating receptors and integrin clustering at the IS.
Defects in lytic granule polarization, activating receptor clustering, and integrin activation in NK cells from patients with pDGS
The experiments described thus far involve CrkL silencing in CD16-KHYG-1 and human primary NK cells. To understand the clinical implications of CrkL deficiency, we turned to primary NK cells from patients with pDGS. Most patients with pDGS carry CrkL gene deletions in chromosome 22q11.2.27 As shown both in Table E1 in this article’s Online Repository at www.jacionline.org and by other groups, lymphocyte phenotyping of patients with pDGS shows variable T-cell depletion with normal NK cell numbers.28 To date, however, there are no published descriptions of NK cell function in these patients. Given the pivotal role of CrkL in NK cells, we hypothesized that NK cells from patients with pDGS would exhibit functional impairments consistent with CrkL silencing. To test this, we first performed standard killing assays, incubating PBMCs from patients with pDGS with 51Cr-labeled K562 (natural cytotoxicity) or rituximab (anti-CD20)–coated Raji target cells (ADCC). Detailed patient information is presented in Table E2 in this article’s Online Repository at www.jacionline.org. As predicted, NK cell cytotoxicity was significantly decreased in patients with pDGS compared with that seen in healthy control subjects (Fig 3). To our knowledge, although NK cell dysfunction is common to many primary immunodeficiencies,29 this is the first demonstration of functional NK deficiency in patients with pDGS.
FIG 3.
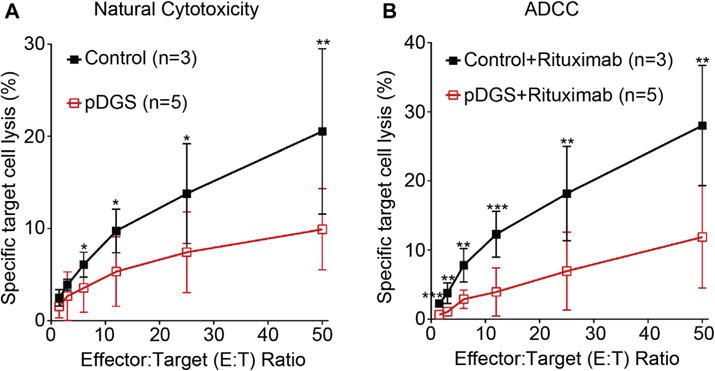
Decreased cytotoxicity of NK cells from patients with pDGS. A, The natural cytotoxicity of human NK cells from 5 patients with pDGS and 3 healthy control subjects was measured with 51Cr-labeled K562 cells at 37°C for 4 hours. B, ADCC was measured with 51Cr-labeled Raji cells in the presence of rituximab. Error bars represent SDs.
To understand the molecular mechanisms underlying functional NK deficiency in patients with pDGS, we investigated the structure, function, and signaling cascades of cytotoxic synapses between NK cells from patients with pDGS and susceptible target cells or lipid bilayers (2 complementary systems). Similar to our findings in CrkL-silenced cells, lytic granules and MTOC did not polarize toward the cytotoxic IS in NK cells from patients with pDGS (Fig 4, A and B). Although granzyme B content was decreased under both resting and activation conditions (see Fig E8, A and B, in this article’s Online Repository at www.jacionline.org), perforin content was normal (see Fig E8, C and D). Although CD107a degranulation in cells from patients with pDGS was marginally decreased (as measured by using flow cytometry) compared with that seen in healthy control subjects (see Fig E9 in this article’s Online Repository at www.jacionline.org), surface CD107a staining correlated positively with CrkL expression in NK cells from both healthy control subjects and patients with pDGS (see Fig E10 in this article’s Online Repository at www.jacionline.org).
FIG 4.
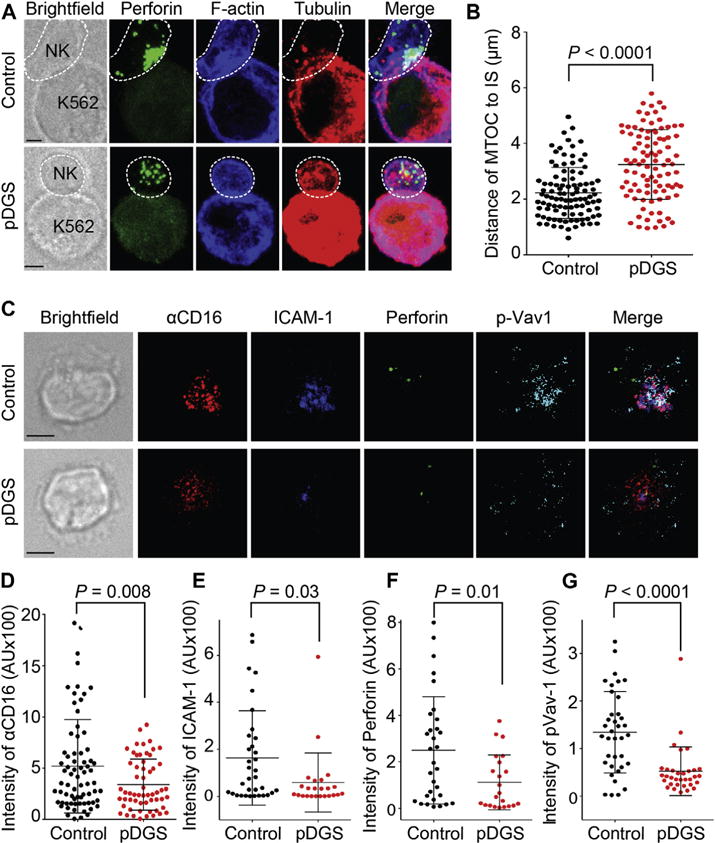
Defects in lytic granule polarization, activating receptor clustering, and signaling in NK cells from patients with pDGS. A, Confocal images of NK cells from healthy control subjects (Control) and patients with pDGS (pDGS) in conjugate with K562 cells. Cells were stained with antibodies against perforin (green), phalloidin (blue), and β-tubulin (red). B, Quantification of the distance between the MTOC and the IS in primary NK cells. Error bars represent SDs. Data are from 3 independent experiments. C, Confocal images of NK cells from control subjects and patients with pDGS stimulated on bilayers carrying anti-CD16–Alexa Fluor 568 (red) and ICAM-1–Alexa Fluor 647 (blue). Cells were stained with anti-perforin–Alexa Fluor 488 (green) and anti–pVav-1–Alexa Fluor 405 (cyan). Data represent 4 independent experiments. D–G, Quantification of total fluorescent intensity of anti-CD16 (Fig 4, D), ICAM-1 (Fig 4, E), perforin (Fig 4, F), and pVav-1 (Fig 4, G) on bilayers carrying ICAM-1 and anti-CD16.
Using our lipid bilayer system, we further demonstrated reduced intensities of anti-CD16, ICAM-1, perforin, and pVav-1 at the lipid bilayer–pDGS NK cell focal plane from patients with pDGS (Fig 4, C to G). Size, total intensity, and numbers of anti-CD16 and ICAM-1 microclusters were significantly decreased compared with those in NK cells from healthy control subjects (see Fig E11 in this article’s Online Repository at www.jacionline.org). Similar results were obtained in NK cells stimulated on bilayers carrying anti-NKG2D and anti-2B4 in the presence of ICAM-1 (see Fig E12 in this article’s Online Repository at www.jacionline.org). Again, overall expressions of CD16, NKG2D, and LFA-1 (MFI by means of flow cytometry) were comparable in patients and control subjects (see Fig E13 in this article’s Online Repository at www.jacionline.org). To summarize, NK cells from patients with pDGS exhibit impaired cytotoxicity, which is characterized by reduced pVav-1, failure of activating receptor and integrin accumulation at the IS, and ultimately impaired lytic granule polarization. We hypothesize that receptor accumulation and downstream defects in cytolytic machinery result from defective integrin activation at the IS.
Movement of activating receptors on NK cells depends on cytoskeletal reorganization and β2-integrin–LFA-1 signaling.20,26 Given that accumulation of activating receptors is impaired in CrkL-silenced and haploinsufficient cells, we hypothesized that CrkL controls LFA-1 activation. To test this hypothesis, we used 2 antibodies (mAb 24 for the high-affinity conformation30 and KIM127 for the intermediate-affinity conformation31) specific for the activated form of LFA-1. NK cells were defined by flow cytometry as live CD45+CD3−CD56+cells (see Fig E14 in this article’s Online Repository at www.jacionline. org). Interestingly, incubation with K562 target cells results in significantly less open and intermediate conformation LFA-1 on NK cells from patients with pDGS (CrkL haploinsufficient) than on NK cells from healthy control subjects, as measured by the percentage and MFI of mAb 24+ and KIM127+ cells over time (Fig 5), although there were no significant differences between resting NK cells from healthy control subjects and patients with pDGS (see Fig E15 in this article’s Online Repository at www.jacionline.org). Percentage and MFI of mAb24 (Fig 5, D) and KIM127 (Fig 5, E) on activated NK cells from 5 control subjects and 4 to 6 patients with pDGS are summarized, illustrating impaired LFA-1 activation in NK cells from patients with pDGS. As an internal control, total CD11a expression on NK cells from patients with pDGS and healthy control subjects was comparable (see Fig E14, B and C). As a positive control, phorbol 12-myristate 13-acetate and ionomycin were able to induce open-conformation LFA-1 (see Fig E14, D). We conclude that CrkL is required for LFA-1 activation in NK cells, providing a mechanism by which activating receptor accumulation and ultimately cytolytic machinery localization and function are impaired in patients with pDGS.
FIG 5.
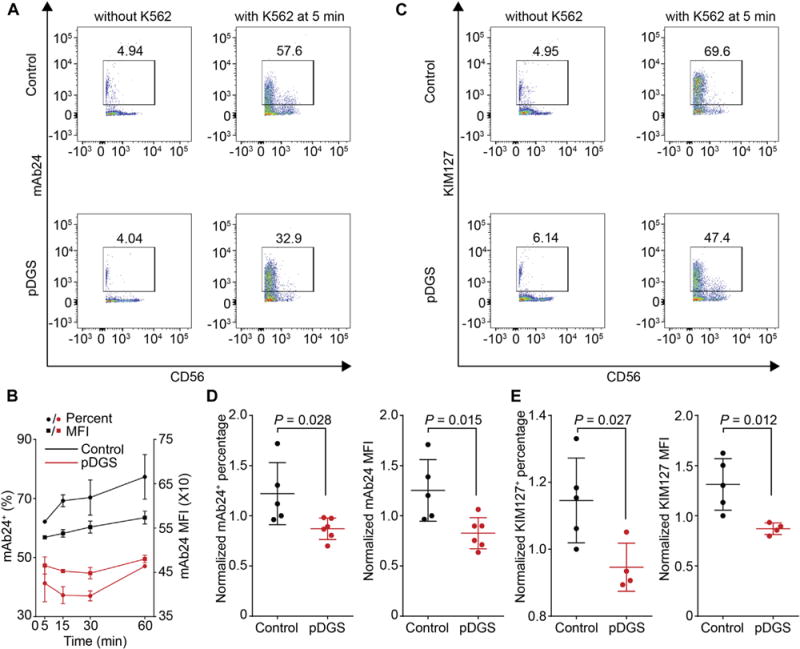
Defective integrin signaling in NK cells from patients with pDGS. A, Flow cytometric analysis of activated LFA-1 (mAb 24+) on primary NK cells with or without K562 stimulation at 5 minutes. B, Kinetics of activated LFA-1 (mAb 24+) at different time points. C, Percentage of intermediate LFA-1 (KIM127+) on primary NK cells with or without K562 stimulation at 5 minutes. D, Percentage (left) and MFI (right) of activated (mAb 24+) LFA-1 after mixing with K562 target cells for 5 minutes. Data are pooled from 5 control subjects and 6 patients with pDGS. E, Percentage (right) and MFI (right) of intermediate (KIM127+) LFA-1 after mixing with K562 target cells for 5 minutes. Data are pooled from 5 control subjects and 4 patients with pDGS.
Reconstitution of CrkL restores NK cell function in patients with pDGS
We next sought to determine whether reconstitution of CrkL expression could restore NK function in cells from patients with pDGS. First, however, we evaluated expression of CrkL and the other commonly deleted genes on chromosome 22q11, Tbx1 and Erk2, using intracellular staining of NK cells from at least 14 patients with pDGS. Antibody specificities were validated by means of Western blotting and confocal microscopy (see Fig E16 in this article’s Online Repository at www.jacionline.org). Although Tbx1 and Erk2 expression levels in patients with pDGS were comparable with those of healthy control cells, CrkL expression was considerably lower, as determined both by means of intracellular staining and Western blotting (see Fig E17 in this article’s Online Repository at www.jacionline.org). Freshly isolated PBMCs from 7 of these patients with pDGS were transduced with lentiviral particles encoding green fluorescent protein (GFP) alone or CrkL-GFP (see Table E1) to reconstitute CrkL in NK cells from patients with pDGS. Transduced NK cells were defined by means of flow cytometry as live CD3−CD56+GFP+cells (see Fig E18 in this article’s Online Repository at www.jacionline.org). We first tested whether CrkL reconstitution correlates with enhanced cytolytic machinery expression (perforin and granzyme B) or cytokine production. Interestingly, the MFI of perforin and granzyme B in NK cells from these 7 patients with pDGS increased significantly (Fig 6, A). Similarly, the percentage of IFN-γ+ and TNF-α+ CrkL-reconstituted NK cells increased significantly in 6 out of 7 patients with pDGS (Fig 6, B). Together, these data demonstrate that CrkL can restore NK cytolytic machinery and cytokine production in patients with pDGS.
FIG 6.
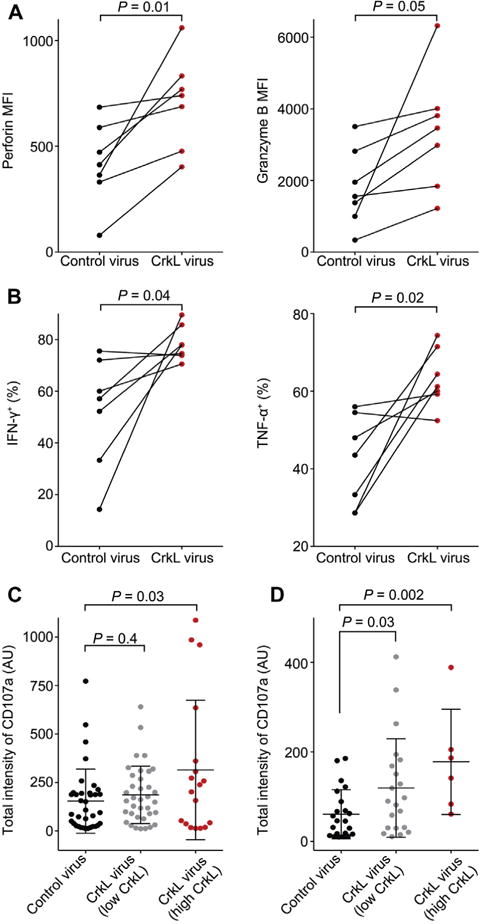
Reconstitution of CrkL restores NK cell function in patients with pDGS. PBMCs from 7 patients with pDGS were transduced with GFP (Control virus) or CrkL-GFP (CrkL virus) lentivirus. A, MFI of perforin and granzyme B in GFP+ NK cells in response to K562 stimulation. B, Percentage of IFN-γ+ and TNF-α+ transduced NK cells in response to phorbol 12-myristate 13-acetate and ionomycin stimulation. C and D, Quantification of CD107a total intensity on the plasma membrane. Transduced NK cells were activated on bilayers carrying anti-CD16–Alexa Fluor 568 and unlabeled ICAM-1 (Fig 6, C) or anti-NKG2D–Alexa Fluor 568 in the presence of unlabeled ICAM-1 and anti-2B4 (Fig 6, D). Data were pooled from 2 independent experiments. P values are for the paired t test (Fig 6, A and B) or the Student t test (Fig 6, C and D).
We further tested restoration by means of imaging degranulation on bilayers carrying ICAM-1 and anti-CD16. We included directly labeled Fab of a CD107a mAb in the bilayer imaging chamber and imaged with 3-dimensional confocal microscopy.20 As expected, when NK cells from healthy control subjects were incubated on bilayers carrying ICAM-1 and anti-CD16, strong CD107a staining accumulated at the center of the synapse (see Fig E19, A, in this article’s Online Repository at www.jacionline.org). When healthy NK cells were incubated on bilayers carrying isotype control mouse IgG1 (mIgG1), no detectable CD107a signal was observed (see Fig E19, B). Strikingly, when CrkL-reconstituted NK cells from patients with pDGS were incubated on bilayers carrying ICAM-1 and CD16 mAb (Fig 6, C) or ICAM-1 and anti-NKG2D (Fig 6, D), CD107a surface intensity increased significantly compared with that seen in NK cells from patients with pDGS transduced with GFP alone. From this, we conclude that CrkL can restore in vitro function in NK cells from patients with pDGS.
DISCUSSION
Immune cell activation, one of the most fundamental processes in immunology and cell biology, is mediated mainly through the IS.32 Although the biology of the IS has been studied extensively over the past 3 decades, the clinical relevance of IS structure and function remains poorly understood. Here we demonstrate, for the first time, functional NK cell deficiency in patients with pDGS. We not only document clinical relevance using NK cells and pDGS as cell and disease models, but also propose a new paradigm for control of NK activation: the adaptor protein CrkL controls NK cell function by influencing integrin activation and activating receptor signaling (Fig 7) through decreased pVav-1 levels. In CrkL-silenced NK cells and pDGS-related CrkL haploinsufficiency, defective integrin activation and defective activating receptor clustering and signaling impair lytic granule polarization and ultimately inhibit target cell killing. Reconstitution of CrkL restores NK cell function.
FIG 7.
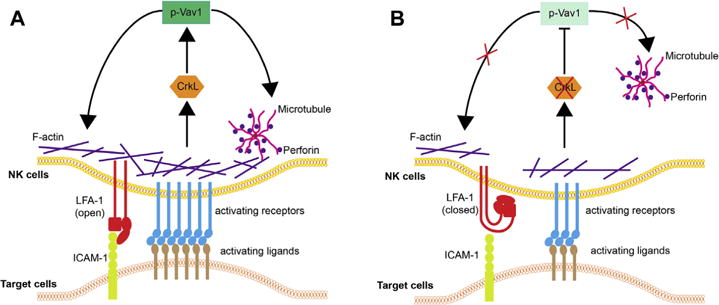
A new model for the adaptor protein CrkL in controlling NK cell activation. A, NK cell activation is triggered by activating receptor (blue bars) signaling. Activating receptors accumulate toward the center of the IS. The adaptor protein CrkL links cytoskeletal scaffold proteins to guanine nucleotide exchange factor Vav-1. Tyrosine phosphorylated Vav1 (p-Vav1) is a central regulator of T-cell and NK cell IS. pVav-1 promotes polarization of MTOC and lytic granules to the IS, as well as activating receptor clustering by promoting F-actin reorganization. Integrin LFA-1 is in open conformation because of activating receptor triggering (red bars). B, After CrkL silencing (or CrkL haploinsufficiency in patients with pDGS, red X), activation strength is reduced (blue bars). In the absence of CrkL, Vav-1 phosphorylation is greatly reduced, less F-actin reorganization occurs, and MTOC and lytic granules cannot polarize to the IS. The majority of integrin remains inactive (closed) because of less activating receptor clustering and signaling.
Consistent with previous studies,10,12 we found that CrkL silencing inhibits both ADCC and natural cytotoxicity. Here CrkL silencing resulted in lower pVav-1 levels in both cell-cell conjugate and lipid bilayer experiments. Given that Vav-1 molecules serve as a signaling integration hub for F-actin reorganization,33 we predict that the low pVav-1 levels seen in our CrkL-silenced cells contribute to impaired IS signaling through decreased IS accumulation of F-actin. Both F-actin networks and integrin signaling play important roles in NK cell lytic granule polarization and activating receptor clustering.26,34 Expectedly, the total amount and number of activating receptor microclusters on bilayers decreased in both CrkL-silenced and haploinsufficient cells. Because comparable overall expression levels of CD16, NKG2D, and LFA-1 were seen in CrkL-KD and WT cell lines, as well as in NK cells from healthy control subjects and patients with pDGS, we believe that defective receptor accumulation truly reflects defective receptor signaling. Indeed, high- and intermediate-affinity conformation LFA-1, markers for LFA-1 activation, which is a prerequisite for integrin signaling and activating receptor localization to the IS, were decreased in NK cells from patients with pDGS. These defects in turn lead to impaired lytic granule polarization and decreased NK cell cytotoxicity. Although our data most strongly support defects in LFA-1 outside-in signaling (lytic granule polarization) and activating receptor signaling, they do not preclude defects in integrin inside-out signaling (conjugate formation). CrkL silencing likely impairs all 3 of these mechanisms, ultimately leading to defective conjugation, granule polarization, and cytotoxicity.
Genetic screens, such as fluorescence in situ hybridization and the chromosome microarray assay, are powerful tools for diagnosing pDGS. However, these techniques do not necessarily reflect protein expression (haploinsufficiency can induce transcriptional compensation35,36), which ultimately determines phenotypic penetrance. Additionally, dose-sensitive interaction of CrkL and Tbx1 also plays a critical role in the penetrance and expressivity of a DGS-like phenotype in mice.37 In our study, although overall significantly decreased in patients with pDGS, CrkL protein levels varied by subject. In contrast, we did not observe reduced Tbx1 levels despite genetic deletion of Tbx1. This might be due to the extremely low expression of Tbx1 in PBMCs or to gene copy number variants.
Our results suggest that NK cell IS quality can directly affect immune status, providing a potential target for diagnosis and therapeutic manipulation. In this study all of the patients with pDGS had recurrent respiratory tract infections, 1 had viral cutaneous syndrome (warts and molluscum), 1 had cutaneous fungal infection with seborrheic dermatitis, and 4 had confirmed metapneumoviral sinopulmonary disease (see the Discussion section in this article’s Online Repository at www.jacionline.org for additional clinical details). Although it is possible that low T-cell numbers could contribute to infectious status, T-cell function (as measured by mitogen-induced proliferation) and overall distribution of lymphocytes were both normal in these patients. Our results suggest a new clinical application for the IS, providing proof of concept for using the IS to assess immune cell function in other disease states. Additionally, given the restoration of NK cell function in patients with pDGS by means of reconstitution of CrkL expression, we propose autologous stem cell CrkL gene therapy as a potential therapeutic strategy for pDGS. Documenting that haploinsufficiency of CrkL impairs integrin and activating receptor signaling and leads to functional NK deficiency lays the preclinical groundwork for this type of novel approach.
Supplementary Material
Key messages.
CrkL silencing inhibits both ADCC and natural cytotoxicity in human NK cells.
Haploinsufficiency of CrkL in patients with pDGS leads to functional NK deficits with impaired β2-integrin activation, key signaling molecule accumulation, and activating receptor localization at the IS.
These deficits can be restored by CrkL reconstitution, highlighting a potential therapeutic approach to functional NK cell deficiency in patients with pDGS.
Acknowledgments
Supported in part by the Baylor–University of Texas at Houston Center for AIDS Research (Core Support Grant AI36211) from the National Institute of Allergy and Infectious Diseases, the Caroline Wiess Law Fund for Research in Molecular Medicine (2531319101), the Texas Children’s Hospital Pediatric Pilot Research Fund (2531319301), and the Lymphoma SPORE Developmental Research Program from Baylor College of Medicine and the Methodist Research Institute (2531319302 and P50 CA126752).
We thank Stephan Kissler (Harvard Medical School) and Marco Herold (Walter and Eliza Hall Institute) for pUGM, psiCheck-2, pH1t-flex, and FH1T-UTG vectors; Eric Long (NIAID) for S2 cell and anti-S2 serum; Bruce Mayer (UConn Health) for CrkL-EYFP plasmid; and Ankita Patel, Samantha Penney, and Asbjorg Stray-Pedersen (Baylor College of Medicine) for pDGS genetic data mining.
Abbreviations
- ADCC
Antibody-dependent cellular cytotoxicity
- Crk
Chicken tumor virus number 10 regulator of kinase
- CrkL
Crk-like
- DGS
DiGeorge syndrome
- Erk2
Extracellular signal-regulated kinase 2
- GFP
Green fluorescent protein
- ICAM-1
Intercellular adhesion molecule 1
- IS
Immunological synapse
- LFA-1
Leukocyte function–associated antigen 1
- MFI
Mean fluorescence intensity
- MTOC
Microtubule organization center
- NK
Natural killer
- pDGS
Partial DiGeorge syndrome
- pVav-1
Phosphorylated Vav-1
- S2
Schneider Drosophila Line 2
- Tbx1
T-box protein 1
- WT
Wild-type
Footnotes
Disclosure of potential conflict of interest: J. S. Orange has received consultancy fees from ADMA Biologics, Walgreens, CSL Behring, Baxter, Atlantic Research Group, and Amerisource Bergen; has received or has grants pending from CSL Behring; has received payment for delivering lectures from Baxter and CSL Behring; and receives royalties from Unimed Publishing and UpToDate. D. Liu’s institution has received funding from Celgene (300011519). The rest of the authors declare that they have no relevant conflicts of interest.
References
- 1.Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet. 1999;8:1157–67. doi: 10.1093/hmg/8.7.1157. [DOI] [PubMed] [Google Scholar]
- 2.Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–9. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- 3.Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–95. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- 4.McLean-Tooke A, Barge D, Spickett GP, Gennery AR. Immunologic defects in 22q112 deletion syndrome. J Allergy Clin Immunol. 2008;122:362–7, e1–4. doi: 10.1016/j.jaci.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 5.Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q112 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–8. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- 6.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–91. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 7.Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, et al. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc Natl Acad Sci U S A. 2008;105:17115–20. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuda M, Mayer BJ, Fukui Y, Hanafusa H. Binding of transforming protein, P47gag-crk, to a broad range of phosphotyrosine-containing proteins. Science. 1990;248:1537–9. doi: 10.1126/science.1694307. [DOI] [PubMed] [Google Scholar]
- 9.Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu D, Peterson ME, Long EO. The adaptor protein Crk controls activation and inhibition of natural killer cells. Immunity. 2012;36:600–11. doi: 10.1016/j.immuni.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D. The adaptor protein Crk in immune response. Immunol Cell Biol. 2014;92:80–9. doi: 10.1038/icb.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segovis CM, Schoon RA, Dick CJ, Nacusi LP, Leibson PJ, Billadeau DD. PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. J Immunol. 2009;182:6933–42. doi: 10.4049/jimmunol.0803840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–58. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8:713–25. doi: 10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng P, Kissler S. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes. 2013;62:896–904. doi: 10.2337/db12-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alpert MD, Harvey JD, Lauer WA, Reeves RK, Piatak M, Jr, Carville A, et al. ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PLoS Pathog. 2012;8:e1002890. doi: 10.1371/journal.ppat.1002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song H, Kim J, Cosman D, Choi I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell Immunol. 2006;239:22–30. doi: 10.1016/j.cellimm.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic granule polarization and degranulation controlled by different receptors in resting NK cells. J Exp Med. 2005;202:1001–12. doi: 10.1084/jem.20051143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, Bryceson YT, Meckel T, Vasiliver-Shamis G, Dustin ML, Long EO. Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity. 2009;31:99–109. doi: 10.1016/j.immuni.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dustin ML, Long EO. Cytotoxic immunological synapses. Immunol Rev. 2010;235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–7. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 23.Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, et al. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science. 2001;293:2263–5. doi: 10.1126/science.1063486. [DOI] [PubMed] [Google Scholar]
- 24.Ritter AT, Angus KL, Griffiths GM. The role of the cytoskeleton at the immunological synapse. Immunol Rev. 2013;256:107–17. doi: 10.1111/imr.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–3. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 26.Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. 2008;224:70–84. doi: 10.1111/j.1600-065X.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gennery AR. Immunological features of 22q11 deletion syndrome. Curr Opin Pediatr. 2013;25:730–5. doi: 10.1097/MOP.0000000000000027. [DOI] [PubMed] [Google Scholar]
- 28.Ferrando-Martinez S, Lorente R, Gurbindo D, De Jose MI, Leal M, Munoz-Fernandez MA, et al. Low thymic output, peripheral homeostasis deregulation, and hastened regulatory T cells differentiation in children with 22q112 deletion syndrome. J Pediatr. 2014;164:882–9. doi: 10.1016/j.jpeds.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 29.Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132:515–26. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Theorell J, Schlums H, Chiang SC, Huang TY, Tattermusch A, Wood SM, et al. Sensitive and viable quantification of inside-out signals for LFA-1 activation in human cytotoxic lymphocytes by flow cytometry. J Immunol Methods. 2011;366:106–18. doi: 10.1016/j.jim.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Robinson MK, Andrew D, Rosen H, Brown D, Ortlepp S, Stephens P, et al. Antibody against the Leu-CAM beta-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J Immunol. 1992;148:1080–5. [PubMed] [Google Scholar]
- 32.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–96. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 33.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–86. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 34.Barber DF, Faure M, Long EO. LFA-1 contributes an early signal for NK cell cytotoxicity. J Immunol. 2004;173:3653–9. doi: 10.4049/jimmunol.173.6.3653. [DOI] [PubMed] [Google Scholar]
- 35.Guidi CJ, Veal TM, Jones SN, Imbalzano AN. Transcriptional compensation for loss of an allele of the Ini1 tumor suppressor. J Biol Chem. 2004;279:4180–5. doi: 10.1074/jbc.M312043200. [DOI] [PubMed] [Google Scholar]
- 36.Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–72. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- 37.Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10:81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.