

Abstract
To examine the intracellular trafficking and translocation of GLUT4 in skeletal muscle, we have generated transgenic mouse lines that specifically express a GLUT4-EGFP (enhanced green fluorescent protein) fusion protein under the control of the human skeletal muscle actin promoter. These transgenic mice displayed EGFP fluorescence restricted to skeletal muscle and increased glucose tolerance characteristic of enhanced insulin sensitivity. The GLUT4-EGFP protein localized to the same intracellular compartment as the endogenous GLUT4 protein and underwent insulin- and exercise-stimulated translocation to both the sarcolemma and transverse-tubule membranes. Consistent with previous studies in adipocytes, overexpression of the syntaxin 4-binding Munc18c isoform, but not the related Munc18b isoform, in vivo specifically inhibited insulin-stimulated GLUT4-EGFP translocation. Surprisingly, however, Munc18c inhibited GLUT4 translocation to the transverse-tubule membrane without affecting translocation to the sarcolemma membrane. The ability of Munc18c to block GLUT4-EGFP translocation to the transverse-tubule membrane but not the sarcolemma membrane was consistent with substantially reduced levels of syntaxin 4 in the transverse-tubule membrane. Together, these data demonstrate that Munc18c specifically functions in the compartmentalized translocation of GLUT4 to the transverse-tubules in skeletal muscle. In addition, these results underscore the utility of this transgenic model to directly visualize GLUT4 translocation in skeletal muscle.
The stimulation of glucose uptake in adipose and muscle tissues primarily occurs through the translocation of the GLUT4 glucose transporter isoform from intracellular storage sites to the cell surface membranes (1–4). Insulin stimulation of this process requires the tyrosine phosphorylation of the insulin receptor substrate family of proteins and subsequent activation of the type 1 phosphatidylinositol (PI)1 3-kinase (5–12). Although the precise signaling steps that occur downstream of the PI 3-kinase ultimately leading to GLUT4 translocation have remained elusive, recent studies have begun to resolve the specific trafficking, docking, and fusions events. In adipocytes, it is well established that GLUT4 storage compartments contain vesicle-localized proteins (v-SNAREs) that specifically interact with cognate cell surface target proteins (t-SNAREs) at the plasma membrane to promote vesicle docking and fusion (13, 14). Insulin-stimulated GLUT4 translocation is dependent upon the interaction of the v-SNARE, VAMP2, with the plasma membrane t-SNAREs, syntaxin 4 and SNAP23 (15–21). Furthermore, the syntaxin 4-binding protein, Munc18c, has been shown to specifically modulate insulin-sensitive GLUT4 translocation in 3T3L1 adipocytes (22–24). However, it is important to recognize that the majority of these studies have been performed in cultured 3T3L1 adipocytes and L6 myotubes due to the inherent technical limitations in the study of GLUT4 trafficking in adipocytes and skeletal muscle in vivo.
Skeletal muscle is the primary depot for insulin-stimulated disposal of blood glucose in vivo and has unique functional and structural characteristics that distinguish it from adipose tissue. Like adipocytes, insulin stimulation of glucose uptake in skeletal muscle also occurs through a PI 3-kinase pathway. But in addition, muscle contraction/exercise also potently stimulates glucose uptake and GLUT4 translocation independently of the PI 3-kinase pathway (25–29). Furthermore, unlike the simpler single plasma membrane of adipocytes, skeletal muscle has two functionally distinct surface membranes, the sarcolemma and transverse-tubule, both of which function as GLUT4 protein acceptor membranes in the translocation process (30–33).
Currently, there is little functional data with regard to the v-SNARE and t-SNARE proteins involved in either insulin- or exercise/contraction-stimulated GLUT4 translocation in skeletal muscle. Thus, to develop a more tractable skeletal muscle system to investigate GLUT4 translocation, we have generated transgenic mice specifically expressing a GLUT4-EGFP fusion protein in skeletal muscle. In this article, we demonstrate that the GLUT4-EGFP fusion protein displays an identical distribution and trafficking pattern as the endogenous GLUT4 protein. These animals provide a model system to investigate skeletal muscle GLUT4 translocation in vivo and can be used to distinguish between sarcolemma and transverse-tubule GLUT4 translocation through the function of the syntaxin 4-binding protein, Munc18c.
Experimental Procedures
Materials
The rabbit polyclonal GLUT4 and Munc18c antibodies were obtained as described previously (24). Monoclonal antibodies 8D5 (specific for β-dystroglycan) and IIID5E1 (specific for the α1 subunit of the L-type calcium channel) were kindly provided by Dr. Kevin P. Campbell, University of Iowa. A rabbit polyclonal antibody against recombinant GFP was obtained from CLONTECH (Palo Alto, CA). A sheep antibody was prepared against the cytosolic domain of syntaxin 4 was obtained as described previously (20). A rabbit Munc18 polyclonal antibody that recognizes Munc18b was purchased from Affinity Bioreagents (Golden, CO). Vectashield and the Monoclonal on Mouse kit were purchased from Vector Laboratories (Burlingame, CA). Adenoviral vectors encoding full-length, Flag-tagged Munc18c and Munc18b were prepared by the University of Iowa Gene Therapy Vector Core according to standard procedures.
Transgenic Mice
A 2.2-kilobase fragment of the GLUT4 cDNA fused in-frame with the EGFP cDNA was ligated into the transgenic vector pStec which contains the SV40 polyadenylation and intron sequences. A 2.2-kilobase fragment of the human skeletal muscle actin (HSA) promoter was isolated from the pHSAaNeor plasmid and ligated upstream of the pStec-GLUT4/EGFP vector. The resulting vector, pStec-HSA-GLUT4/EGFP, was linearized with AflIII, microinjected into the nucleus of pre-implanation embryos, and transferred to the oviduct of psuedo-pregnant mice as described previously by the University of Iowa Transgenic Animal Facility. The resulting pups were screened for the presence of the transgene by polymerase chain reaction of genomic DNA and the positive animals bred onto the C57Bl/6 background.
Tissue Extracts
Various tissues were dissected from carbon dioxide asphixiated mice and frozen in liquid nitrogen. The tissues were diced in a lysis buffer containing 25 mm Hepes, pH 7.4, 1% Nonidet P-40, 10% glycerol, 137 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 μg/ml pepstatin, 5 μg/ml leupeptin, and 5 mm benzamidine and then ground with a handheld pellet pestle (Kontes, Vineland, NJ) for 20 s. The samples were then transferred to a Dounce homogenizer and homogenized with 10 hand strokes. After rocking at 4 °C for 10 min, the samples were centrifuged in a microcentrifuge for 30 min at 13,000 × g and the clear lysate subjected to SDS-polyacrylamide gel electrophoresis and immunoblotting.
Glucose Tolerance Test
Control and GLUT4-EGFP transgenic mice were fasted overnight. A basal glucose level was taken with tail vein blood on a Hemocue Glucometer (Hemocue AB, Angelholm, Sweden). At time point 0, the mice were given an intraperitoneal injection of 2 grams of glucose per kg of body weight. Glucose measurements were then taken from tail vein blood at 30, 60, 90, and 120 min post-glucose injection.
Subcellular Fractionation of Skeletal Muscle
The subcellular fractionation of skeletal muscle was performed as described (34). Briefly, following an overnight fast, the transgenic mice were either left untreated or given an intraperitoneal injection of 21 units of Humulin R per kg of body weight. After 30 min, the mice were sacrificed, and the quadriceps and gastrocnemius muscles were dissected and trimmed of fat and connective tissues. The muscles were put in a homogenization buffer containing 20 mm Hepes, pH 7.4, 250 mm sucrose, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 7μg/ml pepstatin, 5 μg/ml leupeptin, and 5 mm benzamidine, and homogenized with a Polytron PT-10 homogenizer 3 times in 10-s bursts. The samples were then centrifuged at 2,000 × g for 5 min and the supernatant was re-centrifuged at 9,000 × g for 20 min at 4 °C. The resulting pellet (P1) was re-suspended in PBS containing protease inhibitors. The supernatant from the P1 fraction was then centrifuged at 180,000 × g for 90 min, and the pellet was re-suspended in PBS plus protease inhibitors. Equal amounts of protein (1 mg) from each pellet were loaded onto a 4.5-ml 10–30% continuous sucrose gradient and centrifuged at 216,000 × g for 55 min in an SW50.1 rotor (Beckman-Coulter, Palo Alto, CA). The sucrose gradient was fractionated into 500-μl aliquots and the pellet (P2) was re-suspended in PBS plus protease inhibitors. Five μg of protein plus equal volumes of the fractions were resolved by SDS-polyacrylamide gel electrophoresis and subjected to immunoblotting.
Immunofluorescence of Muscle
Transgenic littermates were fasted overnight and either left untreated or given an intraperitoneal injection of 21 units of Humulin R per kg of body weight. After 30 min, the mice were sacrificed, the quadriceps dissected, covered in TissueTek O.C.T. embedding compound, and snap-frozen in liquid nitrogen-cooled isopentane. Frozen muscles were stored at −80 °C, subsequently cut with a Zeiss Cryostat to a thickness of 7 μm and adhered to glass slides. Similarly, mice were exercised as described below and the basal and exercised mice were sacrificed and skeletal muscle tissue prepared as described above.
The tissue slices were incubated in blocking solution containing 3% bovine serum albumin and 5% donkey serum in PBS for 1 h. After rinsing with PBS, the samples were incubated with a 1:50 dilution of the primary antibody in the blocking solution for 90 min. The samples were then washed with PBS three times for 5 min each and then incubated with a 1:50 dilution of the appropriate secondary antibody. The slides were washed with PBS three times for 5 min each, dried, mounted with a drop of Vectashield, and covered with a coverslip. Antibody-labeled tissues slices were then viewed with a Zeiss LSM 510 Confocal Fluorescent Microscope.
Due to the high labeling background of the IIID5E1 mouse monoclonal antibody on mouse tissue, a special antibody labeling protocol was used. In this particular case, the samples were blocked with avidin and biotin blocking buffers, and blocked and labeled using the Monoclonal on Mouse kit according to the manufacturer's instructions.
Exercise Protocol
Mice were trained to run on a Columbus Instruments Rodent Treadmill (Columbus, OH) with 3–4 training sessions consisting of 15 min at a speed of 10–15 m/min at an incline of 10%. The exercise experiment involved the running of fasted mice for 60 min at a speed of 15–20 m/min. Trained but not exercised mice were used as controls for these analyses.
in vivo Adenoviral Infection of Skeletal Muscle
Adenovirus infection of muscle was done according to standard protocols (35, 36). Briefly, 4–6-week-old mice were anesthetized with a dose of 91 mg/kg ketamine and 9.1 mg/kg of xylazine. Purified adenovirus (1 × 109 particles) was injected into the right quadriceps of the mouse just above the kneecap. After 4 days, mice were fasted overnight and either left untreated or stimulated with insulin (21 units/kg for 30 min) and the muscles were isolated as described above.
Results
Protein Expression and Tissue Specificity of the GLUT4-EGFP Transgenic Mice
To develop an in vivo skeletal muscle model in which the trafficking of GLUT4 could be readily visualized, we generated several independent transgenic mouse lines expressing the GLUT4-EGFP fusion protein driven by the HSA promoter. The tissue specificity of GLUT4-EGFP expression was directly compared with that of the endogenous GLUT4 protein in the heart, diaphragm, quadriceps, gastrocnemius, and adipose tissue extracts by immunoblot analysis (Fig. 1). As previously established, control nontransgenic mice displayed strong GLUT4 immunoreactivity in these tissues at a molecular mass of ∼50 kDa (Fig. 1B). In contrast, GLUT4 immunoblots of the transgenic mice show the presence of both the endogenous GLUT4 protein and the GLUT4-EGFP fusion protein with a molecular mass of ∼75 kDa (Fig. 1D). Importantly, the 75-kDa band was only detected in skeletal muscle tissues (diaphragm, quadriceps, and gastrocnemius) but was absent in both cardiac and adipose tissue extracts. As expected, the GFP antibody was unreactive to the tissues from the non-transgenic mice (Fig. 1A), while a 75-kDa band corresponding to GLUT4-EGFP was detected in the skeletal muscle extracts from the transgenic mice (Fig. 1C). Again, this GFP immuno-reactivity was restricted to the skeletal muscle tissues and was not observed in cardiac or adipose tissues. Although the extent of GLUT4-EGFP expression was different among the 4 transgenic lines isolated, all of these lines displayed an identical tissue-specific expression pattern (data not shown). These results demonstrate that GLUT4-EGFP protein expression in these animals occurs to a similar degree as endogenous GLUT4 protein levels and this expression is appropriately restricted to skeletal muscle tissue.
Fig. 1. The human skeletal actin muscle promoter drives the tissue-specific expression of the GLUT4-EGFP fusion protein in transgenic mice.
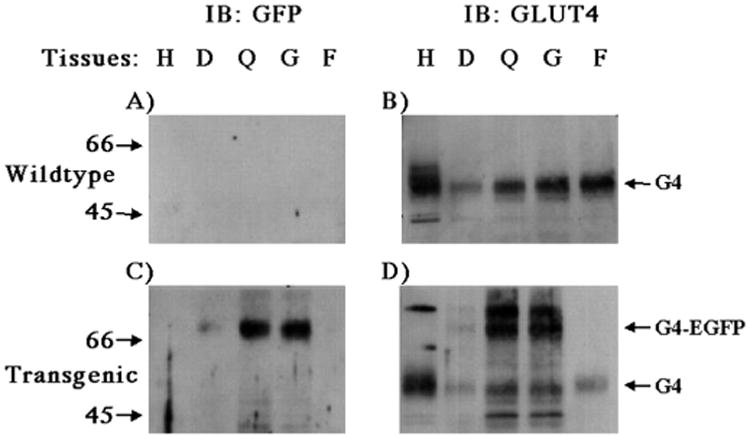
Tissues from wild-type (A and B) and GLUT4-EGFP trangenic (C and D) mice were isolated and subjected to immunoblotting as described under “Experimental Procedures.” Tissue extracts (5 μg) were prepared from the heart (H), diaphragm (D), quadriceps (Q), gastrocnemius (G), and epididymal adipose depot (F) and subjected to SDS-polyacrylamide gel electrophoresis. The samples were then immunoblotted with the GFP antibody (A and C) or the GLUT4 antibody (B and D). These are representative immunoblots performed 6 times from four different independent transgenic mice.
GLUT4-EGFP Transgenic Mice Display Increased Glucose Tolerance
Previous studies have demonstrated that increased expression of GLUT4 in skeletal muscle results in relative fasting hypoglycemia with increased glucose disposal in response to a glucose challenge (37, 38). Therefore, we fasted GLUT4-EGFP transgenic mice and nontransgenic littermates and assessed the changes in plasma glucose levels following an intraperitoneal injection of glucose (Fig. 2). As typically observed, wild-type mice achieved a peak blood glucose level (∼500 mg/dl) at 30 min that subsequently decrease slowly over next 90 min. Although the GLUT4-EGFP transgenic mice also reached a peak glucose level at 30 min, the increase in circulating glucose was markedly reduced averaging only 275 mg/dl. The blood glucose levels were also significantly lower in the transgenic mice over the entire time frame examined. These data demonstrate that the addition of EGFP to the COOH-terminal domain of GLUT4 does not impair its ability to transport glucose and transgenic expression in skeletal muscle results in enhanced glucose tolerance.
Fig. 2. The skeletal muscle specific GLUT4-EGFP transgenic mice display greater insulin sensitivity than wild-type mice.
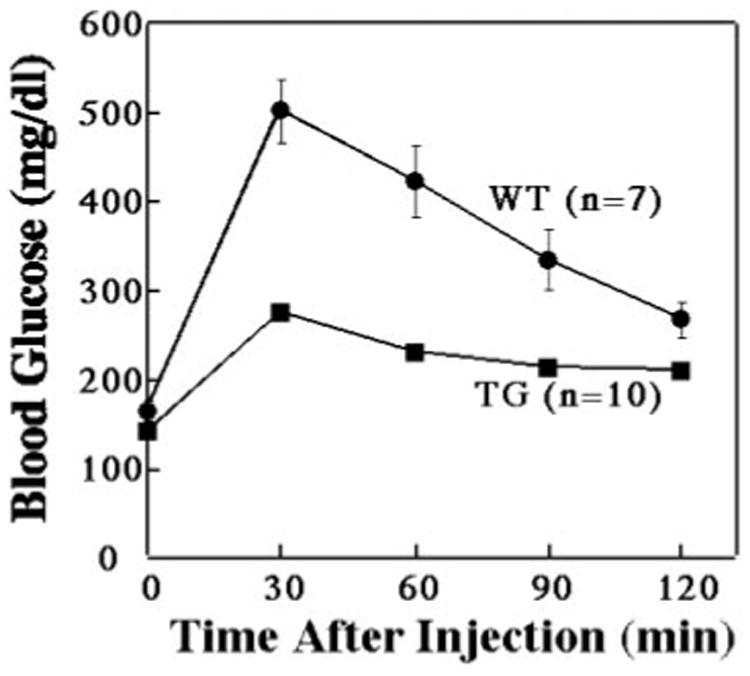
Wild-type and GLUT4-EGFP transgenic mice were fasted overnight and then given an intraperitoneal injection of glucose (2 g/kg). Plasma glucose levels were then determined at the times indicated as described under “Experimental Procedures.” The average and S.E. of the mean are shown for 7 wild-type (WT) and 10 GLUT4-EGFP (TG) transgenic mice.
Insulin-stimulated Translocation of GLUT4-EGFP
The ability of GLUT4-EGFP to enhance glucose disposal could either result from a basal increase in the levels of GLUT4-EGFP at the cell surface and/or an insulin-stimulated translocation of the GLUT4-EGFP from intracellular storage sites to the cell surface. To investigate the subcellular distribution of GLUT4-EGFP, we utilized skeletal muscle homogenization coupled with sucrose velocity sedimentation to separate sarcolemma/transverse-tubule enriched fractions (P1 and P2) from the fractions enriched for intracellular membranes (34). As seen in Fig. 3, in the basal state there is a relatively low level of GLUT4 in the P1 and P2 fractions with the majority distributed in fractions 4–8 of the continuous sucrose velocity gradient indicative of an intracellular localization (Fig. 3A). The distribution of the GLUT4-EGFP fusion protein mirrored the distribution of the endogenous GLUT4 protein. Insulin stimulation resulted in a decreased amount of endogenous GLUT4 localized to the intracellular compartment with a concomitant increase in the P1 and P2 fractions (Fig. 3B). Similarly, the GLUT4-EGFP fusion protein also displayed a marked reduction in the intracellular fractions paralleled with increased levels in the P1 and P2 fractions in response to insulin. These data demonstrate that the transgenic GLUT4-EGFP fusion protein localizes to similar intracellular sites and undergoes an identical pattern of insulin-stimulated translocation as the endogenous skeletal muscle GLUT4 protein.
Fig. 3. Insulin stimulation results in the skeletal muscle surface membrane translocation of both the endogenous GLUT4 and GLUT4-EGFP fusion proteins.
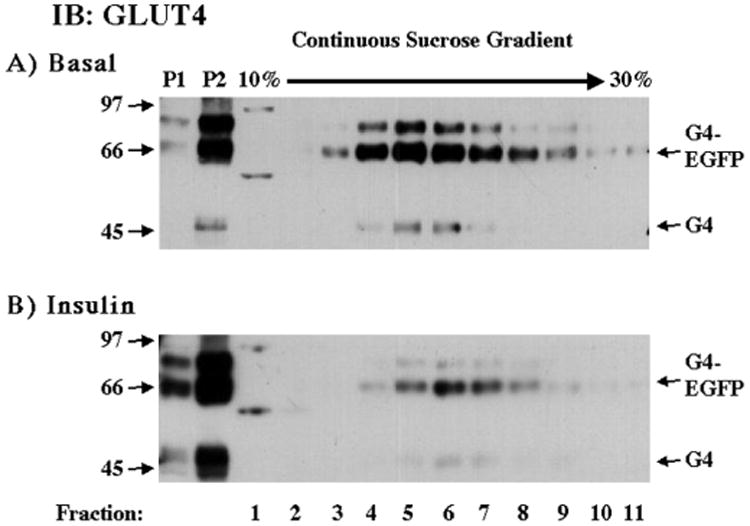
GLUT4-EGFP transgenic mice were fasted overnight and either left untreated (A) or stimulated with insulin (B) as described under “Experimental Procedures.” The hind-quarter skeletal muscle was dissected and subjected to differential and sucrose velocity centrifugation as described under “Experimental Procedures.” The surface membrane fractions (P1 and P2) as well as the intracellular membrane fractions (fractions 1–11) were immunoblotted with the GLUT4 antibody. These are representative immunoblots performed from four different independent pairs of transgenic mice.
To distinguish between sarcolemma and transverse-tubule translocation, we utilized confocal fluorescent microscopy of the GLUT4-EGFP protein in comparison with the established sarcolemma-localized protein (β-dystroglycan) and the transverse-tubule protein marker the L-type voltage-dependent calcium channel (Figs. 4 and 5). In the basal state, GLUT4-EGFP was primarily distributed throughout the interior of the muscle fibers with no evidence of sarcolemma localization (Fig. 4, panel a). Following insulin stimulation, there was a marked redistribution of GLUT4-EGFP resulting in a strong labeling of the fiber surface coupled with a greater organization of the fiber interior (Fig. 4, panel b). The sarcolemma membrane was labeled with the monoclonal β-dystroglycan antibody and did not change following insulin stimulation (Fig. 4, panels c and d). Consistent with the results of the subcellular fractionation, the extent of GLUT4-EGFP and β-dystroglycan colocalization was relatively low in the basal state but greatly increased following insulin stimulation (Fig. 4, panels e and f).
Fig. 4. Insulin stimulation results in the translocation of GLUT4-EGFP to the sarcolemma membrane.
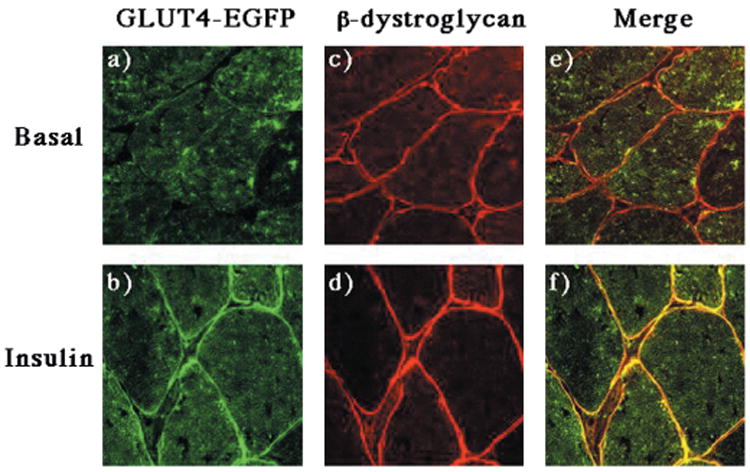
GLUT4-EGFP transgenic mice were fasted overnight and either left untreated (panels a, c, and e) or stimulated with insulin (panels b, d, and f) as described under “Experimental Procedures.” The quadriceps muscle was frozen, sliced, and incubated with the β-dystroglycan monoclonal antibody and secondary donkey anti-mouse IgG conjugated to Texas Red (panels c and d). Sections were visualized by confocal fluorescent microscopy (×63). These are representative fields from three independent experiments.
Fig. 5. Insulin stimulation results in the translocation of GLUT4-EGFP to the transverse-tubule membranes.
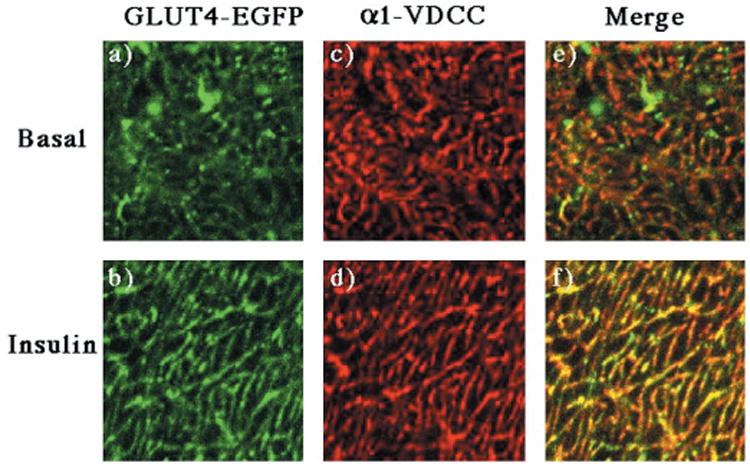
GLUT4-EGFP transgenic mice were fasted overnight and either left untreated (panels a, c, and e) or stimulated with insulin (panels b, d, and f) as described under “Experimental Procedures.” The quadriceps muscle was frozen, sliced, and incubated with the monoclonal antibody directed against the α1 subunit of the α1-VDCC using the Monoclonal on Mouse kit according to the manufacturers instructions (panels c and d). Sections were visualized by confocal fluorescent microscopy (×63) and then further magnified 11 times. The width of each panel is ∼13 μm. These are representative fields from three independent experiments.
In addition to sarcolemma translocation, several studies have demonstrated that GLUT4 protein also translocates to the transverse-tubule membrane (31–33). The transverse-tubules are deep invaginations of surface membrane that originate from and are continuous with the sarcolemma membrane. These invaginations are more difficult to visualize by light microscopy, but can be distinguished by colocalization with an antibody against the transverse-tubule specific α1 subunit of the L-type voltage-dependent calcium channel (α1-VDCC) when visualized at high magnification (Fig. 5). In the basal state, GLUT4-EGFP was randomly distributed within the interior of the fiber and did not colocalize with the α1-VDCC subunit (Fig. 5, panels a, c, and e). In contrast, insulin stimulation resulted in the appearance of GLUT4-EGFP in a striated pattern consistent with localization of the transverse-tubules (Fig. 5, panel b). This was confirmed by the insulin-stimulated colocalization of GLUT4-EGFP with the α1-VDCC subunit (Fig. 5, panels d and f). Together, these data demonstrate that in response to insulin, the GLUT4-EGFP fusion protein translocates to the sarcolemma and transverse-tubule in a manner similar to that of the endogenous GLUT4 protein.
Exercise Also Stimulates the Translocation of GLUT4-EGFP to the Sarcolemma and Transverse Tubules
In addition to insulin, a variety of other stimuli such as exercise, contraction, and hypoxia also enhance glucose uptake by induction of GLUT4 translocation in skeletal muscle (3, 39). These “alternative” pathways of GLUT4 translocation are independent of insulin action and occur in the complete absence of phosphatidylinositol 3-kinase activation (25, 26). Therefore, we determined whether the GLUT4-EGFP protein was appropriately distributed into vesicular compartments responsive to an “alternate” signaling pathway. As shown in Fig. 6, exercise promotes the translocation of the GLUT4-EGFP protein from intracellular storage sites to the sarcolemma membrane (Fig. 6, panels a and b). The sarcolemma translocation was again confirmed by the colocalization of GLUT4-EGFP with β-dystroglycan (Fig. 6, panels c, d, e, and f). Similarly, exercise also induced the redistribution of GLUT4-EGFP to the transverse tubule membranes as detected by colocalization with the α1-VDCC subunit (Fig. 7, panels a-f). It should also be noted that the extent of GLUT4-EGFP translocation was weaker than observed with insulin stimulation, which may reflect the degree of exercise used in these experiments. Regardless, these data further demonstrate that the GLUT4-EGFP protein is also stored in the exercise-responsive population of GLUT4-containing intracellular vesicles.
Fig. 6. Exercise results in the translocation of GLUT4-EGFP to the sarcolemma membrane.
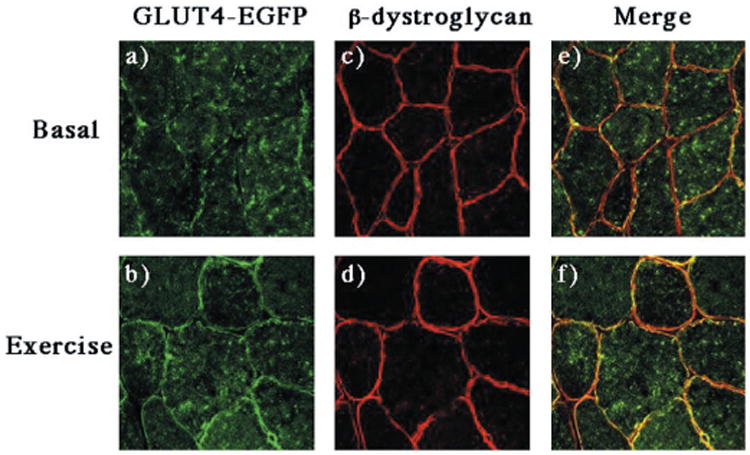
GLUT4-EGFP transgenic mice were fasted overnight and either left untreated (panels a, c, and e) or run for 60 min on a treadmill (panels b, d, and f) as described under “Experimental Procedures.” The quadriceps muscle was frozen, sliced, and incubated with the β-dystroglycan monoclonal antibody and secondary donkey anti-mouse IgG conjugated to Texas Red (panels c and d). Sections were visualized by confocal fluorescent microscopy (×63). These are representative fields from two independent experiments.
Fig. 7. Exercise stimulation results in the translocation of GLUT4-EGFP to the transverse-tubule membranes.
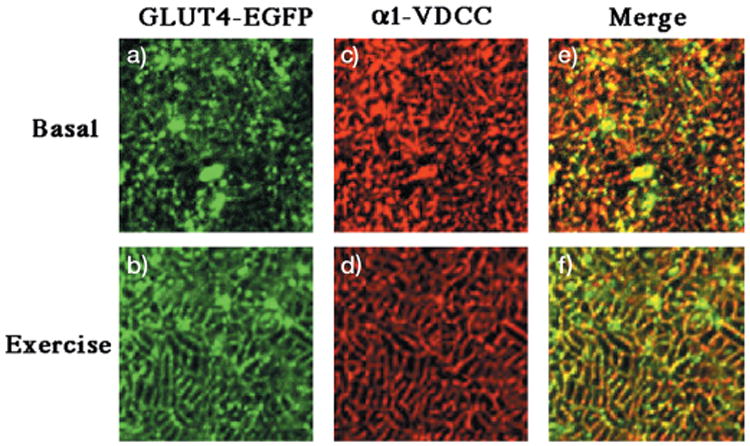
GLUT4-EGFP transgenic mice were fasted overnight and either left untreated (panels a, c, and e) or run for 60 min on a treadmill (panels b, d, and f) as described under “Experimental Procedures.” The quadriceps muscle was frozen, sliced, and incubated with the monoclonal antibody directed against the α1 subunit of the α1-VDCC using the Monoclonal on Mouse kit according to manufacturers instructions (panels c and d). Sections were visualized by confocal fluorescent microscopy (×63) and then further magnified 11 times. The width of each panel is ∼μm. These are representative fields from three independent experiments.
Expression of Munc18c Specifically Inhibits the Insulin-stimulated GLUT4-EGFP Translocation to Transverse Tubule Membranes
Previously, we and others (22–24, 40) have reported that the syntaxin 4-binding protein, Munc18c, but not the Munc18b isoform, is an important regulator of insulin-stimulated GLUT4 translocation in adipocytes. To begin to examine the functional role of Munc18c in skeletal muscle, we compared the level of Munc18c protein expression among several tissues (Fig. 8). Although it was previously reported that the abundance of Munc18c mRNA was relatively low in skeletal muscle (41), we clearly see expression of Munc18c in skeletal muscle is comparable to other tissues examined.
Fig. 8. Munc18c is expressed in a number of different tissues, including skeletal muscle.
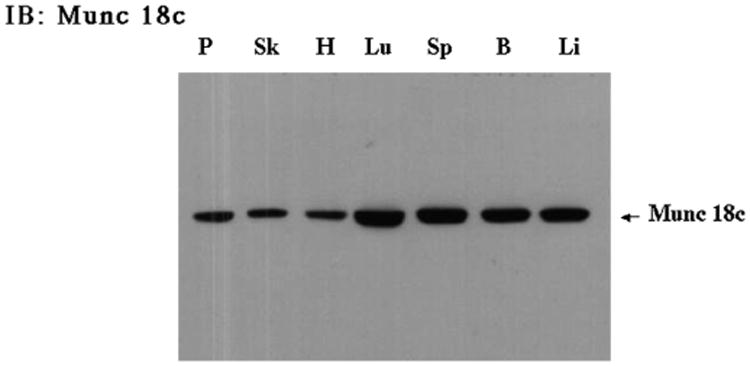
Tissues from a wild-type C57Bl6 mouse were isolated and subjected to immunoblotting as described under “Experimental Procedures.” Tissue extracts (40 μg) were prepared from the following tissues: pancreas (P), skeletal muscle (Sk), heart (H), lung (Lu), spleen (Sp), brain (B), and liver (Li) and subjected to SDS-polyacrylamide gel electrophoresis. The samples were immunoblotted with the Munc18c rabbit polyclonal antibody.
Having confirmed the expression of Munc18c in skeletal muscle, we next assessed the functional role of Munc18c on GLUT4 translocation in vivo via infection with recombinant adenovirus encoding for Munc18c or Munc18b directly into skeletal muscle (Fig. 9). Injection of the Munc18b adenovirus into the quadriceps muscle resulted in several fibers overexpressing the Munc18b protein (Fig. 9, panel a). The overexpression of Munc18b had no significant effect on the basal state distribution of GLUT4-EGFP, which remained dispersed throughout the interior of the fiber (Fig. 9, panels c and e). As typically observed, insulin stimulation resulted in GLUT4-EGFP translocation to the sarcolemma and transverse-tubule membranes in both noninfected fibers as well as those expressing the Munc18b protein (Fig. 9, panels b, d, and f).
Fig. 9. Expression of Munc18b has no effect on the insulin stimulated translocation of GLUT4-EGFP to either the sarcolemma or transverse-tubule membranes.
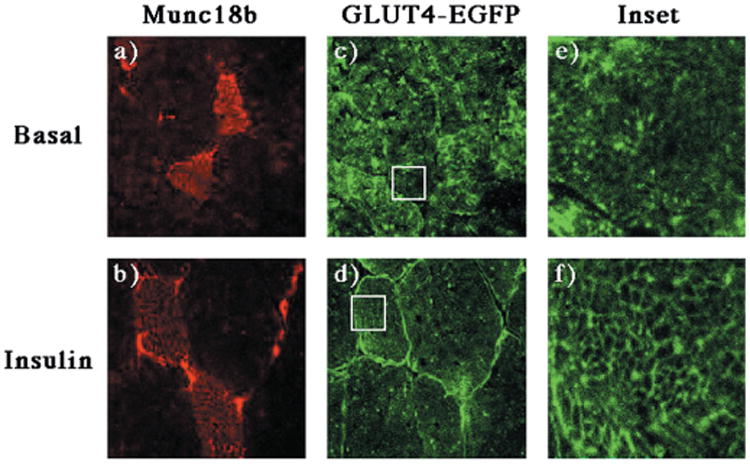
The quadricep muscle of GLUT4-EGFP transgenic mice was injected with a recombinant adenovirus encoding for the Munc18b protein as described under “Experimental Procedures.” Four days following injection, the mice were fasted overnight and either left untreated (panels a, c, and e) or stimulated with insulin (panels b, d, and f). The quadriceps muscle was frozen, sliced, and incubated with the Munc18b antibody and secondary donkey anti-rabbit IgG conjugated to Texas Red (panels a and b). Sections were visualized by confocal fluorescent microscopy (×63) and then further magnified 11 times (insets). The width of the box is ∼μm. These are representative fields from three independent experiments.
Similarly, adenovirus-mediated expression of Munc18c had no effect on the distribution of GLUT4-EGFP in the basal state (Fig. 10, panels a, c, and e). Surprisingly, expression of Munc18c had no effect on the insulin-stimulated translocation of GLUT4-EGFP to the sarcolemma membrane compared with the surrounding noninfected fibers (Fig. 10, panels b and d). In contrast, the infected fibers had a reduced translocation of GLUT4-EGFP to the transverse-tubule membranes (Fig. 10, panels d and f). At low magnification this is observed as a normal translocation to the outer surface membrane with an apparent decrease in the interior signal. At high magnification this is noted by the lack of organized network appearance within the interior of the infected fibers (compare Fig. 9, panel f, with Fig. 10, panel f).
Fig. 10. Expression of Munc18c has no effect on the insulin stimulated translocation of GLUT4-EGFP to the sarcolemma membrane but inhibits GLUT4-EGFP translocation to the transverse-tubules.
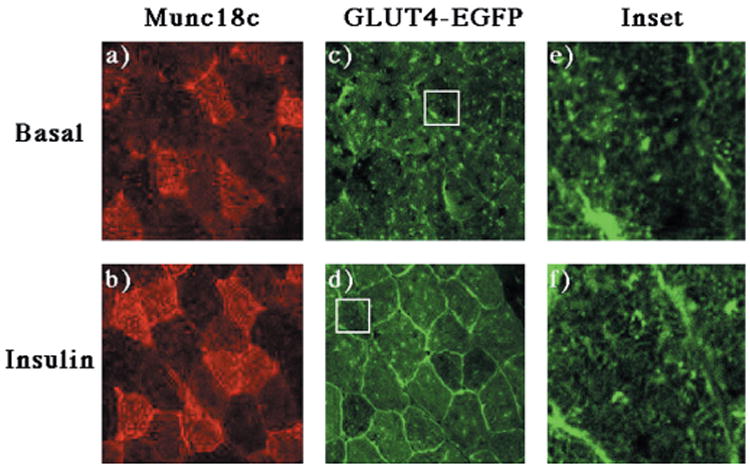
The quadricep muscle of GLUT4-EGFP transgenic mice was injected with a recombinant adenovirus encoding for the Munc18c protein as described under “Experimental Procedures.” Four days following injection, the mice were fasted overnight and either left untreated (panels a, c, and e) or stimulated with insulin (panels b, d, and f). The quadriceps muscle was frozen, sliced, and incubated with the Munc18c antibody and secondary donkey anti-rabbit IgG conjugated to Texas Red (panels a and b). Sections were visualized by confocal fluorescent microscopy (×63) and then further magnified 11 times (insets). The width of the box is ∼μm. These are representative fields from four independent experiments.
Since Munc18c specifically inhibits insulin-stimulated GLUT4 translocation through its binding to syntaxin 4, we next examined the subcellular distribution of syntaxin 4 in skeletal muscle using the subcellular fractionation procedure described previously (Fig. 11). Syntaxin 4 primarily is localized to the P1 membrane fraction, and to a lesser extent the P2 membrane fraction and the sucrose velocity gradient fractions. In addition, syntaxin 4 does not display any insulin-induced translocation.
Fig. 11. Syntaxin 4 localizes primarily to the P1 fraction, and to a lesser extent to the P2 and sucrose gradient fractions, and does not translocate in response to insulin.
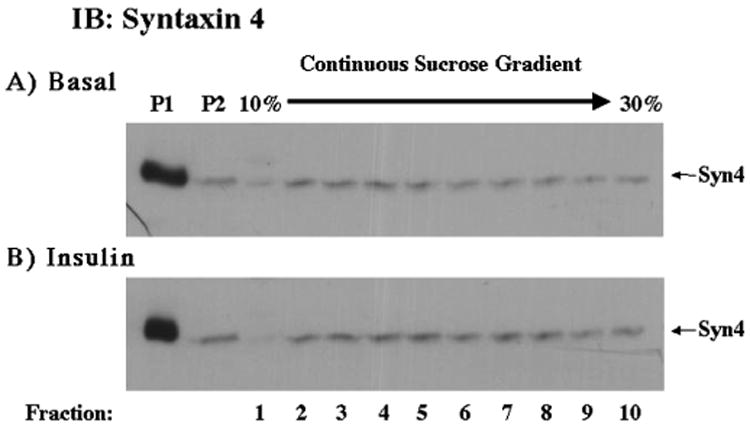
Mice were treated as described under “Experimental Procedures.” 100 μg of the P1 and P2 fractions and 100 μl of each sucrose gradient fraction were subjected to SDS-polyacrylamide gel electrophoresis. The samples were then immunoblotted with a digoxigenin-labeled sheep Syntaxin 4 antibody and anti-digoxigenin POD Fab fragments (Roche Molecular Biochemicals, Germany).
Since this subcellular fractionation protocol does not distinguish between the two surface membranes found in skeletal muscle, we sought to determine the relative surface membrane distribution of syntaxin 4 in skeletal muscle (Fig. 12). Confocal immunofluorescence microscopy indicated that the majority of the endogenous syntaxin 4 protein was localized at the sarcolemma (Fig. 12, panels a, c, and e). Syntaxin 4 was also present in the transverse-tubule membrane compartments as demonstrated by its colocalization with the α1-VDCC subunit (Fig. 12, panels b, d, and f). However, based upon the relative signal intensity, the amount of transverse-tubule localized syntaxin 4 protein was substantially less than that found at the sarcolemma membrane.
Fig. 12. Syntaxin 4 is localized to both the sarcolemma and transverse-tubule membranes in skeletal muscle.
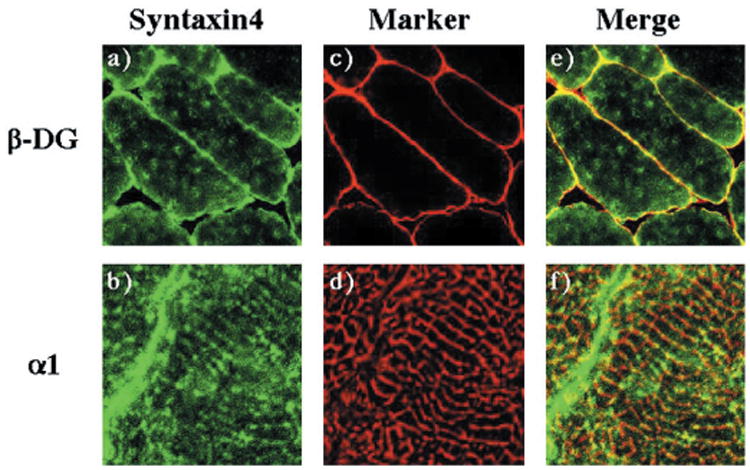
The quadriceps muscle from wild-type mice was frozen, sliced, and labeled with the sheep syntaxin 4 antibody (panels a and b), the β-dystroglycan monoclonal antibody (panel c), or the monoclonal α1-VDCC antibody (panel d) as described under “Experiment Procedures.” Sections were visualized by confocal fluorescent microscopy (×63). The transverse-tubules immunofluorescence was magnified 11 times from the confocal image. The width of each panel is ∼μm. These are representative fields from two independent experiments.
Discussion
The expression of the insulin-responsive glucose transporter GLUT4 is highly restricted to muscle and adipose tissue, the two tissues that undergo insulin-stimulated GLUT4 translocation (3, 42, 43). However, the cellular and molecular analysis of GLUT4 translocation has largely been performed in adipocytes due to the technical limitations associated with skeletal muscle tissue. Nevertheless, insulin-stimulated glucose disposal in skeletal muscle is quantitatively the most important tissue accounting for ∼80% of glucose uptake in the postprandial state (44). In addition, although the general GLUT4 translocation process in skeletal muscle appears similar to adipocytes, there are several tissue-specific differences. Skeletal muscle displays contraction/exercise-stimulated GLUT4 translocation through a pathway independent of but additive with that of insulin (25, 26, 33). Furthermore, skeletal muscle contains two distinct surface membranes, the sarcolemma membrane equivalent to the adipocyte plasma membrane and a unique transverse tubule membrane, both of which function as acceptor sites for GLUT4 translocation (31, 32, 45). Importantly, the transverse-tubule membrane has 1.5-fold more surface area than the sarcolemma membrane and therefore quantitatively accounts for the majority of skeletal muscle glucose uptake (46).
To develop a more tractable system to examine the regulation of GLUT4 translocation in skeletal muscle, we generated transgenic mice expressing the GLUT4-EGFP fusion protein specifically in skeletal muscle. This was based upon our previous observations that expression of the GLUT4-EGFP fusion protein in adipocytes resulted in an identical distribution of basal and insulin-regulated trafficking compared with the endogenous GLUT4 protein (24, 47). As expected, the fidelity of the GLUT4-EGFP transgene was confirmed by its ability to undergo both insulin- and exercise/contraction-stimulated translocation to the sarcolemma and transverse-tubule membranes in vivo. This was confirmed by a more labor intensive subcellular membrane fractionation method and by fluorescent microscopy which provided an easy and rapid visual marker to view the movement of GLUT4-EGFP. In addition, the GLUT4-EGFP protein fluorescence was sensitive enough to distinguish between translocation to the sarcolemma and the transverse-tubules by confocal fluorescence microscopy.
Having established the appropriate in vivo regulation of the GLUT4-EGFP transgene in response to insulin and exercise/contraction, we utilized this system to examine the functional role of Munc18c in these processes. Munc18c is a ubiquitous homologue of the neuronal n-Sec1/Munc18–1/Munc18a protein (41). This family of syntaxin-binding proteins has been postulated to exert both negative and positive roles in modulating the docking and fusion of vesicles with target membranes. For example, overexpression of Munc18c in 3T3L1 adipocytes, or transgenic overexpression of the Drosophila Rop homologue decreases GLUT4 translocation and neurotransmitter release, respectively, consistent with a negative role for Munc18 proteins (24, 48). However, conditional mutants of the yeast homologue Sec1p or null alleles of the Munc18a isoform in mice lead to a complete block of vesicle release in yeast and neurons, respectively, suggesting an essential positive role for Munc18 proteins (49, 50). Furthermore, inhibition of Munc18c binding to syntaxin 4 through the use of a blocking peptide also prevented insulin-stimulated GLUT4 translocation (40).
These apparently contradictory findings can now be partially resolved based upon the functional and structural analysis of the syntaxin 1A-Munc18a complex (51, 52). Munc18a binding appears to maintain syntaxin 1 in a closed conformational state in which the juxtamembrane Hcore domain (directly involved in the SNARE core complex formation) is in an intramolecular complex with the three amino-terminal coiled-coil domains, termed the Habc domains. In this state, the syntaxin 1 Hcore domain is unable to interact with the coiled-coil domains of SNAP25 and VAMP2 responsible for forming the core complex. However, upon alteration of Munc18a binding or a conformational change in Munc18a, syntaxin 1 is able to undergo a transition to an open conformational state. This releases the Hcore domain from the Habc domain allowing the Hcore domain of syntaxin 1 to interact with and form a four-helical, coiled-coil bundle with SNAP25 and VAMP2. According to this model, overexpression of Munc18 would inhibit vesicle fusion by maintaining syntaxin in the closed conformational state, whereas complete displacement of Munc18 would prevent the interconversion between the open and closed conformation, suggesting the necessity of the transition state for the generation of the syntaxin-SNAP25-VAMP2 core complex leading to vesicle docking and fusion.
Consistent with this hypothesis, we and others have previously reported that overexpression of Munc18c in adipocytes inhibit insulin-stimulated GLUT4 translocation (23, 24). This inhibitory effect was fully reversed by increased expression of syntaxin 4, indicating a functional requirement for excess syntaxin 4 over Munc18c (40). Similarly, we have now observed that increased expression of Munc18c in skeletal muscle inhibits insulin-stimulated GLUT4 translocation. However, to our surprise this was specific for the translocation of GLUT4 to the transverse-tubule membrane but without any effect on the translocation of GLUT4 to the sarcolemma membrane. This can be reconciled by the fact that the sarcolemma contains relatively high levels of syntaxin 4 compared with the transverse-tubules. At the level of adenovirus-mediated skeletal muscle gene expression, sufficient Munc18c was generated to saturate the transverse-tubule localized syntaxin 4 protein but not the more abundant sarcolemma-localized syntaxin 4. This would account for the ability of overexpressed Munc18c to specifically prevent GLUT4 translocation to the transverse tubules but not the sarcolemma.
In summary, the data presented in this article demonstrates that the expression of the GLUT4-EGFP fusion protein in skeletal muscle of transgenic mice displays the expected functional properties of the endogenous GLUT4 protein. This includes the ability to translocate to both the sarcolemma and transverse-tubule membranes in response to insulin and exercise/contraction stimulation in vivo. Furthermore, GLUT4 translocation in skeletal muscle also requires a functional SNARE complex with the appropriate stoichiometry between the syntaxin 4 and Munc18c proteins. This mouse model will provide a novel approach to dissect the regulated and dynamic trafficking events controlling GLUT4 translocation in skeletal muscle in vivo.
Acknowledgments
We thank Drs. Kevin P. Campbell and Valerie Allamand for providing skeletal muscle specific antibodies and assistance with the in vivo adenoviral infections. We also thank Diana Boeglin for assistance with the glucose tolerance tests. We further thank the University of Iowa Transgenic Animal Facility.
Footnotes
This work was supported in part by Research Grants DK33823 and DK25295 from the National Institutes of Health.
The abbreviations used are: PI, phosphatidylinositol; SNAP, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; GLUT, glucose transporter; EGFP, enhanced green fluorescent protein; PBS, phosphate-buffered saline; HSA, human skeletal muscle actin; α1-VDCC, L-type voltage-dependent calcium channel.
References
- 1.Kandror KV, Pilch PF. Am J Physiol. 1996;271:E1–E14. doi: 10.1152/ajpendo.1996.271.1.E1. [DOI] [PubMed] [Google Scholar]
- 2.Czech MP, Corvera S. J Biol Chem. 1999;274:1865–1868. doi: 10.1074/jbc.274.4.1865. [DOI] [PubMed] [Google Scholar]
- 3.Goodyear LJ, Kahn BB. Annu Rev Med. 1998;49:235–261. doi: 10.1146/annurev.med.49.1.235. [DOI] [PubMed] [Google Scholar]
- 4.Birnbaum MJ, James DE. Curr Opin Endocrinol. 1995;2:383–391. [Google Scholar]
- 5.Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. Mol Cell Biol. 1994;14:4902–4911. doi: 10.1128/mcb.14.7.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quon MJ, Chen H, Ing BL, Liu ML, Zarnowski MJ, Yonezawa K, Kasuga M, Cushman SW, Taylor SI. Mol Cell Biol. 1995;15:5403–5411. doi: 10.1128/mcb.15.10.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haruta T, Morris AJ, Rose DW, Nelson JG, Mueckler M, Olefsky JM. J Biol Chem. 1995;270:27991–27994. doi: 10.1074/jbc.270.47.27991. [DOI] [PubMed] [Google Scholar]
- 8.Katagiri H, Asano T, Ishihara H, Inukai K, Shibasaki Y, Kikuchi M, Yazaki Y, Oka Y. J Biol Chem. 1996;271:16987–16990. doi: 10.1074/jbc.271.29.16987. [DOI] [PubMed] [Google Scholar]
- 9.Tanti JF, Gremeaux T, Grillo S, Calleja V, Klippel A, Williams LT, Van Obberghen E, Le Marchand-Brustel Y. J Biol Chem. 1996;271:25227–25232. doi: 10.1074/jbc.271.41.25227. [DOI] [PubMed] [Google Scholar]
- 10.Martin SS, Haruta T, Morris AJ, Klippel A, Williams LT, Olefsky JM. J Biol Chem. 1996;271:17605–17608. doi: 10.1074/jbc.271.30.17605. [DOI] [PubMed] [Google Scholar]
- 11.Okada T, Kawano Y, Sakakibara R, Hazeki O, Ui M. J Biol Chem. 1994;269:3568–3573. [PubMed] [Google Scholar]
- 12.Frevert EU, Kahn BB. Mol Cell Biol. 1997;17:190–198. doi: 10.1128/mcb.17.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pessin JE, Thurmond DC, Elmendorf JS, Coker KJ, Okada S. J Biol Chem. 1999;274:2593–2596. doi: 10.1074/jbc.274.5.2593. [DOI] [PubMed] [Google Scholar]
- 14.Rea S, James DE. Diabetes. 1997;46:1667–1677. doi: 10.2337/diab.46.11.1667. [DOI] [PubMed] [Google Scholar]
- 15.Martin LB, Shewan A, Millar CA, Gould GW, James DE. J Biol Chem. 1998;273:1444–1452. doi: 10.1074/jbc.273.3.1444. [DOI] [PubMed] [Google Scholar]
- 16.Rea S, Martin LB, McIntosh S, Macaulay SL, Ramsdale T, Baldini G, James DE. J Biol Chem. 1998;273:18784–18792. doi: 10.1074/jbc.273.30.18784. [DOI] [PubMed] [Google Scholar]
- 17.Volchuk A, Wang Q, Ewart HS, Liu Z, He L, Bennett MK, Klip A. Mol Biol Cell. 1996;7:1075–1082. doi: 10.1091/mbc.7.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamori Y, Hashiramoto M, Araki S, Kamata Y, Takahashi M, Kozaki S, Kasuga M. Biochem Biophys Res Commun. 1996;220:740–745. doi: 10.1006/bbrc.1996.0474. [DOI] [PubMed] [Google Scholar]
- 19.Cheatham B, Volchuk A, Kahn CR, Wang L, Rhodes CJ, Klip A. Proc Natl Acad Sci U S A. 1996;93:15169–15173. doi: 10.1073/pnas.93.26.15169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson AL, Knight JB, Pessin JE. Mol Cell Biol. 1997;17:2425–2435. doi: 10.1128/mcb.17.5.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timmers KI, Clark AE, Omatsu-Kanbe M, Whiteheart SW, Bennett MK, Holman GD, Cushman SW. Biochem J. 1996;320:429–436. doi: 10.1042/bj3200429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tellam JT, Macaulay SL, McIntosh S, Hewish DR, Ward CW, James DE. J Biol Chem. 1997;272:6179–6186. doi: 10.1074/jbc.272.10.6179. [DOI] [PubMed] [Google Scholar]
- 23.Tamori Y, Kawanishi M, Niki T, Shinoda H, Araki S, Okazawa H, Kasuga M. J Biol Chem. 1998;273:19740–19746. doi: 10.1074/jbc.273.31.19740. [DOI] [PubMed] [Google Scholar]
- 24.Thurmond DC, Ceresa BP, Okada S, Elmendorf JS, Coker K, Pessin JE. J Biol Chem. 1998;273:33876–33883. doi: 10.1074/jbc.273.50.33876. [DOI] [PubMed] [Google Scholar]
- 25.Lund S, Holman GD, Schmitz O, Pedersen O. Proc Natl Acad Sci U S A. 1995;92:5817–5821. doi: 10.1073/pnas.92.13.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeh JI, Gulve EA, Rameh L, Birnbaum MJ. J Biol Chem. 1995;270:2107–2111. doi: 10.1074/jbc.270.5.2107. [DOI] [PubMed] [Google Scholar]
- 27.Brozinick JT, Jr, Birnbaum MJ. J Biol Chem. 1998;273:14679–82. doi: 10.1074/jbc.273.24.14679. [DOI] [PubMed] [Google Scholar]
- 28.Lund S, Pryor PR, Ostergaard S, Schmitz O, Pedersen O, Holman GD. FEBS Lett. 1998;425:472–4. doi: 10.1016/s0014-5793(98)00293-2. [DOI] [PubMed] [Google Scholar]
- 29.Sherwood DJ, Dufresne SD, Markuns JF, Cheatham B, Moller DE, Aronson D, Goodyear LJ. Am J Physiol. 1999;276:E870–878. doi: 10.1152/ajpendo.1999.276.5.E870. [DOI] [PubMed] [Google Scholar]
- 30.Rodnick KJ, Slot JW, Studelska DR, Hanpeter DE, Robinson LJ, Geuze HJ, James DE. J Biol Chem. 1992;267:6278–6285. [PubMed] [Google Scholar]
- 31.Marette A, Burdett E, Douen A, Vranic M, Klip A. Diabetes. 1992;41:1562–1569. doi: 10.2337/diab.41.12.1562. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Hansen PA, Marshall BA, Holloszy JO, Mueckler M. J Cell Biol. 1996;135:415–430. doi: 10.1083/jcb.135.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ploug T, van Deurs B, Ai H, Cushman SW, Ralston E. J Cell Biol. 1998;142:1429–1446. doi: 10.1083/jcb.142.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou M, Sevilla L, Vallega G, Chen P, Palacin M, Zorzano A, Pilch PF, Kandror KV. Am J Physiol. 1998;275:E187–E196. doi: 10.1152/ajpendo.1998.275.2.E187. [DOI] [PubMed] [Google Scholar]
- 35.Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holt KH, Lim LE, Straub V, Venzke DP, Duclos F, Anderson RD, Davidson BL, Campbell KP. Mol Cell. 1998;1:841–848. doi: 10.1016/s1097-2765(00)80083-0. [DOI] [PubMed] [Google Scholar]
- 37.Brozinick JT, Jr, Yaspelkis BB, 3rd, Wilson CM, Grant KE, Gibbs EM, Cushman SW, Ivy JL. Biochem J. 1996;313:133–140. doi: 10.1042/bj3130133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsao TS, Burcelin R, Katz EB, Huang L, Charron MJ. Diabetes. 1996;45:28–36. doi: 10.2337/diab.45.1.28. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi T, Wojtaszewski JF, Goodyear LJ. Am J Physiol. 1997;273:E1039–E1051. doi: 10.1152/ajpendo.1997.273.6.E1039. [DOI] [PubMed] [Google Scholar]
- 40.Thurmond DC, Kanzaki M, Khan AH, Pessin JE. Mol Cell Biol. 2000;20:379–388. doi: 10.1128/mcb.20.1.379-388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tellam JT, McIntosh S, James DE. J Biol Chem. 1995;270:5857–5863. doi: 10.1074/jbc.270.11.5857. [DOI] [PubMed] [Google Scholar]
- 42.Mueckler M. Eur J Biochem. 1994;219:713–725. doi: 10.1111/j.1432-1033.1994.tb18550.x. [DOI] [PubMed] [Google Scholar]
- 43.Olson AL, Pessin JE. Annu Rev Nutr. 1996;16:235–256. doi: 10.1146/annurev.nu.16.070196.001315. [DOI] [PubMed] [Google Scholar]
- 44.Katz EB, Burcelin R, Tsao TS, Stenbit AE, Charron MJ. J Mol Med. 1996;74:639–652. doi: 10.1007/s001090050068. [DOI] [PubMed] [Google Scholar]
- 45.Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munoz P, Rosemblatt M, Testar X, Palacin M, Thoidis G, Pilch PF, Zorzano A. Biochem J. 1995;312:393–400. doi: 10.1042/bj3120393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elmendorf JS, Boeglin DJ, Pessin JE. J Biol Chem. 1999;274:37357–37361. doi: 10.1074/jbc.274.52.37357. [DOI] [PubMed] [Google Scholar]
- 48.Schulze KL, Littleton JT, Salzberg A, Halachmi N, Stern M, Lev Z, Bellen HJ. Neuron. 1994;13:1099–1108. doi: 10.1016/0896-6273(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 49.Schekman R. Curr Opin Cell Biol. 1992;4:587–92. doi: 10.1016/0955-0674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- 50.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 51.Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Misura KM, Scheller RH, Weis WI. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]