

INTRODUCTORY PARAGRAPH
Primary sclerosing cholangitis (PSC) is a rare progressive disorder leading to bile duct destruction. We undertook the largest genome-wide association study of PSC (4,796 cases and 19,955 population controls) and identified four novel genome-wide significant loci. The most associated SNP at one locus affects splicing and expression of UBASH3A, with the protective allele (C) predicted to cause non-stop mediated mRNA decay and lower expression of UBASH3A. Although 75% of PSC patients have comorbid inflammatory bowel disease (IBD), our data suggest that the genome-wide genetic correlation (rG) between PSC and ulcerative colitis (UC) (rG=0.29) is significantly greater than that between PSC and Crohn’s disease (CD) (rG=0.04) (P=2.55×10−15). Importantly, UC and CD are genetically more similar to each other (rG=0.56) than either is to PSC (P<1.0×10−15). Our study represents a significant advance in our understanding of the genetics of PSC.
Primary sclerosing cholangitis affects around 1 in 10,000 individuals of European ancestry and is characterised by chronic inflammation and stricturing fibrosis of the biliary tree1. There remains no effective medical therapy and the majority of patients require orthotopic liver transplantation owing to the progressive nature of the disease2. PSC is highly comorbid with IBD, which is ultimately diagnosed in around 75% of patients. The clinical presentation of IBD in PSC is most often consistent with UC (~80%), but CD (~15%) and indeterminate forms of IBD (~5%) do occur in some patients. Time of disease onset and expression of the IBD phenotype in PSC is variable, with an overall trend toward IBD preceding PSC and milder but more extensive intestinal inflammation (pancolitis) compared to classical UC or CD3,4 This tendency, along with other clinical and epidemiological differences, has led to the proposal that IBD in the context of PSC (PSC-IBD) should be considered a disease entity separate from both UC and CD. Elevated risk of PSC and UC in first-degree relatives of PSC patients indicates a strong genetic component to PSC susceptibility and suggests the presence of shared genetic risk factors between PSC and UC5,6. However, the genetic relationship between PSC and UC/CD/IBD remains poorly defined because the low prevalence of PSC has precluded familial studies. Large-scale association studies have identified sixteen loci, including the HLA locus, underlying PSC risk7–12. Here, we undertake the largest genome-wide association study of PSC to date to identify novel PSC risk loci and enable us, for the first time, to estimate the genome-wide genetic correlation between PSC and the common forms of IBD.
Following quality control (Supplementary Tables 1 and 2, Supplementary Figs. 1–3) and imputation using reference haplotypes from the 1000 Genomes (Phase III) and UK10K projects13,14, we tested 7,891,602 SNPs for association in a sample of 2,871 PSC cases and 12,019 population controls using a linear mixed model to account for population stratification (Online Methods, Supplementary Tables 1 and 2). Genome-wide summary statistics are available from the International PSC Study Group website (see URLs). Forty SNPs were tested for association in an independent cohort of 1,925 PSC cases and 7,936 population controls (Online Methods, Supplementary Table 3), including 24 SNPs with P < 5×10−6 in the GWAS that are located outside of known PSC loci. We used an inverse-variance weighted fixed effects meta-analysis, implemented in METAL15, to test the evidence of association across the GWAS and replication cohorts combined and identified four new genome-wide significant loci with P < 5.26 × 10−3 in the replication study and P < 5 × 10−8 in the combined meta-analysis (Table 1, Supplementary Table 4, Supplementary Fig. 4). One of the newly associated loci, tagged by rs80060485 (3:g.71153890T>C) in FOXP1, is associated with immune-mediated disease for the first time. The three other newly associated PSC loci (implicating CCDC88B, CLEC16A and UBASH3A) are in high linkage disequilibrium (LD), defined as (r2 > 0.8) with variants significantly associated to other immune-mediated diseases (Supplementary Table 5). We found consistent evidence of association at fifteen of the sixteen previously established PSC loci and now consider 19 regions of the genome to be associated with PSC risk (Supplementary Table 4, Supplementary Fig. 4).
Table 1. Association summary statistics across four newly associated PSC risk loci.
Base-pair coordinates from build 37, RAF: risk allele frequency in replication controls, OR: odds ratio in the GWAS and replication meta-analysis (Combined), 95%CI: 95% confidence interval of OR estimates. Detailed association results, including those for the 15 loci previously associated with PSC, are given in Supplementary Table 4.
| SNP | Chr:Position (bp) | Risk Allele | RAF | OR | 95% CI | P-value
|
Candidate causal gene | ||
|---|---|---|---|---|---|---|---|---|---|
| GWAS | Replication | Combined | |||||||
| rs80060485 | 3:71153890 | C | 0.07 | 1.44 | 1.32–1.58 | 8.54 × 10−09 | 4.67 × 10−08 | 2.62 × 10−15 | FOXP1 |
| rs663743 | 11:64107735 | G | 0.66 | 1.20 | 1.14–1.26 | 8.42 × 10−08 | 4.44 × 10−07 | 2.24 × 10−13 | CCDC88B |
| rs725613 | 16:11169683 | T | 0.65 | 1.20 | 1.14–1.26 | 5.50 × 10−10 | 9.52 × 10−05 | 3.59 × 10−13 | CLEC16A |
| rs1893592 | 21:43855067 | A | 0.73 | 1.22 | 1.15–1.29 | 1.90 × 10−07 | 2.42 × 10−06 | 2.19 × 10−12 | UBASH3A |
All SNPs in high LD (r2 > 0.8) with the most associated SNP at each PSC locus were evaluated for potential function using SIFT16 and PolyPhen 217, the Genome Wide Annotation of Variants (GWAVA) online tool18, and a number of eQTL databases (Online Methods, Supplementary Tables 6–8). One of the new PSC risk variants (rs1893592, 21:g.43855067A>C) is the most strongly associated eQTL of UBASH3A, a gene involved in regulation of T-cell signalling, in two whole blood-based analyses19,20 and a B-cell only study21. The SNP is located three bases downstream of the 10th exon of UBASH3A, within the splice consensus sequence, and was reported as a splice-QTL in a recent RNA sequencing study19. The C allele, which is associated with reduced risk of PSC and has a frequency of 27.8% in our controls, disrupts the conserved 5′ splice donor sequence at this position in vertebrate introns, which is typically A (71% of sites) or G (24% of sites)22. The predicted consequence of this change is partial retention of the downstream intron possibly leading to non-stop mediated decay. Reanalysis of the gEUVADIS RNA-seq data23 revealed that this SNP was the most strongly associated with increased intron expression (P = 2×10−16, Supplementary Figure 5), with the PSC protective allele causing intron 10 to be retained in the UBASH3A mRNA. Further work is required to determine whether carrying the C allele at this SNP decreases UBASH3A protein levels and if this is the causal mechanism behind the reduced risk of PSC, celiac disease and rheumatoid arthritis (Supplementary Table 5). In addition, another variant within the UBASH3A gene (rs11203203, 21:g.43836186G>A) that is in low-LD (r2 = 0.12) with rs1893592 has been associated with vitiligo24 and type-1 diabetes25, further supporting the role of UBASH3A in immune-mediated disorders. We were unable to identify any current drugs targeting UBASH3A (Supplementary note).
To enable us to address the genetic relationship between PSC and IBD we obtained association summary statistics from the International IBD Genetics Consortium for 20,550 CD cases, 17,647 UC cases and 48,485 controls of European ancestry26. Across each of the eighteen non-HLA PSC risk loci we used a Bayesian test of colocalisation27 to identify loci with strong evidence (posterior probability > 0.8) of either shared or independent causal variants between pairs of traits (Online Methods, Supplementary Table 9). Four of the eighteen PSC risk loci have not been associated at genome-wide significance with IBD (BCL2L11, FOXP1, SIK2 and UBASH3A) although the lead SNPs at two of these loci (rs72837826 – BCL2L11 and rs1893592 – UBASH3A) did demonstrate strong evidence for colocalisation (posterior probability > 0.8) and suggestive evidence of association (P < 10−4) in the UC cohort (Supplementary Table 9, 10). Of the fourteen PSC loci that had been previously associated with IBD (UC, CD or both), four demonstrated strong evidence that the causal variant is independent from that in UC and CD (IL2RA, CCDC88B, CLEC16A and PRKD2), a finding supported by the low linkage disequilibrium (r2 < 0.2) between the lead SNPs in PSC and UC/CD at these loci (Supplementary Tables 9 and 10). Thus, even for highly comorbid diseases, significant association to the same region of the genome will not always be driven by a shared causal variant. This supports similar observations for other related phenotypes such as psoriasis versus psoriatic arthritis28,29. Six of the fourteen loci associated with PSC and IBD displayed strong evidence of a shared causal variant with UC, CD or both (MST1, IL21, HDAC7, SH2B3, CD226 and PSMG1) (Figure 1, Supplementary Tables 9 and 10). We further tested these six SNPs for evidence of heterogeneity of effect using Cochran’s Q test (Online Methods). Four showed significantly increased effect size in PSC relative to both UC and CD (MST1, IL21, SH2B3 and CD226) (P < 2.78×10−3) with an additional locus (PSMG1) showing significantly increased effect size relative to CD only (Figure 1). Simulation studies showed that the observed heterogeneity of effect is unlikely to be driven by the large difference in sample size between the PSC and UC cohorts (Pempirical < 3.00×10−4 at all four SNPs) (Supplementary Note). We did not detect evidence of heterogeneity of effect between PSC patients expressing different IBD phenotypes (PSC-UC, PSC-CD or PSC-NoIBD) (Supplementary Fig. 6). However, our power to detect significant heterogeneity of effect between these PSC subphenotypes was limited by sample size (Supplementary Table 11).
Figure 1. Odds ratios (and their 95% confidence intervals) for PSC, UC and CD across the 6 PSC associated SNPs demonstrating strong evidence for a shared causal variant (maximum posterior probability > 0.8).
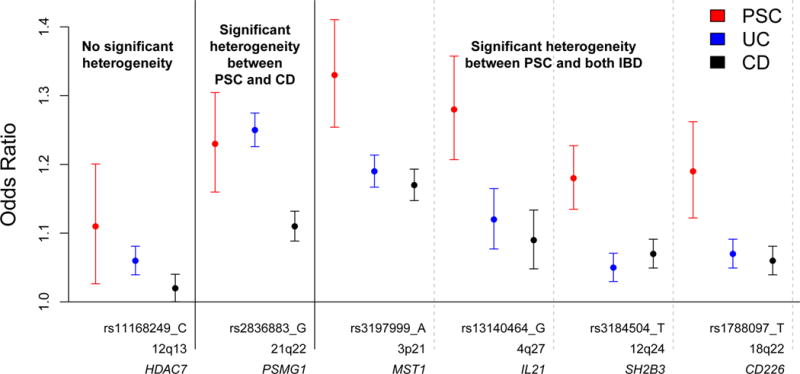
PSC ORs were taken from the GWAS and replication meta-analysis. UC and CD ORs were obtained from the latest association studies conducted by the International IBD Genetics Consortium26. Heterogeneity of odds tests were carried out using Cochran’s Q test. A failure to detect significant heterogeneity of odds does not necessarily indicate that effect sizes are equivalent because power to detect heterogeneity varies across SNPs.
While the much larger size of the UC and CD cohorts gives us power to investigate the effects of PSC risk SNPs in IBD, the PSC cohort is underpowered to do the reverse. Thus, to clarify the pairwise genetic correlation between PSC, UC and CD we obtained genome-wide individual level genotype data from the International IBD Genetics Consortium for 6,247 CD cases, 6,686 UC cases and 34,393 population controls of European descent26 and used GCTA to estimate genome-wide genetic correlations (rG) using a bivariate linear mixed model30,31 (Online Methods, Supplementary Note). This analysis quantified the SNP-heritability (h2SNP) of PSC as 0.148 (95% CI: 0.135–0.161), and showed that in the context of common genetic variation, PSC is significantly more related to UC (rG = 0.29) than CD (rG = 0.04) (P = 2.55×10−15) (Figure 2), consistent with the clinical phenotype most often observed in PSC-IBD patients. Moreover, the genetic correlation between UC and CD (rG = 0.56) is significantly greater than that between PSC and either UC or CD (P < 1.0×10−15). Due to a lack of data regarding the PSC status of individuals in the UC and CD cohorts we could not remove the approximately 5% of patients we would expect to have comorbid PSC. This suggests that, while our estimates of the genome-wide genetic correlation between PSC and both UC and CD may seem surprisingly low, these are likely slight overestimates of the true genetic correlation between the diseases. We validated the GCTA co-heritability estimates using a summary statistics-based genetic correlation analysis (LD score regression32), and found support for the reported genetic relationships (i.e. rGCD.vs.UC = 0.68 > rGPSC.vs.UC = 0.39 > rGPSC.vs.CD = 0.09) (Supplementary Figure 7). The low genome-wide genetic correlation between PSC and the IBDs is also supported by known differences in HLA risk alleles11,33 and our discovery that PSC has both independent causal variants and shared causal variants of heterogeneous effect size compared to both UC and CD. The analyses presented in this study, based on common genetic variants (MAF > 1%), suggest functional studies in both the biliary tree and intestinal tract are required if we are to understand the biological consequences of PSC associated genetic variants, whether or not they are shared with IBD.
Figure 2. Genome-wide genetic correlation between PSC (and its subphenotypes), CD and UC.
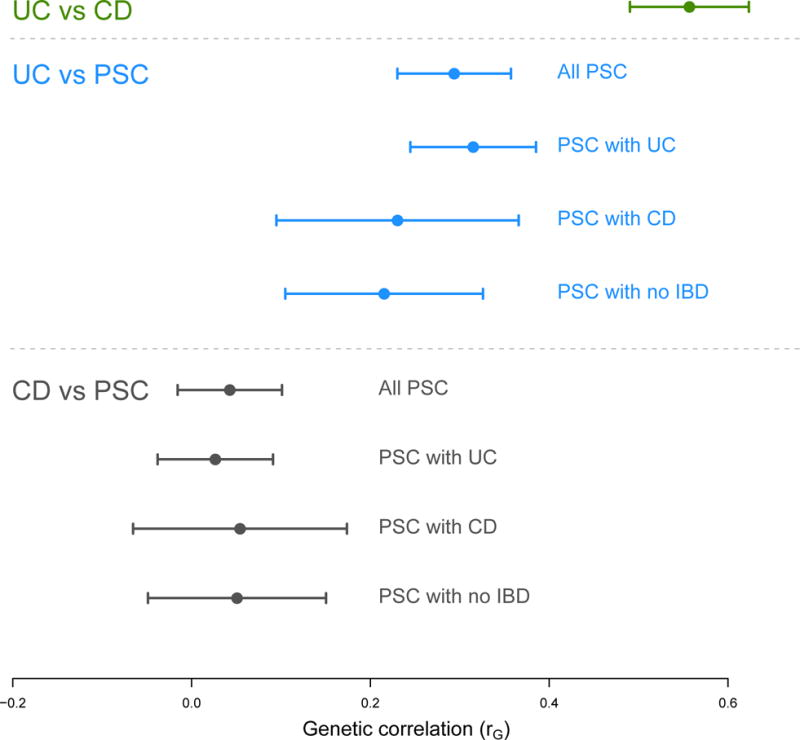
Genetic correlations (and their 95% confidence intervals) were calculated using a bivariate extension of the linear mixed model30 implemented in GCTA (Online Methods). PSC has a lower genetic correlation with both CD and UC than the two inflammatory bowel diseases have to each other. PSC is genetically more correlated to UC than it is to CD and this is consistent across the PSC subphenotypes.
While it is clear that a substantial component of the genetic architecture of PSC is not shared with either CD or UC, our data also show that shared genetic risk factors do certainly exist and likely play some role in disease comorbidity. However, under a purely additive genetic liability threshold model, the genetic covariance between the two diseases would need to be greater than 0.76 to fully explain the fact that 60% of PSC cases have comorbid UC (Supplementary Figure 8). In contrast, the observed genetic correlation (rG = 0.29) would generate a PSC-UC comorbidity rate of only 1.6% under this model. This demonstrates that the observed extent of comorbidity between PSC and UC is not fully explained by shared additive genetic effects of common variants and that other factors must play a role, such as shared environmental effects or shared rare variants not captured by our GWAS and imputation data.
In summary, we have performed the largest genome-wide association study of PSC to date and identified four new PSC risk loci. We now consider 23 regions of the genome to be associated with disease risk, including four loci only recently associated with PSC in a cross-disease meta-analysis34. One of our new associations suggests that decreased UBASH3A is associated with a lower risk of PSC through a common NMD variant. We have also shown that, even for highly comorbid phenotypes such as PSC and IBD, significant association to the same region of the genome will not always be driven by a common causal variant. Furthermore, by conducting genome-wide comparisons with CD and UC we have, for the first time, shown that the comorbid gastrointestinal inflammation seen in the majority of PSC patients cannot be fully explained by shared genetic risk. Thus, the biliary and intestinal inflammation seen specifically in PSC should be studied to advance our understanding of the disease and improve clinical outcome for patients with this devastating disorder.
Online Methods
Ethical Approval
The ethics committees or institutional review boards of all participating centers approved the studies and the recruitment of participants. Written informed consent was obtained from all participants.
GWAS cohort
Cohorts and genotyping
731 PSC cases and 3,202 population controls from Scandinavia and Germany were ascertained and genotyped using the Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA) at three different centers7. A cohort of 1,227 UK PSC cases was recruited from across more than 150 UK National Health Service Trusts or Health Boards, including all transplant centers in the UK, by the UK-PSC consortium. A cohort of 904 US PSC patients were enrolled in the PSC Resource of Genetic Risk, Environment and Synergy Studies (PROGRESS), a multicenter collaboration between eight academic research institutions across the US and Canada. PROGRESS ascertained additional DNA samples from established PSC cohorts from Canada (N=259) and Poland (N=43). The UK and US GWAS cohorts were genotyped using the Illumina HumanOmni2.5-8 BeadChip (Illumina, San Diego, CA, USA) and called using the GenCall algorithm implemented in GenomeStudio. UK samples were genotyped at the Wellcome Trust Sanger Institute (Hinxton, UK) and the US samples at the Mayo Clinic Medical Genome Facility (Rochester, MN, USA). A diagnosis of PSC was based on standard clinical, biochemical, cholangiographic and histological criteria35, with exclusion of secondary causes of sclerosing cholangitis. Commonly accepted clinical, radiological, endoscopic and histological criteria were also used for diagnosis and classification of IBD36. Genetic data from 12,595 individuals genotyped on the Illumina HumanOmni2.5-4v1 array (Omni2.5-4) as part of The University of Michigan Health Retirement Study were downloaded from the Database of Genotypes and Phenotypes (dbGaP37). Genotyping was performed at the Center for Inherited Disease Research (CIDR) and genotypes called using GenomeStudio version 2011.e, (see the HRS website for more details).
Quality control
All SNPs were aligned to NCBI build 37 (hg19). Genotype data were quality controlled independently across 6 batches defined by genotyping centre (AffySF: N = 2,205, AffyHZ: N = 1,256; AffyAB: N = 472; IlluminaWTSI: N= 1,227;IlluminaMAYO: N= 1,206; IlluminaCIDR: N = 12,595). Initially, SNPs out of Hardy-Weinberg equilibrium (HWE: P < 1×10−6) in controls (excluding those in the HLA region) or with a call rate less than 80% were removed. SNPs failing in at least one batch were removed from all cohorts genotyped using the same chip. For sample QC, individuals whose sex determined using the X chromosome homozygosity rate (F) and Y chromosome call rate differed from that in our patient database (or could not be genetically determined, F or Y-chromosome call rate between 0.3–0.7) were removed. Next, Abberant38 was used to identify samples with outlying heterozygosity or genotype call rate. Samples with a call rate less than 90% for an individual chromosome were also removed. A set of 82,085 independent SNPs (pairwise r2 <0.2) genotyped on all arrays was identified for the purpose of estimating sample relatedness and ancestry, excluding SNPs that a) were within regions of high linkage disequilibrium, b) had a MAF < 10% or c) were A/T or C/G SNPs. Pairwise identity by descent was estimated for all individuals in the study using PLINK, and the sample with the lowest genotype call rate was removed for all pairs with IBD > 0.9. Both samples were excluded if case/control status was discordant between duplicates. To maximize power to detect association, related samples (0.1875 < IBD < 0.9) were retained and a mixed model used for association testing. Sample ancestry was inferred via principal components analysis implemented in EIGENSTRAT39. Population principal components were calculated using genotype data from the CEU, YRI and CHB/JPT samples from the 1000 Genomes Project. Factor loadings from these principal components were then used to project these principal components for our cases and controls. Samples of non-European ancestry were identified using Aberrant38. The number of samples failing each QC step is shown in Supplementary Table 1. In total, 2,871 cases and 12,019 controls passed sample QC. Next, a more thorough marker QC was conducted within batches by excluding, genotyping platform-wide, SNPs with a) different probe sequences on the Omni2.5-4 and Omni2.5-8 array, b) a call rate < 98%, c) MAF<1%, d) significant evidence of deviation from HWE (P < 1×10−5) in controls and e) a significant difference in call rate between cases and controls (P < 1×10−5), in at least one of the genotyping batches. Outside of the HLA region, markers only present on one of the two Illumina arrays were also removed. After SNP QC, 1,207,121 Omni2.5-4 SNPs, 1,215,097 Omni2.5-8 SNPs and 528,496 Affymetrix 6 SNPs were available.
Genotype Imputation
Only 322,807 SNPs feature on both the Affy6 and Omni2.5 arrays so the samples genotyped on these arrays were phased and imputed separately. For computational efficiency, the genome was split into 3Mbp batches and those spanning the centromere were split and joined to the last complete batch either side of the centromere. Batches of less than 200 SNPs were merged with an adjacent batch. Pre-phasing was performed using the SHAPEIT2 algorithm40 and imputation using IMPUTE241. We used a combined reference panel of the 1000 Genomes Phase 1 integrated version 3 and the UK10K cohort, consisting of 4,873 individuals and 42,359,694 SNPs (k_hap=2,000, Ne=20,000). Post-imputation, SNPs with a posterior probability less than 0.9 or info score less than 0.5 were removed. The QC steps outlined above for directly genotyped SNPs were applied to the imputed genotype data. SNPs with r2 < 0.8 between directly genotyped and imputed genotypes were removed and phasing and imputation repeated. Following QC (as outlined above), a total of 7,891,602 SNPs available for association testing across 2,871 PSC cases and 12,019 population controls (Supplementary Table 2).
Association Analysis
A linear mixed model implemented in the MMM software42 was used to test association between genetic variants and case/control status. To reduce compute time the relationship matrix was constructed using the 82,085 quasi-independent SNPs previously used in the PCA. To prevent the association analyses being biased by informed missingness across our genotyping batches, linear mixed model association tests were conducted across three different batches of directly-genotyped and imputed SNPs, defined on their availability for only the Omni2.5 genotyped samples (N = 2,015,514), only the Affy6 genotyped samples (N = 114,935), or across all genotyped samples (N = 5,761,153).
Stepwise conditional regression analysis (excluding the extended MHC region) was undertaken in MMM to identify independent association signals (P < 5.0 × 10−6) within PSC associated loci. The previously reported lead SNP within each of the 15 known PSC loci was selected for replication, though we also took forward the most associated SNP in our study if it was a poor tag (r2 < 0.8) of previously reported SNP. In addition, 24 SNPs outside of established PSC risk loci with P < 5 ×10−6 were also included in the replication experiment. All cluster plots were manually inspected prior to SNP selection.
Validation and replication cohorts
Cohorts and genotyping
An independent replication cohort of 2,011 PSC cases from Europe and North America was ascertained following the diagnostic criteria outlined above. A total of 8,784 population controls of European descent were ascertained, including 515 from the Mayo Clinic Biobank43 and 1000 from the INTERVAL study44. British and Canadian samples were genotyped at the Wellcome Trust Sanger Institute in Cambridge, UK (N = 2,366) and all other samples at the Institute of Clinical Molecular Biology in Kiel, Germany (N = 11,152) using the same Agena Biosciences iPLEX design. To reduce the risk of false-positive associations being driven by imputation errors we undertook a substantial validation experiment, genotyping the 40 SNPs in our replication experiment across 2,723 cases in the GWAS study.
Quality control
Two SNPs yielded poor genotype clusters and were removed from further study. Four SNPs with a call rate less than 95% or Hardy Weinberg equilibrium P < 1.25 × 10−3 (Bonferroni correction for 40 SNPs) within controls were excluded (Supplementary Table 12). Samples with a call rate less than 92%, or where the genetically determined sex differed from that in our patient database, were removed. The sample with the lowest call rate in duplicate pairs was removed from duplicate pairs (IBS > 0.9) (Supplementary Table 3). Post-QC, one SNP had an r2 less than 0.90 between the discovery and validation genotyping and, following manual inspection of cluster plots, was removed from the replication study.
Replication and Combined association analyses
For the replication analysis, logistic regression tests of association were performed separately for samples from six geographic regions (Supplementary Table 3) using SNPTEST v2 (Marchini et al., 2007). Inverse-variance weighted fixed effects meta-analyses implemented in METAL16 were then used to a) test for association across all replication samples and b) test the evidence of association across the GWAS and replication cohorts combined. To classify a region as newly associated with PSC we required both significant evidence of association in the replication cohort (P < 5.26 × 10−3, Bonferroni correction for 19 one-tailed tests) and genome-wide significance (P < 5 × 10−8) in the combined meta-analysis.
Candidate gene prioritization
Functional annotation
All SNPs in high LD (r2 > 0.8) with lead SNPs at PSC associated loci were annotated for potential function using the Genome Wide Annotation of Variants (GWAVA) online tool19. In addition, all coding SNPs from this set were also annotated using SIFT16 and PolyPhen218.
Pathway analysis
To quantify the functional relationship between genes within PSC risk loci, we conducted a GRAIL pathway analysis. GRAIL evaluates the degree of functional connectivity between genes based on the extent they co-feature in published abstracts (we used all PubMed abstracts prior to 2006 to avoid biasing our analysis due to results from large-scale GWASs). All PSC associated loci were included in the analysis and only genes with GRAIL P < 0.05 and edges with a score of > 0.5 were included in the connectivity map.
Expression quantitative trait loci (eQTL)
eQTL analysis focused on published cis-eQTLs due to the lower reproducibility caused by smaller effect sizes and context-specificity of trans-eQTL45. Eight eQTL datsets were included in the analysis: eQTL data from 12 studies collated in the Chicago eQTL browser, eQTL results from 1,421 samples of 13 different tissue types by the genotype-tissue expression (GTEx) project46, 462 lymphoblastoid cell lines24, 922 whole blood samples20, 8,086 whole blood samples21, purified B cells and monocytes from 283 individuals22, activated monocytes from 432 individuals47, and activated monocyte-derived dendritic cells from diverse populations48. The most significant variant-gene associations were extracted from each eQTL dataset and were reported as overlapping if that variant was in high LD (r2 > 0.8) with any of the lead SNPs in the PSC GWAS meta-analysis.
Modelling PSC and IBD genetic risk
Association summary statistics from the European arm of the latest International IBD Genetics Consortium study27 were downloaded. Where available we used results from their combined GWAS plus Immunochip follow-up study and otherwise used those from the GWAS analysis. Definition of the 231 significantly associated loci as CD, UC or both (IBD) was taken from Liu and van Sommeren et al27. Due to the limited availability of relevant subphenotype data within the IIBDGC data, we were unable to identify the 3–5% of IBD cases that we expect to have PSC. Including these individuals as IBD cases in our comparisons lowers our power to detect differences between the two diseases.
Causal variant co-localisation analysis
To identify causal variants within disease associated loci that are shared between diseases we used a summary statistic based Bayesian test of colocalisation (COLOC), implemented in R28. Briefly, COLOC generates posterior probabilities for five different hypotheses: 1) no association to either disease, 2) association to disease 1 but not disease 2, 3) association to disease 2 but not disease 1, 4) association to both disease 1 and 2 but independent causal variants and 5) association to both disease 1 and 2 with a common causal variant. Only SNPs present in all the cohorts (PSC, CD, UC and IBD) were included in the analysis and associated regions were defined as 1MB regions with the most associated SNP at the centre. Within each region we calculated the r2 between the PSC lead SNP and the SNP most associated with each of the other three diseases. Default priors were used for the probability of a SNP being a) associated to an individual disease (1×10−4) and b) causally associated to both diseases (1×10−5). This prior probability of colocalisation is more conservative in declaring distinct causal variants compared to a recent colocalisation analysis across six immune-mediated disorders49.
Heterogeneity of effects analysis
A formal heterogeneity of odds test was performed between PSC and IBD using the Cochran’s Q test implemented in METAL16 for all 18 PSC risk loci. The odds ratios and standard errors were obtained from our current PSC GWAS and the IIBDGC analysis27. A locus was declared to have significant heterogeneity of effects based on a threshold of P = 2.78×10−3 to account for multiple testing (Bonferroni correction applied to 5% significance threshold, N=18 tests). In order to test whether the significant heterogeneity of effects are due to an overestimation of effect sizes in the smaller PSC cohort, we undertook a simulation study which demonstrated that the observed degree of heterogeneity is unlikely to occur by chance (Supplementary Note).
Genetic correlation analysis
Genome-wide SNP data from 12,933 IBD cases and 34,393 population controls of European descent was made available to us by the International IBD Genetics Consortium (IIBDGC). The quality control and imputation of these data using 1000 Genomes haplotypes has been previously described27. See Supplementary Note for details of the SNP and sample quality control (Supplementary Table 13) undertaken across the IIBDGC and PSC data to ensure compatibility and remove duplicated individuals. Individual level genotype data for PSC, CD, UC and IBD were used to estimate the proportion of variance in liability explained by SNPs genome-wide under a multiplicative model using the linear mixed model based restricted maximum likelihood (REML) method implemented in the GCTA software32,50,51. Ancestry principal components were calculated using genotype data from the 1000 Genomes project and were projected for all our cases and controls. The first twenty principals components were included as covariates in the linear mixed model. We assumed a prevalence of 0.0001 for PSC, 0.005 for CD and 0.0025 for UC. A bivariate extension of the linear mixed model31, again implemented in GCTA32, was used to estimate the additive covariance component and estimate the genetic correlation (rG) between PSC and either CD, UC, or IBD.
In addition, we undertook an alternative genetic correlation analysis that uses summary statistics and LD score regression33. Of the 7,458,430 SNPs that were shared between PSC and both IBDs, 1,102,210 HapMap3 SNPs were selected for the analysis as recommended. Then, pre-computed LD scores from the 1000 Genomes European data were used to run LD score regression to estimate genetic correlation.
Calculating comorbidity under a purely pleiotropic genetic model
Under a bivariate liability threshold model, where all disease risk is explained by additive genetics, the probability that an individual has disease 1, given that he has disease 2, is given by
where Ki is the prevalence of disease i, Ti =Φ−1(1−Ki) is the liability threshold of disease i, is the heritability of disease i, rg is the genetic correlation and F(.) is the multivariate cumulative distribution function for normal distribution.
Supplementary Material
Acknowledgments
We thank the patients and healthy controls for their participation, and are grateful to the physicians, scientists and nursing staff who recruited individuals whose data is used in our study. We acknowledge the use of DNA or genotype data from a number of sources, including a) The Health and Retirement Study (HSR) conducted by the University of Michigan, funded by the National Institute on Aging (grant numbers U01AG009740, RC2AG036495, and RC4AG039029) and accessed via dbGAP, b) Popgen 2.0, supported by a grant from the German Ministry for Education and Research (01EY1103), c) The Mayo Clinic Biobank, supported by the Mayo Clinic Center for Individualized Medicine, d) The INTERVAL study, undertaken by the University of Cambridge with funding from the National Health Service Blood and Transplant (NHSBT) – the views expressed in this publication are those of the authors and not necessarily those of the NHSBT, e) The FOCUS biobank. We thank the investigators of the 1000 genomes and UK10K projects for generating and sharing the population haplotypes and Dr. Jie Huang for advice regarding imputation. We thank all members of the International IBD Genetics Consortium for sharing genetic data vital to the success of our study.
This study was supported by NoPSC, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK RO1DK084960, KNL), the Wellcome Trust (098759/Z/12/Z: LJ; 098051: S-GJ, JZL, TS, JG-A, NK, DJG and CAA), the Kwanjeong Educational Foundation (S-GJ), the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (SysInflame grant 01ZX1306A) and the Chris M. Carlos and Catharine Nicole Jockisch Carlos Endowment in PSC. This project received infrastructure support from the DFG Excellence Cluster No. 306 “Inflammation at Interfaces” and the PopGen Biobank (Kiel, Germany), an endowment professorship (AF) by the Foundation for Experimental Medicine (Zurich, Switzerland). The recruitment of patients in Hamburg was supported by the YAEL-Foundation and the DFG (SFB841). BA Lie and the Norwegian Bone Marrow Donor Registry at Oslo University Hospital, Rikshospitalet in Oslo are acknowledged for sharing the healthy Norwegian controls.
Participants in the INTERVAL randomised controlled trial were recruited with the active collaboration of NHS Blood and Transplant England (www.nhsbt.nhs.uk), which has supported field work and other elements of the trial. DNA extraction and genotyping was funded by the National Institute of Health Research (NIHR), the NIHR BioResource(http://bioresource.nihr.ac.uk/) and the NIHR Cambridge Biomedical Research Centre (www.cambridge-brc.org.uk). The academic coordinating centre for INTERVAL was supported by core funding from: NIHR Blood and Transplant Research Unit in Donor Health and Genomics, UK Medical Research Council (G0800270), British Heart Foundation (SP/09/002), and NIHR Research Cambridge Biomedical Research Centre.
We thank K Cloppenborg-Schmidt, I Urbach, I Pauselis, T Wesse, T Henke, R Vogler, V Pelkonen, K Holm, H Dahlen Sollid, B Woldseth, J Andreas A and L Wenche Torbjørnsen for expert help. RKW is supported by a clinical fellowship grant (90.700.281) from the Netherlands Organization for Scientific Research. BE receives support from Medical Research Council, United Kingdom. TM and DG are supported by Deutsche Forschungsgemeinschaft, Grant. AP is supported by Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), grant PI071318 Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación, and grant PI12/01448, from Ministerio de Economía y Competitvidad, Spain. PD is supported by Canadian Institutes of Health research (CIHR) and Genome Canada. CW is supported by grants from the Celiac Disease Consortium (BSIK03009) and Netherlands Organization for Scientific Research (NWO, VICI grant918.66.620).
Finally, we acknowledge the members of the International PSC Study Group, the NIDDK Inflammatory Bowel Disease Genetics Consortium (IBDGC), and the UK-PSC Consortium for their participation. We thank Jenn Rud for secretarial support.
Footnotes
URLS
http://www.genome.gov/gwastudies/
http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/
http://hrsonline.isr.umich.edu/
https://www.sanger.ac.uk/sanger/StatGen_Gwava
http://sift.jcvi.org/www/SIFT_chr_coords_submit.html
http://genetics.bwh.harvard.edu/pph2/
http://www.broadinstitute.org/mpg/grail/
http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/
Data Availability
Genome-wide summary statistics are available at www.ipscsg.org/downloads. The University of Michigan HRS data is available from dbGap under accession number phs000428.
Author contribution
S-G.J., B.D.J., N.K., T.S., J.G-A. and C.A.A. performed statistical data analysis, S-G.J., B.D.J., S.M., T.F., E.M., E.J.A. and C.A.A performed initial quality control and sample identification, L.J., J.Z.L., D.J.G., M.d.A. and C.A.A. provided statistical and analytical advice, T.H.K., K.N.L. and C.A.A. coordinated the project and supervised the analyses, S-G.J., B.D.J., T.H.K., K.N.L. and C.A.A. drafted of the manuscript. E.M.S., K.M.B., A.B., S.V., B.E., P.R., M.F., T.M., C.S., M.S., T.J., D.N., D.E., F.B., A.T., M.L., W.L., G.J., U.B., R.K.W., C.W., H-U.M., P.M., A.P., K.K., O.C., P.I., E.G., K.S., C.M., J.S., W.H.O., D.J.R., J.D., A.F., A.F.G., J.E.E., S.S., C.C., C.L.B., V.A.L., J.A.O., K.B.C., K.V.K., N.C., M.P.M., B.S., G.M., R.N.S., G.A., R.W.C., G.M.H., S.M.R., A.F., K.N.L., C.A.A., The UK-PSC Consortium, The International IBD Genetics Consortium, and The International PSC Study Group collected the samples, performed clinical ascertainment, or coordinated sample logistics. All authors read and approved the final version of the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Boonstra K, et al. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflammatory Bowel Diseases. 2012;18:2270–2276. doi: 10.1002/ibd.22938. [DOI] [PubMed] [Google Scholar]
- 2.Tischendorf JJW, Hecker H, Kruger M, Manns MP, Meier PN. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: A single center study. American Journal of Gastroenterology. 2007;102:107–114. doi: 10.1111/j.1572-0241.2006.00872.x. [DOI] [PubMed] [Google Scholar]
- 3.Karlsen TH, Kaser A. Deciphering the Genetic Predisposition to Primary Sclerosing Cholangitis. Seminars in Liver Disease. 2011;31:188–207. doi: 10.1055/s-0031-1276647. [DOI] [PubMed] [Google Scholar]
- 4.Karlsen TH, Schrumpf E, Boberg KM. Update on primary sclerosing cholangitis. Digestive and Liver Disease. 2010;42:390–400. doi: 10.1016/j.dld.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Bergquist A, et al. Increased risk of primary sclerosing cholangitis and ulcerative colitis in first-degree relatives of patients with primary sclerosing cholangitis. Clinical gastroenterology and hepatology. 2008;6:939–943. doi: 10.1016/j.cgh.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 6.de Vries AB, Janse M, Blokzijl H, Weersma RK. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World Journal of Gastroenterology. 2015;21:1956–1971. doi: 10.3748/wjg.v21.i6.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melum E, et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nature Genetics. 2011;43:17–19. doi: 10.1038/ng.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellinghaus D, et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology. 2013;58:1074–1083. doi: 10.1002/hep.25977. [DOI] [PubMed] [Google Scholar]
- 9.Folseraas T, et al. Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. Journal of hepatology. 2012;57:366–375. doi: 10.1016/j.jhep.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlsen TH, et al. Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology. 2010;138:1102–1111. doi: 10.1053/j.gastro.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 11.Liu JZ, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature Genetics. 2013;45:670–675. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava B, et al. Fine mapping and replication of genetic risk loci in primary sclerosing cholangitis. Scandinavian journal of gastroenterology. 2012;47:820–826. doi: 10.3109/00365521.2012.682090. [DOI] [PubMed] [Google Scholar]
- 13.1000 Genomes Project. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–09. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 17.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ritchie GRS, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nature Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Battle A, et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Research. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westra HJ, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature Genetics. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fairfax BP, et al. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nature Genetics. 2012;44:502–510. doi: 10.1038/ng.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang MQ. Statistical features of human exons and their flanking regions. Human Molecular Genetics. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 23.Lappalainen T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin Y, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nature genetics. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nature genetics. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu JZ, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nature Genetics. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giambartolomei C, et al. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genetics. 2014;10(5):e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuart PE, et al. Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. American Journal of Human Genetics. 2015;97:816–836. doi: 10.1016/j.ajhg.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowes J, et al. Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nature Communications. 2015;6046 doi: 10.1038/ncomms7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2542. doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JA, Lee SH, Goddard ME, Visscher PM. GCTA: A Tool for Genome-wide Complex Trait Analysis. American Journal of Human Genetics. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature Genetics. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goyette P, et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nature Genetics. 2015;47:172–9. doi: 10.1038/ng.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellinghaus D, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nature Genetics. 2016;48:510–518. doi: 10.1038/ng.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 35.Chapman RWG, et al. Primary Sclerosing Cholangitis - a Review of Its Clinical-Features, Cholangiography, and Hepatic Histology. Gut. 1980;21:870–877. doi: 10.1136/gut.21.10.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yimam KK, Bowlus CL. Diagnosis and classification of primary sclerosing cholangitis. Autoimmunity Reviews. 2014;13:445–450. doi: 10.1016/j.autrev.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 37.Mailman MD, et al. The NCBI dbGaP database of genotypes and phenotypes. Nature Genetics. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bellenguez C, et al. A robust clustering algorithm for identifying problematic samples in genome-wide association studies. Bioinformatics. 2012;28:134–135. doi: 10.1093/bioinformatics/btr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 40.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nature Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 41.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pirinen M, Donnelly P, Spencer CCA. Efficient Computation with a Linear Mixed Model on Large-Scale Data Sets with Applications to Genetic Studies. Annals of Applied Statistics. 2013;7:369–390. [Google Scholar]
- 43.Olson JE, et al. The Mayo Clinic Biobank: A Building Block for Individualized Medicine. Mayo Clinic Proceedings. 2013;88:952–962. doi: 10.1016/j.mayocp.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore C, et al. The INTERVAL trial to determine whether intervals between blood donations can be safely and acceptably decreased to optimise blood supply: study protocol for a randomised controlled trial. Trials. 2014;15:363. doi: 10.1186/1745-6215-15-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaffney DJ. Global properties and functional complexity of human gene regulatory variation. PLoS Genetics. 2013;9:e1003501. doi: 10.1371/journal.pgen.1003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lonsdale J, et al. The Genotype-Tissue Expression (GTEx) project. Nature Genetics. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fairfax BP, et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science. 2014;343 doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee MN, et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;343 doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fortune MD, et al. Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nature genetics. 2015;47:839–846. doi: 10.1038/ng.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. American journal of human genetics. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang JA, et al. Common SNPs explain a large proportion of the heritability for human height. Nature Genetics. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.