

Abstract
Background
Microbleeds in the brain have been shown to occur in Alzheimer’s disease (AD), affecting approximately a third of subjects that present with typical dementia of the Alzheimer’s type (DAT). However, little is known about the frequency or distribution of microbleeds in subjects with AD that present with atypical clinical presentations.
Objective
To determine whether the frequency and regional distribution of microbleeds in atypical AD differs from that observed in subjects with DAT, and to determine whether microbleeds in atypical AD are associated with age, demographics or cognitive impairment.
Methods
Fifty-five subjects with Aβ deposition on Pittsburgh compound B (PiB) PET who presented with predominant language (n=37) or visuospatial/perceptual (n=18) deficits underwent T2*weighted MRI. These subjects were compared to 41 PiB-positive subjects with DAT. Microbleeds were identified and assigned a lobar location.
Results
The proportion of subjects with microbleeds did not differ between atypical AD (42%) and DAT (32%). However, atypical AD had larger numbers of microbleeds than DAT. In addition, the topographic distribution of microbleeds differed between atypical AD and DAT, with atypical AD showing the highest density of microbleeds in the frontal lobes. Among atypical AD, number of microbleeds was associated with age, but not gender or cognition. Microbleeds were more common in subjects with language (51%) versus visuospatial/perceptual deficits (22%).
Conclusions
Microbleeds are relatively common in both DAT and atypical AD, although atypical AD subjects appear to be at particular risk for developing large numbers of microbleeds and for developing microbleeds in the frontal lobe.
Keywords: Alzheimer disease, Alzheimer Dementia, microbleeds, beta-amyloid, magnetic resonance imaging, positron emission tomography, early-onset Alzheimer’s disease, logopenic, posterior cortical atrophy
Introduction
Alzheimer’s disease (AD) is associated with amyloid-related imaging abnormalities [1], including microbleeds (MBs). Microbleeds located in cortical areas are thought to result from deposition of the protein beta-amyloid (Aβ) in cerebral vessels [2], while subcortical and infratentorial MBs from cerebrovascular disease [3]. Microbleeds can be identified as small hemosiderin deposits on T2*-weighted MRI. Microbleeds occur in 12–40% of patients with dementia of the Alzheimer’s type (DAT) [4–11] which is characterized by early memory loss and hippocampal atrophy [12]. The topographic distribution of MBs in DAT has been postulated to match the typical distribution of cerebral amyloid angiopathy, with greatest burden in the occipital lobes[5, 13], with MBs also associated with older age [6, 13]. It is less clear whether MBs are associated with cognitive impairment, with some studies finding an association in healthy populations[14, 15] but others failing to find an association within DAT cohorts [5, 16].
Approximately 25% of AD subjects do not complain of early memory loss [17], but instead present with other cognitive complaints, including difficulty with language [18–21] or visuospatial/perceptual function [22–24], and show relative sparing of the hippocampi [25, 26]. These subjects are considered to have atypical AD. We have previously shown that MBs do occur in atypical AD subjects [27], although the small number of subjects (n=13) at that time did not allow us to investigate whether MBs are associated with cognitive, demographic and imaging features of atypical AD, and our study was limited to only subjects with a language presentation of AD. The aim of the current study was first, to utilize a larger cohort of atypical AD subjects to determine whether the frequency and distribution of MBs differs from that observed in subjects with DAT. Secondly, we aimed to determine whether MBs in atypical AD are associated with age, cognitive impairment or Aβ burden, and whether the frequency of MBs differs in subjects presenting with a language versus visuospatial/perceptual presentation.
Materials and Methods
Subject selection
We identified 55 atypical AD subjects who had Aβ deposition on Pittsburgh compound B (PiB) PET, had undergone T2*weighted MRI and presented with predominant language (n=37) or visuospatial/perceptual (n=18) deficits. All subjects had been prospectively recruited from the Department of Neurology between September 1st 2011 and March 3rd 2014. The subjects with language impairment all met clinical criteria for the logopenic variant of primary progressive aphasia (lvPPA) [18], and all subjects with visuospatial/perceptual deficits met criteria for posterior cortical atrophy (PCA) [22]. Subjects were excluded if they had a chief complaint of memory impairment or if memory impairment was identified on cognitive testing as a prominent deficit at disease onset or presentation. All subjects underwent a neurological assessment and speech/language assessments as previously described [28].
We also identified a comparison cohort of subjects that presented with memory loss and met established criteria for DAT [12] that had also completed similar MRI sequencing. None of the DAT subjects met criteria for lvPPA or PCA. We selected all subjects from the Mayo Clinic Alzheimer’s Disease Research Center (ADRC) that met criteria for DAT, that had Aβ deposition on PiB PET, and had undergone T2*-weighted imaging (n=41). All subjects had been recruited into the ADRC from the Department of Neurology between September 1st 2011 and March 3rd 2014 and underwent standardized neurological and cognitive assessments.
The study was approved by the Mayo Clinic IRB. All patients consented for enrolment into the study. If the patient was deemed not to have the capacity to give informed consent, then permission was obtained from a proxy with both actual capacity and legal authority to give it, and assent was obtained from the participant.
MRI acquisition
All 96 subjects (55 atypical AD and 41 DAT) underwent a standardized MRI imaging protocol at 3T that included a 3D magnetization prepared rapid acquisition gradient echo (MPRAGE) sequence, a fluid-attenuated inversion recovery (FLAIR) sequence, and a T2*-weighted sequence to detect the presence of microbleeds. The majority of cases underwent a 2D GRE T2*-weighted sequence (TR/TE=200/20ms; flip angle=12°; FOV=20cm; in-plane matrix=256×224; phase FOV=1.00; slice thickness=3.3mm; bandwidth=15.63 kHz), although a couple of subjects underwent a 3D Swan T2*-weighted sequence (TR/TE=min/24.8ms; flip angle=30°; FOV=22cm; in-plane matrix=320×224; phase FOV=1.00; slice thickness=2.0mm; bandwidth=62.50 kHz).
MRI analysis
Each T2*-weighted MRI sequence was reviewed by an experienced image analyst and the location of each MB was marked using in-house software. Each marked MB was secondarily confirmed by one of two radiologists (CRJ or KK). Both the image analyst and the radiologists were blinded to clinical diagnosis. Microbleeds were defined as homogeneous hypointense lesions up to 10 mm in diameter in the grey or white matter. Occasionally it was not possible to make a definitive decision, such as distinguishing a MB from a vascular flow void. In such instances, the MB was labeled as “possible MB”. Possible MBs were not included in the analysis. As previously reported, interrater reliability for MB classification was excellent [13].
An in-house modified version of the automated anatomical labelling (AAL) atlas with 22 regions, including bilateral frontal, parietal, temporal, occipital lobar regions and deep/infratentorial grey and white matter regions, was created and registered into the space of each MPRAGE. Each MPRAGE with its atlas images were registered and resampled into the space of the T2*weighted image. Each MB was then assigned a location in the coordinate system of the T2*weighted image and hence also assigned an atlas location.
Maps of MB locations were built by transforming T2*weighted image locations into the MPRAGE image space and applying the discrete cosine transformation to the template space derived during statistical parametric mapping unified segmentation of the MPRAGE image. A sphere was placed around each MB to make it visible on 3D rendering; overlapping spheres were allowed to merge, forming clusters.
In addition, in order to assess potential vascular risk factors across groups, the burden of white matter hyperintensities (WMH) was assessed for each subject using the FLAIR sequence and an automated intensity-based algorithm, as previously described [27]. The burden of WMH was divided by total white matter volume and multiplied by 100, resulting in a WMH proportion (WMHp) that reflects the percentage of white matter affected by WMH. Total hippocampal volume was also measured for each subject to corroborate the clinical diagnoses using FreeSurfer software version 5.3 [29]. Hippocampal volumes were divided by total intracranial volume to correct for differences in head size.
PiB-PET analysis
All 96 subjects underwent Pittsburgh Compound B (PiB) PET imaging using a PET/CT scanner (GE Healthcare, Milwaukee, Wisconsin) operating in 3D mode. Subjects were injected with PiB (average, 614 MBq; range, 414–695 MBq) and after a 40–60 minute uptake period a 20 minute PiB scan was obtained consisting of four 5-minute dynamic frames following a low dose CT transmission scan. Standard corrections were applied. Individual frames of the PiB dynamic series were realigned if motion was detected. Each PiB scan was co-registered to the MPRAGE for each subject using 6 degrees-of-freedom affine registration. The AAL atlas was then transformed into the native space of each subject. A global PiB retention summary was generated by calculating median uptake for 6 ROIs: temporal, parietal, posterior cingulate/precuneus, anterior cingulate, prefrontal, and orbitofrontal cortex (left and right were combined for all ROIs). These regions were originally chosen to reflect the distribution of Aβ in the brain in AD[30]. Median PiB uptake in each region was divided by median uptake in cerebellar grey matter to create uptake ratios, and a global PiB SUV ratio (SUVR) was formed by calculating median uptake ratio values across all 6 regions. The global PiB SUVR was used to classify each subject as PiB-positive using a global cortical-to-cerebellar ratio cut-point of 1.5 [30]. Visual assessment of all PiB-PET scans was also performed (VJL and JLW) to verify PiB status. Regional PiB SUVRs were also calculated for frontal, temporal, parietal and occipital lobe using the AAL atlas.
Statistical analysis
Group comparisons were performed using Kruskal-Wallis tests for continuous variables and chi-squared or Fisher’s exact tests for categorical variables. Chi-squared tests were also used to evaluate the association between clinical group and absence/presence of microbleeds. When all expected cell counts from the 2×2 cross-classification table were greater than one we used the “N−1” method, and used Fisher’s exact test otherwise [31].
We used negative binomial regression models to evaluate whether the number of microbleeds differed by group, region, or other clinical factors. The negative binomial regression model is a widely used generalization of the Poisson regression model which incorporates “overdispersion” or variability that is greater than the mean often indicated by right-skewed counts[32]. We modeled cognitive performance measured using the Montreal Cognitive Assessment Battery (MoCA) [33], age, disease duration, and global PiB SUVR on a continuous linear scale.
We used a generalized estimating equations (GEE) approach with negative binomial models to examine regional differences by treating each subject as a “cluster” with four repeated measurements consisting of MB counts for the frontal, temporal, parietal, and occipital lobes [32]. This GEE approach allowed us to take into account the dependence among the repeated regional measurements from the same subject and examine both between-group and within-group regional differences in a single model. We estimated microbleed density, defined as microbleeds per L of tissue, using a GEE model with log volume included as an offset. Tissue volume was based on an atlas-based segmentation of a reference cohort and not estimated separately for each person, as previously described [13]. We assumed an exchangeable correlation structure for GEE models. To evaluate PiB deposition as a potential confounder contributing to increased MBs, we fit total and regional MB models adjusted for global and regional PiB SUVR (frontal, temporal, parietal and occipital lobe), respectively. We used the GENMOD procedure from SAS version 9.2 (SAS Institute Inc., Cary NC) to fit the negative binomial models and used R version 3.0.2 (R Foundation for Statistical Computing, Vienna, Austria) for other analyses.
We performed sensitivity analyses to examine the extent to which our findings were influenced by three atypical AD cases having >50 MBs. We examined the model estimates three ways: (a) by omitting these three cases entirely; (b) by omitting only two cases with >100 MB, and (c) by including these three cases in the analysis but replacing their observed MB count with the maximum observed across all other cases. This approach was used for total MBs and regional MBs.
Results
Demographics
The atypical AD and DAT cohorts did not differ in education, age at examination, age at onset, PiB SUVR or in clinical vascular risk factors (hypertension, hypercholesterolemia, diabetes, history of strokes and smoking) (Table 1). However, the atypical AD group did have a higher proportion of females, shorter disease duration, better performance on the Mini-Mental State Examination, and lower WMHp than the DAT cohort. Consistent with the clinical diagnoses, the atypical AD cohort had larger hippocampal volumes than the DAT cohort. Within atypical AD, gender and PiB SUVR differed according to the presenting syndrome (i.e. lvPPA versus PCA).
Table 1.
Subject demographics and clinical features
| DAT (n=41) | Atypical AD (n=55) | P value | lvPPA (n=37) | PCA (n=18) | P value | |
|---|---|---|---|---|---|---|
| Female, no. (%) | 14 (34) | 31 (56) | 0.03 | 17 (46) | 14 (78) | 0.03 |
| Education, yrs | 16 (14, 18) | 16 (14, 18) | 0.90 | 16 (14, 18) | 16 (13, 18) | 0.84 |
| Age at exam, yrs | 70 (60, 76) | 68 (60, 72) | 0.09 | 68 (62, 72) | 65 (60, 70) | 0.24 |
| Age at onset, yrs | 63 (56, 72) | 62 (56, 67) | 0.45 | 64 (58, 69) | 60 (54, 63) | 0.05 |
| Disease duration, yrs | 5.0 (4.0, 8.0) | 4.0 (2.5, 5.0) | 0.01 | 3.5 (3.0, 5.0) | 5.0 (2.2,7.0) | 0.07 |
| MMSE (/30) | 20 (15, 23) | 24 (16, 28) | 0.006 | 24.5 (15, 28) | 24 (17.8, 27.5) | 0.81 |
| MoCA (/30) | NA | 17 (9, 20) | NA | 16 (9, 20) | 18 (10, 21) | 0.55 |
| Hypertension, no. (%)* | 20 (49%) | 14 (41%) | 0.51 | 9 (43%) | 5 (38%) | 0.80 |
| Hypercholesterolemia, no. (%)* | 17 (42%) | 11 (32%) | 0.42 | 6 (29%) | 5 (38%) | 0.55 |
| Diabetes, no. (%)* | 2 (5%) | 2 (6%) | 0.85 | 1 (5%) | 1 (8%) | 0.72 |
| History of stroke, no. (%)* | 0 | 2 (6%) | 0.12 | 2 (10%) | 0 | 0.25 |
| History of smoking, no. (%)* | 20 (49%) | 12 (35%) | 0.23 | 7 (33%) | 5 (38%) | 0.76 |
| Global PiB SUVR | 2.28 (2.13, 2.40) | 2.18 (2.00, 2.36) | 0.12 | 2.10 (1.97, 2.32) | 2.27 (2.16, 2.60) | 0.01 |
| Total WMHp | 4.70 (2.72, 6.60) | 2.93 (1.75, 4.76) | 0.02 | 3.04 (1.71, 4.66) | 2.62 (1.87, 4.79) | 0.96 |
| Hippo/TIV*100 | 0.36 (0.31, 0.39) | 0.46 (0.42, 0.48) | <0.001 | 0.46 (0.42, 0.48) | 0.45 (0.41, 0.48) | 0.43 |
Data shown as median (inter-quartile range); MMSE = Mini-Mental State Examination; MoCA = Montreal Cognitive Assessment Battery; MoCA was not performed on the DAT subjects; WMHp = White matter hyperintensities proportion, shown as percentage of regional white matter volume; Hippo = hippocampus; TIV = total intracranial volume.
A total of 41 DAT and 34 atypical AD subjects (21 lvPPA and 13 PCA) had data available
Comparison of atypical AD and DAT
Of the 55 atypical AD subjects, 23 (42%) had at least one MB in the brain. This frequency did not differ significantly from the frequency observed in DAT (32%, p=0.31). Within the subjects with MBs, 61% of the atypical AD subjects and 46% of the DAT subjects had more than one MB (p=0.40). The atypical AD group had a larger number of MBs than DAT (p<0.001, Figure 1A), with a mean (95% CI) of 8 (4 to 17) MBs per subject (range 0–152) in atypical AD compared to only 1 (0.4 to 2) (range 0–12) MBs per subject in DAT.
Figure 1. Comparison of microbleed counts and density between atypical AD and DAT.
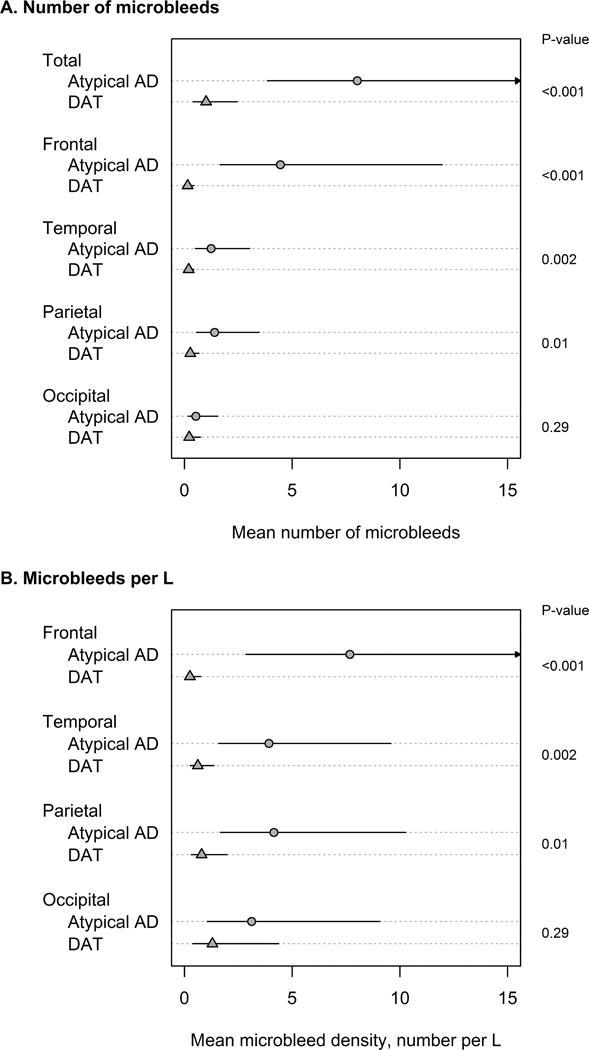
Panel A shows estimated mean number of microbleeds with 95% confidence intervals (CIs). Total counts were estimated from a negative binomial model. Regional counts were estimated with a negative binomial generalized estimating equations approach to account for the correlation among a subject’s four regional counts. Panel B shows regional microbleed density, defined as microbleeds per L of tissue, based on negative binomial models with log tissue volume included as an offset term. P-values in the right margin are from Wald test of group-wise differences. P-values in panels A and B are the same because regional densities were calculated using the same atlas volume estimates for all subjects.
The regional location of each MB was categorized as lobar (frontal, temporal, parietal or occipital), deep grey/white matter, or infratentorial (cerebellum or brainstem) for each subject. The majority of microbleeds were lobar in both atypical AD (419/441=95%) and DAT (34/41=83%). Both the number of MBs and the density of MBs were greater in the frontal, temporal and parietal lobes in atypical AD compared to DAT (Figure 1A and B). Significant differences between atypical AD and DAT in the total number of MBs and the number of frontal MBs remained after the sensitivity analyses accounting for outliers (Table 2). Furthermore, in our models adjusted for PiB, we did not find evidence that increased global or regional PiB deposition was associated with increased MBs nor did we find evidence that adjusting for PiB had a meaningful impact on the group-wise relative difference in number of regional microbleeds.
Table 2.
Estimated mean number of microbleeds based on main analysis and several sensitivity analyses looking at how three atypical AD subjects with a large number of microbleeds affect the results.
| Total | Frontal | Temporal | Parietal | Occipital | |
|---|---|---|---|---|---|
| DAT | 1.0 (0.4, 2.4) | 0.1 (0.0, 0.4) | 0.2 (0.1, 0.4) | 0.3 (0.1, 0.7) | 0.2 (0.1, 0.7) |
| Atypical AD | |||||
| All subjects | 8.0 (3.9, 17)*** | 4.5 (1.7, 12)*** | 1.2 (0.5, 3.0)** | 1.4 (0.6, 3.5)* | 0.5 (0.2, 1.5) |
| Omitting three outliers | 1.5 (0.8, 2.9) | 0.7 (0.2, 1.8)* | 0.3 (0.1, 0.8) | 0.3 (0.1, 0.6) | 0.1 (0.0, 0.3) |
| Omitting two outliers | 3.1 (1.6, 6.1)* | 1.5 (0.5, 5.1)** | 0.5 (0.2, 1.4) | 0.6 (0.2, 1.6) | 0.2 (0.1, 0.5) |
| Giving outliers the highest value among non-outliers | 2.7 (1.5, 5.0)* | 1.3 (0.6, 2.8)** | 0.5 (0.3, 1.1) | 0.4 (0.2, 0.8) | 0.2 (0.1, 0.4) |
P < 0.05;
P < 0.01;
P <0.001 versus Dementia of the Alzheimer’s Type (DAT)
Within atypical AD, the frontal lobe showed a greater density and number of MBs than all other lobes (all p<0.001), and the occipital lobe showed a lower number of MBs than the temporal (p=0.003) and parietal (p=0.006) lobes, although MB density was similar across the temporal, parietal and occipital lobes. Three-dimensional renders of the regional location of MBs in the atypical AD subjects are shown in Figure 2. Each of the five subjects with greater than 15 MBs (subjects 1–5) showed a relatively symmetric frontal predominance of MBs. In contrast to atypical AD, within DAT, the mean number of MBs was similar across regions, while MB density was generally lowest in frontal lobes (versus occipital, p<0.001; versus temporal, p=0.06, and versus parietal, p=0.07).
Figure 2. Transparent brain renders showing the regional location of microbleeds.
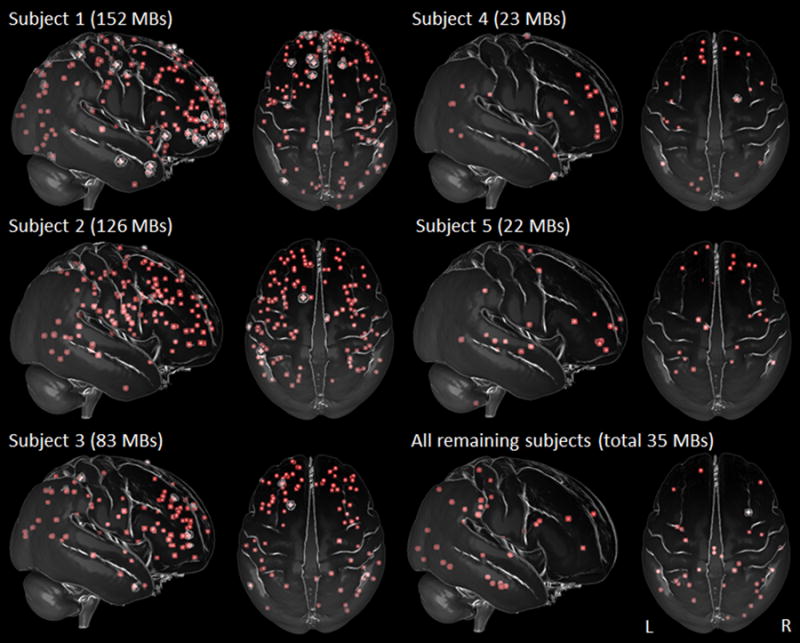
Maps show lateral and superior view of each of the five atypical AD subjects that had more than 15 microbleeds (MBs) (subjects 1–5), with the MBs from all the remaining atypical AD subjects (n=18) shown in a composite map. Subjects 1–5 show a predominance of frontal lobe MBs. Subjects 1 was diagnosed with PCA and subjects 2–5 were diagnosed with lvPPA. Renders were created using ImageVis3D (http://www.sci.utah.edu/software/imagevis3d.html).
No differences were observed across atypical AD and DAT in the proportion of subjects with deep (15% versus 5%, p=0.13) or infratentorial (7% versus 10%, p=0.66) MBs.
MB associations within atypical AD
Within atypical AD, the number of MBs was associated with both age at onset and age at examination, with a greater number of MBs observed with older age (Figure 3). There was also an association between the number of MBs and the global PiB SUVR, with a greater number of microbleeds associated with lower global PiB SUVR (Figure 3). No associations were observed between the number of microbleeds and gender or performance on the MoCA.
Figure 3. Relationship between microbleeds and clinical and demographic features in atypical AD.
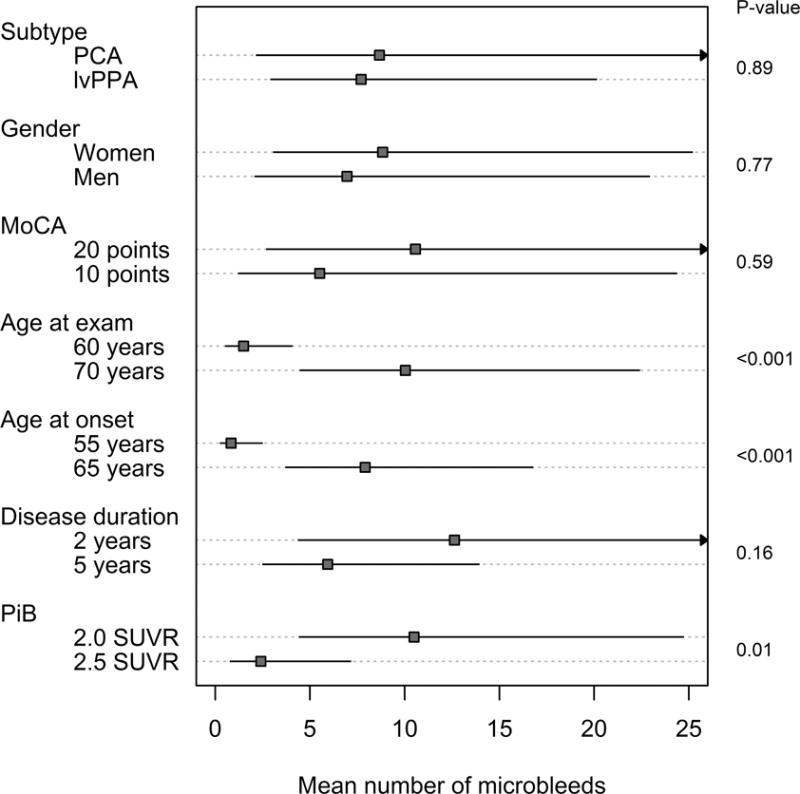
Estimated mean number of microbleeds with 95% confidence intervals based on negative binomial regression models. MoCA, age, disease duration, and PiB were modeled as continuous variables. To facilitate interpretation, we chose two clinically meaningful reference values for which to show the estimates.
While the number of MBs did not differ according to the presenting syndrome (i.e. lvPPA versus PCA) (Figure 3), the proportion of subjects with MBs was greater in lvPPA compared to PCA (51% versus 22%, p=0.04). The regional distribution of MBs was similar for both lvPPA and PCA, with highest density and number of MBs observed in the frontal lobe for both syndromes.
Discussion
This is the largest study to date to examine the frequency and distribution of MBs in subjects with atypical presentations of AD. Our findings show that MBs affect approximately 40% of these subjects, with many subjects showing multiple MBs. A striking finding of the study was that the distribution of MBs differs in atypical compared to typical presentations of AD, with an abundance of MBs observed in the frontal lobes in atypical AD.
The MB frequency of 42% observed in atypical AD was relatively high, yet was similar to the frequency of 32% observed in our DAT cohort, as well as other DAT cohorts reported in the literature [3, 10, 13]. This is consistent with the fact that both syndromes are caused by AD pathology which is strongly associated with cerebral amyloid angiopathy [34]. Indeed, the majority of the MBs in both syndromic groups were found in lobar regions, consistent with a presumed cerebral amyloid angiopathy etiology [2]. Despite this common lobar predominance, there was a tendency for atypical AD subjects to have a larger number of MBs per subject compared to DAT. Five (9%) of our atypical AD subjects had greater than 15 MBs, with three of these subjects having over 50 MBs, while none of the DAT subjects in our cohort had more than 12 MBs. While DAT subjects have been observed with large numbers of MBs [4], another study similarly only observed a range of 0–16 MBs per subject in a cohort of 40 DAT subjects [10] suggesting that subjects with large numbers of MBs are relatively rare. Subjects with an atypical presentation of AD seem to be at particularly high risk of developing large numbers of MBs. While older age was associated with a greater number of MBs within atypical AD, it cannot explain the observed difference between groups because the DAT cohort was slightly older on average than the atypical AD cohort. Disease duration was also shorter in atypical AD and hence could not explain these differences. Vascular risk factors were also not more prevalent in atypical AD compared to DAT. The global PiB SUVR values did not differ between atypical AD and DAT suggesting that the greater number of MBs was not related to the degree of Aβ deposition.
There was also evidence that the distribution of lobar MBs differed between atypical AD and DAT. The atypical AD subjects showed a tendency for MBs in the frontal lobe, with higher numbers of MBs and a higher density of MBs compared to temporal, parietal and occipital lobes. Conversely, the DAT subjects showed lowest counts in the frontal lobes, similar to previous studies [4, 5, 13], and showed lower frontal counts than atypical AD. The frontal predominance was clear in the atypical AD subjects that had a large number of MBs, although, importantly, our sensitivity analyses showed that the differences between atypical AD and DAT were not driven by these outliers. A previous study that has reported DAT subjects with large numbers of MBs (range 8–104) showed that the MBs were the least common in the frontal lobe [4], suggesting that even if we had observed DAT subjects with multiple MBs, the topographic differences with atypical AD would likely persist. It is unclear why such a topographic difference exists or why the frontal lobe was particularly vulnerable in atypical AD. It is possible that the distribution of vascular Aβ may differ between atypical AD and DAT and hence may be related to the development of local MBs. However, we did not find that the regional distribution of MBs was associated with the regional distribution of PiB uptake in our subjects. Pathological studies will be needed to investigate this hypothesis.
The number of MBs was not associated with performance on the MoCA within the atypical AD subjects, suggesting that the presence of MBs was not related to the degree of general cognitive impairment in these subjects. We did, however, observe a trend for the frequency of MBs to be higher in atypical AD subjects who presented with the lvPPA clinical syndrome, compared to those who presented with the PCA clinical syndrome. Approximately 50% of our lvPPA cohort had one or more MBs, a frequency that was higher than both PCA and DAT, suggesting these subjects are at particular risk. The number of PCA subjects in the study was, however, relatively small and so the estimates for this group will need to be confirmed in a larger study.
There was no evidence that the number of MBs was associated with higher global PiB SUVR in the atypical AD subjects. In fact, the opposite association was observed, with lower PiB SUVR associated with a higher number of MBs. It is likely that this relationship was artifactual and somewhat driven by the fact that the lvPPA group, which showed a higher proportion of MBs, had on average slightly lower PiB SUVR values than the PCA group. One previous study found that PiB retention was increased at the site of MBs, suggesting that MBs preferentially occur in regions of concentrated Aβ [35], and another group found evidence that the presence of MBs in DAT are associated with lower CSF Aβ42 levels [36, 37]. We did not, however, observe any association between regional PiB SUVR and regional MB density. While studies have found associations between the presence of MBs and PiB or Florbetapir retention across mixed cohorts consisting of both cognitively normal and DAT subjects [13, 38], none have yet reported any relationship within DAT subjects and none have assessed this relationship within atypical presentations of AD. Our findings suggest that higher PiB SUVR is not a risk factor for MBs in AD.
These findings demonstrate that a fairly large proportion of subjects with atypical presentations of AD have underlying MBs, although it should be stressed that the majority of these subjects had only one MB present in the brain. Nevertheless, these MBs are typically lobar and have some predilection for the frontal lobe. The high proportion and number of MBs in atypical AD should be an important consideration in the use of anti-coagulation treatment approaches in these subjects. This should also be considered if these subjects are included in future Aβ immunotherapy trials in AD which can exacerbate amyloid angiopathy and MBs [39] and lead to life-threatening vasogenic edema.
Acknowledgments
The study was funded by Alzheimer’s Association grant NIRG-12-242215 and NIH-funded grant R01 DC010367, R01-AG11378 and P50-AG16574. We would like to acknowledge Dr.’s Bradley Boeve, David Knopman, Jon Graff-Radford and David Jones, Mayo Clinic Rochester, MN, for referring subjects with atypical AD for the study and for performing some of the clinical evaluations on some of the DAT subjects.
References
- 1.Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C, Vinters HV, Haacke EM, Holshouser B, Kido D, Kirsch WM. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol. 2010;119:291–302. doi: 10.1007/s00401-009-0615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain. 2011;134:335–344. doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- 4.Goos JD, Kester MI, Barkhof F, Klein M, Blankenstein MA, Scheltens P, van der Flier WM. Patients with Alzheimer disease with multiple microbleeds: relation with cerebrospinal fluid biomarkers and cognition. Stroke. 2009;40:3455–3460. doi: 10.1161/STROKEAHA.109.558197. [DOI] [PubMed] [Google Scholar]
- 5.Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, Rogaeva E, Black SE. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008;65:790–795. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 6.Cordonnier C, van der Flier WM, Sluimer JD, Leys D, Barkhof F, Scheltens P. Prevalence and severity of microbleeds in a memory clinic setting. Neurology. 2006;66:1356–1360. doi: 10.1212/01.wnl.0000210535.20297.ae. [DOI] [PubMed] [Google Scholar]
- 7.Nakata Y, Shiga K, Yoshikawa K, Mizuno T, Mori S, Yamada K, Nakajima K. Subclinical brain hemorrhages in Alzheimer’s disease: evaluation by magnetic resonance T2*-weighted images. Ann N Y Acad Sci. 2002;977:169–172. doi: 10.1111/j.1749-6632.2002.tb04813.x. [DOI] [PubMed] [Google Scholar]
- 8.Hanyu H, Tanaka Y, Shimizu S, Takasaki M, Abe K. Cerebral microbleeds in Alzheimer’s disease. J Neurol. 2003;250:1496–1497. doi: 10.1007/s00415-003-0245-7. [DOI] [PubMed] [Google Scholar]
- 9.Nakata-Kudo Y, Mizuno T, Yamada K, Shiga K, Yoshikawa K, Mori S, Nishimura T, Nakajima K, Nakagawa M. Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr Cogn Disord. 2006;22:8–14. doi: 10.1159/000092958. [DOI] [PubMed] [Google Scholar]
- 10.Yates PA, Desmond PM, Phal PM, Steward C, Szoeke C, Salvado O, Ellis KA, Martins RN, Masters CL, Ames D, Villemagne VL, Rowe CC, Group AR. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology. 2014;82:1266–1273. doi: 10.1212/WNL.0000000000000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryan NS, Bastos-Leite AJ, Rohrer JD, Werring DJ, Fox NC, Rossor MN, Schott JM. Cerebral microbleeds in familial Alzheimer’s disease. Brain. 2012;135:e201. doi: 10.1093/brain/awr126. author reply e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 13.Kantarci K, Gunter JL, Tosakulwong N, Weigand SD, Senjem MS, Petersen RC, Aisen PS, Jagust WJ, Weiner MW, Jack CR, Jr, Alzheimer’s Disease Neuroimaging I Focal hemosiderin deposits and beta-amyloid load in the ADNI cohort. Alzheimers Dement. 2013 doi: 10.1016/j.jalz.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke. 2004;35:1415–1420. doi: 10.1161/01.STR.0000126807.69758.0e. [DOI] [PubMed] [Google Scholar]
- 15.Werring DJ, Frazer DW, Coward LJ, Losseff NA, Watt H, Cipolotti L, Brown MM, Jager HR. Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain. 2004;127:2265–2275. doi: 10.1093/brain/awh253. [DOI] [PubMed] [Google Scholar]
- 16.van der Vlies AE, Goos JD, Barkhof F, Scheltens P, van der Flier WM. Microbleeds do not affect rate of cognitive decline in Alzheimer disease. Neurology. 2012;79:763–769. doi: 10.1212/WNL.0b013e3182661f91. [DOI] [PubMed] [Google Scholar]
- 17.Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Jack CR, Jr, Josephs KA. Neuroimaging correlates of pathologically-defined atypical Alzheimer’s disease. Lancet Neurol. 2012;11:868–877. doi: 10.1016/S1474-4422(12)70200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain. 2014;137:1176–1192. doi: 10.1093/brain/awu024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chare L, Hodges JR, Leyton CE, McGinley C, Tan RH, Kril JJ, Halliday GM. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2013-306948. [DOI] [PubMed] [Google Scholar]
- 21.Whitwell JL, Lowe VJ, Duffy JR, Strand EA, Machulda MM, Kantarci K, Wille SM, Senjem ML, Murphy MC, Gunter JL, Jack CR, Jr, Josephs KA. Elevated occipital beta-amyloid deposition is associated with widespread cognitive impairment in logopenic progressive aphasia. J Neurol Neurosurg Psychiatry. 2013;84:1357–1364. doi: 10.1136/jnnp-2013-305628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook R, Caselli RJ, Knopman DS, Petersen RC. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63:1168–1174. doi: 10.1212/01.wnl.0000140289.18472.15. [DOI] [PubMed] [Google Scholar]
- 23.Formaglio M, Costes N, Seguin J, Tholance Y, Le Bars D, Roullet-Solignac I, Mercier B, Krolak-Salmon P, Vighetto A. In vivo demonstration of amyloid burden in posterior cortical atrophy: a case series with PET and CSF findings. J Neurol. 2011;258:1841–1851. doi: 10.1007/s00415-011-6030-0. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann M, Ghosh PM, Madison C, Laforce R, Jr, Corbetta-Rastelli C, Weiner MW, Greicius MD, Seeley WW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ, Rabinovici GD. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136:844–858. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madhavan A, Whitwell JL, Weigand SD, Duffy JR, Strand EA, Machulda MM, Tosakulwong N, Senjem ML, Gunter JL, Lowe VJ, Petersen RC, Jack CR, Jr, Josephs KA. FDG PET and MRI in logopenic primary progressive aphasia versus dementia of the Alzheimer’s type. PLoS One. 2013;8:e62471. doi: 10.1371/journal.pone.0062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitwell JL, Jack CR, Jr, Kantarci K, Weigand SD, Boeve BF, Knopman DS, Drubach DA, Tang-Wai DF, Petersen RC, Josephs KA. Imaging correlates of posterior cortical atrophy. Neurobiol Aging. 2007;28:1051–1061. doi: 10.1016/j.neurobiolaging.2006.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitwell JL, Jack CR, Jr, Duffy JR, Strand EA, Gunter JL, Senjem ML, Murphy MC, Kantarci K, Machulda MM, Lowe VJ, Josephs KA. Microbleeds in the logopenic variant of primary progressive aphasia. Alzheimers Dement. 2014;10:62–66. doi: 10.1016/j.jalz.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Master AV, Lowe VJ, Jack CR, Jr, Whitwell JL. Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain: a journal of neurology. 2012;135:1522–1536. doi: 10.1093/brain/aws032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 30.Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell I. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat Med. 2007;26:3661–3675. doi: 10.1002/sim.2832. [DOI] [PubMed] [Google Scholar]
- 32.Hilbe JM. Negative binomial regression. Cambridge University Press; Cambridge, UK: 2011. [Google Scholar]
- 33.Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. Journal of the American Geriatrics Society. 2005;53:695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- 34.Attems J, Jellinger KA, Lintner F. Alzheimer’s disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol. 2005;110:222–231. doi: 10.1007/s00401-005-1064-y. [DOI] [PubMed] [Google Scholar]
- 35.Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA, Kumar A, Neal KL, Betensky RA, Frosch MP, Rosand J, Johnson KA, Viswanathan A, Salat DH, Greenberg SM. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol. 2010;68:545–548. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goos JD, Teunissen CE, Veerhuis R, Verwey NA, Barkhof F, Blankenstein MA, Scheltens P, van der Flier WM. Microbleeds relate to altered amyloid-beta metabolism in Alzheimer’s disease. Neurobiol Aging. 2012;33:1011 e1011–1019. doi: 10.1016/j.neurobiolaging.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 37.Benedictus MR, Goos JD, Binnewijzend MA, Muller M, Barkhof F, Scheltens P, Prins ND, van der Flier WM. Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer’s disease. Neurobiol Aging. 2013;34:2488–2494. doi: 10.1016/j.neurobiolaging.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 38.Park JH, Seo SW, Kim C, Kim GH, Noh HJ, Kim ST, Kwak KC, Yoon U, Lee JM, Lee JW, Shin JS, Kim CH, Noh Y, Cho H, Kim HJ, Yoon CW, Oh SJ, Kim JS, Choe YS, Lee KH, Lee JH, Ewers M, Weiner MW, Werring DJ, Na DL. Pathogenesis of cerebral microbleeds: In vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann Neurol. 2013;73:584–593. doi: 10.1002/ana.23845. [DOI] [PubMed] [Google Scholar]
- 39.Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, Nicoll JA. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain. 2008;131:3299–3310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]