

SUMMARY
Here we report the identification and verification of a β-hydroxybutyrate-derived protein modification, lysine β-hydroxybutyrylation (Kbhb), as a new type of histone mark. Histone Kbhb marks are dramatically induced in response to elevated β-hydroxybutyrate levels in cultured cells, and in livers from mice subjected to prolonged fasting or streptozotocin-induced diabetic ketoacidosis. In total, we identified 44 histone Kbhb sites, a figure comparable to the known number of histone acetylation sites. By ChIP-seq and RNA-seq analysis, we demonstrate that histone Kbhb is a mark enriched in active gene promoters, and that the increased H3K9bhb levels that occur during starvation are associated with genes up-regulated in starvation-responsive metabolic pathways. Histone β-hydroxybutyrylation thus represents a new epigenetic regulatory mark that couples metabolism to gene expression, offering a new avenue to study chromatin regulation and the diverse functions of β-hydroxybutyrate in the context of important human pathophysiological states, including diabetes, epilepsy, and neoplasia.
INTRODUCTION
Emerging evidence suggests an important role of histone marks in metabolic control of epigenetics (Kaelin and McKnight, 2013, Katada et al., 2012, Lu and Thompson, 2012). Under physiological conditions, cells are constantly exposed to variations in nutrient availability and environmental stress. It is believed that histone PTMs, such as lysine acetylation and methylation, provide a means of adapting to these challenges to maintain physiological homeostasis (Wellen et al., 2009). In addition, the activity of histone PTM-modifying enzymes can be regulated by cellular metabolites, such as NAD+ and 2-hydroxyglutarate (Dang et al., 2009, Guarente, 2011). Recently, several types of new lysine acylation modifications were identified on histones. These lysine acylation reactions appear to use their corresponding short-chain CoAs as cofactors in the acylation reactions, suggesting additional mechanisms that link cellular metabolism and epigenetic regulation (Huang et al., 2015, Huang et al., 2014, Hirschey and Zhao, 2015). Despite tremendous progress in the past decade in the field of epigenetics and metabolism, it remains elusive how histone PTMs are influenced by cellular energy status to in turn control gene expression.
Ketone bodies, consisting of β-hydroxybutyrate, acetoacetate, and acetone, provide energy source to heart and brain during starvation. In humans, it is estimated that ketone bodies provide 30-40% of the energy supply after 3 days of starvation (Robinson and Williamson, 1980, Cahill, 2006, Laffel, 1999). In untreated diabetes, serum concentrations of β-hydroxybutyrate can be elevated up to 20 fold (~25 mM concentration or higher) (Laffel, 1999). Besides serving as an energy source, β-hydroxybutyrate has been used to treat epilepsy (McNally and Hartman, 2012) and plays a neuro-protective role in models of neurodegenerative diseases, such as Parkinson’s disease and Alzheimer’s disease (Kashiwaya et al., 2000, Lim et al., 2011). Moreover, it has been reported that β-hydroxybutyrate contributes to cancer cell ‘stemness’ (Martinez-Outschoorn et al., 2011). Ketogenic diets are under evaluation as adjunctive treatments for patients with brain tumors and other malignancies (Allen et al., 2014). These lines of evidence suggest a regulatory role for ketone bodies beyond serving as an energy source. However, the mechanisms underlying these physiological and pharmacological effects of ketone bodies remain largely unknown.
Here we describe identification and confirmation of lysine β-hydroxybutyrylation (Kbhb) as a novel, β-hydroxybutyrate-derived, histone modification. We observe that histone Kbhb levels are significantly induced under conditions of starvation or STZ-induced diabetic ketosis, physiologic states where β-hydroxybutyrate levels are drastically elevated. In fasted mouse liver, we demonstrate that the increased Kbhb on histone H3 lysine 9 (H3K9bhb) is associated with up-regulation of genes involved in starvation-responsive pathways. Importantly, H3K9bhb distinguishes a set of up-regulated genes from others that bear H3K9ac and H3K4me3 marks, suggesting histone Kbhb has different functions from histone acetylation and methylation. Our findings identify histone β-hydroxybutyrylation as a novel mechanism by which ketone bodies regulate cellular physiology, and support a model in which a shift in cellular utilization of energy source alters gene expression through metabolite-directed histone modifications.
RESULTS
Identification and Validation of Kbhb Residues in Histones
To search for possible novel histone marks, we analyzed a tryptic digest of core histones from HEK293 cells by HPLC/MS/MS. These MS/MS data were subjected to non-restrictive sequence alignment (Chen et al., 2009), searching for amino acid residues bearing a mass shift distinct from those of known PTMs. The analysis detected a mass shift of +86.0376 Da (monoisotopic mass) at lysine residues of a histone H3 peptide, K+86.0376QLATKacAAR. The most likely elemental composition for this modification moiety is C4H7O2 (formula of mass shift plus one proton, with theoretical mass shift of +86.0368 Da) based on this accurately determined mass shift (Huang et al., 2015). There are eight possible structural isomers corresponding to this molecular formula: R- and S- isoforms of 3-hydroxybutyryl (3hb) (also named β-hydroxybutyryl (bhb)), R- and S- isoforms of 2-hydroxybutyryl (2hb), R- and S- isoforms of 3-hydroxyisobutyryl (bhib), 2-hydroxyisobutyryl (2hib), and 4-hydroxybutyryl (4hb) (Figure 1A).
Figure 1. Identification and Verification of Lysine β-hydroxybutyrylation.
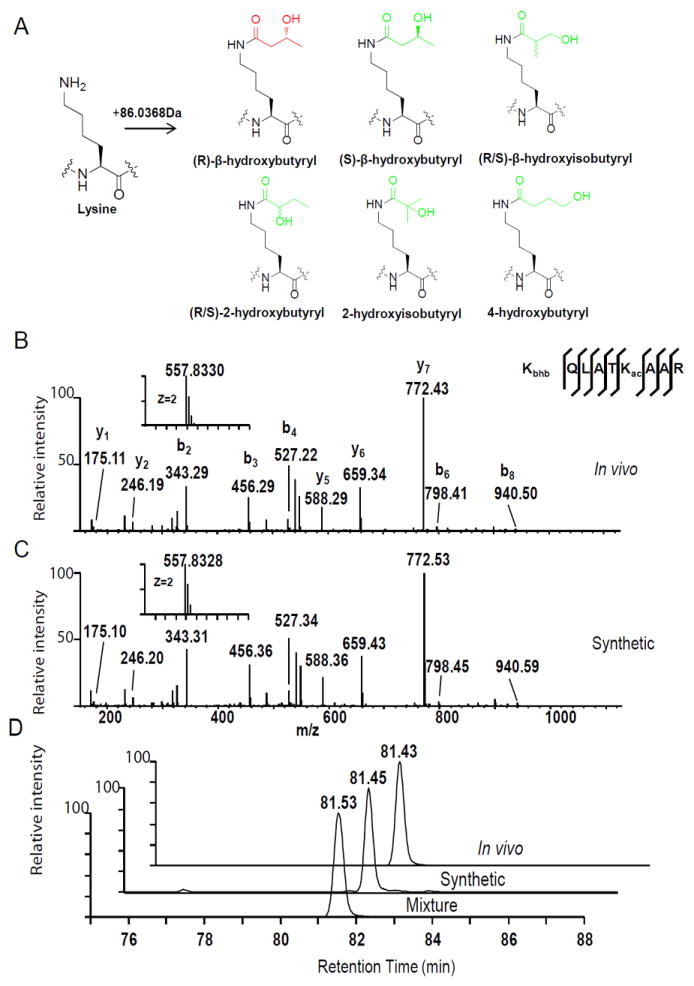
(A) Chemical structures of eight possible isomers that can cause a mass shift of +86.0368 Da.
(B, C) MS/MS spectra of a tryptic peptide derived from HEK293 core histones (B) and the synthetic peptide (C). Kac indicates acetyllysine and Kbhb indicates β-hydroxybutyryllysine. The insets show the mass-to-charge ratios (m/z) of the doubly charged precursor peptide ions. The x and y axis represents m/z and relative ion intensity, respectively.
(D) Reconstructed ion chromatograms from HPLC/MS/MS analyses of the in vivo-derived peptide, its synthetic Kbhb counterpart, and their mixture, showing co-elution of the two peptides. The x and y axis represent retention time of HPLC/MS analysis and the MS signal, respectively. See also Figure S1.
To determine which structural isomer is responsible for the +86.0368 Da mass shift, we synthesized the histone peptide, K+86.0368QLATKacAAR, incorporating each of the eight possible isomers at the lysine site of the mass shift. We then compared them with the peptides detected in vivo by HPLC/mass spectrometric analysis. If two peptides are indistinguishable in HPLC profiling and have identical mass spectrometric fragmentation patterns (or mass spectrometry (MS)/MS spectra), they are considered to have the same peptide sequence and modification moiety. Because enantiomers (R/S) are impossible to separate in a non-chiral HPLC column, and R-β-hydroxybutyrate is a major component of ketone bodies, we chose lysine R-β-hydroxybutyrylation instead of S-β-hydroxybutyrylation as the relevant candidate. In the remainder of this paper, the term β-hydroxybutyrylation is used to refer specifically to the R isoform.
Among the five synthetic peptides, only KbhbQLATKacAAR co-eluted with the corresponding in vivo-derived peptide on HPLC, and showed the same fragmentation pattern by high-resolution MS/MS (Figures 1B-1D). In contrast, synthetic peptides containing one of the other modification isomers, KbhibQLATKacAAR, K2hb QLATKacAAR, K2hibQLATKacAAR or K4hbQLATKacAAR, showed HPLC retention times distinct from that of the in vivo-derived peptide K+86.0368QLATKacAAR (Figures S1A and S1B). These results suggest that the +86.0368 Da mass shift derives from β-hydroxybutyrylation.
The same approach was used to confirm another peptide, PEPAK+86.0364SAPAPK as Kbhb-modified histone peptide (Figures S1C and S1D). Taken together, we conclude that the +86 Da mass shift is caused by lysine β-hydroxybutyrylation, and not by the presence of other structural isomers.
Metabolic Labelling of Histone Kbhb Marks by Isotopic β-Hydroxybutyrate
Short-chain acyl-CoAs are cofactors for a variety of lysine acylation reactions (Allis et al., 1985, Tan et al., 2011, Huang et al., 2015). Thus, β-hydroxybutyryl-CoA may represent the cofactor for lysine β-hydroxybutyrylation (Figure 2A). We speculated that β-hydroxybutyryl-CoA may be generated from cellular β-hydroxybutyrate, possibly by short-chain-Coenzyme A synthetase (Frenkel and Kitchens, 1977), analogous to the way that acetate and crotonate can be converted to their corresponding CoA derivatives. To test this possibility, we treated human HEK293 cells with 10 mM isotopically labelled sodium β-hydroxybutyrate [2, 4-13C2]. Histones were extracted, trypsin-digested, and analyzed by HPLC/MS/MS. As expected, we detected histone peptides modified by the isotopic β-hydroxybutyryl group (Figure 2B) and identified a total of 28 Kbhb-bearing histone peptides with an additional mass shift of 2 Da due to the isotopic labelling (Data S1). These peptides showed the same fragmentation patterns as the corresponding in vivo-derived and synthetic Kbhb-containing peptides (compare Figure 2B with Figures 1B and 1C). Furthermore, we detected a dose-dependent increase of isotopic bhb-CoA by treating HEK293 cells with isotopic sodium β-hydroxybutyrate (Figures 2C and S2A-S2B), suggesting that sodium β-hydroxybutyrate (Nabhb) can be converted into bhb-CoA in cells. These results demonstrate that β-hydroxybutyryl-CoA is the cofactor for lysine β-hydroxybutyrylation.
Figure 2. Metabolic Labelling of Histone Kbhb Marks by Isotopic β-Hydroxybutyrate.
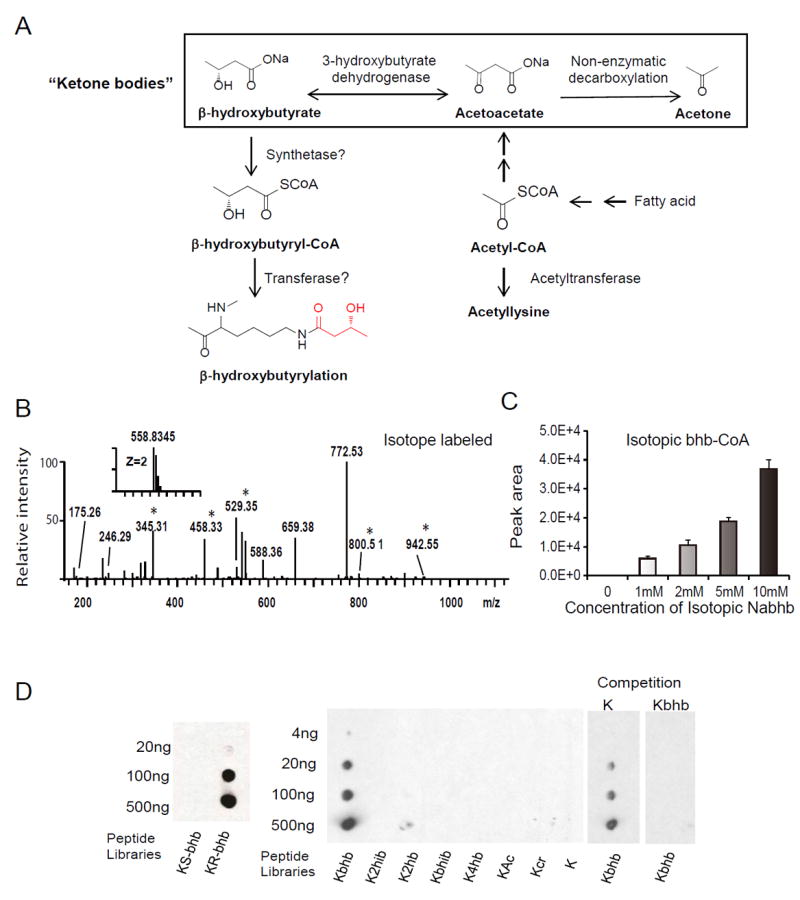
(A) Biosynthetic pathways for β-hydroxybutyrate and β-hydroxybutyryl-CoA. Also depicted are the three ketone bodies: β-hydroxybutyrate, acetoacetate, and acetone. (B) MS/MS spectra of a tryptic peptide identified from (R/S)-β-hydroxybutyrate-[2, 4-13C2]-treated HEK293 cells. An asterisk indicates a mass shift induced by isotopic labelling.
(C) MS detection of isotopic β-hydroxybutyryl-CoA from HEK293 cells treated with the indicated concentration of isotopic R-sodium β-hydroxybutyrate (13C4). Data are represented as means (±SEM) of three independent experiments.
(D) Specificity of pan anti-Kbhb antibody revealed by dot-blot and competition assay. Dot-blot assay was carried out using pan anti-Kbhb antibody and indicated amount of modified peptide libraries. The libraries were composed of mixtures of CXXXXKXXXX peptides, where C is cysteine, X is a mixture of all 19 amino acids except for cysteine, and the 6th residual is a lysine: R- or S-β-hydroxybutyryl lysine (Kbhb) (left panel); R-β-hydroxybutyryl lysine, 2-hydroxyisobutyryl lysine (K2hib), 2-hydroxybutyryl lysine (K2hb), β-hydroxyisobutyryl lysine (Kbhib), 4-hydroxybutyryl lysine (K4hb), acetyl lysine (Kac), crotonyl lysine (Kcr), or unmodified lysine (K) (middle panel). Competition was carried out by incubation of pan anti-Kbhb antibodies with 10-fold excess of unmodified or Kbhb containing peptides (right panel). See also Figure S2.
Histone Lysine β-Hydroxybutyrylation is Regulated by Cellular β-Hydroxybutyrate
To further characterize histone Kbhb, we generated a pan antibody against lysine β-hydroxybutyrylation (pan anti-Kbhb). This antibody showed robust specificity as demonstrated by dot blot assay and competition experiments (Figure 2D). Using this pan anti-Kbhb antibody, we detected histone Kbhb in yeast S. cerevisiae cells, Drosophila S2 cells, mouse embryonic fibroblast (MEF) cells and human HEK293 cells (Figure 3A), indicating that histone Kbhb is an evolutionarily conserved PTM among diverse eukaryotic species.
Figure 3. Histone β-Hydroxybutyrylation is Metabolically Regulated by Cellular β-Hydroxybutyrate Levels.
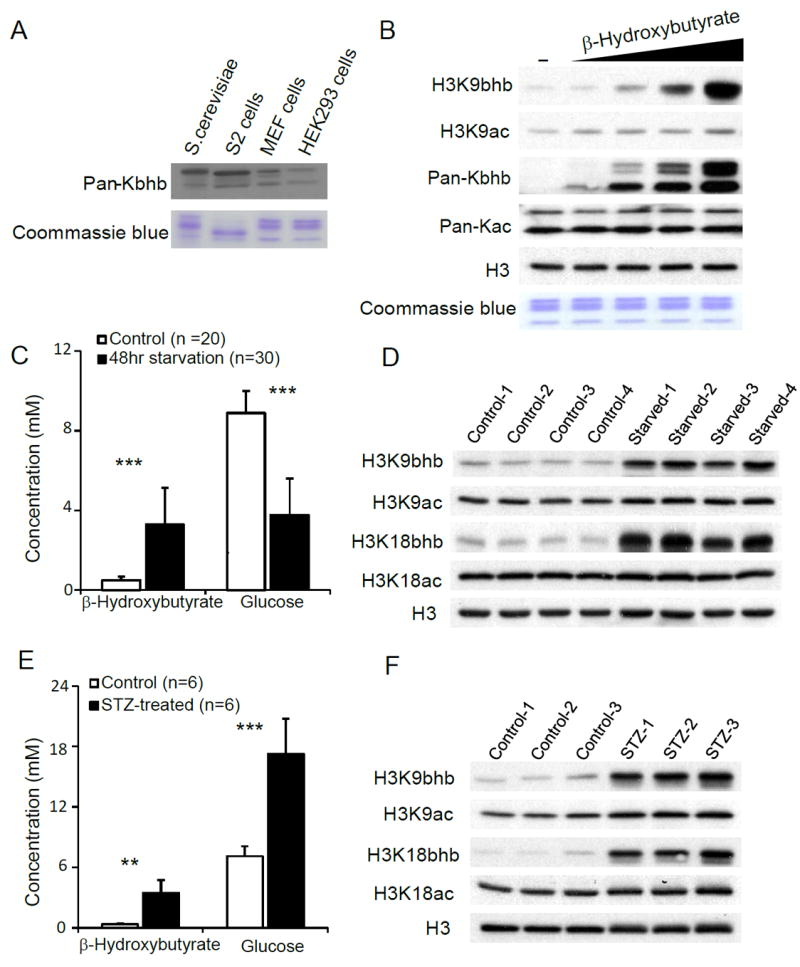
(A) Immunoblot analysis of histones from S. cerevisiae, D. melanogaster S2 cells, MEF cells, and HEK293 cells using pan anti-Kbhb antibody.
(B) Immunoblot analysis of histones from HEK293 cells treated with dose-increased sodium β-hydroxybutyrate.
(C) Blood glucose and β-hydroxybutyrate concentrations measured by a glucose-ketone meter from “fed” or “fasted” mice. Data are represented as means (±SEM). Twenty “fed” and 30 “fasted” mice were used. **P < 0.01, ***P < 0.001.
(D) Histone Kbhb and Kac levels in livers from “fed” or “fasted” mice were detected by Western blot using indicated antibodies.
(E) Blood glucose and β-hydroxybutyrate concentrations measured from “healthy” or “STZ-treated” mice. Data are represented as means (±SEM). Six pairs of healthy and streptozotocin (STZ) treated mice were used. **P < 0.01, ***P < 0.001.
(F) Histone Kbhb and Kac levels in livers from “healthy” or “STZ-treated” mice were detected by Western blot using indicated antibodies. See also Figure S3.
Next, we asked if cellular β-hydroxybutyrate concentrations can impact levels of histone Kbhb. Immunoblot analysis of protein lysates from HEK293 cell treated with sodium β-hydroxybutyrate revealed that histone Kbhb levels are induced in a dose-dependent manner, whereas no change was observed in overall histone acetylation (Figure 3B). We further tested histone Kbhb levels with site-specific histone Kbhb antibodies (Figures S3A-S3F). Our results showed that H3K9bhb, H3K18bhb, H4K8bhb, and H3K4bhb were all induced in a β-hydroxybutyrate dose-dependent manner, whereas a marginal change in H3K9ac was observed and no change in H3K18ac and H4K8ac levels were observed (Figures 3B and S3F). These results suggest that histone Kbhb levels are regulated by cellular β-hydroxybutyrate concentrations.
We hypothesized that cellular histone Kbhb levels might drastically increase in response to the high levels of β-hydroxybutyrate. To test this hypothesis, we assessed histone Kbhb status from livers of C57BL/6 mice that were either fed normal chow or subjected to 48 h of fasting (supplied with water only). Under fasting conditions, serum β-hydroxybutyrate concentrations increased 7-fold compared to normal fed controls (Figure 3C). Immunoblot analysis of liver histones showed that histone Kbhb was elevated greatly after a prolonged fasting, while histone acetylation showed no significant change (Figure 3D). We also did immunoblot analysis of Kbhb and Kac levels in kidney, showing that histone Kbhb levels (H3K9bhb and H4K8bhb) were induced significantly during prolonged fasting, while H4K8ac increased very modestly and H3K9ac showed no change (Figure S3G).
Next we examined histone Kbhb levels in the streptozotocin (STZ)-induced T1DM mouse model. Compared to healthy mice, we detected 2.4-fold and 10-fold elevations in blood glucose and β-hydroxybutyrate levels, respectively, in diabetic mice compared to littermate controls (Figure 3E). As expected, histone Kbhb levels are dramatically elevated in the livers of STZ-treated mice, while histone acetylation shows little change (Figure 3F). Taken together, our results demonstrate that increased histone Kbhb levels are tightly associated with elevated β-hydroxybutyrate concentrations occurring in both prolonged fasting and a T1DM model.
Quantification of Histone Kbhb Sites in Fasted versus Fed Mouse Liver
To gain further insight into how histone Kbhb and acetylation respond to starvation, we performed mass spectrometry-based quantification of histone Kbhb and Kac marks in livers from fasted and fed mice, to identify Kbhb- and Kac-containing histone peptides and determine their ratios between “fasted”- and “fed”-derived histone samples. We detected 11 histone Kbhb sites and 18 histone Kac sites in both “fed” and “fasted” liver samples. Kbhb levels at major histone sites were elevated by 4-40 fold under the fasted condition, whereas most of the acetylation sites showed less than 2-fold changes (Table 1 and Table S1). Taken together, both quantitative mass spectrometry analysis and immunoblotting demonstrate that liver histone Kbhb marks, but not histone Kac marks, are highly sensitive to the higher levels of serum β-hydroxybutyrate associated with prolonged fasting.
Table 1.
Quantification of Kbhb on Histones from Control and Starved Mice Livers
| Modification | Protein name | Modification site | Score | Modified sequence | Normalized ratios (fasted/fed) |
|---|---|---|---|---|---|
| Kbhb | Histone H1.0 | H1 K168 | 50.04 | _(pr)VK(pr)PVK(bhb)ASK(pr)_ | 10.23 |
| Histone H2A type 2-A | H2A K5 | 119.7 | _(pr)GK(bhb)QGGK(pr)_ | 33.73 | |
| Histone H2A type 2-A | H2A K125 | 130.41 | _(pr)K(pr)TESHHK(bhb)AK(pr)_ | 22.25 | |
| Histone H2B type 2-B | H2B K20 | 136.11 | _(pr)AVTK(bhb)VQK(pr)_ | 13.09 | |
| Histone H3.1 | H3 K4 | 84.025 | _(pr)TK(bhb)QTAR_ | 25.30 | |
| Histone H3.1 | H3 K9 | 87.298 | _K(bhb)STGGK(pr)APR_ | 14.44 | |
| Histone H3.1 | H3 K14 | 97.431 | _(pr)K(pm)STGGK(bhb)APR_ | 3.57 | |
| Histone H3.1 | H3 K18 | 130.41 | _K(bhb)QLATK(pr)AAR_ | 9.53 | |
| Histone H3.1 | H3 K23 | 93.429 | _(pr)QLATK(bhb)AAR_ | 31.21 | |
| Histone H4 | H4 K8 | 123.67 | _(pr)GGK(bhb)GLGK(ac)GGAK(ac)R_ | 20.67 | |
| Histone H4 | H4 K12 | 157.91 | _(pr)GLGK(bhb)GGAK(ac)R_ | 39.05 |
Note:
Pr- propionylation (lysine or peptide N-terminal)
Pm- propmethyl (lysine)
Ac- acetylation (lysine)
Bhb- β-hydroxybutyrylation (lysine)
Mapping Histone Kbhb Sites in Mouse and Human Cells
Given that histone Kbhb levels are elevated in fasted and diabetic mouse livers, we carried out an experiment to identify major histone sites bearing Kbhb in human HEK293 cells treated with 10 mM sodium β-hydroxybutyrate and in livers from either fasted or STZ-induced diabetic mice. Altogether, we identified 44 histone Kbhb sites, including 38 sites from β-hydroxybutyrate-treated human HEK293 cells (Figure 4 and Data S1-S2) and 26 sites from mouse liver (Figure 4 and Data S3), suggesting that β-hydroxybutyrylation is a widespread histone mark. The identified histone Kbhb sites include those lysine residues, such as H4K8, H4K12, H3K4, H3K9, H3K56, whose acetylation and methylation are important to chromatin structure and function (Bannister and Kouzarides, 2011). Thus, it is highly likely that lysine β-hydroxybutyrylation at these histone lysine residues also affects chromatin functions.
Figure 4. Proteomic Screening of Histone Kbhb Sites in Human and Mouse Cells.

Modified Lysine residues are highlighted in red. See also Data S1-S3 for the corresponding MS/MS spectra.
Genomic Localization of Histone Kbhb in Mouse Liver
To study the genome-wide distribution of histone Kbhb marks, we carried out ChIP-seq experiments. Histone H3K9bhb, H3K4bhb, and H4K8bhb were chosen for this experiment because they show robust responses to starvation in mouse liver and the antibodies against these sites are highly specific (Figures S3A-S3E). H3K9ac and H3K4me3 were also included as active promoter-specific marks (Wang et al., 2008, Heintzman et al., 2007). Our analysis of the ChIP-seq data showed that all three histone Kbhb marks are preferentially associated with gene promoters (defined as ± 2 kb around the transcription start sites [TSSs]). Among these Kbhb marks, the enrichment of H3K9bhb in promoter regions is comparable to H3K9ac and H3K4me3 (Figure 5A). We focused on H3K9bhb for further analysis because this mark is most highly enriched in gene promoters and its ChIP-signal intensities are strongest at the TSSs (Figures S4A and S4B) among the three Kbhb marks. By close inspection of H3K9bhb modified chromatin at TSSs, we found strong enrichment of H3K9bhb at the TSSs of active genes (Figure 5B). A comparison of H3K9bhb distribution with other histone modifications (using our H3K9bhb, H3K9ac and H3K4me3 ChIP-seq data and published H3K9me3 and H3K27me3 ChIP-seq data in mouse liver) (Sugathan and Waxman, 2013) revealed a strong positive correlation between H3K9bhb and two active marks, H3K9ac and H3K4me3, but a negative correlation with two repressive marks, H3K9me3 and H3K27me3 (Figure 5C). Consistently, we found a positive correlation between H3K9bhb and RNA Pol II (Ser5p) occupancy at TSSs (Figure S4C), when comparing H3K9bhb ChIP-seq data and published Pol II (Ser5p) ChIP-seq data in mouse liver (Koike et al., 2012). Furthermore, we showed that the enrichment of H3K9bhb at TSSs is indicative of gene expression in mouse liver (Figure 5D). ChIP-qPCR analysis on the representative Hnf4a gene using primers targeting multiple genomic locations validated that H3K9bhb is enriched in the promoter region, displaying a similar pattern to H3K9ac, H3K4me3 and Pol II Ser5p (Figure 5E).
Figure 5. Genome-Wide Localization of Histone Kbhb Marks.
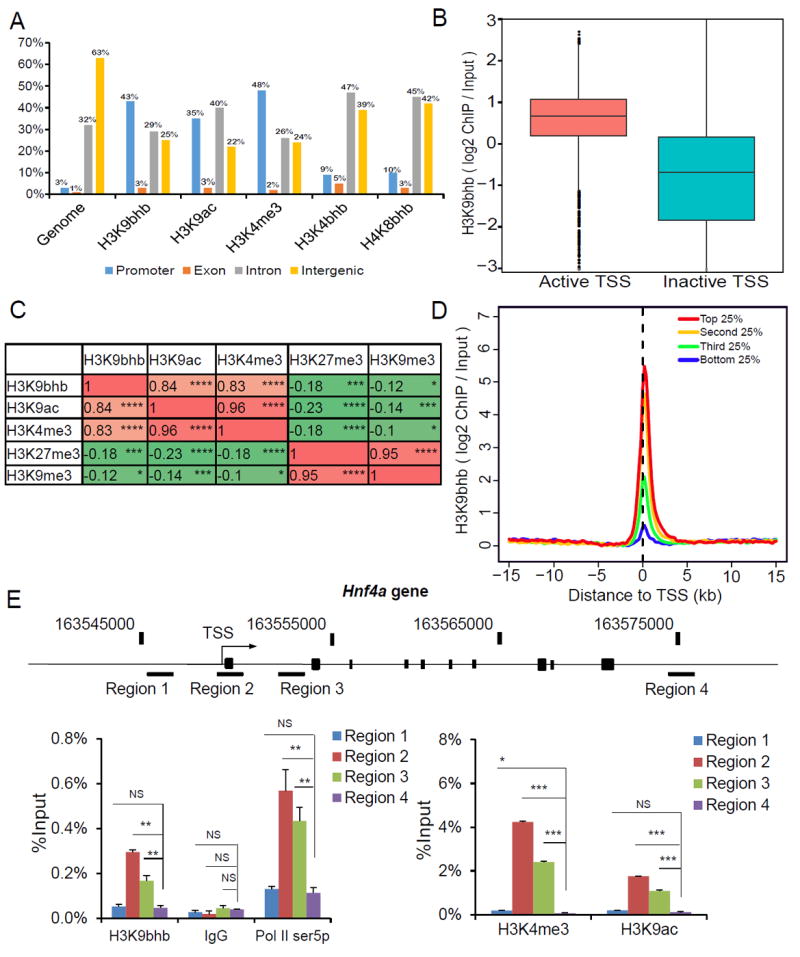
(A) Genome wide distribution of histone Kbhb in liver cells. Bar plot showing histone H3K4me3, H3K9ac, H3K9bhb, H3K4bhb, H4K8bhb and a random distribution of peaks of similar size (genome) over promoter (blue), intron (red), exon (purple) and intergenic regions (green). The promoter is defined as regions ± 2kb around known transcription start sites (TSSs).
(B) H3K9bhb signal (log2 ChIP/ input) in active and inactive TSSs. Active TSSs were defined as TSSs corresponding to the top 20% most expressed genes. Similarly, inactive TSSs were defined as TSSs corresponding to 20% lowest expressed genes.
(C) Pearson correlation coefficient estimates of H3K9bhb enrichment with either active histone marks or repressive marks, calculated using log10-transformed ChIP read counts in either promoter regions (H3K9bhb, H3K9ac, and H3K4me3) or gene bodies (H3K27me3 and H3K9me3). * (p <0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001).
(D) H3K9bhb signal intensity (Scaled log2 ChIP/ input) in promoter regions with the top 25%, the second 25%, the third 25%, and the bottom 25% RNA-seq counts.
(E) ChIP-qPCR analysis of histone H3K9bhb, H3K9ac, H3K4me3 and RNA Pol II (Ser5p) distribution on the Hnf4a gene. Data are represented as means (±SEM) of three independent experiments. NS (p > 0.05), * (p <0.05), ** (p < 0.01), and *** (p < 0.001). See also Figure S4.
Starvation-Induced H3K9bhb Marks Metabolic Pathways
We hypothesized that histone Kbhb is induced at specific genomic regions in response to starvation, based on two lines of evidence: 1) histone Kbhb marks are dramatically elevated in mouse liver during starvation; and 2) histone Kbhb marks, especially H3K9bhb, are localized in gene promoters. To test this hypothesis, we performed ChIP-seq using chromatin isolated from the livers of mice that were either fed ad libitum (AL condition) or fasted for 48 hours (ST condition). The ChIP-seq read counts of each histone modification in each gene promoter region (defined as +/-2kb around TSS) were first extracted. We then calculated the log2-transformed ratios of ChIP-seq read counts, log2 (ST/AL (ST counts divided by the AL counts)), following normalization of ChIP-seq data between ST and AL conditions (see Supplemental Experimental Procedure). These data were used for subsequent analyses.
To visualize the responses of H3K9bhb, H3K9ac and H3K4me3 marks to starvation, we generated a heat map in which all the genes were ordered by ratios of H3K4me3 ChIP counts between the ST and AL conditions. The H3K4me3 mark increases around the TSS regions in a large proportion of genes (Figure 6A). Interestingly, H3K9bhb displayed an overall similar pattern to that of H3K4me3, while displaying a different pattern with H3K9ac (Figure 6A). To further investigate possible associations among the three histone marks, we carried out pair-wise correlation analysis. We detected a strong positive correlation between the change of H3K9bhb and the change of H3K4me3 (Coef. Est. = 0.57), but no overall correlation between those of H3K9bhb and H3K9ac (Coef. Est. = -0.03), or between those of H3K9ac and H3K4me3 (Coef. Est. = -0.05) (Figures 6B, 6C and S5A). When considering only the genes with induced H3K4me3 (for which H3K4me3 log2 (ST/AL)>0 in Figure S5A), we detected a weak positive correlation between the change of H3K9ac and the change of H3K4me3 (Coef. Est. = 0.34). To annotate the genes associated with increased H3K9bhb during 48h fasting, we tested the enrichment of these genes with respect to the KEGG pathways using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). We extracted disease-related pathways in the database for a separate analysis in order to focus on physiological processes. The top pathways marked by increased H3K9bhb involve amino acid catabolism (alanine, aspartate and glutamate metabolism), circadian rhythm; redox balance (selenoamino acid metabolism; cysteine and methionine metabolism), PPAR signaling pathway, and oxidative phosphorylation (Figure 6D) (see the list of genes in Table S4). This result therefore reveals the relevance of K3K9bhb to metabolic response to starvation. In a separate analysis of disease-related pathways, we found that the pathways related to neurodegenerative disorders, cancer, infection and diabetes are marked by increased H3K9bhb (Figure S5B). ChIP-seq results for a small group of selected genes from top pathways were validated by ChIP-qPCR (Figures 6E-6G).
Figure 6. Starvation-Induced H3K9bhb increase is Associated with Active Gene Expression.
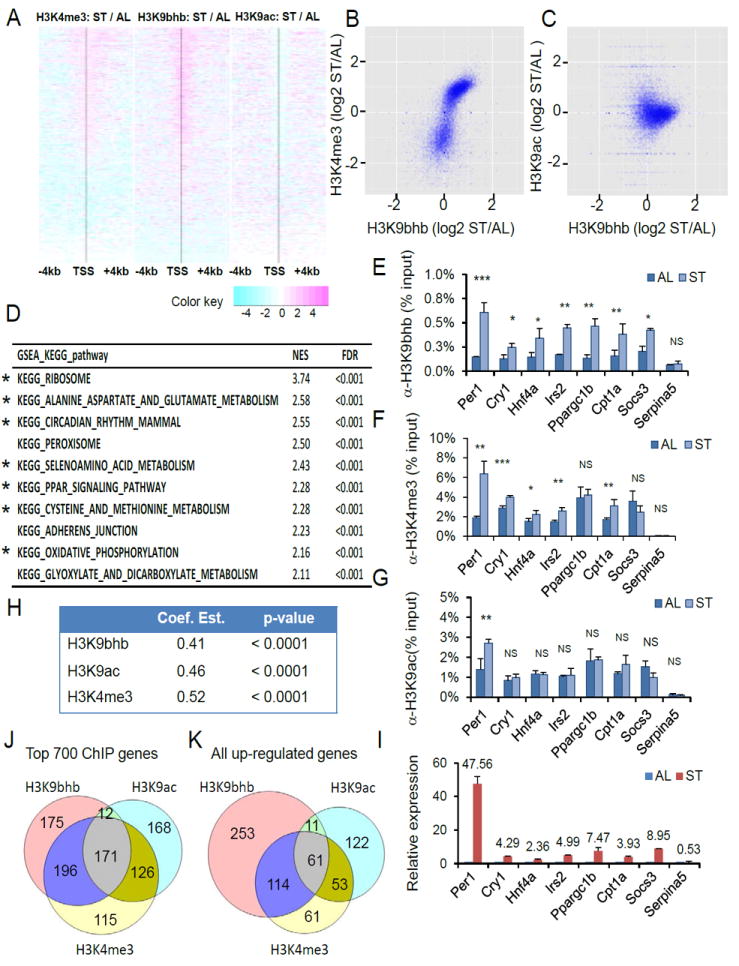
(A) Heat map showing changes of H3K9bhb-, H3K9ac-, and H3K4me3- ChIP-seq signals around promoter regions (± 4kb around TSS). The order of genes are same in all the three heat maps, which were based on the ChIP-seq read counts of H3K4me3 marks in starvation (ST) condition. The black vertical line in each heat map indicates location of TSS and changes in signals were presented in logarithm scale.
(B) Correlation between changes in ChIP-seq signals of H3K9bhb and those of H3K4me3 upon starvation. Library sizes of ChIP-seq data were normalized between “starvation” (ST) and “fed” (AL) conditions, for each modification. Coef. Est. = 0.57, p-value < 0.0001.
(C) Correlation between changes in ChIP-seq signals of H3K9bhb and those of H3K9ac upon starvation. Library sizes of ChIP-seq data were normalized between “starvation” (ST) and “fed” (AL) conditions, for each modification. Coef. Est. = -0.03, p-value = 0.5274.
(D) KEGG pathway analysis of H3K9bhb ChIP-seq data using Gene Set Enrichment Analysis (GSEA). The top 10 H3K9bhb-enriched pathways in response to starvation were listed, in the order of normalized enriched score (NES). The asterisk marked the same pathways appeared in Figure S5D. See also the list of genes and their rankings in Table S4.
(E-G) qPCR analysis of H3K9bhb-, H3K4me3-, and H3K9ac- ChIP products from mouse livers either fed normally or fasted for 48 hours. Data are represented as means ±SEM (n=3). NS (p > 0.05), * (p <0.05), ** (p < 0.01), and *** (p < 0.001).
(H) Pearson correlation coefficient estimates representing the correlation between induced gene expression and increased histone marks of interest. P-values were calculated using t-distribution.
(I) RT-qPCR analysis of gene expression during 48h of fasting. Relative expression was normalized to Actin. Data were represented as mean fold change ± standard deviation in relative expression (fasted vs fed) from three mice each group. Data are represented as means ±SEM (n=3).
(J) Venn diagram of up-regulated genes marked by each of the increased histone modifications during starvation. The top 700 genes were chosen based on their fold change of ChIP-seq signals for H3K9bhb, H3K9ac, and H3K4me3 modification during starvation, respectively. The Venn diagram shows the numbers of the top 700 genes with increased gene expression in response to starvation.
(K) Starvation-induced genes are associated with increased histone marks. All the 1742 up-regulated genes (log2-transformed fold change in expression of ≥ 0.5) were used to generate this Venn diagram. The number in each circle represents the up-regulated genes in which promoter regions have at least 2-fold increases of ChIP-seq signals of H3K9bhb, H3K9ac, or H3K4me3, respectively. See also Figures S5-S7.
H3K9bhb is Associated with Genes Up-Regulated during Prolonged Fasting
To explore the effect of elevated H3K9bhb marks on gene expression during prolonged fasting, we next performed RNA-seq on 5 pairs of liver samples from mice that were either fed ad libitum (AL condition) or fasted for 48 hours (ST condition). We extracted differentially expressed genes between “AL” and “ST” conditions and created a heat map (Figure S5C). Using the criterion of absolute values of log2-transformed fold change ≥ 0.5 and FDR=0.05, we found that 1742 genes were up-regulated while 2104 genes were down-regulated after 48h fasting (Tables S2 and S3). KEGG pathway analysis was carried out on up-regulated and down-regulated genes using GSEA (Figures S5D and S5E), in which disease-related pathways were manually removed in order to focus on physiological processes and compare with ChIP-seq pathways.
We first compared the gene pathways marked by increased H3K9bhb in ChIP-seq data with the up- or down-regulated gene pathways in RNA-seq data. This analysis showed that among the top ten H3K9bhb-enriched pathways, seven of them appear in the top pathways for up-regulated gene expression (marked by asterisk in Figures 6D and S5D), while none of them appear in the down-regulated pathways (compare Figures 6D and S5E), suggesting that increased H3K9bhb is indicative for active gene expression. To further test this, we carried out the following analysis. First, we ranked the genes in both H3K9bhb ChIP-seq data and RNA-seq data by fold change values (ST condition over AL condition). Second, we averaged the ranks of genes between H3K9bhb ChIP-seq and RNA-seq. Third, we used this averaged ranking as an order of genes for GSEA KEGG pathway analysis. The rationale for this analysis is that if a gene ranks high in both lists (e.g., H3K9bhb significantly increases and the transcript is significantly up-regulated), the gene will still rank high in the combined gene list. If a gene ranks high in one list but low in the other list (e.g., H3K9bhb significantly increases but expression is down-regulated), then the ranking of the gene will decrease significantly and have only minor effects on enrichment analysis. The results showed that the top pathways listed by this ranking method are consistent with those generated from H3K9bhb ChIP-seq data, including amino acid catabolism (alanine, aspartate and glutamate metabolism), circadian rhythm; redox balance (selenoamino acid metabolism; cysteine and methionine metabolism), PPAR signaling pathway, and oxidative phosphorylation (marked by asterisk in Figure S6A). This result again supports the association of H3K9bhb with active gene expression. Furthermore, we carried out correlation analysis between changes of each histone mark in ChIP-seq data and corresponding changes of gene expression from RNA-seq data. The result demonstrates that the increase of H3K9bhb is positively correlated with up-regulated gene expression during starvation, with a correlation coefficient comparable to the two established active marks, H3K4me3 and H3K9ac (Figure 6H).
We chose 7 genes to validate the correlation between H3K9bhb and gene expression. The genes were derived from circadian rhythm, PPAR signaling, and insulin signaling pathways, which are affected by starvation, including: Per1, Cry1, HNF4a, Irs2, Ppargc1b, Cpt1a, and Socs3. We also included Serpina5, whose promoter does not show any changes in H3K9bhb, H3K4me3 or H3K9ac, as a negative control. RT-qPCR results showed that the genes marked by increased H3K9bhb, regardless of H3K9ac, were up-regulated during starvation, while the genes without increases in H3K9bhb, H3K9ac or H3K4me3 were not up-regulated (Figure 6I). Taken together, our data suggest that H3K9bhb is a histone mark for active gene expression during starvation.
H3K9bhb Distinguishes a Set of Up-Regulated Genes from Others that Bear H3K9ac and H3K4me3 Marks
To examine whether H3K9bhb indicates a distinct role in the regulation of gene expression during starvation, we used the multiple regression model in which regression of gene expression on all three marks (H3K9bhb, H3K4me3, and H3K9ac) was examined. If H3K9bhb provides non-redundant information, H3K9bhb will still be statistically correlated with gene expression after removing the influences of H3K4me3 and H3K9ac, which can be detected by multiple regression analysis. Our result showed that the correlation between H3K9bhb and gene expression is statistically significant (Figure S7A). Thus our data implies that the fold change of gene expression can be more accurately predicted by considering H3K9bhb in addition to H3K9ac and H3K4me3.
To further determine the different impacts of H3K9bhb, H3K9ac, and H3K4me3 on gene expression in response to starvation, we carried out two parallel analyses. In the first experiment, we chose the genes that showed the most dramatic changes in each histone mark upon starvation and then correlated them with the changes in gene expression. We picked the top 700 genes that are marked by H3K9bhb, H3K9ac, and H3K4me3, respectively, based on ranking of fold change in the ChIP-seq data in response to starvation. This analysis showed that among the 700 genes, 554, 477, and 638 genes with increased H3K9bhb, H3K9ac, and H3K4me3, respectively, are up-regulated in gene expression during starvation (defined by an absolute value of log2-transformed fold change in expression of ≥ 0.5 and FDR = 0.05) (Figure 6J). Notably, more genes are shared by increased H3K9bhb and H3K4me3 than those shared by increased H3K9bhb and H3K9ac, or H3K9ac and H3K4me3 (Figure 6J). In addition, 175, 168, and 145 genes are marked by a specific increase in H3K9bhb, H3K9ac and H3K4me3, respectively (Figure 6J). We then performed the DAVID analysis of KEGG pathways for these exclusive genes. The pathways marked exclusively by H3K9bhb are spliceosome, selenoamino acid metabolism, PPAR signaling pathway, fatty acid metabolism, and proteasome (Figure S7B), which is distinct from H3K9ac-specific pathways (Figure S7C). The H3K4me3-specific pathways are proteasome and MAPK signaling pathway (Figure S7D). These results are consistent with the non-redundant function of H3K9bhb in gene expression indicated by multiple-regression models (Figure S7A).
In the second experiment, we chose the genes up-regulated by starvation and study their association with the changes in the three histone marks. We chose the set of 1742 genes up-regulated by starvation (defined by an absolute value of log2-transformed fold change in expression of ≥ 0.5 and FDR = 0.05) and asked how many of them are marked by an increase in H3K9bhb, H3K9ac, and H3K4me3, respectively. Our analysis showed that out of 1742 up-regulated genes, 439, 247, and 289 genes are associated with at least a two-fold increase in the ChIP-seq signals for H3K9bhb, H3K9ac, and H3K4me3, respectively (the sum of numbers in each cycle in Figure 6K). Similarly, the number of genes with concurrent increases in H3K9bhb and H3K4me3 marks is greater than that with concurrent increases in H3K9bhb and H3K9ac, or H3K9bhb and H3K9ac (Figure 6K). In addition, 253, 122, and 61 up-regulated genes are marked by a unique increase in H3K9bhb, H3K9ac and H3K4me3 marks, respectively (Figure 6K), suggesting the non-redundant role for each histone modification. Together, our analysis revealed that H3K9bhb is an active gene mark that is induced during prolonged fasting, which has an overlapping but non-redundant function as compared with the active H3K4me3 and H3K9ac marks.
DISCUSSION
Increasing evidence implicates altered histone modifications in translating cellular metabolic states into changes in gene expression (Kaelin and McKnight, 2013, Katada et al., 2012, Lu and Thompson, 2012). This occurs, at least in part, because chromatin modifying enzymes use cellular intermediary metabolites as cofactors to either add or remove chromatin modifications. Thus, fluctuations in levels of these metabolites may up- or down-regulate gene expression by modulating chromatin states (Love et al., 2010, Ruan et al., 2013, Ruan et al., 2012). Given the dynamic nature of β-hydroxybutyrate and histone Kbhb levels, and the association of histone Kbhb marks with gene promoters, we propose that Kbhb serves as an important mechanism by which cells adapt to changes in cellular energy sources by rewriting epigenetic programs and modulating gene expression.
Differential Regulation of Histone Kbhb and Kac during Starvation
During prolonged fasting, carbohydrates are scarce, and consequently energy is derived to a greater extent from fatty acid metabolism. Under these conditions, large amounts of acetyl-CoA generated from fatty acid oxidation are converted into ketone bodies (Laffel, 1999). We observed a dramatic increase in histone Kbhb after 48 hours of fasting, by both immunoblotting and quantitative mass spectrometric analysis. Moreover, during prolonged fasting, ketone body-derived histone Kbhb is associated with amino acid catabolism, redox balance, and circadian rhythm. Specifically, alanine, aspartate and glutamate metabolism are known to provide the substrates for gluconeogenesis upon starvation, which is accompanied by increased oxidative phosphorylation capacity. Selenoamino acid metabolism and cysteine/methionine metabolism are involved in maintaining redox balance, a process that is also associated with peroxisome activity. It has been well established that PPAR signaling induces expression of genes involved in mitochondrial and peroxisomal β-oxidation in fasted liver. Finally, all the above pathways have been known to display circadian rhythm. As compared to other histone marks, histone Kbhb appears to coordinate an overlapping but distinct genetic program in adaption to starvation. Both immunoblot and mass spectrometric analysis suggest that β-hydroxybutyrate has only a marginal impact on lysine acetylation both in liver and kidney. Recently, β-hydroxybutyrate was reported to repress oxidative stress in mouse kidney by inhibiting endogenous class I HDAC activity (Shimazu et al., 2013). However, when we treated human HEK293 cells with up to 20 mM sodium β-hydroxybutyrate, only marginal increases in histone acetylation were detected. In contrast, a dramatic increase in histone Kbhb occurred in a dose-dependent manner (Figure 3B). Quantitative mass spectrometry demonstrated that the levels of most histone Kbhb sites were elevated by 10-40 fold in response to prolonged fasting, under conditions where serum β-hydroxybutyrate levels increased roughly 7-fold (Figure 3C and Table 1). In contrast, levels of all the major histone acetylation sites in histones H3 and H4 showed only marginal changes (Table S1). Together, our data support the notion that β-hydroxybutyrate has a much more profound impact on histone β-hydroxybutyrylation than on histone acetylation. Moreover, changes in histone Kbhb levels likely play a much more significant role in altered gene expression occurring in response to prolonged fasting or T1DM than do changes in Kac.
β-Hydroxybutyrate and Its Association with Physiology and Diseases
Several lines of evidence suggest that β-hydroxybutyrate has diverse functions, instead of simply serving as an energy source. A ketogenic diet and β-hydroxybutyrate have been used successfully to treat epilepsy (McNally and Hartman, 2012). They also show potential for treating several neurological conditions (Lim et al., 2011, Kashiwaya et al., 2000). The ketogenic diet can also lower blood glucose and enhance glycemic control in diabetics, and is useful clinically in management of patients with certain inborn errors of metabolism (Allen et al., 2014). Moreover, a ketogenic diet inhibits tumor growth in xenograft models, and potentially in human patients (Allen et al., 2014). It is tempting to speculate that these interventions, which would both be predicted to elevate histone Kbhb levels, may work in part by altering chromatin structure to induce a more tumor suppressive gene expression pattern.
At the cellular level, β-hydroxybutyrate has been reported to modulate sperm motility, receptor signaling pathways, and autophagy (Taggart et al., 2005, Finn and Dice, 2005), as well as global gene expression profiles associated with cancer cell “stemness” (Martinez-Outschoorn et al., 2011). Despite this progress, the molecular mechanisms by which β-hydroxybutyrate exerts these diverse effects remain unclear. Therefore, the discovery of the lysine β-hydroxybutyrylation pathway not only reveals a new mechanism of epigenetic regulation, but also illuminates a new direction in studying the diverse physiological functions of β-hydroxybutyrate and its pharmacological significance.
EXPERIMENTAL PROCEDURES
Identification of histone Kbhb-peptides
Core histone proteins were extracted and tryptically digested. Immunoprecipitation of Kbhb-containing peptides, HPLC/MS/MS analysis, sequence alignment of the MS/MS data for identifying and quantifying the Kbhb peptides were carried out as previously described (Dai et al., 2014). Detailed experimental procedures are described in the Supplemental Experimental Procedures.
Cell Culture and Animal Experiments
HEK293 cells were grown in complete DMEM medium, supplemented with 10% FBS, and 1% GlutaMAX (Gibco, Thermo Fisher Scientific Inc., Waltham, MA). C57BL/6 mice were either fed with standard chow diet or fasted (with free access to water) for a specified number of hours as detailed in the text. C57BLS/J db/db littermates (licensed by the Jackson Laboratory, Bar Harbor, Maine) were given single-dose intraperitoneal injections of either streptozotocin (STZ, 200 mg/kg body weight) or the sodium citrate buffer vehicle for 48 hours. Blood samples were collected for determination of the concentrations of glucose and ketone bodies. Animal experiments are described in detail in Supplemental Experimental Procedures. All animal experiments were done following Dr. Zhao’s approved animal protocol (ACUP#72296) at the University of Chicago or following Dr. Li’s approved animal protocol (2012-04-LJ-09) by the Animal Ethics Committee of the Shanghai Institute of Materia Medica, China.
RNA-Seq and ChIP-Seq
Total RNA was extracted from frozen mouse livers using the RNeasy Mini Kit (QIAGEN INC, Valencia, CA). RNA-seq libraries were constructed according to manufacturer’s instructions using TruSeq Stranded Total Sample Preparation kit (Illumina, San Diego, CA). ChIP-seq experiments for histone marks were carried out according to the instructions from the ChIP-IT® High Sensitivity kit (Active Motif, Carlsbad, CA). Thirty ug of chromatin DNA from “control” or “starved” mice liver sample and 3 ug of anti-H3K4bhb, -H3K9bhb, -H4K8bhb, -H3K9ac or -H3K4me3 antibodies were used for each reaction. ChIP-seq libraries were prepared following the Ovation Ultralow DR Multiplex System protocols (NuGEN Technologies Inc., San Carlos, CA), followed by a size selection (200-700bp) step using SPRIselect beads (Beckman Coulter, Brea, CA). The libraries were sequenced by Illumina HiSeq 2500 at the University of Chicago Genomics Core Facility. Sequencing data were processed and analyzed as described in detail in the Supplemental Experimental Procedures.
Detailed descriptions of the materials and methods are provided in the supplemental information.
Supplementary Material
Acknowledgments
We are grateful to Saadi Khochibin, Wei Gu, Lev Becker, Mindian Li, and Benjamin R. Sabari for valuable discussion. This work was supported by the following grants and agencies: NIH DK089098, P01 DK057751, CT DPH 2014-0139 and Ellison Medical Foundation to X.Y.; NIH DK71900 and the Starr Foundation Tri-Institutional Stem Cell Initiative (2014-021) to R.G.R.; NIH R01GM101171 and R21CA177925 to D.L.; NIH GM59507 to H. Z.; NIH R01AG030593 and R01AG023166 to S.D.P.; National Natural Science Foundation of China (81125023) and Shanghai Commission of Science and Technology (14431902800) to J.L and J.L.; National Basic Research Program of China (973 Program) (No. 2014CBA02004) and the Shanghai Municipal Science and Technology Commission (No. 15410723100) to M.T.. Y.Z. is supported by the Nancy and Leonard Florsheim Family Fund and is a shareholder of PTM BioLabs, Co., Ltd (Chicago, IL).
Footnotes
ACCESSION NUMBERS
The Gene Expression Omnibus (GEO) accession number for the data sets reported in this paper is GSE69617. The following link has been created to allow review of record while it remains in private status: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ajerswoefbyzrut&acc=GSE69617
AUTHOR CONTRIBUTIONS
Y.Z. initiated and conceived the study; Y.Z.,X.Y., D.Z., and D.C. designed and performed the experiments; Z.X., D.Z., D.C., and Y.Z. wrote the manuscript; Z.X. verified the Kbhb modification; D.Z. characterized the functions of Kbhb in cells and in mouse; D.C. did the bioinformatics and biostatistics analysis under the guidance of H.Z. and X.Y.; Z.T carried out in vitro transcription assay under the guidance of R.G.R; H.H., Y.C., O.K.K., S.L., and M.T. contributed to mass spectrometric analysis; L.Z. synthesized the modified compound; S.Q. constructed the library for ChIP-seq and RNA-seq; J.L., G.C., C.X., D.B,L. and J.L. contributed to animal models and animal analysis; M.K., and K.P.W. helped with next generation sequencing; C.P., H.R., D.W., L.M.J and S.D.P provided important feedback to the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental information includes Supplemental Experimental Procedures, seven figures, four tables, and three Supplemental data files and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ALLEN BG, BHATIA SK, ANDERSON CM, EICHENBERGER-GILMORE JM, SIBENALLER ZA, MAPUSKAR KA, SCHOENFELD JD, BUATTI JM, SPITZ DR, FATH MA. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biology. 2014;2:963–970. doi: 10.1016/j.redox.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLIS CD, CHICOINE LG, RICHMAN R, SCHULMAN IG. Deposition-related histone acetylation in micronuclei of conjugating Tetrahymena. Proc Natl Acad Sci U S A. 1985;82:8048–52. doi: 10.1073/pnas.82.23.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANNISTER AJ, KOUZARIDES T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAHILL GF. Fuel metabolism in starvation. Annual Review of Nutrition. 2006;26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- CHEN Y, CHEN W, COBB MH, ZHAO Y. PTMap--a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc Natl Acad Sci U S A. 2009;106:761–6. doi: 10.1073/pnas.0811739106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAI LZ, PENG C, MONTELLIER E, LU ZK, CHEN Y, ISHII H, DEBERNARDI A, BUCHOU T, ROUSSEAUX S, JIN FL, SABARI BR, DENG ZY, ALLIS CD, REN B, KHOCHBIN S, ZHAO YM. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nature Chemical Biology. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- DANG L, WHITE DW, GROSS S, BENNETT BD, BITTINGER MA, DRIGGERS EM, FANTIN VR, JANG HG, JIN S, KEENAN MC, MARKS KM, PRINS RM, WARD PS, YEN KE, LIAU LM, RABINOWITZ JD, CANTLEY LC, THOMPSON CB, HEIDEN MGV, SU SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINN PF, DICE JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005;280:25864–70. doi: 10.1074/jbc.M502456200. [DOI] [PubMed] [Google Scholar]
- FRENKEL EP, KITCHENS RL. Purification and properties of acetyl coenzyme A synthetase from bakers’ yeast. J Biol Chem. 1977;252:504–7. [PubMed] [Google Scholar]
- GUARENTE L. Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:81–90. doi: 10.1101/sqb.2011.76.010629. [DOI] [PubMed] [Google Scholar]
- HEINTZMAN ND, STUART RK, HON G, FU Y, CHING CW, HAWKINS RD, BARRERA LO, VAN CALCAR S, QU C, CHING KA, WANG W, WENG Z, GREEN RD, CRAWFORD GE, REN B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- HIRSCHEY MD, ZHAO Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol Cell Proteomics. 2015;14:2308–15. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG H, LIN S, GARCIA BA, ZHAO Y. Quantitative proteomic analysis of histone modifications. Chem Rev. 2015;115:2376–418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG H, SABARI BR, GARCIA BA, ALLIS CD, ZHAO Y. SnapShot: histone modifications. Cell. 2014;159:458–458 e1. doi: 10.1016/j.cell.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAELIN WG, MCKNIGHT SL. Influence of Metabolism on Epigenetics and Disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KASHIWAYA Y, TAKESHIMA T, MORI N, NAKASHIMA K, CLARKE K, VEECH RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5440–5444. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATADA S, IMHOF A, SASSONE-CORSI P. Connecting Threads: Epigenetics and Metabolism. Cell. 2012;148:24–28. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- KOIKE N, YOO SH, HUANG HC, KUMAR V, LEE C, KIM TK, TAKAHASHI JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–54. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAFFEL L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev. 1999;15:412–26. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- LIM S, CHESSER AS, GRIMA JC, RAPPOLD PM, BLUM D, PRZEDBORSKI S, TIEU K. D-beta-Hydroxybutyrate Is Protective in Mouse Models of Huntington’s Disease. Plos One. 2011;6:e24620. doi: 10.1371/journal.pone.0024620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVE DC, GHOSH S, MONDOUX MA, FUKUSHIGE T, WANG P, WILSON MA, ISER WB, WOLKOW CA, KRAUSE MW, HANOVER JA. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc Natl Acad Sci U S A. 2010;107:7413–8. doi: 10.1073/pnas.0911857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU C, THOMPSON CB. Metabolic Regulation of Epigenetics. Cell Metabolism. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTINEZ-OUTSCHOORN UE, PRISCO M, ERTEL A, TSIRIGOS A, LIN Z, PAVLIDES S, WANG CW, FLOMENBERG N, KNUDSEN ES, HOWELL A, PESTELL RG, SOTGIA F, LISANTI MP. Ketones and lactate increase cancer cell “stemness”, driving recurrence, metastasis and poor clinical outcome in breast cancer. Cell Cycle. 2011;10:1271–1286. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNALLY MA, HARTMAN AL. Ketone bodies in epilepsy. J Neurochem. 2012;121:28–35. doi: 10.1111/j.1471-4159.2012.07670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON AM, WILLIAMSON DH. Physiological Roles of Ketone-Bodies as Substrates and Signals in Mammalian-Tissues. Physiological Reviews. 1980;60:143–187. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- RUAN HB, HAN X, LI MD, SINGH JP, QIAN K, AZARHOUSH S, ZHAO L, BENNETT AM, SAMUEL VT, WU J, YATES JR, 3RD, YANG X. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012;16:226–37. doi: 10.1016/j.cmet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUAN HB, SINGH JP, LI MD, WU J, YANG X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab. 2013;24:301–9. doi: 10.1016/j.tem.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMAZU T, HIRSCHEY MD, NEWMAN J, HE WJ, SHIRAKAWA K, LE MOAN N, GRUETER CA, LIM H, SAUNDERS LR, STEVENS RD, NEWGARD CB, FARESE RV, DE CABO R, ULRICH S, AKASSOGLOU K, VERDIN E. Suppression of Oxidative Stress by beta-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBRAMANIAN A, TAMAYO P, MOOTHA VK, MUKHERJEE S, EBERT BL, GILLETTE MA, PAULOVICH A, POMEROY SL, GOLUB TR, LANDER ES, MESIROV JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUGATHAN A, WAXMAN DJ. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol. 2013;33:3594–610. doi: 10.1128/MCB.00280-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAGGART AK, KERO J, GAN X, CAI TQ, CHENG K, IPPOLITO M, REN N, KAPLAN R, WU K, WU TJ, JIN L, LIAW C, CHEN R, RICHMAN J, CONNOLLY D, OFFERMANNS S, WRIGHT SD, WATERS MG. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–52. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- TAN MJ, LUO H, LEE S, JIN FL, YANG JS, MONTELLIER E, BUCHOU T, CHENG ZY, ROUSSEAUX S, RAJAGOPAL N, LU ZK, YE Z, ZHU Q, WYSOCKA J, YE Y, KHOCHBIN S, REN B, ZHAO YM. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell. 2011;146:1015–1027. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Z, ZANG C, ROSENFELD JA, SCHONES DE, BARSKI A, CUDDAPAH S, CUI K, ROH TY, PENG W, ZHANG MQ, ZHAO K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WELLEN KE, HATZIVASSILIOU G, SACHDEVA UM, BUI TV, CROSS JR, THOMPSON CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.