

Abstract
Background
Members of the Bifidobacteriaceae family represent both dominant microbial groups that colonize the gut of various animals, especially during the suckling stage of their life, while they also occur as pathogenic bacteria of the urogenital tract. The pan-genome of the genus Bifidobacterium has been explored in detail in recent years, though genomics of the Bifidobacteriaceae family has not yet received much attention. Here, a comparative genomic analyses of 67 Bifidobacteriaceae (sub) species including all currently recognized genera of this family, i.e., Aeriscardovia, Alloscardovia, Bifidobacterium, Bombiscardovia, Gardnerella, Neoscardovia, Parascardovia, Pseudoscardovia and Scardovia, was performed. Furthermore, in order to include a representative of each of the 67 (currently recognized) (sub) species belonging to the Bifidobacteriaceae family, we sequenced the genomes of an additional 11 species from this family, accomplishing the most extensive comparative genomic analysis performed within this family so far.
Results
Phylogenomics-based analyses revealed the deduced evolutionary pathway followed by each member of the Bifidobacteriaceae family, highlighting Aeriscardovia aeriphila LMG 21773 as the deepest branch in the evolutionary tree of this family. Furthermore, functional analyses based on genome content unveil connections between a given member of the family, its carbohydrate utilization abilities and its corresponding host. In this context, bifidobacterial (sub) species isolated from humans and monkeys possess the highest relative number of acquired glycosyl hydrolase-encoding genes, probably in order to enhance their metabolic ability to utilize different carbon sources consumed by the host.
Conclusions
Within the Bifidobacteriaceae family, genomics of the genus Bifidobacterium has been extensively investigated. In contrast, very little is known about the genomics of members of the other eight genera of this family. In this study, we decoded the genome sequences of each member of the Bifidobacteriaceae family. Thanks to subsequent comparative genomic and phylogenetic analyses, the deduced pan-genome of this family, as well as the predicted evolutionary development of each taxon belonging to this family was assessed.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3955-4) contains supplementary material, which is available to authorized users.
Keywords: Bifidobacteriaceae, Genomics, Phylogenomics, Bifidobacterium, Bifidobacteria
Background
The Bifidobacteriaceae is the sole family member of the Bifidobacteriales order, and has been shown to represent the deepest branch within the Actinobacteria phylum [1]. Currently, the Bifidobacteriaceae family includes 55 (sub) species of the genus Bifidobacterium [1, 2] and members of eight additional genera, i.e., Aeriscardovia, Alloscardovia, Bombiscardovia, Gardnerella, Neoscardovia, Parascardovia, Pseudoscardovia and Scardovia, which together encompass 12 species [1, 3]. Furthermore, a novel, yet unculturable species was identified from termites and included in the Bifidobacteriaceae family with the taxonomic denomination of ‘Candidatus Ancillula trichonymphae’ [4]. The name of this latter organism originates from a variety of flagellates of the genus Trichonympha, of which this strain is symbiont [4].
Bifidobacteriaceae are chemoorganotrophs with a fermentative type of metabolism, Gram-positive, non-spore-forming, non-motile, and anaerobic or facultative anaerobic bacteria [5]. They reside in different ecological niches, such as the human and animal gastrointestinal tract (GIT), oral cavity and the (social) insect gut [6], while they may also be found in blood and sewage, possibly due to environmental contamination. Many bifidobacteria are appreciated for their purported health-promoting activities as well as their relevance in early life colonization and contributions to the infant gut glycobiome [7]. Conversely, members of the other genera of the Bifidobacteriaceae family are generally associated with human and animal dental caries, and are commonly isolated from human clinical samples of tonsil abscesses and bacterial vaginosis [8, 9]. Furthermore, in contrast to bifidobacteria, which mainly include strict anaerobes with some exceptions, such as Bifidobacterium animalis subsp. lactis and Bifidobacterium asteroides [10, 11], other members of the family can grow under aerobic conditions and possess DNA with a lower G + C content [12–14].
The most controversial species belonging to this family is Gardnerella vaginalis [9], originally described by Leopold in 1953 and named Haemophilus vaginalis [15]. Subsequently, taxonomic studies mixed with data obtained from biochemical analyses and electron microscopic examinations, supported the need for its re-classification as a new genus [16, 17]. Currently, G. vaginalis is described as an opportunistic pathogen whose presence is tightly associated with bacterial vaginosis [18, 19]. Furthermore, Parascardovia denticolens and Scardovia inopinata, which were classified in 1996 as Bifidobacterium denticolens and Bifidobacterium inopinatum [20], respectively, together with Scardovia wiggsiae and Bifidobacterium dentium are associated with human dental caries [21]. While members of these species are present at high numbers in the saliva of adults, their presence strongly correlates with other caries-associated organisms [22, 23].
Notably, comparative genome analyses of the genus Bifidobacterium have been targeting the entire genus [2, 24] or one specific bifidobacterial taxa, i.e., B. bifidum [25], B. adolescentis [26], B. breve [27], B. longum [28] or the B. animalis subsp. lactis taxon [11]. In contrast, the genomics of the other eight genera belonging to the Bifidobacteriaceae family have not yet been investigated in detail. Here, we decoded the genomes of 11 species belonging to the Bifidobacteriaceae family for which there was no prior genomic data. Furthermore, we performed an in depth comparative genomic analysis, as well as a phylogenetic reconstruction of the 67 (sub) species currently assigned to the Bifidobacteriaceae family.
Methods
Bifidobacteriaceae strains
We retrieved the complete and partial genome sequences of 56 Bifidobacteriaceae strains from the National Center for Biotechnology Information (NCBI) public database (Table 1). Additionally, we sequenced and analyzed the genome sequences of 11 Bifidobacteriaceae strains deposited in the GenBank sequence database (Table 2).
Table 1.
General features of Bifidobacteriaceae genomes
| Taxon number | Bifidobacteriaceae strains | Genome statusa | Genome size | GC content | ORFs number | rRNA loci | tRNA number | GHs number | GH index | Isolation | Accession number |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 01 | B. actinocoloniiforme DSM 22766 | Draft (4) | 1,823,388 | 62.71 | 1484 | 2 | 46 | 41 | 0.0276 | Bumblebee digestive tract | JGYK00000000 |
| 02 | B. adolescentis ATCC 15703 | Complete | 2,089,645 | 59.18 | 1649 | 5 | 54 | 81 | 0.0491 | Intestine of adult | AP009256.1 |
| 03 | B. aesculapii DSM 26737 | Draft (118) | 2,794,396 | 64.58 | 2172 | 6 | 60 | 82 | 0.0378 | Faeces of baby common marmosets | BCFK00000000 |
| 04 | B. angulatum LMG 11039 | Draft (6) | 2,003,806 | 59.41 | 1523 | 4 | 48 | 63 | 0.0414 | Human faeces | JGYL00000000 |
| 05 | B. animalis subsp. animalis LMG 10508 | Draft (13) | 1,915,007 | 60.47 | 1527 | 3 | 52 | 48 | 0.0314 | Rat feces | JGYM00000000 |
| 06 | B. animalis subsp. lactis DSM 10140 | Complete | 1,938,606 | 60.48 | 1518 | 4 | 52 | 53 | 0.0349 | Fermented milk | CP001606.1 |
| 07 | B. aquikefiri LMG 28769 | Draft (18) | 2,408,364 | 52.29 | 2000 | 2 | 45 | 56 | 0.0280 | Household water kefir | MWXA00000000 |
| 08 | B. asteroides LMG 10735 (PRL2011) | Complete | 2,167,304 | 60.05 | 1653 | 2 | 44 | 58 | 0.0351 | Honeybee hindgut | CP003325.1 |
| 09 | B. biavatii DSM 23969 | Draft (56) | 3,252,147 | 63.1 | 2557 | 5 | 61 | 137 | 0.0536 | Feces of tamarin | JGYN00000000 |
| 10 | B. bifidum LMG 11041 | Draft (2) | 2,208,468 | 62.67 | 1704 | 3 | 53 | 66 | 0.0387 | Brest-feed Infant feaces | JGYO00000000 |
| 11 | B. bohemicum DSM 22767 | Draft (5) | 2,052,470 | 57.45 | 1632 | 2 | 47 | 56 | 0.0343 | Bumblebee digestive tract | JGYP00000000 |
| 12 | B. bombi DSM 19703 | Draft (4) | 1,895,239 | 56.08 | 1454 | 2 | 48 | 44 | 0.0303 | Bumblebee digestive tract | ATLK00000000 |
| 13 | B. boum LMG 10736 | Draft (18) | 2,171,356 | 59.31 | 1726 | 4 | 49 | 45 | 0.0261 | Bovine rumen | JGYQ00000000 |
| 14 | B. breve LMG 13208 | Draft (31) | 2,263,780 | 58.88 | 1887 | 2 | 53 | 67 | 0.0355 | Infant intestine | JGYR00000000 |
| 15 | B. callitrichos DSM 23973 | Draft (33) | 2,887,313 | 63.52 | 2364 | 3 | 58 | 105 | 0.0444 | Feces of common marmoset | JGYS00000000 |
| 16 | B. catenulatum LMG 11043 | Draft (11) | 2,082,756 | 56.11 | 1664 | 5 | 55 | 91 | 0.0547 | Adult intestine | JGYT00000000 |
| 17 | B. choerinum LMG 10510 | Draft (20) | 2,096,123 | 65.53 | 1672 | 3 | 55 | 54 | 0.0323 | Piglet faeces | JGYU00000000 |
| 18 | B. commune R-52791 | Dreaft (4) | 1,633,662 | 53.93 | 1303 | 1 | 47 | 31 | 0.0238 | Bumble bee gut | FMBL00000000 |
| 19 | B. coryneforme LMG 18911 | Complete | 1,755,151 | 60.51 | 1364 | 3 | 56 | 43 | 0.0315 | Honeybee hindgut | CP007287 |
| 20 | B. crudilactis LMG 23609 | Draft (6) | 2,362,816 | 57.72 | 1883 | 2 | 45 | 51 | 0.0271 | Raw cow milk | JHAL00000000 |
| 21 | B. cuniculi LMG 10738 | Draft (41) | 2,531,592 | 64.87 | 2194 | 4 | 63 | 70 | 0.0319 | Rabbit faeces | JGYV00000000 |
| 22 | B. dentium LMG 11045 (Bd1) | Complete | 2,636,367 | 58.54 | 2129 | 4 | 55 | 113 | 0.0531 | Oral cavity | CP001750.1 |
| 23 | B. eulemuris DSM 100216 | Draft (34) | 2,913,389 | 62.2 | 2331 | 2 | 53 | 126 | 0.0541 | Faeces of the black lemur | MWWZ00000000 |
| 24 | B. gallicum LMG 11596 | Draft (12) | 2,004,594 | 57.61 | 1507 | 2 | 58 | 45 | 0.0299 | Adult intestine | JGYW00000000 |
| 25 | B. gallinarum LMG 11586 | Draft (10) | 2,160,836 | 64.22 | 1654 | 2 | 53 | 78 | 0.0472 | Chicken caecum | JGYX00000000 |
| 26 | B. hapali DSM 100202 | Draft (76) | 2,834,308 | 54.5 | 2253 | 3 | 54 | 121 | 0.0537 | Faeces of baby common marmosets | MWWY00000000 |
| 27 | B. indicum LMG 11587 | Complete | 1,734,546 | 60.49 | 1352 | 3 | 47 | 41 | 0.0303 | Insect | CP006018 |
| 28 | B. kashiwanohense DSM 21854 | Draft (30) | 2,307,960 | 56.2 | 1948 | 5 | 53 | 91 | 0.0467 | Infant feaces | JGYY00000000 |
| 29 | B. lemurum DSM 28807 | Draft (38) | 2,944,293 | 62.64 | 2321 | 3 | 49 | 122 | 0.0526 | Faeces of the ring-tailed lemur | MWWX00000000 |
| 30 | B. longum subsp. infantis ATCC 15697 | Complete | 2,832,748 | 59.86 | 2500 | 4 | 79 | 71 | 0.0284 | Intestine of infant | AP010889.1 |
| 31 | B. longum subsp. longum LMG 13197 | Draft (8) | 2,384,703 | 60.33 | 1899 | 3 | 71 | 73 | 0.0384 | Adult intestine | JGYZ00000000 |
| 32 | B. longum subsp. suis LMG 21814 | Draft (36) | 2,335,832 | 59.96 | 1955 | 3 | 55 | 74 | 0.0379 | Pig faeces | JGZA00000000 |
| 33 | B. magnum LMG 11591 | Draft (13) | 1,822,476 | 58.72 | 1507 | 5 | 56 | 46 | 0.0305 | Rabbit faeces | JGZB00000000 |
| 34 | B. merycicum LMG 11341 | Draft (16) | 2,280,236 | 60.33 | 1741 | 3 | 53 | 66 | 0.0379 | Bovine rumen | JGZC00000000 |
| 35 | B. minimum LMG 11592 | Draft (18) | 1,892,860 | 62.73 | 1590 | 2 | 53 | 41 | 0.0258 | Sewage | JGZD00000000 |
| 36 | B. mongoliense DSM 21395 | Draft (43) | 2,170,490 | 62.78 | 1798 | 2 | 47 | 65 | 0.0362 | Fermented mare’s milk | JGZE00000000 |
| 37 | B. moukalabense DSM 27321 | Draft (12) | 2,515,335 | 59.87 | 2046 | 4 | 56 | 105 | 0.0513 | Feces of wild western lowland gorilla | AZMV00000000 |
| 38 | B. myosotis DSM 100196 | Draft (58) | 2,944,195 | 62.55 | 2168 | 4 | 56 | 101 | 0.0466 | Faeces of baby common marmosets | MWWW00000000 |
| 39 | B. pseudocatenulatum LMG 10505 | Draft (10) | 2,283,767 | 56.36 | 1771 | 6 | 53 | 85 | 0.0480 | Infant faeces | JGZF00000000 |
| 40 | B. pseudolongum subsp. globosum LMG 11569 | Draft (26) | 1,935,255 | 63.39 | 1574 | 4 | 52 | 53 | 0.0337 | Bovine rumen | JGZG00000000 |
| 41 | B. pseudolongum subsp. pseudolongum LMG 11571 | Draft (11) | 1,898,684 | 63.06 | 1495 | 3 | 52 | 57 | 0.0381 | Swine faeces | JGZH00000000 |
| 42 | B. psychraerophilum LMG 21775 | Draft (11) | 2,615,078 | 58.75 | 2122 | 1 | 45 | 80 | 0.0377 | Pig caecum | JGZI00000000 |
| 43 | B. pullorum DSM 20433 | Draft (38) | 2,100,948 | 64.31 | 1678 | 2 | 51 | 81 | 0.0479 | Faeces of chicken | JDUI00000000 |
| 44 | B. reuteri DSM 23975 | Draft (28) | 2,847,572 | 60.45 | 2149 | 4 | 53 | 85 | 0.0396 | Feces of common marmoset | JGZK00000000 |
| 45 | B. ruminantium LMG 21811 | Draft (23) | 2,249,807 | 59.18 | 1832 | 4 | 50 | 62 | 0.0338 | Bovine rumen | JGZL00000000 |
| 46 | B. saeculare LMG 14934 | Draft (14) | 2,263,283 | 63.75 | 1857 | 2 | 48 | 82 | 0.0442 | Rabbit faeces | JGZM00000000 |
| 47 | B. saguini DSM 23967 | Draft (33) | 2,787,036 | 56.35 | 2321 | 5 | 59 | 104 | 0.0448 | Feces of tamarin | JGZN00000000 |
| 48 | B. scardovii LMG 21589 | Draft (34) | 3,141,793 | 64.63 | 2480 | 3 | 55 | 128 | 0.0516 | Blood | JGZO00000000 |
| 49 | B. stellenboschense DSM 23968 | Draft (40) | 2,812,864 | 65.34 | 2202 | 6 | 59 | 80 | 0.0363 | Feces of tamarin | JGZP00000000 |
| 50 | B. subtile LMG 11597 | Draft (27) | 2,790,088 | 60.92 | 2260 | 1 | 47 | 56 | 0.0248 | Sewage | JGZR00000000 |
| 51 | B. thermacidophilum subsp. porcinum LMG 21689 | Draft (3) | 2,079,368 | 60.2 | 1738 | 3 | 40 | 46 | 0.0265 | Piglet faeces | JGZS00000000 |
| 52 | B. thermacidophilum subsp. thermacidophilum LMG 21395 | Draft (8) | 2,233,072 | 60.38 | 1823 | 4 | 48 | 47 | 0.0258 | Anaerobic digester | JGZT00000000 |
| 53 | B. thermophilum DSM 20212 | Draft (50) | 2,252,351 | 60.07 | 1756 | 3 | 49 | 58 | 0.0341 | Bovine rumen | JHWM00000000 |
| 54 | B. tissieri DSM 100201 | Draft (38) | 2,873,483 | 61.05 | 2260 | 2 | 60 | 79 | 0.0350 | Faeces of baby common marmosets | MWWV00000000 |
| 55 | B. tsurumiense JCM 13495 | Draft (25) | 2,164,426 | 52.84 | 1629 | 3 | 46 | 85 | 0.0522 | Hamster dental plaque | JGZU00000000 |
| 56 | Aeriscardovia aeriphila LMG 21773 | Draft (12) | 1,631,097 | 54.03 | 1288 | 3 | 47 | 53 | 0.0411 | Pig caecum | MWWU00000000 |
| 57 | Alloscardovia criceti DSM 17774 | Draft (11) | 1,884,654 | 50.06 | 1524 | 4 | 45 | 58 | 0.0381 | Dental plaque, golden hamster | AQXR00000000 |
| 58 | Alloscardovia macacae DSM 24762 | Draft (20) | 1,891,581 | 55.82 | 1552 | 4 | 48 | 50 | 0.0322 | Milk of a female macaque bred | MWWT00000000 |
| 59 | Alloscardovia omnicolens DSM 21503 | Draft (43) | 1,847,146 | 46.65 | 1564 | 3 | 47 | 56 | 0.0358 | Human tonsil | ATVB00000000 |
| 60 | Bombiscardovia coagulans DSM 22924 | Draft (15) | 1,741,326 | 47.33 | 1441 | 2 | 45 | 36 | 0.0250 | Bumblebee digestive tract | MWWS00000000 |
| 61 | Gardnerella vaginalis ATCC 14018 | Complete | 1,667,406 | 41.36 | 1271 | 2 | 45 | 41 | 0.0323 | Vaginal secretions | AP012332 |
| 62 | Neoscardovia arbecensis 1879 | Draft (21) | 1,971,875 | 54.25 | 1641 | 2 | 45 | 56 | 0.0341 | Feces of rabbit | n.d. |
| 63 | Parascardovia denticolens DSM 10105 | Complete | 1,890,857 | 58.31 | 1528 | 2 | 45 | 56 | 0.0366 | Human dental caries | AP012333 |
| 64 | Pseudoscardovia radai DSM 24742 | Draft (35) | 2,436,770 | 65.03 | 1779 | 3 | 47 | 41 | 0.0230 | Digestive tract of wild pig Sus scrofa | MWWR00000000 |
| 65 | Pseudoscardovia suis DSM 24744 | Draft (26) | 2,270,618 | 60.57 | 1736 | 3 | 48 | 45 | 0.0259 | Digestive tract of wild pig Sus scrofa | MWWQ00000000 |
| 66 | Scardovia inopinata JCM 12537 | Complete | 1,797,862 | 48.63 | 1465 | 2 | 46 | 49 | 0.0334 | Human dental caries | AP012334 |
| 67 | Scardovia wiggsiae F0424 | Complete | 1,550,817 | 52.93 | 1244 | 2 | 45 | 32 | 0.0257 | Human dental caries | AKCI00000000 |
aNumbers in brackets indicate the numbers of assembled contigs
Table 2.
Sequencing data of the Bifidobacteriaceae genomes
| Bifidobacteriaceae Strains | Genome statusa | Sequence coverage | Genome size | GC content | ORFs number | rRNA loci | tRNA number | Isolation | Accession number |
|---|---|---|---|---|---|---|---|---|---|
| Aeriscardovia aeriphila LMG 21773 | Draft (12) | 259.66 | 1,631,097 | 54.03 | 1288 | 3 | 47 | Pig caecum | MWWU00000000 |
| Alloscardovia macacae DSM 24762 | Draft (20) | 173.37 | 1,891,581 | 55.82 | 1552 | 4 | 48 | Milk of a female macaque bred | MWWT00000000 |
| Bifidobacterium aquikefiri LMG 28769 | Draft (18) | 96.25 | 2,408,364 | 52.29 | 2000 | 2 | 45 | Household water kefir | MWXA00000000 |
| Bifidobacterium eulemuris DSM 100216 | Draft (34) | 75.23 | 2,913,389 | 62.2 | 2331 | 2 | 53 | Faeces of the black lemur | MWWZ00000000 |
| Bifidobacterium hapali DSM 100202 | Draft (76) | 109.83 | 2,834,308 | 54.5 | 2253 | 3 | 54 | Faeces of baby common marmosets | MWWY00000000 |
| Bifidobacterium lemurum DSM 28807 | Draft (38) | 71.87 | 2,944,293 | 62.64 | 2321 | 3 | 49 | Faeces of the ring-tailed lemur | MWWX00000000 |
| Bifidobacterium myosotis DSM 100196 | Draft (58) | 57.03 | 2,944,195 | 62.55 | 2168 | 4 | 56 | Faeces of baby common marmosets | MWWW00000000 |
| Bifidobacterium tissieri DSM 100201 | Draft (38) | 66.77 | 2,873,483 | 61.05 | 2260 | 2 | 60 | Faeces of baby common marmosets | MWWV00000000 |
| Bombiscardovia coagulans DSM 22924 | Draft (15) | 220.35 | 1,741,326 | 47.33 | 1441 | 2 | 45 | Bumblebee digestive tract | MWWS00000000 |
| Pseudoscardovia radai DSM 24742 | Draft (35) | 145.91 | 2,436,770 | 65.03 | 1779 | 3 | 47 | Digestive tract of wild pig Sus scrofa | MWWR00000000 |
| Pseudoscardovia suis DSM 24744 | Draft (26) | 140.27 | 2,270,618 | 60.57 | 1736 | 3 | 48 | Digestive tract of wild pig Sus scrofa | MWWQ00000000 |
aNumbers in brackets indicate the numbers of assembled contigs
Bacterial strains and growth condition
Bifidobacteriaceae pure cultures were inoculated in de Man-Rogosa-Sharpe (MRS) medium (Scharlau Chemie) supplemented with 0.05% (wt/vol) L-cysteine hydrochloride and were grown in an anaerobic atmosphere (2.99% H2, 17.01% CO2, and 80% N2) in a chamber (Concept 400, Ruskin) at 37 °C for 16 h. DNA was extracted as described previously [29] and subjected to further phenol-chloroform purification using a previously described protocol [30].
Genome sequencing and assemblies
DNA extracted from the various Bifidobacteriaceae strains was subjected to whole genome sequencing using MiSeq (Illumina, UK) at GenProbio srl (Parma, Italy) following the supplier’s protocol (Illumina, UK). Fastq files of the paired-end reads obtained from targeted genome sequencing of the isolated strains were used as input for genome assemblies through the MEGAnnotator pipeline [31]. The MIRA program (version 4.0.2) was used for de novo assembly of each Bifidobacteriaceae genome sequence [32].
Sequence annotation
Protein-encoding open reading frames (ORFs) were predicted using Prodigal [33]. Transfer RNA genes were identified using tRNAscan-SE v1.4 [34], while ribosomal RNA genes were detected using RNAmmer v1.2 [35]. Results of the gene-finder program were combined with data from RAPSearch2 analysis (Reduced Alphabet based Protein similarity Search) [36] of a non-redundant protein database provided by the National Center for Biotechnology Information (NCBI) and Hidden Markov Model profile (HMM) search (http://hmmer.org/) in the manually curated Pfam-A protein family database [37]. The combined results were inspected by Artemis [38], which was used for manual editing purposes aimed at verifying and, where necessary, redefining the start of each predicted coding region, and to remove or add coding regions.
Bifidobacteriaceae pan-genome analysis
For the 67 genome sequences of each member of the Bifidobacteriaceae family, a pan-genome calculation was performed using the PGAP pipeline [39]. The ORF contents from all genomes used in this study were organized in functional clusters using the GF (Gene Family) method involving comparison of each protein to all other proteins using BLAST analysis (cutoff E-value of 1 X 10−5 and 50% identity over at least 50% of both protein sequences), followed by clustering into protein families, named Bifidobacteriaceae-specific clusters of orthologous groups (BaeCOGs), using MCL (graph-theory-based Markov clustering algorithm) [40]. A pan-genome profile was built using an optimized algorithm incorporated in PGAP software, based on a presence/absence matrix that included all identified BaeCOGs in the analyzed genomes. Following this, unique protein families for each of the 67 Bifidobacteriaceae genomes were classified. Protein families shared between all genomes, named core BaeCOGs, were defined by selecting the families that contained at least one protein member for each genome.
Phylogenetic comparison
The concatenated core genome sequence of the family (core BaeCOGs), was aligned using MAFFT [41], and phylogenetic trees were constructed using the neighbor-joining method in Clustal W, version 2.1 [42]. The core genome supertree was built using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Values of average nucleotide identity (ANI) were calculated using the program JSpecies, version 1.2.1 [43].
Functional analysis
The prediction of genes encoding enzymes that possess structurally-related catalytic and carbohydrate-binding modules catalyzing hydrolysis, modification or synthesis of glycoside bounds was performed by means of the CAZy database [44]. Functional annotation of each gene was performed employing the eggNOG database [45]. A survey of complete pathways involved in both primary and secondary metabolism was performed by means of the MetaCyc metabolic pathways database [46]. Gene function was predicted using a cutoff E-value of 1 × 10−10 to identify the best hit from each database.
Gene gain/loss through evolution reconstruction
Predicting gene acquisition or gene loss as a result of evolution of the bacterial species with at least four available genomes was performed with Count software [47] using Dollo’s parsimony.
Results and discussion
General genome features of Bifidobacteriaceae genomes
Genome sequences of six bifidobacterial species, i.e., Bifidobacterium aquikefiri LMG 28769, Bifidobacterium eulemuris DSM 100216, Bifidobacterium hapali DSM 100202, Bifidobacterium lemurum DSM 28807, Bifidobacterium myosotis DSM 100196 and Bifidobacterium tissieri DSM 100201, as well as five chromosomes belonging to different genera of the Bifidobacteriaceae family, including Aeriscardovia aeriphila LMG 21773, Alloscardovia macacae DSM 24762, Bombiscardovia coagulans DSM 22924 and Pseudoscardovia radai DSM 24742 and Pseudoscardovia suis DSM 24744, were decoded through shotgun sequencing. Genome features and sequencing data of these 11 Bifidobacteriaceae genomes are summarized in Table 2. In order to provide a complete genome analysis of the Bifidobacteriaceae family, a representative genome sequence for each of the currently described 67 (sub) species belonging to this family, was retrieved from the NCBI public database (Table 1). Due to the incomplete genome sequences of Candidatus Ancillula trichonymphae ImTpAt recovered from the NCBI database, and the impossibility to retrieve this strain from any public bacterial culture collection, we decided to exclude the genome sequences of ImTpAt from our analyses. The combination of genomic data of 56 previously characterized bifidobacterial taxa [2, 3, 48–51] with the chromosome sequences of the 11 Bifidobacteriaceae species reported here, resulted in the most comprehensive database of genome sequences of representative members of the Bifidobacteriaceae family. The Bifidobacteriaceae genomes have an average genome length of 2.25 Mb, and range in size from 1.55 Mb for Scardovia wiggsiae F0424 to 3.25 Mb for Bifidobacterium biavatii DSM 23969, corresponding to 1244 and 2557 predicted protein-encoding open reading frames (ORFs), respectively (Table 1). Average genomic GC content ranges from 41.36% for Gardnerella vaginalis ATCC 14018 to 65.53% for Bifidobacterium choerinum LMG 10510, and revealed a higher average for the bifidobacterial strains (60.24%) as compared to the other taxa of the Bifidobacteriaceae genera (52.91%) [52]. The average genome size of bifidobacterial strains is also higher than that observed for the other family members, being 2.33 Mb and 1.88 Mb, respectively, highlighting a gene ratio of 1.24 (obtained by dividing the average gene number of bifidobacterial (sub) species with that of the non-bifidobacterial taxa of the Bifidobacteriaceae family) in favor of the bifidobacterial strains. The larger gene complement possessed by members of the Bifidobacterium genus may reflect an increased genetic variability of this genus, endowing bifidobacteria with an enhanced ability to adapt to a broad range of ecological niches as compared to other members of the Bifidobacteriaceae family, which, as is listed in Table 1, were isolated from a very limited number of environments. Further analyses involving mobile elements of the different species belonging to the Bifidobacteriaceae family reveal varying percentages of such mobile elements (calculated as a proportion of the total number of genes within these genomes), ranging from 0.07% for Bifidobacterium indicum LMG 11587 to 5.02% for B. hapali DSM 100202. Furthermore, the overall Bifidobacterium genus contains a percentage of 1.5% mobile elements, while non-bifidobacterial species reveal a percentage of 0.9%, highlighting an approximate mobile element ratio of 2.1 (obtained by dividing the average contents of the predicted mobile elements of bifidobacterial (sub) species with that of the non-bifidobacterial taxa belonging to the Bifidobacteriaceae family). Thus, the larger abundance of predicted mobile elements in bifidobacterial genomes reflects the above mentioned increased genomic variability of these strains. Interestingly, the number of tRNA genes in bifidobacteria ranges from 40 for Bifidobacterium thermacidophilum subsp. porcinum LMG 21689 to 79 for Bifidobacterium longum subsp. infantis ATCC 15697, while in other members of the Bifidobacteriaceae the tRNA abundance seems to be much less variable, ranging from 45 to 48 (Table 1). Nevertheless, the Bifidobacteriaceae strains that do not belong to the Bifidobacterium genus possess at least one tRNA gene for each of the 20 amino acids (Additional file 1: Table S1). Additionally, a deeper screening of the anticodon sequences for each (sub) species does not display major differences, except for a lower abundance of the anticodon GGG in Proline tRNA of the Bifidobacteriaceae strains that do not belong to the Bifidobacterium genus (Additional file 1: Table S1). Consequently, it may be argued that the lower number of tRNA genes among certain members of the Bifidobacteriaceae family is not associated with a simplification of the codon usage of these strains. Furthermore, while the bifidobacterial genomes contain between one and six rRNA loci, with an average of 3.2 per genome [2], the genomes of the Aeriscardovia, Alloscardovia, Bombiscardovia, Gardnerella, Neoscardovia, Parascardovia, Pseudoscardovia and Scardovia genera exhibit a lower average number of rRNA loci, i.e., 2.6, which is consistent with the less extensive ORFome and tRNA arsenal identified in the corresponding genomes (Table 2).
Furthermore, in silico analyses of the 12 genomes of the Bifidobacteriaceae family based on the Virulence Factor Database (VFDB) [53], did not reveal the occurrence of any virulence genetic determinants. Such results confirm previously reported findings for the genome of B. dentium Bd1 [23].
Pan-genome, core genome and unique genes of the Bifidobacteriaceae family
Comparative genome analyses involving the 67 (sub) species belonging to the Bifidobacteriaceae family were performed to unveil the corresponding pan-genome, core genome and unique genes of this bacterial family. All genomes were subjected to identical ORFs finding and annotation protocols [31] in order to generate comparable data sets for each Bifidobacteriaceae taxa. A total of 25,744 BaeCOGs (Bifidobacteriaceae-specific clusters of orthologous genes) were identified in the 67 Bifidobacteriaceae (sub) species, of which 8359 had members present in at least two genomes. The pan-genome size, when plotted versus the number of included genomes, clearly shows that the power trend line has yet to reach a plateau (Fig. 1). Actually, the number of new genes discovered by sequential addition of genome sequences was reduced from 839 to 636 BaeCOGs in the first three genomes additions to a number that ranged from 274 to 272 BaeCOGs in the final three additions, demonstrating the existence of an open pan-genome within Bifidobacteriaceae family. This finding suggests that more members of the Bifidobacteriaceae family have yet to be identified, especially members of the family that do not belong to the Bifidobacterium genus, as these remain poorly characterized in various environments compared to the currently recognized bifidobacterial (sub)species.
Fig. 1.
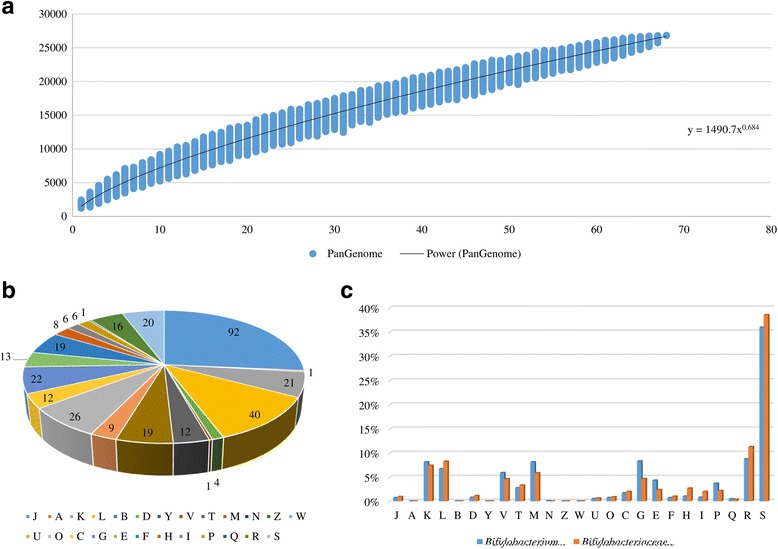
Pan-genome and functional classification of genes of the Bifidobacteriaceae family. Panel a shows the pan-genome represented as variations of the sizes of the resulting gene pool upon sequential addition of the 67 Bifidobacteriaceae genomes. The x axis represents the number of included genomes, whereas the y axis represents the number of genes in the generated pan-genome. Panel b exhibits the number of core BaeCOGs associated with the predicted EggNOG classification. Panel c displays the percentages of TUGs associated with functional categories as classified through the EggNOG database between bifidobacterial and members of the Bifidobacteriaceae family that do not belong to the Bifidobacterium genus. COG families are identified by a one-letter abbreviation: A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control and mitosis; E, amino acid metabolism and transport; F, nucleotide metabolism and transport; G, carbohydrate metabolism and transport; H, coenzyme metabolism; I, lipid metabolism; J, translation; K, transcription; L, replication and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, post translational modification, protein turnover, and chaperone functions; P, inorganic ion transport and metabolism; Q, secondary structure; T, signal transduction; U, intracellular trafficking and secretion; Y, nuclear structure; V, defense mechanisms; Z, cytoskeleton; R, general functional prediction only; S, function unknown
Pan-genome analysis of the Bifidobacteriaceae family allowed the identification of 353 COGs shared by all 67 (sub) species, representing the core genome of currently sequenced Bifidobacteriaceae representatives (core BaeCOGs). An examination of the functional annotation of Core BaeCOGs employing the eggNOG database [45] shows that the most conserved core genes specify housekeeping functions such as replication, transcription and translation, or functions related to adaptation such as carbohydrate, nucleotide and amino acid metabolism as well as cell envelope biogenesis (Fig. 1).
The pan-genome analysis also allowed the identification of truly unique genes (TUGs) of the Bifidobacteriaceae family, i.e., those genes that are presented in one particular strain yet absent in any of the other examined representative of the Bifidobacteriaceae family. The number of TUGs range from 42 for B. indicum LMG 11587 to 585 for Bifidobacterium cuniculi LMG 10738. EggNOG analysis showed that the majority of TUGs (59%) have no functional annotation (Additional file 1: Table S2). Nevertheless, taking into account the classified genes through the eggNOG analysis excluding the hypothetical and no-function genes, the highest number of genes fall in carbohydrate metabolism and cell envelope biogenesis together with replication and transcription. As mentioned above, these are the same four categories identified as containing the highest numbers of core BaeCOGs. Interestingly, the functional annotation of TUGs revealed that bifidobacterial genomes exhibit a higher abundance of TUGs involved in carbohydrate metabolism and cell envelope biogenesis compared to those of other members of the Bifidobacteriaceae family, reflecting a 38% and 78% of additional TUGs based on the average numbers between groups, respectively [2, 54] (Fig. 1 and Additional file 1: Table S2). These results are consistent with previous genomic and functional analyses based on the reference strains for each (sub) species of the Bifidobacterium genus indicating that bifidobacteria are under strong selective pressure to acquire and retain accessory genes for carbohydrate utilization in order to be competitive in the specific ecological niches in which they reside [2, 54].
Phylogenomic analyses of members of the Bifidobacteriaceae family
The availability of genome sequences for each member of the Bifidobacteriaceae family allowed an in-depth analysis of the evolutionary development followed by each member of this extensive family. A phylogenetic supertree was constructed based on the concatenation of 314 protein sequences that represent the core BaeCOGs with the exclusion of paralogs identified in each genome (Fig. 2).
Fig. 2.
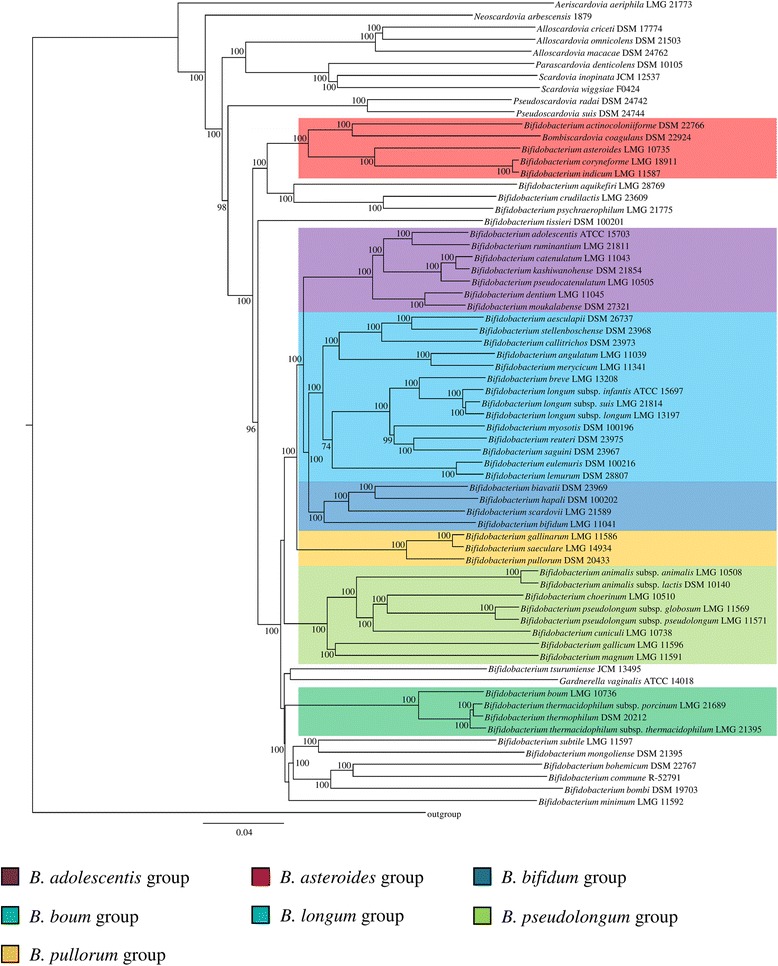
Supertree of the Bifidobacteriaceae family based on the concatenation of the amino acid sequences deduced from 314 core genes. Bootstrap values higher than 70 are marked near the respective nodes. Bifidobacterial groups are highlighted in different colors
The generated phylogenetic supertree showed that ten strains of the Bifidobacteriaceae species that do not belong to the Bifidobacterium genus represent the deepest branches of this supertree, being separated from the 55 bifidobacterial (sub) species (Fig. 2). Consequently, the bifidobacterial species were positioned at the top of the supertree, reflecting their highest average gene ratio (1.24) as compared to the non-bifidobacterial members of the Bifidobacteriaceae family. Thus, these data clearly indicate that evolution of currently known bifidobacterial species involved a relatively limited number of ancestral gene loss incidences, yet an extensive number of gene acquisition events, corroborating previously published data [2]. Interestingly, two members of the Bifidobacteriaceae family that do not belong to the Bifidobacterium genus, i.e., G. vaginalis ATCC 14018 and Bo. coagulans DSM 22924, appear to possess a higher level of phylogenetic relatedness with bifidobacterial strains as compared to other, non-bifidobacterial members of the Bifidobacteriaceae family (Fig. 2). While G. vaginalis ATCC 14018 shares the same phylogenetic branch as that of Bifidobacterium subtile LMG 11597, Bo. coagulans DSM 22924 is positioned within the deepest branch of the Bifidobacterium genus, i.e., within the B. asteroides group [2, 10], exhibiting higher genome relatedness with Bifidobacterium actinocoloniiforme DSM 22766, which was also isolated from Bombus. In order to validate the branching of these two Bifidobacteriaceae strains, a phylogenetic tree based on the 16S rRNA genes was constructed, as well as a tree based on five housekeeping genes including hsp60, rpoB, dnaJ, dnaG and clpC (Additional file 2: Figure S1). While Bo. coagulans DSM 22924 shares the same phylogenetic branch with B. actinocoloniiforme DSM 22766 in both trees, G. vaginalis ATCC 14018 occupies different positions within these phylogenetic trees. Nonetheless, the housekeeping-based tree confirmed the position of G. vaginalis ATCC 14018 within the Bifidobacterium genus, while in the 16S rRNA-based tree it is placed between bifidobacterial species and the other non-bifidobacterial species. These findings cast doubts on the correct taxonomical classification of Bo. coagulans DSM 22924, and reinforce the importance of a phylogenomic approach as a tool for taxonomic validation [55].
Furthermore, we investigated the occurrence of genes predicted to encode enzymes for anaerobic respiration in the pangenome of the members of the Bifidobacteriaceae as previously described for the genome of B. asteroides PRL2011 [10]. Such in silico analyses highlight the presence of a cytochrome bd oxidase-encoding complex in the genome of Bo. coagulans DSM 22924 (Additional file 1: Table S3), including the cydA and cydB, which code for structural subunit of the cytochrome, as well as cydC and cydD, encoding a transporter required for the cytochrome assembly [10]. Furthermore, these four genes were identified in 11 genomes of the Bifidobacteriaceae family, including six strains isolated from insects, i.e., B. actinocoloniiforme DSM 22766, Bifidobacterium bohemicum DSM 22767, Bifidobacterium bombi DSM 19703, Bifidobacterium commune R-52791, Bifidobacterium coryneforme LMG 18911 and B. indicum LMG 11587, highlighting a correlation between the presence of the cytochrome bd oxidase complex and the ecological niche of isolation (Additional file 1: Table S3).
Based on the Bifidobacteriaceae supertree reconstruction, five out of the six bifidobacterial species that were newly sequenced as part of this study are positioned within one of the previously identified bifidobacterial groups [24, 56]. Interestingly, four strains appear to belong to the Bifidobacterium longum group, i.e., B. myosotis DSM 100196, B. reuteri DSM 23975, B. eulemuris DSM 100216 and B. lemurum DSM 28807 (Fig. 2). This finding is in line with the particular ecological origin of these strains. In fact, each one of these four species was isolated from feces of monkeys, similar to the other two members that had previously been assigned to the B. longum group, i.e., Bifidobacterium stellenboschense DSM 23968 and Bifidobacterium callitrichos DSM 23973. These findings therefore highlight that the B. longum group includes bifidobacterial (sub) species isolated from humans and other related primates (Fig. 2). Furthermore, B. hapali DSM 100202 exhibits a genetic relatedness with Bifidobacterium biavatii DSM 23969, which belongs to the B. bifidum group [24]. Notably, B. tissieri DSM 100201 was shown to occupy a unique position within the Bifidobacteriaceae supertree. This observation was confirmed through an average nucleotide identity (ANI) analysis showing the highest value of 88.02% with the most related Alloscardovia criceti DSM 17774 strain. Notably, an ANI value below to 95% is assumed to be sufficient to classify that taxon as a distinct species [43]. B. tissieri DSM 100201 is therefore an interesting strain for further investigation due to its genetic divergence from other bifidobacterial taxa. In order to validate the species assignment for the other 10 sequenced members of the Bifidobacteriaceae family (Table 2) from a genomic prospective, the decoded genomes were subjected to ANI comparisons with the other 56 (sub) species that had been sequenced. The sequenced genomes of the 10 Bifidobacteriaceae strains showed ANI values below 95%, confirming their status as distinct species, with a relatively high value of 93.8% observed between B. eulemuris DSM 100216 and B. lemurum DSM 28807 (Additional file 1: Table S4).
Furthermore, the Bifidobacteriaceae supertree confirmed the phylogenetic relatedness of the recently decoded genome sequences of B. commune R-52791 and Bifidobacterium aesculapii DSM 26737, which occupy positions within the same branches with B. bohemicum DSM 22767 and B. stellenboschense DSM 23968, respectively [49, 57]. Such data further confirms the existence of a direct relatedness between the ecological origin of both strains, i.e., insects of the Bombus genus and monkeys of the Callitrichidae family.
Enzymatic profiling and evolutionary development of the Bifidobacteriaceae family
A functional profiling analysis was performed to assess the presence of genes encompassing carbohydrate, amino acid and fatty acid degradation pathways in each (sub) species of the Bifidobacteriaceae family. Normalizing the obtained number of gene matches with the overall genetic arsenal (i.e. total amount of genes) of each strain, non-bifidobacterial strains retrieved percentages that were slightly lower compared to the bifidobacterial strains, i.e., 7%, 11% and 9%, respectively (Additional file 1: Table S5). Interestingly, all members of the Bifidobacteriaceae family possess the Bifidobacterium shunt pathway [6, 58], including the gene xfp that encodes the enzyme D-xylulose 5-phosphate phosphoketolase/D-fructose 6-phosphate phosphoketolase, expanding the notion of this signature metabolic trait of bifidobacteria to the whole family.
To further investigate the carbohydrate utilization abilities encoded by the genomes of the 67 Bifidobacteriaceae (sub) species, an enzyme classification toward glycans was performed. This enzyme classification was based on the Carbohydrate Active Enzymes (CAZy) database [44], which encompasses all currently known genetic determinants involved in the breakdown and utilization of carbohydrates, and revealed that the pan-genome of the Bifidobacteriaceae family includes 9742 genes predicted to encode carbohydrate-active enzymes, i.e., glycosyl hydrolases (GHs), glycosyl transferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs) and carbohydrate binding modules (CBMs), present at 43.4%, 43.8%, 0.2% 5.9% and 12.6%, respectively. This very substantial number of retrieved enzymes reflects the findings of a previous analysis conducted on the 47 type-strains of the Bifidobacterium genus, where the glycan-breakdown potential of bifidobacteria in the mammalian gut was subjected to an extensive scrutiny [54].
Focusing on GH identification, 3989 genes were predicted on the analyzed genomes of 55 bifidobacterial (sub) species, while the remaining 12 Bifidobacteriaceae genomes contain 573 such genes. The Bifidobacteriaceae genomes specify a large arsenal of GH families, were GH13, GH3, GH43, GH23, GH32 and GH25 outnumber the other identified families (Additional file 1: Table S6). Despite the higher average amount of predicted GH-encoding genes for a given bifidobacterial genome, i.e., 72, as compared to that for a non-bifidobacterial member of the Bifidobacteriaceae family, i.e., 48, normalization of GH counts against the total amount of predicted genes provided similar GH indexes, i.e., 0.039 and 0.032, respectively (Additional file 1: Table S6). Nonetheless, genomes of the 20 strains that exhibit the highest GH index all belong to the Bifidobacterium genus, and are nearly all isolated from fecal samples of humans and monkeys, chief among them being Bifidobacterium catenulatum LMG 11043, B. eulemuris DSM 100216, B. hapali DSM 100202 and B. biavatii DSM 23969 (Table 1). These results suggest that bifidobacteria that reside in the primate/human gut have enjoyed a relatively high number of adaptive events related to carbohydrate metabolism to benefit from a wider source of different nutrients present in this particular environment. In contrast, Bifidobacteriaceae species that exhibit a relatively low GH index originate from a broad spectrum of environments, such as the gut of insects or other animals, and sewage (Table 1). Furthermore, genomes with a low GH index correspond with the smaller genomic complement of certain members of the Bifidobacteriaceae family, probably due to gene decay characteristic of those microbes that are considered harmful for human health, such as G. vaginalis, A. omnicolens, S. inopinata and S. wiggsiae [59].
While gene acquisition events that occur during evolution of microbial genomes support adaptation to new ecological niches, gene loss on the other hand contributes to genome simplification in order to preserve energy and biological compounds [59, 60]. Prediction of the complete Bifidobacteriaceae glycobiome content allowed us to estimate the acquisition and loss rates of genes encoding carbohydrate-active enzymes within this saccharolytic family. In order to depict gain and loss events of genes with a predicted function in carbohydrate metabolism, we collected the BaeCOGs that include GH-encoding genes obtained from the pan-genome analysis. The resulting 846 BaeCOGs were allocated among the Bifidobacteriaceae supertree showing that evolution of the current bifidobacterial (sub) species involved only a limited number of ancestral gene loss events, yet a substantial number of GH-encoding gene acquisitions (Fig. 3). Thus, genes encoding GHs appear to have been acquired early in the evolution of bifidobacteria, followed by a simplification of the GH-associated gene arsenal that has resulted in or followed specialization toward those ecological niches in which current bifidobacterial species have been identified. Interestingly, members of the B. bifidum group possess the highest number of GH-encoding gene acquisitions compared to those of other groups, probably in order to expand its metabolic ability towards different carbon sources present in the host, similar to what was mentioned above regarding GH index discrepancies (Fig. 3 and Table 1). Taking into account only those BaeCOGs that include members of GH families known to be involved in host-glycan degradation, i.e., GH20, GH29, GH33, GH38, GH95, GH101, GH112, GH125 and GH129 [54], the B. bifidum group, once again, displays the highest number of GH-encoding gene acquisitions (up to seven BaeCOGs). In this context B. bifidum exhibits the highest host-glycan degradation BaeCOGs acquisition number of the Bifidobacteriaceae family, thereby highlighting the capability of this bifidobacterial species to feed on host-glycan [25, 61, 62].
Fig. 3.
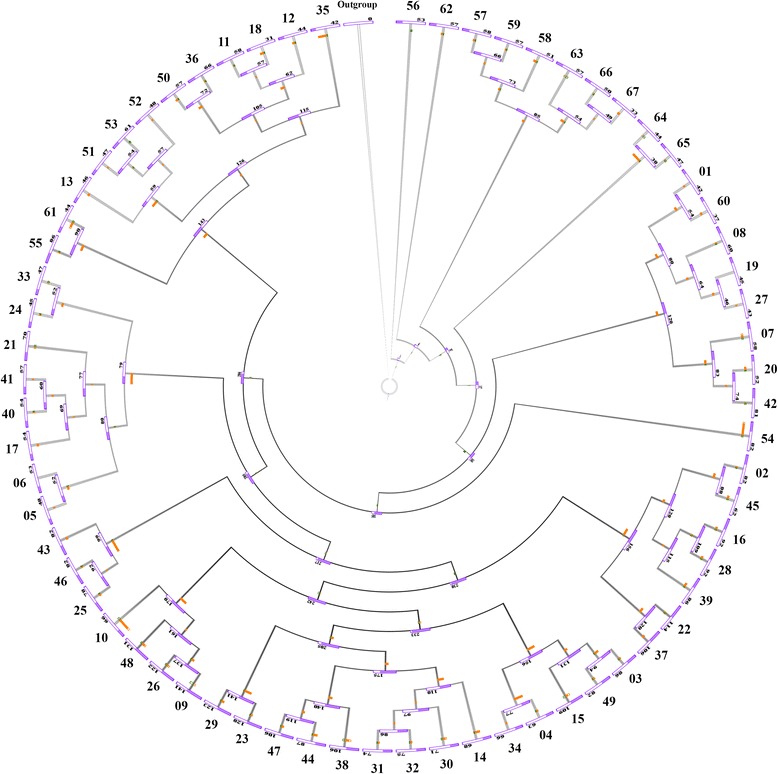
Analysis of evolutionary gain and loss events as based on predicted GHs within the Bifidobacteriaceae family. The image displays the core gene-based supertree of the Bifidobacteriaceae family, where each node reports the number of predicted GHs identified for each strain. Furthermore, gain and loss events are indicated by green and orange bars on the edge leading to each node, while the numbers placed at the supertree leaves represent the related bifidobacterial taxa presented in Table 1
Conclusions
A lot of effort has been invested in the dissection and characterization of the genomic content from different members of the Bifidobacterium genus [2, 11, 24–28]. In contrast, very little is known about the genomics of other members of the Bifidobacteriaceae family, which include apart from the Bifidobacterium genus eight additional genera. In this study, genome sequencing allowed us to explore the genome content of known members across the Bifidobacteriaceae family, as represented by 67 (sub) species, and to scrutinize the phylogenetic relatedness between each taxon belonging to this family. Bifidobacteria exhibit a higher number of genes per genome compared to other members of the Bifidobacteriaceae family, perhaps reflecting an increased competitiveness based on a broad spectrum of ecological niches from which they were isolated. The more complex genome content of bifidobacteria is also reflected by the capability of these microorganisms to degrade multiple carbon sources [54, 63]. Such findings were further validated in this study by the analyses of the pan-genome of the members of the Bifidobacteriaceae family, which highlights the occurrence of a higher abundance of TUGs in bifidobacterial genomes dedicated to the carbohydrate metabolism and cell envelope biogenesis, when compared to such numbers for other members of the Bifidobacteriaceae family. Enzymatic gene profiling revealed that the 20 strains that showed the highest GH indexes belong to bifidobacterial strains that are nearly all isolated from humans or monkeys. Moreover, a gene gain/loss analysis shows that members of the Bifidobacterium genus isolated from such primates possess the highest number of GH-encoding gene acquisitions, probably in order to expand their ability to metabolize different carbon sources. These results highlight a relatively large number of adaptive events related to carbohydrate metabolism among members of the Bifidobacterium genus that reside in omnivorous organisms that consume a wide variety of nutrients.
Furthermore, our phylogenomic analysis revealed possible taxonomic inconsistencies in the classification of G. vaginalis and Bo. coagulans, which displayed a close phylogenetic relatedness with other bifidobacterial strains, i.e., B. subtile LMG 11597 and B. actinocoloniiforme DSM 22766. Such findings further corroborate the strengths of genome-based analyses as an essential approach to be incorporated in phylogenome-based taxonomic studies.
Additional files
Tables S1, S2, S3, S4, S5 and S6. (XLSX 3628 kb)
Phylogenetic trees of the Bifidobacteriaceae family. Panel a display the 16S rRNA gene-based tree of the current recognized (sub) species of the family. Panel b shows the phylogenetic tree based on the concatenation of the amino acid sequences of five housekeeping genes including hsp60, rpoB, dnaJ, dnaG and clpC. For each tree, bootstrap values higher than 70 are marked near the respective nodes. (TIFF 2260 kb)
Acknowledgements
Part of this research is conducted using the High Performance Computing (HPC) facility of the University of Parma.
Funding
This work was funded by the EU Joint Programming Initiative – A Healthy Diet for a Healthy Life (JPI HDHL, http://www.healthydietforhealthylife.eu/) to DvS (in conjunction with Science Foundaion Ireland [SFI], Grant number 15/JP-HDHL/3280) and to MV (in conjunction with MIUR, Italy). We thank GenProbio srl for financial support of the Laboratory of Probiogenomics. LM is supported by Fondazione Cariparma, Parma, Italy. DvS is a member of The APC Microbiome Institute funded by Science Foundation Ireland (SFI), through the Irish Government’s National Development Plan (Grant number SFI/12/RC/2273). The authors declare that they have no competing interests.
Availability of data and materials
Genome sequences reported in this article have been deposited in GenBank under the following accession numbers: MWWQ00000000, MWWR00000000, MWWS00000000, MWWT00000000, MWWU00000000, MWWV00000000, MWWW00000000, MWWX00000000, MWWY00000000, MWWZ00000000 and MWXA00000000. The version described in this paper is version MWWQ01000000, MWWR01000000, MWWS01000000, MWWT01000000, MWWU01000000, MWWV01000000, MWWW01000000, MWWX01000000, MWWY01000000, MWWZ01000000 and MWXA01000000.
Abbreviations
- ANI
Average nucleotide identity
- BaeCOGs
Bifidobacteriaceae-specific clusters of orthologous groups
- CAZy
Carbohydrate active enzymes
- CBMs
Carbohydrate binding modules
- CEs
Carbohydrate esterases
- GF
Gene family
- GHs
Glycosyl hydrolases
- GIT
Gastrointestinal tract
- GTs
Glycosyl transferases
- HMM
Hidden Markov Model
- MRS
Man-Rogosa-Sharpe
- NCBI
National Center for Biotechnology Information
- ORFs
Open reading frames
- PLs
Polysaccharide lyases
- TUGs
Truly unique genes
Authors’ contributions
GAL designed the study, performed analyses and wrote the manuscript. GAL, CM and LM performed bioinformatic analyses. SD, MMa, and CF performed genome sequencing of the bacterial strains. MMo, PM and KJ provided bacterial strains. DVS and FT participated and supervised the study. MV conceived the study, participated in its design and coordination, and contributed to the manuscript preparation. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3955-4) contains supplementary material, which is available to authorized users.
Contributor Information
Gabriele Andrea Lugli, Email: gabrieleandrea.lugli@unipr.it.
Christian Milani, Email: christian.milani@unipr.it.
Francesca Turroni, Email: francesca.turroni@unipr.it.
Sabrina Duranti, Email: sabrina.duranti@unipr.it.
Leonardo Mancabelli, Email: leonardo.mancabelli@genprobio.com.
Marta Mangifesta, Email: marta.mangifesta@genprobio.com.
Chiara Ferrario, Email: chiara.ferrario@unipr.it.
Monica Modesto, Email: monica.modesto@unibo.it.
Paola Mattarelli, Email: paola.mattarelli@unibo.it.
Killer Jiří, Email: Killer.Jiri@seznam.cz.
Douwe van Sinderen, Email: d.vansinderen@ucc.ie.
Marco Ventura, Phone: +39-521-905666, Email: marco.ventura@unipr.it.
References
- 1.Bergey DHGM, Whitman WB, Parte AC. The actinobacteria. Bergey’s manual of systematic bacteriology. 2012;5:171-224.
- 2.Milani C, Lugli GA, Duranti S, Turroni F, Bottacini F, Mangifesta M, Sanchez B, Viappiani A, Mancabelli L, Taminiau B, et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl Environ Microbiol. 2014;80:6290–6302. doi: 10.1128/AEM.02308-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang G, Gao B, Adeolu M, Khadka B, Gupta RS. Phylogenomic analyses and comparative studies on genomes of the Bifidobacteriales: identification of molecular signatures specific for the order Bifidobacteriales and its different subclades. Front Microbiol. 2016;7:978. doi: 10.3389/fmicb.2016.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strassert JF, Kohler T, Wienemann TH, Ikeda-Ohtsubo W, Faivre N, Franckenberg S, Plarre R, Radek R, Brune A. 'Candidatus Ancillula trichonymphae', a novel lineage of endosymbiotic Actinobacteria in termite gut flagellates of the genus Trichonympha. Environ Microbiol. 2012;14:3259–3270. doi: 10.1111/1462-2920.12012. [DOI] [PubMed] [Google Scholar]
- 5.Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E. The prokaryotes. New York: Springer; 2006.
- 6.Ventura M, van Sinderen D, Fitzgerald GF, Zink R. Insights into the taxonomy, genetics and physiology of bifidobacteria. Antonie Van Leeuwenhoek. 2004;86:205–223. doi: 10.1023/B:ANTO.0000047930.11029.ec. [DOI] [PubMed] [Google Scholar]
- 7.Turroni F, Ventura M, Butto LF, Duranti S, O'Toole PW, Motherway MO, van Sinderen D. Molecular dialogue between the human gut microbiota and the host: a lactobacillus and Bifidobacterium perspective. Cell Mol Life Sci. 2014;71:183–203. doi: 10.1007/s00018-013-1318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantzourani M, Fenlon M, Beighton D. Association between Bifidobacteriaceae and the clinical severity of root caries lesions. Oral Microbiol Immunol. 2009;24:32–37. doi: 10.1111/j.1399-302X.2008.00470.x. [DOI] [PubMed] [Google Scholar]
- 9.Catlin BW. Gardnerella vaginalis: characteristics, clinical considerations, and controversies. Clin Microbiol Rev. 1992;5:213–237. doi: 10.1128/CMR.5.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bottacini F, Milani C, Turroni F, Sanchez B, Foroni E, Duranti S, Serafini F, Viappiani A, Strati F, Ferrarini A, et al. Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect gut. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milani C, Duranti S, Lugli GA, Bottacini F, Strati F, Arioli S, Foroni E, Turroni F, van Sinderen D, Ventura M. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl Environ Microbiol. 2013;79:4304–4315. doi: 10.1128/AEM.00984-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huys G, Vancanneyt M, D'Haene K, Falsen E, Wauters G, Vandamme P. Alloscardovia omnicolens gen. Nov., sp. nov., from human clinical samples. Int J Syst Evol Microbiol. 2007;57:1442–1446. doi: 10.1099/ijs.0.64812-0. [DOI] [PubMed] [Google Scholar]
- 13.Okamoto M, Benno Y, Leung KP, Maeda N. Metascardovia criceti gen. Nov., Sp. Nov., from hamster dental plaque. Microbiol Immunol. 2007;51:747–754. doi: 10.1111/j.1348-0421.2007.tb03964.x. [DOI] [PubMed] [Google Scholar]
- 14.Simpson PJ, Ross RP, Fitzgerald GF, Stanton C. Bifidobacterium psychraerophilum sp. nov. and Aeriscardovia aeriphila gen. Nov., sp. nov., isolated from a porcine caecum. Int J Syst Evol Microbiol. 2004;54:401–406. doi: 10.1099/ijs.0.02667-0. [DOI] [PubMed] [Google Scholar]
- 15.Leopold S. Heretofore undescribed organism isolated from the genitourinary system. U S Armed Forces Med J. 1953;4:263–266. [PubMed] [Google Scholar]
- 16.Piot P, van Dyck E, Goodfellow M, Falkow S. A taxonomic study of Gardnerella vaginalis (Haemophilus vaginalis) Gardner and Dukes 1955. J Gen Microbiol. 1980;119:373–396. doi: 10.1099/00221287-119-2-373. [DOI] [PubMed] [Google Scholar]
- 17.Piot P, Van Dyck E, Peeters M, Hale J, Totten PA, Holmes KK. Biotypes of Gardnerella vaginalis. J Clin Microbiol. 1984;20:677–679. doi: 10.1128/jcm.20.4.677-679.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeoman CJ, Yildirim S, Thomas SM, Durkin AS, Torralba M, Sutton G, Buhay CJ, Ding Y, Dugan-Rocha SP, Muzny DM, et al. Comparative genomics of Gardnerella vaginalis strains reveals substantial differences in metabolic and virulence potential. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsum U, Hallen A, Larsson PG. Bacterial vaginosis--a laboratory and clinical diagnostics enigma. APMIS. 2005;113:153–161. doi: 10.1111/j.1600-0463.2005.apm1130301.x. [DOI] [PubMed] [Google Scholar]
- 20.Jian W, Dong X. Transfer of Bifidobacterium inopinatum and Bifidobacterium denticolens to Scardovia inopinata gen. Nov., comb. nov., and Parascardovia denticolens gen. Nov., comb. nov., respectively. Int J Syst Evol Microbiol. 2002;52:809–812. doi: 10.1099/00207713-52-3-809. [DOI] [PubMed] [Google Scholar]
- 21.Modesto M, Biavati B, Mattarelli P. Occurrence of the family bifidobacteriaceae in human dental caries and plaque. Caries Res. 2006;40:271–276. doi: 10.1159/000092237. [DOI] [PubMed] [Google Scholar]
- 22.Beighton D, Gilbert SC, Clark D, Mantzourani M, Al-Haboubi M, Ali F, Ransome E, Hodson N, Fenlon M, Zoitopoulos L, Gallagher J. Isolation and identification of bifidobacteriaceae from human saliva. Appl Environ Microbiol. 2008;74:6457–6460. doi: 10.1128/AEM.00895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ventura M, Turroni F, Zomer A, Foroni E, Giubellini V, Bottacini F, Canchaya C, Claesson MJ, He F, Mantzourani M, et al. The Bifidobacterium dentium Bd1 genome sequence reflects its genetic adaptation to the human oral cavity. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lugli GA, Milani C, Turroni F, Duranti S, Ferrario C, Viappiani A, Mancabelli L, Mangifesta M, Taminiau B, Delcenserie V, et al. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl Environ Microbiol. 2014;80:6383–6394. doi: 10.1128/AEM.02004-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duranti S, Milani C, Lugli GA, Turroni F, Mancabelli L, Sanchez B, Ferrario C, Viappiani A, Mangifesta M, Mancino W, et al. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ Microbiol. 2015;17:2515–2531. doi: 10.1111/1462-2920.12743. [DOI] [PubMed] [Google Scholar]
- 26.Duranti S, Milani C, Lugli GA, Mancabelli L, Turroni F, Ferrario C, Mangifesta M, Viappiani A, Sanchez B, Margolles A, et al. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci Rep. 2016;6:23971. doi: 10.1038/srep23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bottacini F, O'Connell Motherway M, Kuczynski J, O'Connell KJ, Serafini F, Duranti S, Milani C, Turroni F, Lugli GA, Zomer A, et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genomics. 2014;15:170. doi: 10.1186/1471-2164-15-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Callaghan A, Bottacini F, O'Connell Motherway M, van Sinderen D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC Genomics. 2015;16:832. doi: 10.1186/s12864-015-1968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ventura M, Reniero R, Zink R. Specific identification and targeted characterization of Bifidobacterium lactis from different environmental isolates by a combined multiplex-PCR approach. Appl Environ Microbiol. 2001;67:2760–2765. doi: 10.1128/AEM.67.6.2760-2765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.JFSaDW R. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; 2001.
- 31.Lugli GA, Milani C, Mancabelli L, van Sinderen D, Ventura M. MEGAnnotator: a user-friendly pipeline for microbial genomes assembly and annotation. FEMS Microbiol Lett. 2016;363 [DOI] [PubMed]
- 32.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, Tang H, Ye Y. RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics. 2012;28:125–126. doi: 10.1093/bioinformatics/btr595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. PGAP: pan-genomes analysis pipeline. Bioinformatics. 2012;28:416–418. doi: 10.1093/bioinformatics/btr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 43.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2014;42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Csuros M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- 48.Lugli GA, Duranti S, Milani C, Turroni F, Viappiani A, Mangifesta M, van Sinderen D, Ventura M. The genome sequence of Bifidobacterium moukalabense DSM 27321 highlights the close phylogenetic relatedness with the Bifidobacterium dentium taxon. Genome Announc. 2014;2. [DOI] [PMC free article] [PubMed]
- 49.Praet J, Meeus I, Cnockaert M, Aerts M, Smagghe G, Vandamme P. Bifidobacterium commune sp. nov. isolated from the bumble bee gut. Antonie Van Leeuwenhoek. 2015;107:1307–1313. doi: 10.1007/s10482-015-0425-3. [DOI] [PubMed] [Google Scholar]
- 50.Toh H, Yamazaki Y, Tashiro K, Kawarai S, Oshima K, Nakano A, Kim CN, Mimura I, Arakawa K, Iriki A, et al. Draft genome sequence of Bifidobacterium aesculapii DSM 26737T, isolated from feces of baby common marmoset. Genome Announc. 2015;3. [DOI] [PMC free article] [PubMed]
- 51.Garcia-Aljaro C, Balleste E, Rossello-Mora R, Cifuentes A, Richter M, Blanch AR. Neoscardovia arbecensis gen. Nov., sp. nov., isolated from porcine slurries. Syst Appl Microbiol. 2012;35:374–379. doi: 10.1016/j.syapm.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 52.Sun Y, Tamarit D, Andersson SG. Switches in Genomic GC Content Drive Shifts of Optimal Codons under Sustained Selection on Synonymous Sites. Genome Biol Evol. 2016; [DOI] [PMC free article] [PubMed]
- 53.Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: Hierarchical and refined dataset for big data analysis--10 years on. Nucleic Acids Res. 2016;44:D694–D697. doi: 10.1093/nar/gkv1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milani C, Lugli GA, Duranti S, Turroni F, Mancabelli L, Ferrario C, Mangifesta M, Hevia A, Viappiani A, Scholz M, et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci Rep. 2015;5:15782. doi: 10.1038/srep15782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chun J, Rainey FA. Integrating genomics into the taxonomy and systematics of the bacteria and archaea. Int J Syst Evol Microbiol. 2014;64:316–324. doi: 10.1099/ijs.0.054171-0. [DOI] [PubMed] [Google Scholar]
- 56.Ventura M, Canchaya C, Del Casale A, Dellaglio F, Neviani E, Fitzgerald GF, van Sinderen D. Analysis of bifidobacterial evolution using a multilocus approach. Int J Syst Evol Microbiol. 2006;56:2783–2792. doi: 10.1099/ijs.0.64233-0. [DOI] [PubMed] [Google Scholar]
- 57.Modesto M, Michelini S, Stefanini I, Ferrara A, Tacconi S, Biavati B, Mattarelli P. Bifidobacterium aesculapii sp. nov., from the faeces of the baby common marmoset (Callithrix Jacchus) Int J Syst Evol Microbiol. 2014;64:2819–2827. doi: 10.1099/ijs.0.056937-0. [DOI] [PubMed] [Google Scholar]
- 58.Scardovi V. LDT: The fructose-6-phosphate shunt as a peculiar pattern of hexos degradation in the genus Bifidobacterium. Annali di Microbiologia ed Enzimologia. 1965;15:19–23. [Google Scholar]
- 59.Albalat R, Canestro C. Evolution by gene loss. Nat Rev Genet. 2016;17:379–391. doi: 10.1038/nrg.2016.39. [DOI] [PubMed] [Google Scholar]
- 60.Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/S0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- 61.Turroni F, Bottacini F, Foroni E, Mulder I, Kim JH, Zomer A, Sanchez B, Bidossi A, Ferrarini A, Giubellini V, et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci U S A. 2010;107:19514–19519. doi: 10.1073/pnas.1011100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turroni F, Milani C, van Sinderen D, Ventura M. Genetic strategies for mucin metabolism in Bifidobacterium bifidum PRL2010: an example of possible human-microbe co-evolution. Gut Microbes. 2011;2:183–189. doi: 10.4161/gmic.2.3.16105. [DOI] [PubMed] [Google Scholar]
- 63.Pokusaeva K, Fitzgerald GF, van Sinderen D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011;6:285–306. doi: 10.1007/s12263-010-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1, S2, S3, S4, S5 and S6. (XLSX 3628 kb)
Phylogenetic trees of the Bifidobacteriaceae family. Panel a display the 16S rRNA gene-based tree of the current recognized (sub) species of the family. Panel b shows the phylogenetic tree based on the concatenation of the amino acid sequences of five housekeeping genes including hsp60, rpoB, dnaJ, dnaG and clpC. For each tree, bootstrap values higher than 70 are marked near the respective nodes. (TIFF 2260 kb)
Data Availability Statement
Genome sequences reported in this article have been deposited in GenBank under the following accession numbers: MWWQ00000000, MWWR00000000, MWWS00000000, MWWT00000000, MWWU00000000, MWWV00000000, MWWW00000000, MWWX00000000, MWWY00000000, MWWZ00000000 and MWXA00000000. The version described in this paper is version MWWQ01000000, MWWR01000000, MWWS01000000, MWWT01000000, MWWU01000000, MWWV01000000, MWWW01000000, MWWX01000000, MWWY01000000, MWWZ01000000 and MWXA01000000.