

Summary
Clearance of bacteria by macrophages involves internalization of the microorganisms into phagosomes, which are then delivered to endolysosomes for enzymatic degradation. These spatiotemporally segregated processes are not known to be functionally coupled. Here, we show that lysosomal degradation of bacteria sustains phagocytic uptake. In Drosophila and mammalian macrophages, lysosomal dysfunction due to loss of the endolysosomal Cl− transporter, ClC-b/CLCN7, delayed degradation of internalized bacteria. Unexpectedly, defective lysosomal degradation of bacteria also attenuated further phagocytosis, resulting in elevated bacterial load. Exogenous application of bacterial peptidoglycans restored phagocytic uptake in the lysosomal degradation-defective mutants via a pathway requiring cytosolic pattern recognition receptors and NF-κB. Mammalian macrophages that are unable to degrade internalized bacteria also exhibit compromised NF-κB activation. Our findings reveal a role for phagolysosomal degradation in activating an evolutionarily conserved signaling cascade, which ensures that continuous uptake of bacteria is preceded by lysosomal degradation of microbes.
Keywords: ClC-b, CLCN7, cytosolic pattern recognition receptors, lysosomal degradation, Relish, TRPML, vesicular trafficking
Graphical Abstract
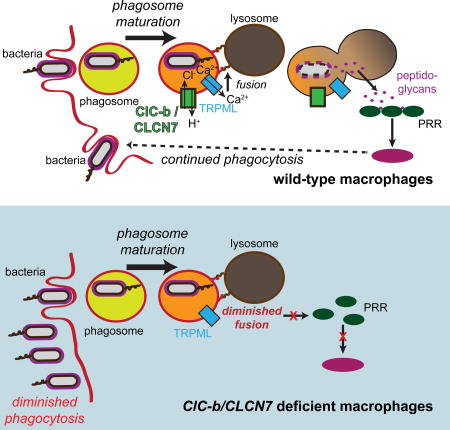
Macrophages clear bacteria by phagosomal internalization and subsequent lysosomal degradation of the microbes. Wong et al. find that defective lysosomal degradation attenuates further phagocytosis resulting in elevated bacterial load. Cytosolic pattern recognition receptors and NF-κB signal to couple phagolysosomal degradation to sustained phagocytic bacterial clearance.
Introduction
Macrophages are sentinel cells that require a series of vesicular trafficking events to orchestrate the elimination of bacteria (Fairn and Grinstein, 2012). Extracellular bacteria are internalized into phagosomes, which are delivered to endolysosomes resulting in enzymatic degradation of the cargo (Fairn and Grinstein, 2012; Haas, 2007). Effective clearance of bacteria relies on both initial phagocytosis and the subsequent lysosomal degradation (Fairn and Grinstein, 2012; Haas, 2007). Although it is obvious that lysosomal degradation of bacteria will proceed only after the internalized microorganisms are delivered to the lysosomes, it is not known whether the ongoing phagocytic uptake of bacteria at the cell surface requires the complete degradation of the previously internalized bacteria. Given that certain strains of bacteria are able to escape from phagosomes and endosomes (Flannagan et al., 2009), it stands to reason that macrophages minimize the time bacteria spend within endolysosomes prior to degradation. Coupling phagocytosis to lysosomal degradation would ensure that internalization of bacteria matches the ability of the lysosomes to handle the additional load. However, no mechanism linking phagocytosis to lysosomal degradation has ever been described.
The degradative capacity of endolysosomes is achieved by the presence of degradative enzymes and a luminal environment that is conducive for these enzymes to degrade the internalized cargo. The features of the endolysosomal lumen that encourage the activity of these enzymes are a low pH and the presence of several other important cations such as Ca2+ (Xu and Ren, 2015). To maintain electro-neutrality, the endolysosomal lumen takes up Cl−, which is the main anion in these vesicles (Stauber and Jentsch, 2013; Xu and Ren, 2015). Several members of the chloride channel (CLC) gene family, CLCN-3, -4, -5, -6, and -7, encode Cl−/H+ antiporters that localize to intracellular membranes and regulate vesicular Cl− concentration ([Cl−]) (Stauber and Jentsch, 2013). A conserved gating glutamate residue in these vesicular CLC transporters couples the transport of 2 Cl− ions to the exchange of 1 H+ ion in opposite directions (Stauber and Jentsch, 2013). Consistent with a critical role of the CLCs in regulating endolysosomal function, loss of function mutations in CLCN7, the gene encoding a late-endosomal Cl−/H+ antiporter, results in endolysosomal dysfunction in multiple tissues in affected humans (Weinert et al., 2010). However, the molecular mechanism of luminal [Cl−] in endolysosomal function remains unknown.
To elucidate the biological roles of endolysosomal Cl−, we sought to examine the functions of the sole Drosophila late-endosomal/lysosomal Cl− transporter, ClC-b, which is required for maintaining [Cl−] within these organelles (Saha et al., 2015). Our studies revealed that ClC-b is expressed in circulating macrophages, and is necessary for maintaining endolysosomal [Ca2+] in those cells. Given the critical importance of endolysosomal Ca2+ in the delivery of endocytosed and phagocytosed cargo to lysosomes (Lloyd-Evans and Platt, 2011), degradation of phagocytosed bacteria is diminished within macrophages lacking ClC-b. Remarkably, attenuated degradation of internalized bacteria prevented the further phagocytic uptake of bacteria by the mutant macrophages. These data show that endolysosomal degradation of bacteria is required to maintain the continued phagocytic uptake of bacteria. Furthermore, we demonstrate that sensing of bacterial peptidoglycans generated from lysosomal degradation by cytosolic pattern-recognition receptors, and downstream NF-κB activation are necessary for sustaining phagocytosis. Importantly, we show that our observations are evolutionarily conserved since mammalian macrophages that are unable to degrade internalized bacteria also exhibit compromised NF-κB activation. In summary, we define and elucidate the mechanistic basis of an evolutionarily conserved signaling cascade involving phagolysosomal degradation, which ensures that continuous uptake of bacteria is preceded by the lysosomal degradation of the microbes.
Results
Drosophila ClC-b is a late-endosomal/lysosomal protein enriched in immune cells
ClC-b is the homolog of mammalian late-endosomal/lysosomal Cl− transporter, CLCN7 (Figure 1A) (Cabrero et al., 2014; Saha et al., 2015). Since the tissue distribution of ClC-b was unknown, we used CRISPR/Cas9-mediated gene editing to generate an in-frame GFP knock-in line (GFP::ClC-b) (Figure 1B), which expresses ClC-b tagged with GFP at the N-terminus. We found that GFP::ClC-b was expressed in many tissues including the nervous system, Malpighian tubules (the Drosophila equivalent of a kidney), the gut, skeletal muscle, and pericardial cells (components of the Drosophila heart) (Figure 1C). We observed a particularly strong enrichment of GFP::ClC-b in lymph glands, macrophages (also called plasmatocytes), and fat bodies (Figure 1D). In both fat bodies and macrophages, we found that GFP::ClC-b was localized to late-endosomes/lysosomes, as evidenced by colocalization with LysoTracker and YFP::Rab7 (Figures 1E and 1G). Together, these data show that Drosophila ClC-b is a late-endosomal/lysosomal membrane protein in several tissues including cells of the immune system.
Figure 1. Ubiquitous expression of ClC-b and its localization to endolysosomes in macrophages.

(A) Phylogeny of human and Drosophila chloride transporters proteins. Numbers shown are bootstrap test values (%). ClC-a, -b, and -c are annotated Drosophila chloride channel family subunits according to Flybase (http://flybase.org). Highlighted in red are the genes of interest of the current study. (B) The ClC-b genomic locus and alleles used in the study. (C) GFP::ClC-b expression in larval tissues. Larval tissues were fixed and stained with anti-GFP (green), phalloidin for actin (magenta), and DAPI for nuclear DNA (blue). Scale bars represent 20 µm. (D) GFP::ClC-b expression in larval lymph glands and fat bodies. GFP single was not amplified by using primary or secondary antibodies. Scale bar represents 50 µm. (E and F) LysoTracker staining in fat body (E) and macrophages (F) isolated from GFP::ClC-b larvae. Scale bars represent 10 µm. (G) Macrophages isolated from larvae expressing ClC-b::myc and YFP::Rab7 with Cg-GAL4 driver. Scale bar represents 10 µm.
ClC-b is required in macrophages for the clearance of bacteria
The enrichment and subcellular localization of ClC-b to the degradative organelles in macrophages led us to ask whether the transporter could affect the well-known function of macrophages in the clearance of injected bacteria (Buchon et al., 2014). Indeed, following isolation of macrophages and incubation with heat-killed fluorescent E. coli, we found that the bacteria internalized into macrophage were localized to GFP::ClC-b positive vesicles (Figure 2A). These data indicate that in macrophages, ClC-b localizes to phagolysosomes, which also contain internalized bacteria. To directly examine the functional role of ClC-b in macrophage-mediated clearance of bacteria, we generated two ClC-b null alleles, ClC-b1 and ClC-b2 (Figure 1B), and examined the ability of these animals to clear a systemic bacterial infection (experimental scheme shown in Figure S1A) (Pham et al., 2007). ClC-b deficient animals did not exhibit a significant alteration in survival after injection of the non-pathogenic bacteria, E. coli (data not shown). However, we found that ClC-b1 mutants were unable to clear the injected E. coli. This defect led to elevated number of colonies formed (colony forming units, CFUs) beginning at 3 hours after E. coli injection, and lasting for several days when compared to ClC-b1 flies carrying a genomic wild-type ClC-b transgene (gen-res) (Figures 2B, 2C and S1B). The elevated CFU in ClC-b1 was observed across a 100-fold range of injected E. coli concentration (Figure S1C). We also observed diminished clearance of E. coli in flies carrying the ClC-b1 allele in trans with a deficiency uncovering the ClC-b locus, and in ClC-b1/ClC-b2 transheterozygous flies (Figure 2C). These data suggest that the Drosophila late-endosomal/lysosomal Cl− transporter mediates the clearance of bacteria.
Figure 2. ClC-b mutants show defective clearance of septic bacteria.

(A) Confocal image of fluorescent E. coli containing macrophages isolated from GFP::ClC-b larvae. Scale bar represents 10 µm. (B) CFU count from flies of the indicated genotypes at the indicated time points after E. coli injection. The dashed line reflects the amount of E. coli injected into flies. (C) CFU counts from flies of the indicated genotypes 1 day after E. coli injection. Values shown are normalized to the genomic rescue (gen-res) (ClC-b1;P[ClC-b+]) means. (D) CFU counts from ClC-b1 flies expressing ClC-b in macrophages (He>ClC-b). Values shown are normalized to genomic rescue (gen-res) means. (E) CFU counts from wild-type flies expressing RNAi in macrophages (He>ClC-b-IR). Values normalized to control (UAS-ClC-b-IR) means. (F) CFU counts from wild-type flies overexpressing ClC-b in macrophages. Values normalized to control (UAS-ClC-b) means. All values shown represent mean ±SEM. See also Figures S1 and S2.
Next, we assessed the cellular requirement of ClC-b in the clearance of bacteria. Expression of UAS-ClC-b in ClC-b1 macrophages was sufficient to fully rescue the defects in E. coli clearance (Figure 2D). Furthermore, macrophage-specific RNAi-mediated knockdown of ClC-b, but not of the early-endosomal Cl−transporter, ClC-c (Cabrero et al., 2014; Saha et al., 2015) (Figure S1D), resulted in diminished clearance of injected E. coli (Figures 2E, S1E and S1F). Interestingly, we found that overexpression of UAS-ClC-b in wild-type macrophages led to even lower CFUs compared to control animals when the flies were challenged with 4x bacterial loads (Figure 2F). Together, these data indicate that expression of ClC-b is necessary in macrophages for clearance of septic bacterial infection, and increased expression of ClC-b is sufficient to elevate the ability of macrophages to clear bacteria.
Diminished clearance of bacteria in ClC-b1 flies is not due to developmental defects or diminished antimicrobial response
The blood cells in Drosophila circulation consist mainly of macrophages (also called plasmatocytes), and <5% of crystal cell (Meister and Lagueux, 2003), which are responsible for mediating melanization during injuries or infections. We did not observe any alterations in melanization in ClC-b1 upon wounding or bacterial infection, indicating that the role of crystal cells in melanization remains unaltered in the mutants. We also examined whether the defective clearance of bacteria in ClC-b1 animals arises from alterations in macrophage development or survival. We found that heat shock-mediated ClC-b knock-down in adult flies recapitulated the ClC-b1 phenotype (Figure S2A), demonstrating post-developmental requirement of CIC-b in the clearance of bacterial infection. ClC-b1 did not exhibit changes in levels of a marker expressed in larval and adult macrophages, number of circulating larval hemocytes, or hemocyte size (Figure S2B and S2C). Furthermore, expression of an apoptosis inhibitor in ClC-b1 macrophages did not rescue the phenotype (Figure S2D). Together, these data indicate that the impaired clearance of bacteria in ClC-b1 is not a consequence of diminished macrophage number or survival.
The Drosophila immune system also consists of a humoral component that is mediated by fat-body-derived anti-microbial peptides (AMPs) (Buchon et al., 2014). To evaluate whether the ClC-b1 animals exhibit compromised humoral immune response, we compared the expression levels of AMP genes after infection. We found that AMP gene expression is not diminished in the ClC-b1 mutants (Figure S2E), indicating that the immune phenotype in ClC-b mutants is likely not due to a defective humoral response.
Defects in Cl− and Ca2+ transport underlie the ClC-b1 phenotype
The mammalian homolog of ClC-b, CLCN7, is a late endosomal/lysosomal Cl−/H+ transporter (Leisle et al., 2011), and the amino acid residues responsible for the transporter activity in human CLCN7 are conserved in ClC-b (Figure S3A) (Weinert et al., 2014; Weinert et al., 2010). Macrophage-specific expression of a ClC-b variant, ClC-btd, analogous to the human transporter-dead variant (Weinert et al., 2014) failed to rescue the E. coli clearance defects observed in ClC-b1 despite its normal localization to E. Coli-containing vesicles (Figure S3B and S3C). Another CLCN7 variant, CLCN7unc, is a purely Cl− conducting transporter (Weinert et al., 2010). To examine the relative contributions of Cl− and H+ in the ClC-b null phenotypes, we expressed ClC-bunc, which is analogous to CLCN7unc, in the ClC-b1 macrophages and observed a robust, albeit not complete, suppression of the E. coli clearance phenotype (Figure S3A and 3A). Thus, loss of Cl− transport across the endolysosomal membrane may contribute to the phenotype observed in ClC-b deficient Drosophila macrophages.
Endolysosomal Ca2+ promotes heterotypic fusion of late-endosomes and lysosomes (Luzio et al., 2007a). Since Cl− is the major counter-ion for endolysosomal cations such as Ca2+, we hypothesized that in the absence of Cl− transport by ClC-b, late-endosomes cannot effectively accumulate luminal Ca2+, thus leading to diminished Ca2+ release and a defect in the fusion of phagosomes/endosomes with lysosomes. To measure [Ca2+] release from endolysosomes, we expressed the Ca2+ sensor, GCaMP5G, fused with the endolysosomal cation channel TRPML (Venkatachalam et al., 2015) in macrophages (Figure 3B). TRPML, which is the Drosophila ortholog of mucolipin subfamily of TRP channels (Venkatachalam et al., 2008), is a non-selective endolysosomal cation channel that releases luminal cations such as Na+, Ca2+, and Fe2+ (Feng et al., 2014a). The Ca2+ released by TRPML is required for the Ca2+- and SNARE-dependent heterotypic fusion of late-endosomes and lysosomes (Wong et al., 2012). Consequently, late-endosomal contents are not degraded in Drosophila cells lacking TRPML (Wong et al., 2012). As expected, TRPML::GCaMP5G colocalized with ClC-b::Myc on endolysosomal membranes (Figure 3C). We then evaluated the relative levels of Ca2+ mobilized from endolysosomes upon application of either the TRPML agonist MLSA1 (Feng et al., 2014b) or the lysosomolytic agent, glycyl-L-phenylalanine-2-naphthylamide (GPN) (Berg et al., 1994). Application of either MLSA1 or GPN to isolated wild-type macrophages expressing TRPML::GCaMP5G revealed robust Ca2+ elevations (Figures 3D and 3E). In contrast, Ca2+ elevation with either MLSA1 or GPN was significantly smaller in ClC-b1 macrophages (Figures 3D and 3E). Therefore, endolysosomal [Ca2+] release is diminished in the absence of ClC-b.
Figure 3. Chloride transporter ClC-b regulates endolysosome luminal Ca2+ levels.

(A) Schematic description of the “uncoupled” mutation and CFU counts from ClC-b1 flies expressing ClC-bunc in macrophages. Values shown are normalized to genomic rescue (gen-res) means. (B) Schematic description of the experiment using TRPML::GCaMP5G to assess endolysosomal Ca2+. (C) Confocal image showing colocalization of the expressed TRPML::GCaMP5G with ClC-b::myc in an isolated larval macrophage. Scale bar represents 5 µm. (D) Representative traces showing changes in TRPML::GCaMP5G fluorescence in larval macrophages isolated from wild-type (blue) and ClC-b1 (red) larvae. Each trace represents mean values from ≥10 macrophages measured in a single experiment. Concentrations of MLSA1 and GPN were 40 µM and 200 µM respectively. (E) Quantification of the endolysosomal Ca2+ measurement experiments. (F) CFU counts from flies of the indicated genotypes. trpml1 flies expressed the trpml+transgene only in neurons to circumvent the pupal lethality observed in trpml-deficient flies. Values shown are normalized to trpml1/+ means. (G) CFU counts from flies overexpressing ClC-b in wild-type trpml1/+ genetic backgrounds. Values shown are normalized to UAS-ClC-b;trpml1/+ means. (H) CFU counts from ClC-b1 flies with or without overexpression of trpml in macrophages. Values are normalized to ClC-b1;UAS-trpml means. All values shown represent mean ±SEM. See also Figure S3.
We found that flies lacking trpml in macrophages exhibited compromised clearance of E. coli similar to the phenotype observed in ClC-b mutants (Figure 3F). Moreover, the enhanced clearance of E. coli achieved by ClC-b overexpression was abrogated in trpml1/+ heterozygotes (Figure 3G), indicating that TRPML functions downstream of ClC-b. In concordance with this notion, overexpression of trpml in ClC-b1 macrophages partially rescued the E. coli clearance phenotype (Figure 3H). The limited suppression following trpml overexpression suggests that enhancing lysosomal Ca2+ release cannot fully restore vesicular fusion defects caused by decreased luminal [Ca2+]. Together, these data indicate that ClC-b-mediated Cl− transport into endolysosomes is necessary for the accumulation of luminal Ca2+, which when released through TRPML drives the delivery of phagocytic cargo to lysosomes for degradation.
Degradation of internalized E. coli is delayed in ClC-b1 macrophages
If the delivery of phagocytic cargo to lysosomes is delayed, degradation of bacteria could be diminished in ClC-b1 macrophages. To evaluate the ability of ClC-b1 macrophages to degrade internalized bacteria, we isolated circulating naïve macrophages (not previously exposed to E. coli) from larvae and subjected these cells to fluorescently-labeled heat-killed E. coli. Neither the percentage of macrophages engulfing E. coli nor the number of E. coli particles internalized by naïve macrophages were significantly different in ClC-b1 macrophages (Figures 4A and 4B). Next, we monitored the clearance of internalized fluorescent E. coli over time. In control macrophages, the E. coli fluorescence decreased by ~60 % and ~75 % following chases of 4-hours and 2-days respectively (Figures 4C and 4D). However, clearance of fluorescent E. coli was significantly delayed in ClC-b1 macrophages (fluorescence decreased by <10% and ~20% following chases of 4-hours and 2-days respectively) (Figures 4C and 4D). A similar delay in degradation was also observed in macrophages isolated from ClC-b1 adult flies after injecting fluorescent E. coli (Figure S4A and S4B). Together, these data indicate that ClC-b-deficient macrophages are characterized by diminished lysosomal degradation of the phagocytic cargo.
Figure 4. Impaired degradation of bacteria in ClC-b mutant macrophages prevents persistent clearance of bacteria.

(A) Confocal images showing isolated larval macrophages (HmlD>EGFP; green) at indicated time points after encountering fluorescent E. coli (magenta) during the “short time-course ex vivo phagocytosis assay”. Scale bar represents 5 µm. (B) Quantification of fluorescent E. coli uptake in the “short time-course ex vivo phagocytosis assay”. Data shown are from 3 independent replicates. (C) Z-stack projections of confocal images showing isolated larval macrophages with internalized fluorescent E. coli (green) at indicated time points in the “ex vivo degradation assay”. Nucleus was stained by DAPI (blue) and F-actin was stained by phalloidin (magenta). Scale bar represents 5 µm. (D) Quantification of the “ex vivo degradation assay” shown in (C). All values shown are normalized to genomic rescue (gen-res) means at time 0. (E and F) Schematic representation and quantification of the “ex vivo sequential phagocytosis assay” performed on isolated larval macrophages. (G and H) Schematic representation and quantification of the “in vivo sequential E. coli injection assay”. Values in (H) were normalized to the mean values of the genomic rescue (gen-res) flies with the relevant pre-injection. All values shown represent mean ±SEM. See also Figure S4.
Although the initial uptake of bacteria into naïve macrophages does not require ClC-b (Figures 4A and 4B), we asked whether impaired lysosomal degradation of phagocytosed bacteria could disrupt the uptake and continued clearance of bacteria during protracted infections. To address this question, we performed an ex vivo sequential phagocytosis assay (schematic shown in Figure 4E) on isolated naïve macrophages from larvae. We pulsed these cells with Texas Red labeled (red) E. coli and, after 4 hours of chase to allow lysosomal degradation, we pulsed them with Alexa Fluor 488 labeled (green) E. coli. We then assessed the number of green E. coli particles adhered to or internalized into the macrophages. Whereas wild-type and ClC-b1 macrophages showed no difference in the initial uptake of red E. coli, ClC-b1 macrophages showed significantly diminished uptake of green E. coli (Figure 4F). Furthermore, the phenotype of diminished uptake of E. coli during the 2nd bacterial challenge was also observed in ClC-b1 hemocytes co-cultured with GFP-expressing wild-type hemocytes (Figure S4C and S4E), which argues against a potential role for diminished paracrine signaling between macrophages in the described ClC-b1 phenotype. Together, these results indicate that the phagocytic capacity of ClC-b-deficient macrophages declines upon multiple bacterial encounters.
Since ClC-b1 macrophages that previously internalized bacteria exhibit a decline in phagocytic uptake, we asked whether clearance of systemic bacterial infection in ClC-b1 flies deteriorates when the macrophages accumulate undegraded bacteria. To address this question, we first injected flies with either PBS (controls) or heat-killed E. coli to allow phagocytic uptake and delivery to lysosomes within the macrophages (schematic shown in Figure 4G). Two days later, we injected both cohorts with live E. coli and performed the CFU assay to assess the clearance of the live E. coli. We found that pre-injection with dead E. coli enhanced the clearance of the subsequent live E. coli in wild-type flies (Figure S4F). When flies were pre-injected with PBS, the CFU count in ClC-b1 flies was ~7-fold higher than control flies (Figure 4H). However, if flies were pre-injected with dead E. coli, the difference in CFU counts between ClC-b1 and control flies was ~20-fold (Figure 4H). These data show that lysosomal degradation of internalized bacteria in macrophages provides a feedforward signal to sustain continued phagocytic clearance of bacteria and prevent a decline in phagocytic capacity.
ClC-b-dependent lysosomal degradation is required for the activation of cytosolic pattern recognition receptors and NF-κB
Cytosolic pattern recognition receptors detect pathogen-associated molecular patterns (PAMPs) that enter the cytosol, and elicit an immune response (Takeuchi and Akira, 2010). The Drosophila genome encodes a cytosolic pattern recognition receptor, PGRP-LE, which oligomerizes upon binding diaminopimelic acid (DAP)-type peptidoglycans derived from gram-negative bacteria such as E. coli, and induces NF-κB (Relish) activation (Takehana et al., 2002; Takeuchi and Akira, 2010). Although known to regulate the Drosophila humoral immune response, it is not known whether PGRP-LE-Relish signaling also regulates the cellular response mediated by macrophages. Compared to the heterozygotes, the PGRP-LE null flies exhibited diminished clearance of injected E. coli, which was rescued by expression of UAS-PGRP-LE in macrophages (Figures 5A and 5B). We found that PGRP-LE knockdown in macrophages also resulted in elevated CFU counts (Figure 5C). Similarly, knockdown of Relish in macrophages also caused diminished clearance of bacteria (Figure 5D). To examine whether Relish is required for sustaining phagocytosis after internalization of bacteria, we performed sequential phagocytosis assay on primary isolated larval macrophages from Relish null mutants (RelE20). Similar to our findings with the ClC-b1 macrophages, whereas we observed no difference in the initial uptake of red E. coli, the uptake of green E. coli during the 2nd bacterial challenge was significantly diminished in RelE20 macrophages (Figures 5E and 5F).
Figure 5. PGRP-LE and Relish signaling are necessary for macrophage mediated clearance of bacteria.

(A to D) CFU counts from flies of the indicated genotypes. (A) PGRP-LE null (PGRP-LE112) and control (PGRP-LE112/+) flies. (B) Macrophage overexpression of PGRP-LE (He>LE) in PGRP-LE null flies. (C and D) RNAi mediated knockdown of PGRP-LE (C) or Relish (D) in macrophages of wild-type flies. All CFU values (mean ±SEM) are normalized to means of the respective control genotype (blue bar). (E) Z-stack projections of confocal images of isolated larval macrophages with internalized/adhered fluorescent E. coli (red and green) from the “ex vivo sequential phagocytosis assay”. Shown are single larval macrophages isolated from RelE20 mutants and RelE20/+ control animals. Cells were stained with DAPI (blue). Scale bar represents 10 µm. (F) Quantification of data shown in (E). All values shown represent mean ±SEM.
Next, we investigated whether the PGRP-LE-Relish signaling axis operates downstream of ClC-b. We found that macrophage-specific overexpression of PGRP-LE, which potentiates Relish signaling (Takehana et al., 2002), was sufficient to enhance E. coli clearance in wild-type flies (Figure 6A). The enhanced clearance of bacteria is likely due to elevated phagocytosis since macrophages overexpressing PGRP-LE exhibited increased E. coli uptake (Figure S5A and S5B). Interestingly, macrophage-specific overexpression of PGRP-LE also increased expression of receptor genes associated with phagocytosis, including eater and NimC1 (Kocks et al., 2005; Kurucz et al., 2007), in both wild-type and ClC-b1 adult flies (Figure S5C and S5D). Importantly, overexpression of PGRP-LE in ClC-b1 macrophages restored E. coli clearance (Figure 6A). This rescue was completely abolished in the ClC-b1;RelE20 double mutants (Figure 6D) indicating that PGRP-LE couples phagolysosomal degradation to bacterial clearance via a process requiring Relish. We also performed sequential phagocytosis assay on ClC-b1 macrophages overexpressing PGRP-LE, and found that uptake of green E. coli during the 2nd challenge was significantly enhanced compared to control macrophages (Figures 6B and 6C). PGRP-LE activates the transcription factor Relish through the Imd signaling pathway (Takehana et al., 2002). Rel68 encodes the N-terminal fragment of Relish that is cleaved from the full-length protein and translocates to the nucleus to upregulate the transcription of some, albeit not all, Relish targets (Wiklund et al., 2009). Whereas macrophage-specific knockdown of Relish did not aggravate the ClC-b1 E. coli clearance phenotype (Figure S5E), we found that expression of Rel68 partially rescued the phenotype (Figure 6E). In contrast, the diminished clearance of E. coli following macrophage-specific knockdown of Relish was not rescued by overexpression of ClC-b (Figure S5F), indicating that Relish signaling functions downstream of ClC-b.
Figure 6. ClC-b facilitates lysosomal degradation of bacteria to activate PGRP-LE and Relish in macrophages.

(A) CFU counts from flies overexpressing PGRP-LE in macrophages (He>PGRP-LE). Values normalized to wild-type (WT) UAS-PGRP-LE. (B) Z-stack projections of confocal images of isolated larval macrophages with internalized/adhered fluorescent E. coli (red and green) from the “ex vivo sequential phagocytosis assay”. Shown are single larval ClC-b1 macrophages with (He>LE) or without (UAS-LE) PGRP-LE overexpression. (C) Quantification of data shown in (B). (D) CFU counts from ClC-b1 flies overexpressing (O.E.) PGRP-LE in macrophages. Values normalized to ClC-b1;RelE20 PGRP-LE O.E.. (E) CFU counts from ClC-b1 flies expressing N-terminal fragment of Relish (Rel68) in macrophages. Values are normalized to control (ClC-b1;UAS-Rel68/+). (F) CFU counts from flies pre-injected with PBS or TCT (16 µM) 1 day before E. coli injection. Values shown are normalized to PBS controls. All values shown represent mean ±SEM. (G) Model showing the ClC-b mediated endolysosome-Relish signaling axis in Drosophila macrophages. See also Figure S5.
Phagolysosomal degradation leads to release of bacterial peptidoglycans (Girardin et al., 2003; Nakamura et al., 2014), which can induce oligomerization and activation of cytosolic pattern recognition receptors such as PGRP-LE (Kaneko et al., 2006; Lim et al., 2006). Injecting the ClC-b1 flies with DAP-type peptidoglycan tracheal cytotoxin (TCT), a ligand for PGRP-LE (Lim et al., 2006), before E. coli injection led to a significant reduction in CFU counts (Figure 6F). However, muramyl dipeptide (MDP), a molecule that lacks the DAP chemical moiety, had no effect on the ClC-b1 phenotype (Figure S5G). The TCT-dependent rescue of the ClC-b1 phenotype was diminished in ClC-b1 mutants with macrophage-specific knockdown of Relish or the ClC-b1;RelE20 double mutants (Figure 6F). Collectively, these results demonstrate that ClC-b-mediated lysosomal degradation of bacteria activates the PGRP-LE-Relish pathway via bacterial peptidoglycans to promote phagocytic clearance of septic bacteria (Figure 6G).
Mammalian CLCN7 is required in macrophages for degradation of internalized bacteria and subsequent NF-κB activation
To determine whether the role of the endolysosomal Cl− transporters in macrophage is conserved across evolution, we examined the role of CLCN7 in mammalian macrophages. We first knocked down CLCN7 by stably expressing shRNAs in the RAW 264.7 macrophage cell line (Figure 7A). Subsequently, we exposed these macrophages to E. coli, and performed the gentamicin protection assay (schematic shown in Figure S6) to determine the number of viable E. coli inside the cells after 4 hours. Consistent with the diminished E. coli degradation in ClC-b1 hemocytes, mouse macrophages with CLCN7 knockdown showed a ~65% increase in viable internalized E. coli (Figure 7B). Thus, degradation of phagocytosed bacteria in mammalian macrophages requires endolysosomal Cl− transport mediated by CLCN7.
Figure 7. CLCN7 mediates the conserved endolysosome-NF-κB signaling axis in mammalian macrophages.

(A) Western blots performed of RAW 264.7 cell lysates probed with anti-CLCN7 and anti-α-tubulin antibodies. Cells were transduced with lentivirus carrying control shRNA (Ctrl ShR) or shRNAs against CLCN7 (CLCN7 ShR1/2). (B) Quantification of viable internalized E. coli extracted from RAW 264.7 cells in the gentamicin protection assay. (C to E) IL-1β and TNF-α mRNA levels in the shRNA expressing RAW 264.7 cell lines. Values are normalized to Ctrl ShR (naïve) (C and D) and to Ctrl ShR (+ E. coli) (E). (F) Effect of pharmacological inhibition of endosome maturation on IL-1β mRNA levels in the wild-type RAW 264.7 cell lines. Values are normalized to Control (naïve). Abbreviations: naïve, without E. coli uptake; + E. coli, with E. coli uptake; + MDP, 10 µg/mL MDP treatment after E. coli uptake; Apilimod, 200 nM apilimod treatment after E. coli uptake; Control, 0.1% DMSO vehicle control. All values shown represent mean ±SEM. (G) Model showing the endolysosome-NF-κB signaling axis in Drosophila and mammalian macrophages. See also Figure S6.
Next, we asked whether CLCN7-mediated lysosomal degradation of bacteria is required for NF-κB signaling. In mammalian macrophages, NF-κB regulates the expression of several cytokines including IL-1β and TNF-α (Lawrence, 2009). We measured the expression levels of IL-1β and TNF-α in control and CLCN7-knockdown RAW 264.7 cells. After uptake of heat-killed E. coli, upregulation of both IL-1β and TNF-α in the CLCN7 knockdown cells was significantly dampened (Figures 7C and 7D), indicating diminished NF-κB signaling. The expression of anti-inflammatory cytokine, IL-10, which is induced by ERK signaling (Saraiva and O’Garra, 2010), was not significantly altered by either E. coli challenge or CLCN7 knockdown (Figure S6B). Mammalian NF-κB signaling can be activated by the cytosolic pattern recognition receptor NOD2, which recognizes the bacterial cell wall-derived peptidoglycan muramyl dipeptide (MDP) in the cytosol (Girardin et al., 2003; Nakamura et al., 2014). Since we found that CLCN7 is required for lysosomal degradation of internalized bacteria, the dampened NF-κB signaling upon CLCN7 knockdown could be due to insufficient MDP released from endolysosomes. Indeed, when supplemented with exogenous MDP after E. coli uptake, the upregulation of IL-1β in CLCN7 knockdown cells was restored (Figure 7E). We then examined whether blocking phagolysosomal function using an alternate approach would also decrease NF-κB signaling. The PIKfyve-generated endolysosomal lipid, phosphatidylinositol-3,5-biphosphate, is required for phagosome maturation and phagosome-lysosome fusion in a TRPML1-dependent manner (Cai et al., 2013; Dayam et al., 2015; Kim et al., 2014). Treatment of wild-type RAW 264.7 cells with the PIKfyve inhibitor, apilimod, after E. coli uptake drastically suppressed the induction of IL-1β (Figure 7F). Again, co-treatment with MDP restored IL-1β upregulation (Figure 7F). Together with the CLCN7 knockdown experiments, these data indicate the conserved function of endolysosomes in regulating NF-κB signaling axis in macrophages (Figure 7G).
Discussion
Our study makes several advances in the understanding of endolysosomal function in macrophage-dependent innate immunity. First, we find a hitherto unappreciated role for endolysosomal Cl− transporters in regulating the release of luminal Ca2+, which is essential for phagosome-lysosome fusion in macrophages (Luzio et al., 2007b). Absence of ClC-b in Drosophila macrophages leads to diminished endolysosomal Ca2+ release and an attendant delay in the fusion of phagosomes/endosomes with lysosomes. The defects in vesicle fusion observed in the ClC-b1 macrophages is likely a consequence of attenuated Cl− transport because ClC-bunc, which is analogous to the solely Cl− transporting, CLCN7unc (Weinert et al., 2014), largely suppressed the ClC-b1 phenotype. Further supporting the notion that diminished degradation of phagocytosed bacteria in ClC-b1 animals arises from decreased endolysosomal Ca2+ release, loss of trpml, the predominant endolysosomal Ca2+ release channel in Drosophila (Wong et al., 2012), phenocopied ClC-b1. Furthermore, overexpression of trpml in ClC-b1 macrophages partially suppressed the mutant phenotype.
Another important conceptual advance with respect to the role of endolysosomal degradation in phagocytosis and macrophage-dependent inflammation is depicted in Figure 7G. We show that phagolysosomal degradation of bacteria is necessary for the generation of bacteria-derived peptidoglycans that activate NF-κB in Drosophila and mammalian macrophages respectively. Our findings indicate that in Drosophila macrophages, bacteria peptidoglycans derived by lysosomal degradation of bacteria activate the cytosolic pattern recognition receptor, PGRP-LE, which in turn activates NF-κB (Figure 7G, left). Although PGRP-LE is not conserved in mammals, our data suggest that the MDP-sensitive NOD2 receptor is likely the functional equivalent of PGRP-LE in mammalian macrophages (Figure 7G, right) (Kaneko et al., 2006; Nakamura et al., 2014).
Although the role of NF-κB in innate immunity has been studied extensively, we show that NF-κB is required for the phagocytosis of bacteria in macrophages. Our results are consistent with the notion that NF-κB activation following the phagolysosomal degradation of bacteria triggers the transcription of genes encoding phagocytic receptors. Thus, the signaling cascade we have identified matches the rate of phagocytosis with the flux of lysosomal degradation, allowing further bacterial uptake only upon completion of degradation of the previously internalized bacteria. Concatenating the rates of degradation and uptake would diminish the stress associated with overloading the endolysosomal system. This coupling could also prevent the further uptake of specific pathogens that are able to escape phagolysosomal degradation and dwell within macrophages (Pryor and Raines, 2010).
Our results show that PGRP-LE and Relish overexpression in ClC-b deficient macrophages robustly enhanced bacterial clearance despite the persistent loss of vesicular function attributed to the absence of ClC-b. Another well-established function of Relish/NF-κB signaling in Drosophila immunity is the transcriptional upregulation of anti-microbial peptide (AMP) genes in fat body cells. Although we observed ClC-b expression in fat body, our results suggest that ClC-b expression in that tissue is expendable for bacterial clearance. This conclusion is consistent with the notion that fat body cells do not require phagocytosis of bacteria to activate Relish signaling. Rather, plasma membrane receptors, such as PGRP-LC and Toll, serve as the pattern recognition receptor to signal AMP production via NF-κB (Buchon et al., 2014). It is, however, possible that Relish signaling in Drosophila macrophage upregulates both phagocytosis and cell autonomous synthesis of AMPs in response to lysosomal degradation of bacteria and liberation of cell wall peptidoglycans. AMPs produced by macrophage could also mediate in situ killing of bacteria that internalized into phagosomes, which is consistent with the findings that AMPs localize to intracellular granules in mammalian immune cells such as neutrophils (Flannagan et al., 2009; Haas, 2007). If so, Relish signaling in macrophages could perform a dual role in the clearance of bacteria by triggering the potential synergy of phagocytosis and AMP biosynthesis.
Our study also identifies endolysosomes as upstream regulators of inflammation in mammalian macrophages as evidenced by diminished expression of several cytokines including IL-1β and TNF-α following CLCN7 knockdown. It is also important to note that the role of CLCN7 in NF-κB activation and cytokine expression reflects a general requirement for endolysosomal degradation since apilimod-induced inhibition of PIKfyve—a manipulation that prevents vesicular fusion with lysosomes (Kim et al., 2014), robustly attenuates cytokine expression. These findings raise the intriguing possibility that targeting any one of a host of endolysosomal proteins could serve as potential interventions for treating inflammatory diseases such as Crohn’s disease which are associated with elevated expression of pro-inflammatory cytokines (Lawrence, 2009).
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kartik Venkatachalam (kartik.venkatachalam@uth.tmc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All Drosophila melanogaster flies were reared at 23 °C in a 12-hour light-dark cycle on standard fly food (1 L of food contained: 95 g agar, 275 g Brewer’s yeast, 520 g of cornmeal, 110 g of sugar, 45 g of propionic acid, and 36 g of Tegosept). For heat shock mediated knockdown of ClC-b in adult flies, flies were reared at 18 °C throughout development, and one day before ba cteria injection, the animals were heat shocked at 37 °C for 1 hour, and then transferred to 29 °C till the experiments were conducted. In-house generated transgenic flies: ClC-b1, ClC-b2, GFP::ClC-b, ClC-b1;P[ClC-b+], UAS-ClC-b, UAS-ClC-b::myc, UAS-ClC-btd::myc, and UAS-ClC-bunc::myc. The following lines were obtained from Bloomington Drosophila Stock Center (BDSC): P{EP}ClC-bG2453, He-GAL4, HmlΔ-GAL4, lsp2-GAL4, Cg-GAL4, hs-GAL4, elav-GAL4, HmlΔ-GAL4, UAS-2xEGFP (III), UAS-p35, RelE20, PGRP-LE112, UAS-PGRP-LE, and UAS-Rel68. UAS-ClC-b-IR, UAS-ClC-c-IR, UAS-PGRP-LE-IR, and UAS-Rel-IR are TRiP.JF01844, TRiP.JF02360, TRiP.HMC05031, and TRiP.HM05154 respectively, and were also obtained from BDSC. UAS-Vha36-1-IR and UAS-VhaSFD-IR are P{GD17846}v49888 and P{GD8795}v47471 respectively, and were from Vienna Drosophila Resource Center. For E. coli infection experiment, adult males and female virgins (1–3 days-old) were collected and raised in two separate groups to prevent mating. E. coli strain DH5 alpha transformed with pCDNA3 was used for in vivo infection. E. coli was cultured in lysogeny broth containing 50 µg/mL ampicillin. RAW 264.7 cells were cultured with Dulbecco’s Modified Eagle’s medium (DMEM; Sigma-aldrich D5796) supplemented with 10% FBS and 100 U/mL penicillin-streptomycin, and were maintained in 5% CO2 supplemented 37 °C incubator.
METHOD DETAILS
Generation of transgenic Drosophila lines
ClC-b1 was generated by imprecise excision of the P{EP}ClC-bG2453 transposon insertion line. The deletion spanned the region corresponding to +519 to +2389 base pairs relative to the ClC-b translation start site. ClC-b2 and GFP::ClC-b were generated by CRISPR-Cas9 mediated gene editing as described below. The genomic rescue line P[ClC-b+] was generated by inserting a 20 kb BAC clone (CH322-146D02), which encompasses the entire ClC-b genomic locus, into the 3rd chromosomal docking site carrying PBac{y+-attP-3B}VK00033. Genomic rescue of ClC-b1 was achieved by crossing P[ClC-b+] into the ClC-b1 background (ClC-b1;P[ClC-b+]) (gen-res). The deficiency line for ClC-b (Df) is Df(2R)Exel7121, which was obtained from the Bloomington Drosophila Stock Center (BDSC). UAS-ClC-b was generated by subcloning the ClC-b cDNA from the EST clone RE63672 (Berkeley Drosophila Genome Project) into the pUAS-attB vector followed by insertion into the 3rd chromosomal docking site carrying PBac{y+-attP-9A}VK00019. UAS-ClC-b::myc was generated using a similar approach except that a myc tag was added to the ClC-b sequence. UAS-ClC-btd::myc and UAS-ClC-bunc::myc were generated by site-directed mutagenesis of the pUAS-attB-ClC-b vector using In-Fusion Cloning (Clontech). ClC-btd carries 1018GAA to 1018GCA mutation and ClC-bunc carries 817GAG to 817GCG mutation.
CRISPR-Cas9 mediated gene editing
Two gRNA sequences were used to target Cas9 to the ClC-b coding sequence Fig. 1a). gRNA1: 5’-CGAGTAACCATACTCTCATTCGG-3’; gRNA2: 5’-GCTGATAATGGAGGCCACTAAGG-3’. The gRNA-coding oligos were subcloned into the pCFD3-dU6:3gRNA vector. Two donor constructs for homology directed repair (HDR) were generated. Donor construct 1 consisted of the pCDNA3.1 vector carrying the genomic sequence from 1 kb upstream to 1kb downstream of ClC-b gene, and was PCR amplified using the following primers: 5’-AAGCGAGATGTCGACATCTCTCAC-3’ and 5’-TACTTCGAAGACACCGTATCGCATC-3’. A codon optimized EGFP with a C-terminal Ser-Ser-Gly-Ser linker coding sequence was PCR amplified from pBS-KS-attB1–2-PT-SA-SD-0-EGFP-FlAsH-StrepII-TEV-3xFlag. The EGFP construct was then inserted in-frame after the start codon of ClC-b using In-Fusion cloning kit (Clontech). Donor construct 2 consisted of an A→C point mutation at coding nucleotide position 1748, resulting in a Glu to Ala change at 583. During HDR with construct 2, a deletion at the gRNA2 site (1793T) created frame shift and introduced a premature stop codon. To generate the transgenic flies, 10 ng/µL gRNAs, 100 ng/µL pnos-Cas9-nos, and ~100 ng/µL donor construct were mixed and injected to the embryos of ClC-bG2453. Flies positive for CRISPR-Cas9 mediated gene editing were screened by the loss of the red-eye marker of the P{EP}ClC-bG2453 line.
Phylogenetic analysis of CLC proteins
We used CLUSTALW (http://www.genome.jp/tools/clustalw/) to perform protein sequence alignment, and construct a tree dendrogram. Bootstrap tests were performed using PhyLM (Guindon and Gascuel, 2003).
Drosophila E. coli infection and CFU assay
Adult males and female virgins (1–3 days-old) were collected and raised separately to prevent mating. E. coli (DH5 alpha strain) transformed with pCDNA3 (E. coliAmp) was cultured in lysogeny broth containing 50 µg/mL ampicillin (LBA) till the culture reached optical density of ~1.0 at 600 nm (OD600nm). E. coli cultures were then spun down and resuspended in fresh LBA for infection. A glass needle of 0.5 mm lumenal diameter was pulled using a P-30 puller (Sutter Instrument), and 1 mm of the tip was clipped off to generate an opening. The glass needle was loaded with the E. coli suspension and mounted on an Eppendorf Transjector 5246 microinjector. Injection pressure, duration, and compensation pressure were set at 2.5 Psi, 0.2 s, and 0.4 Psi respectively. Approximately 60 nL of the E. coli suspension was injected into the antero-ventral-lateral abdominal cavity of individual flies that were anesthetized on a CO2 pad. E. coliamp equivalent to approximately 3134 CFUs were injected into each fly. For pre-injection of peptidoglycan derivatives, flies were injected with ~60 nL of TCT (16 µM) (Cookson et al., 1989), MDP (10 mg/mL; Invivogen), or vehicle 1 day before the E. coliamp was injected into the contralateral side of the abdomen. For sequential injections of E. coli, heat-killed (80 °C, 2 hours) E. coli at OD600nm~1 was resuspended in PBS (1:1 v/v). Flies were injected with either ~60 nL of the dead E. coli suspension or PBS, 2 days before the injection of live E. coliamp.
After injection of live E. coliamp, flies were kept in regular husbandry conditions for 24 hours or as indicated. Each fly was then anesthetized and crushed in a 1.5-mL microcentrifuge tube with 200 µL PBS to release the circulating E. coli and 10 µL of the extract was spread onto LBA agar in a 10-cm petri dish. After 16 hours of incubation at 37 °C, the colonies formed on each plate (CFUs) were counted by individuals who were blinded to the fly genotypes. For each experiment, CFUs from 5–20 flies of a genotype were normalized to the average CFU of the controls (ClC-b genomic rescue or respective genetic control) that were injected on the same day using the same bacterial culture and needle.
Western blot analysis of GFP expressing macrophages
Wild-type and ClC-b1 flies expressing GFP in macrophages (HmlΔ>EGFP) were used to assess relative macrophage levels. Three 3rd instar wandering larvae or 10 adult flies were collected in 1.5 mL tube containing 50 µL ice cold lysis buffer (in mM: 150 NaCl, EGTA, 0.1% triton X-100, 10 HEPES, pH 7.4). Following an incision on the bodies of the larvae/flies using a pointed scalpel, the animals were ground in lysis buffer using a homogenizer. After centrifugation at 1000 rpm for 2 minutes, the supernatant of the lysate was mixed with Laemmli sample buffer (Bio-Rad) and loaded onto 4–20% gradient Tri-glycine gel (Bio-Rad) for SDS-PAGE. After transfer onto nitrocellulose, the blot was incubated with rabbit anti-GFP (Molecular Probes) and mouse anti-α-tubulin (Developmental Studies Hybridoma Bank). Primary antibody signals were then probed by multiplex secondary antibodies, IRDye 680LT anti-rabbit and IRDye 800CW anti-mouse (LI-COR Biosciences). The blot was imaged by Odyssey imaging platform (LI-COR Biosciences). Band intensities were quantified using ImageJ (NIH).
Circulating larval hemocyte cell count
Four wandering 3rd instar larvae were bled in 80 µL PBS on a glass slide by making a tear dorsally with a pair of forceps. 20 µL of the cell suspension was injected into SD100 counting chambers (Nexcelom Bioscience). Hemocyte cell size and concentration were measured by Cellometer Auto 2000 (Nexcelom Bioscience).
Quantification of AMP expression by qRT-PCR
3–4 day-old adult flies were injected with ~60 nL of E. coliamp suspension (OD600nm ~ 1) or plain LBA. Six hours after injection, 7–9 flies from each treatment group were collected in a 1.5 mL tube and quick-frozen on dry ice. Total RNA from each tube was prepared using the RNeasy Mini Kit (Qiagen), followed by cDNA reverse transcription using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). SYBR green based quantitative real time PCR using primers targeting AMP genes and the house keeping gene rp49 were performed using Eppendorf Realplex2 Mastercycler (Eppendorf). Each primer set for individual sample was ran in triplicate. The ΔCt value for each AMP was calculated by subtracting the Ct value of the rp49 set from that of the AMP gene from the same treatment group (E. coliamp or LBA). The ΔΔCt values were the ΔCt difference between the E.coliamp group and the control LBA group. AMP gene expression level was calculated by 2−ΔΔCt.
Sequences of the primers used for qRT-PCR:
| attacin A-forward: | TGCAGAACACAAGCATCCTAA |
| attacin A-reverse: | TAAGGAACCTCCGAGCACCT |
| defensin-forward: | GATGTGGATCCAATTCCAGA |
| defensin-reverse: | CTTTGAACCCCTTGGCAAT |
| diptericin-forward: | ACCGCAGTACCCACTCAATC |
| diptericin-reverse: | CCATATGGTCCTCCCAAGT |
| drosocin-forward: | TTCACCATCGTTTTCCTGCT |
| drosocin-reverse: | GGCAGCTTGAGTCAGGTGAT |
| drosomycin-forward: | GTACTTGTTCGCCCTCTTCG |
| drosomycin-reverse: | ACTGGAGCGTCCCTCCTC |
| eater-forward: | GGAAGTGGCTTCTGCACGAAAC |
| eater-reverse: | CGACTACATCCCTTGCAGTAGGG |
| NimC1-forward: | GTTTGTAACCGATCGCAGGTGG |
| NimC1-reverse: | TCGTAGCCCTCACAGCAACTG |
| rp49-forward: | ATCGGTTACGGATCGAACAA |
| rp49-reverse: | GACAATCTCCTTGCGCTTCT |
Macrophage cultures and ex vivo E. coli phagocytosis assay
Schneider’s medium (SM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin was used for culturing macrophages. Wandering 3rd instar larvae were first washed with water and 70% ethanol. Next, 8 larvae immersed in 100 µL SM on a glass depression slide were bled by making a tear dorsally with a pair of forceps. To prevent macrophage melanization, 1 µM N-Phenylthiourea (Sigma-Aldrich) was added to SM on some occasions. When larger quantities of macrophage suspensions were needed, this process was repeated in multiple glass depression slides.
For the ex vivo phagocytosis assays, fluorescently labeled heat-killed E. coli (Molecular Probes) washed with PBS and resuspended in SM were used. For determining E. coli uptake over a short time course (≤120 min), 4 µg/mL (~1.2 ×106 particles/mL) of fluorescent E. coli was added to the macrophage culture chilled on ice. 40 µL of the mixture was then dispensed onto concanavalin A coated glass-bottom dishes (MatTek), followed by incubation on ice for 15 minutes to allow particle adhesion. The dishes were then brought back to room temperature and the E. coli-hemocyte mixture was fixed with 4% paraformaldehyde at desired time points. At least 30 minutes of fixation was allowed before immunostaining.
For determining degradation of internalized E. coli within macrophages (ex vivo degradation assay), 20 µg/mL (~6 ×106 particles/mL) of fluorescent E. coli was added to the macrophage culture for 15 minutes at room temperature. The mixture was then chilled on ice, centrifuged (100 g, 3 min), and washed thrice in fresh SM to remove the E. coli particles that were not internalized. 30 µL of the washed macrophage suspension was dispensed onto concanavalin A coated glass-bottom dishes and incubated at 25 °C in a moist chamber, followed by fixation at desired time points. For time point 0, the macrophages were fixed immediately after being dispensed onto the dish. The macrophage culture was replenished with fresh SM daily. For determining acute phagocytic capacity, the procedures were the same as the degradation assay, except that the macrophage suspension was fixed 15 minutes after being dispensed onto the dish.
For the ex vivo sequential phagocytosis assay, 5 larvae were bled in glass depression slides containing 75 µL SM (+1 µM N-Phenylthiourea) before addition of 20 µg/mL (~6 ×106 particles/mL) Texas Red labeled E. coli. After 15 minutes of incubation on a rocking platform at room temperature, the macrophages were centrifuged (100 g, 3 min) and washed in serum-free SM for 2 times, before being resuspended in 50 µL serum-free SM and plated on uncoated glass-bottom dishes. After 4-hours of incubation at 25 °C, 10 µL of SM containing 120 µg/mL Alexa Fluor 488 E. coli was added to the culture, resulting in 20 µg/mL final concentration of E. coli. After 30 minutes of incubation on a rocking platform at room temperature, the macrophage culture was washed with PBS and fixed with 4% paraformaldehyde. For sequential phagocytosis assay performed on He>GFP and ClC-b1 macrophage co-culture, 3 larvae of each genotype were bled in a glass depression slide before sequential challenge with bacteria using the protocol described above, except that we substituted Texas Red labeled E. coli and Alexa Fluor 488 E. coli with heat-killed E. coli and Texas Red labeled E. coli respectively. After fixation, confocal imaging was performed after 2-hours incubation with Alexa Fluor 647 conjugated Phalloidin (Molecular Probes).
Z-series confocal imaging at 1 µm interval of individual macrophages was done by Nikon A1 Confocal Laser Microscope System (Nikon). Immuno-fluorescently labeled vesicles/E. coli particles were manually counted by surveying individual Z-series images of the whole cell.
Immunohistochemistry
For labeling acidic endosomes, primary macrophages and fat bodies were incubated with LysoTracker Red (Molecular Probes; 10 µM for hemocytes and 2 µM for fat bodies) in SM for 30 minutes at room temperature, followed by a single wash with PBS before fixation. Primary macrophages and fat bodies were fixed in PBS+4% paraformaldehyde for at least 30 minutes at room temperature, followed by 3 washes with PBS+0.1% Triton X-100 (PBST). The samples were then incubated overnight with primary antibodies in PBST+5% donkey serum at 4 °C. GFP::ClC-b, YFP::Rab7, and TRPML::GCaMP5G were detected using rabbit anti-GFP (Molecular Probes). ClC-b::myc was detected using mouse anti-myc (Sigma-Aldrich). After incubation with primary antibodies, the samples were washed thrice with PBST, before further incubation with the appropriate Alexa Fluor 488/568 conjugated secondary antibodies (Molecular Probes) at room temperature for 2.5 hours. To fluorescently mark the macrophages we either drove the expression of cytosolic GFP in macrophages (signal enhanced in fixed samples with GFP-booster (ChromoTek)) or labeled the macrophages with Alexa Fluor 647 phalloidin (Molecular Probes). The samples were finally washed thrice with PBST before being mounted in DAPI-containing VECTASHIELD (Vector Labs). Immunofluorescence images were acquired by Nikon A1 Confocal Laser Microscope System (Nikon) using 60× or 100× oil immersion objectives.
Lysosomal Ca2+ measurement in Drosophila macrophages
Three 3rd instar wandering larvae were bled on concanavalin A coated glass-bottom dishes with 60 µL SM at ~23 °C. Hemocytes were allowed to adhere to the glass for 15 minutes, before being washed twice with fresh SM. The macrophage cultures were then incubated for 1–2 hours at 25 °C before live-cell Ca2+ imaging. During imaging, the culture medium was replaced with 150 µL of HL-3 saline (70 mM NaCl, 5 mM KCl, 20 mM MgCl2, 10 mM NaHCO3, 1mM CaCl2, 115 mM sucrose, 5 mM trehalose, and 5 mM HEPES; pH7.2.). Next, dishes containing the cells were mounted onto a Nikon TiE wide-field fluorescence imaging system. After base-line fluorescence images were acquired for ~1 minute, approximately 30 µL of bath solution was pipetted out to dilute MLSA-1 or GPN, and added back to the bath. GCaMP5G signals excited by 490 nm, which represent lysosomal [Ca2+] release, were recorded by the Nikon TiE Wide-Field Fluorescence Imaging System (Nikon). The background subtracted GCaMP5G signals were expressed as a real-time fluorescence (Ft) relative to the intensity at the beginning of the experiment (F0).
CLCN7 knockdown in RAW 264.7 macrophage
The shRNA expression constructs (TRC2-pLKO-puro) carrying the shRNA sequence against mouse CLCN7 were purchased from Sigma-Aldrich. Target sequence of ShR1: GCCTACACCATCCATGAGATT. Target sequence of ShR2: CAGACGTGAGCTACTACTTAC. Control ShR was the non-mammalian control shRNA expression vector SHC002 from Sigma-Aldrich. Lentiviral particles containing the ShR expression vector were generated from HEK293T cells. RAW 264.7 cells were transduced with the lentiviral particles for 16 hours, before recovery in fresh culture medium (DMEM+10% FBS; D5796 Sigma-Aldrich) for one day. The transduced cells were then selected with 5 g/mL puromycin for 2 weeks with multiple passages, before being used for experiments. Knockdown efficiency in the stable cell lines was verified by Western blotting of the cell lysates with anti-CLCN7 antibodies (Thermo Scientific).
Gentamicin protection assay using RAW 264.7 cells
RAW 264.7 cells were seeded onto 12-well culture plates with Dulbecco’s Modified Eagle’s medium (DMEM; Sigma-aldrich D5796) supplemented with 10% FBS and 100 U/mL penicillin-streptomycin. When ~70% confluency was reached, the cells were serum starved for 16 hours. E. coliAmp was cultured to reach OD600 nm of ~1.0 in LBA. Subsequently, the E. coliAmp culture was spun down and resuspended in PBS and added to DMEM+10% FBS to make a 2% E. coliAmp infection medium, with estimated multiplicity of infection (MOI) of ~20. The serum-starved macrophages were incubated with the infection medium at 37 °C for 30 minutes, followed by washing with PBS containing 1 mM CaCl2. One plate of the cells was taken for assessing the amount of phagocytosed E. coli immediately after washing. Another plate of cells was then cultured with DMEM+10% FBS containing 200 µg/mL gentamicin for the desired length of time. To release the viable E. coliAmp inside the cells, the cells were incubated with 500 µL 0.1% saponin per well for 10 minutes on a rocking platform. 10 µL of the extract from each well was then spread onto LBA agar in a 10-cm petri dish, followed by the CFU quantification as described above.
Quantification of cytokine production in RAW 264.7 cells by qRT-PCR
Either wild-type RAW 264.7 cells or shRNA expressing RAW 264.7 cells (ShR1, ShR2, or control shRNA) were seeded onto 6-well culture plates and cultured as described in the gentamicin protection assay. The cells were serum starved for 16 hours before incubation with DMEM+10% FBS with or without 1% v/v heat-killed E. coli (OD600 nm = 1) for 30 minutes. Cells were then washed thrice with PBS containing 1 mM CaCl2 before incubation in fresh DMEM+10% FBS for 4 hours. When needed, 0.1% v/v DMSO, 10 µg/mL MDP (Invivogen), or 200 nM apilimod (Cayman Chemical) was added to the culture medium during the 4-hour period. The cells were then washed thrice with ice-cold PBS before being scraped in lysis buffer for total RNA preparation using RNeasy Mini Kit (Qiagen). cDNA was generated using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). SYBR green based quantitative real time PCR using primers targeting cytokine genes and actin gene were performed using Eppendorf Realplex2 Mastercycler (Eppendorf). Each primer set for individual sample was ran in triplicate. The ΔCt value for each cytokine was calculated by subtracting the Ct value of the actin set from that of the cytokine gene from the same treatment group (with or without E. coli). Cytokine gene expression level for each sample was calculated by 2−ΔCt. The 2−ΔCt values were then normalized to the appropriate controls indicated in the figure legends.
Sequences of the primers used for the quantification of cytokine production:
| IL-1β | forward: CAACCAACAAGTGATATTCTCCATG |
| IL-1β reverse: | GATCCACACTCTCCAGCTGCA |
| TNF-α forward: | TGGAGTCATTGCTCTGTGAAGGGA |
| TNF-α reverse: | AGTCCTTGATGGTGGTGCATGAGA |
| IL-10 forward: | GTGAAGACTTTCTTTCAAACAAAG |
| IL-10 reverse: | CTGCTCCACTGCCTTGCTCTTATT |
| β-actin forward: | TACTGCCCTGGCTCCTAGCA |
| β-actin reverse: | TGGACAGTGAGGCCAGGATAG |
QUANTIFICATION AND STATISTICAL ANALYSIS
Student’s t-test was used for used for all dataset analyses. Excel (Microsoft) was used for mean and SEM calculations, and Student’s t-test analysis. Statistical significance was defined as a p-value of 0.05. p-values were shown on the figures as asterisks: * p<0.05; ** p<0.01; *** p<0.001.
Data and Software Availability
Mean and SEM values, sample numbers, and numbers of experiments are shown in Supplemental Information Table S1. Raw data (e.g. CFU count of individual flies) are available upon request to the lead contact author. No proprietary software was used in the data analysis.
Supplementary Material
Highlights.
Vesicular Cl− transporters drive phagolysosomal degradation of internalized bacteria
Lysosomal degradation is required for sustained bacteria phagocytosis by macrophages
Lysosomal degradation activates cytosolic PRRs and NF-κB to sustain phagocytosis
NF-κB couples phagolysosomal degradation to sustained clearance of bacteria
Acknowledgments
We thank the Bloomington Drosophila Stock Center (NIH P40OD018537) for fly stocks. We also thank Drs. Thomas Jentsch, Tobias Stauber, Stefanie Weinert, and Neal Silverman for reagents and useful discussions; Drs. Siu Ling Wong, Herman Dierick, Hamed Jafar-Nejad, Danielle Garsin, and Marco Sardiello for reading the manuscript and providing valuable feedback; Dr. Jefferey Frost for allowing the use of the microinjector; and Meera Namireddy for assisting with counting CFUs. Confocal microscopy was performed at the Center for Advanced Microscopy, Department of Integrative Biology & Pharmacology at McGovern Medical School, UTHealth. H.J.B. is an investigator of the HHMI and D.L.K. is supported by the Belfer Family Foundation. This work was supported by the NINDS grants, R21NS094860 and R01NS081301 (to K.V.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
C.W. and K.V. designed the study, interpreted the results and wrote the paper. C.W. made the genetic constructs and performed most of the experiments. H.H. and S.G. performed real-time PCR experiment. H.H. and Y.C. assisted in CFU assay and performed CFU counting. V.E.S. and W.E.G. purified TCT. Y.H. performed embryo injection of the CRISPR constructs. D.L-K. and H.J.B. assisted in construct design and provided reagents.
References
- Berg TO, Stromhaug E, Lovdal T, Seglen O, Berg T. Use of glycyl-L-phenylalanine 2-naphthylamide, a lysosome-disrupting cathepsin C substrate, to distinguish between lysosomes and prelysosomal endocytic vacuoles. Biochem J. 1994;300(Pt 1):229–236. doi: 10.1042/bj3000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchon N, Silverman N, Cherry S. Immunity in Drosophila melanogaster--from microbial recognition to whole-organism physiology. Nat Rev Immunol. 2014;14:796–810. doi: 10.1038/nri3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrero P, Terhzaz S, Romero MF, Davies SA, Blumenthal EM, Dow JA. Chloride channels in stellate cells are essential for uniquely high secretion rates in neuropeptide-stimulated Drosophila diuresis. Proc Natl Acad Sci U S A. 2014;111:14301–14306. doi: 10.1073/pnas.1412706111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcazar-Roman A, Murphy L, Billich A, Zhang B, Feng Y, Klumpp M, et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem Biol. 2013;20:912–921. doi: 10.1016/j.chembiol.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson BT, Cho HL, Herwaldt LA, Goldman WE. Biological activities and chemical composition of purified tracheal cytotoxin of Bordetella pertussis. Infect Immun. 1989;57:2223–2229. doi: 10.1128/iai.57.7.2223-2229.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayam RM, Saric A, Shilliday RE, Botelho RJ. The Phosphoinositide-Gated Lysosomal Ca(2+) Channel, TRPML1, Is Required for Phagosome Maturation. Traffic. 2015;16:1010–1026. doi: 10.1111/tra.12303. [DOI] [PubMed] [Google Scholar]
- Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. 2012;33:397–405. doi: 10.1016/j.it.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Feng X, Huang Y, Lu Y, Xiong J, Wong CO, Yang P, Xia J, Chen D, Du G, Venkatachalam K, et al. Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J Biol Chem. 2014a;289:4262–4272. doi: 10.1074/jbc.M113.506501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Xiong J, Lu Y, Xia X, Zhu MX. Differential mechanisms of action of the mucolipin synthetic agonist, ML-SA1, on insect TRPML and mammalian TRPML1. Cell Calcium. 2014b;56:446–456. doi: 10.1016/j.ceca.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Haas A. The phagosome: compartment with a license to kill. Traffic. 2007;8:311–330. doi: 10.1111/j.1600-0854.2006.00531.x. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Yano T, Aggarwal K, Lim JH, Ueda K, Oshima Y, Peach C, Erturk-Hasdemir D, Goldman WE, Oh BH, et al. PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat Immunol. 2006;7:715–723. doi: 10.1038/ni1356. [DOI] [PubMed] [Google Scholar]
- Kim GH, Dayam RM, Prashar A, Terebiznik M, Botelho RJ. PIKfyve inhibition interferes with phagosome and endosome maturation in macrophages. Traffic. 2014;15:1143–1163. doi: 10.1111/tra.12199. [DOI] [PubMed] [Google Scholar]
- Kocks C, Cho JH, Nehme N, Ulvila J, Pearson AM, Meister M, Strom C, Conto SL, Hetru C, Stuart LM, et al. Eater, a transmembrane protein mediating phagocytosis of bacterial pathogens in Drosophila. Cell. 2005;123:335–346. doi: 10.1016/j.cell.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Kurucz E, Markus R, Zsamboki J, Folkl-Medzihradszky K, Darula Z, Vilmos P, Udvardy A, Krausz I, Lukacsovich T, Gateff E, et al. Nimrod, a putative phagocytosis receptor with EGF repeats in Drosophila plasmatocytes. Curr Biol. 2007;17:649–654. doi: 10.1016/j.cub.2007.02.041. [DOI] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T. ClC-7 is a slowly voltage-gated 2Cl(−)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 2011;30:2140–2152. doi: 10.1038/emboj.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Kim MS, Kim HE, Yano T, Oshima Y, Aggarwal K, Goldman WE, Silverman N, Kurata S, Oh BH. Structural basis for preferential recognition of diaminopimelic acid-type peptidoglycan by a subset of peptidoglycan recognition proteins. J Biol Chem. 2006;281:8286–8295. doi: 10.1074/jbc.M513030200. [DOI] [PubMed] [Google Scholar]
- Lloyd-Evans E, Platt FM. Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium. 2011;50:200–205. doi: 10.1016/j.ceca.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Bright NA, Pryor PR. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem Soc Trans. 2007a;35:1088–1091. doi: 10.1042/BST0351088. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007b;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- Meister M, Lagueux M. Drosophila blood cells. Cell Microbiol. 2003;5:573–580. doi: 10.1046/j.1462-5822.2003.00302.x. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, Klumperman J, Schlatter M, Delamarre L, Mellman I. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature. 2014;509:240–244. doi: 10.1038/nature13133. [DOI] [PubMed] [Google Scholar]
- Pham LN, Dionne MS, Shirasu-Hiza M, Schneider DS. A specific primed immune response in Drosophila is dependent on phagocytes. PLoS Pathog. 2007;3:e26. doi: 10.1371/journal.ppat.0030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor PR, Raines SA. Manipulation of the host by pathogens to survive the lysosome. Biochem Soc Trans. 2010;38:1417–1419. doi: 10.1042/BST0381417. [DOI] [PubMed] [Google Scholar]
- Saha S, Prakash V, Halder S, Chakraborty K, Krishnan Y. A pH-independent DNA nanodevice for quantifying chloride transport in organelles of living cells. Nat Nanotechnol. 2015;10:645–651. doi: 10.1038/nnano.2015.130. [DOI] [PubMed] [Google Scholar]
- Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- Stauber T, Jentsch TJ. Chloride in vesicular trafficking and function. Annu Rev Physiol. 2013;75:453–477. doi: 10.1146/annurev-physiol-030212-183702. [DOI] [PubMed] [Google Scholar]
- Takehana A, Katsuyama T, Yano T, Oshima Y, Takada H, Aigaki T, Kurata S. Overexpression of a pattern-recognition receptor, peptidoglycan-recognition protein-LE, activates imd/relish-mediated antibacterial defense and the prophenoloxidase cascade in Drosophila larvae. Proc Natl Acad Sci U S A. 2002;99:13705–13710. doi: 10.1073/pnas.212301199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Long AA, Elsaesser R, Nikolaeva D, Broadie K, Montell C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135:838–851. doi: 10.1016/j.cell.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Wong CO, Zhu MX. The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium. 2015;58:48–56. doi: 10.1016/j.ceca.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Hohensee S, Chan WL, Kornak U, Jentsch TJ. Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 2014;15:784–791. doi: 10.15252/embr.201438553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U, Jentsch TJ. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science. 2010;328:1401–1403. doi: 10.1126/science.1188072. [DOI] [PubMed] [Google Scholar]
- Wiklund ML, Steinert S, Junell A, Hultmark D, Stoven S. The N-terminal half of the Drosophila Rel/NF-kappaB factor Relish, REL-68, constitutively activates transcription of specific Relish target genes. Dev Comp Immunol. 2009;33:690–696. doi: 10.1016/j.dci.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Wong CO, Li R, Montell C, Venkatachalam K. Drosophila TRPML is required for TORC1 activation. Curr Biol. 2012;22:1616–1621. doi: 10.1016/j.cub.2012.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. doi: 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.