

Abstract
Objective:
Despite the enormous advancements made in deciphering the genetic architecture of Parkinson disease (PD), the majority of PD is idiopathic, with single gene mutations explaining only a small proportion of the cases.
Methods:
In this study, we clinically evaluated 2 unrelated Spanish families diagnosed with PD, in which known PD genes were previously excluded, and performed whole-exome sequencing analyses in affected individuals for disease gene identification.
Results:
Patients were diagnosed with typical PD without relevant distinctive symptoms. Two different novel mutations were identified in the CSMD1 gene. The CSMD1 gene, which encodes a complement control protein that is known to participate in the complement activation and inflammation in the developing CNS, was previously shown to be associated with the risk of PD in a genome-wide association study.
Conclusions:
We conclude that the CSMD1 mutations identified in this study might be responsible for the PD phenotype observed in our examined patients. This, along with previous reported studies, may suggest the complement pathway as an important therapeutic target for PD and other neurodegenerative diseases.
Parkinson disease (PD; MIM# 168600) is the second most common neurodegenerative disease affecting more than 4 million people worldwide.1,2 PD is thought to be caused by a combination of genetic and environmental risk factors, with only 5%–10% of patients reporting to have a mendelian pattern of inheritance.3–5 To date, 3 different genes, SNCA (MIM# 163890), LRRK2 (MIM# 609007), and VPS35 (MIM# 601501), have been linked to autosomal dominant PD,6–8 with 2 of these (SNCA and LRRK2) being well established as susceptibility loci for sporadic PD.9–11 In addition, common single nucleotide variations within GBA, MAPT, MCCC1, STK39, GAK, BST1, SYT11, ACMSD, and the HLA region have been reported to be associated with the risk of PD through genome-wide association studies performed by numerous different scientific groups.9,12–16 However, in the majority of these associated genes, no pathogenic (or rare) mutation has been identified in familial or sporadic PD, except for the ACMSD gene, in which pathogenic mutations have been reported in complex parkinsonism and idiopathic PD.17
In this study, we clinically evaluated 2 different families suffering from late-onset PD (LOPD) without mutations in the known genes18 and performed whole-exome sequencing (WES) analyses in 3 affected family members to identify the genetic causes of disease to enhance our knowledge of the genetic architecture of PD. We identified 2 different mutations in a novel gene, previously reported to be associated with the risk of PD, as possible disease-causing mutations in both unrelated PD families.
METHODS
Standard protocol, approvals, registrations, and patient consents.
Two individual families with LOPD and of Basque origin and 1 isolated familial PD (fPD) case were clinically examined. Patients of 3 families were diagnosed and treated by a group of movement disorder specialists (J.R.-M., A.B., and J.F.M.-M.) at the University Hospital Donostia and were included as having fPD without mutations in the known genes. Mutations in known genes were previously excluded through custom targeted sequencing.18 Patients were diagnosed according to the UK PD Brain Bank Society and Gelb criteria.19,20 Written informed consent, fully approved by the local ethics committee of the Hospital Universitario Donostia, was obtained from all participants.
DNA samples from 115 Spanish patients with PD and 94 DNA samples belonging to ethnicity-matched neurologically normal individuals (45 men and 49 women) without a family history of any movement disorders were also available for genetic screening. The age at sample collection of the control individuals ranged from 60 to 93 years with an average of 69.1 years.17 The NDPT102 Parkinson panel from the Coriell Institute for Medical Research (coriell.org/) that contains DNA from 92 unique and unrelated Caucasian individuals with idiopathic PD (59 men and 33 women) was also used for mutational screening.
All available DNA samples were isolated from whole blood using standard procedures.
Whole-exome sequencing.
Four DNA samples belonging to 2 different families with LOPD (cases A1, B1, and B2) and 1 isolated familial case were subject to WES analyses (figure, A), which were conducted as previously described.21 The SureSelect Human All Exon 50Mb exon-capture kit was used for library enrichment (Agilent Technologies Inc., Santa Clara, CA), and captured libraries were sequenced on the HiSeq2000 according to the manufacturer's instructions for paired-end 100-bp reads (Illumina Inc., San Diego, CA), using a single flow cell lane per sample. Sequencing data were put through a computational pipeline for WES data processing and analysis following the general workflow adopted by the 1000 Genomes Project,22 where raw sequence reads were aligned to the human reference genome sequence (NCBI GRCh37.p13) using the fast lightweight Burrows-Wheeler Alignment Tool (BWA),23 followed by a base-quality recalibration and local realignment through the Genome Analysis Toolkit (GATK v1.5-16-g58245bf). Single nucleotide substitutions (single nucleotide polymorphism/single nucleotide variant) and short insertions/deletions (INDELs) were called using the GATK Unified Genotyper tool, where calls were filtered based on mapping quality (q30 or higher) and depth of coverage (d10 or higher). Last, the AnnTools kit was used for annotation of the resulting calls,24 and PICARD was used to conduct exomes' statistics (picard.sourceforge.net/).
Figure. CSMD1 mutations in Parkinson disease.

(A) Pedigree structures of 2 families with LOPD and CSMD1 mutations; family A is shown on the left side, whereas family B is shown on the right side. Wt/p.R1962H: heterozygous carriers for the CSMD1 p.R1962H mutation; Wt/p.G2987R: heterozygous carriers for the CSMD1 p.G2987R mutation; Wt/Wt: noncarriers. (B) Sanger chromatogram sequences for both CSMD1 mutations identified in this study are shown at the top, while conservation of both mutations across different species is shown at the bottom. The exact position of the mutations is highlighted with a red arrow. (C) CSMD1 encoding protein structure and the localization of both mutations identified in this study. LOPD = late-onset Parkinson disease.
Genomic variations observed as common mutations (frequency > 5%) in the latest dbSNP149 build, 1000 Genomes Project Phase 3, the Exome Variant Server of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (evs.gs.washington.edu/EVS/),25 and the Exome Aggregation Consortium (exac.broadinstitute.org/) were excluded from subsequent analyses, as were genomic variations mapped to intragenic, intronic, and noncoding exonic regions.
Given the low frequency of pathogenic autosomal dominant PD mutations in public databases, which are either not present (SNCA, VPS35) or identified with very low frequency (LRRK2, 1E-06), with the exception of LRRK2 p.Gly2019Ser mutation that is identified with a higher frequency (1E-04) because of its prevalence in European population, novel and genomic variations with very low frequency were prioritized in follow-up analyses.
The pathogenicity of the identified mutations was predicted using the following computational methods: MutPred (mutpred.mutdb.org/), SNPs&Go (snps-and-go.biocomp.unibo.it/snps-and-go/), MutationTaster (mutationtaster.org/), and CADD tool.26 We also used the HomoloGene database (ncbi.nlm.nih.gov/homologene/) to examine the conservation of the mutated CSMD1 protein across different species, as well as the professional Human Gene Mutation Database (HGMD) (portal.biobase-international.com/hgmd/pro/start.php) and the NCBI ClinVar database (ncbi.nlm.nih.gov/clinvar/) to determine whether CSMD1 mutations were known to be associated with a disease phenotype.
Candidate gene screening.
To amplify all coding CSMD1 exons, genomic primers were designed using the ExonPrimer script (ihg.gsf.de/ihg/ExonPrimer.html) (primer sequences available on request), and PCR products were purified using the ExoSap-IT reagent (Applied Biosytems, Foster City, CA). These were sequenced in both forward and reverse directions using Applied Biosystems BigDye terminator v3.1 sequencing chemistry as per the manufacturer's instructions, resolved on an ABI3730 genetic analyzer (Applied Biosytems), and analyzed using Sequencher 5.4.1 software (Gene Codes Corporation, Ann Arbor, MI).
RESULTS
Clinical history.
Family A: Patient A1.
This patient began feeling tired, clumsy, and very slow in movements at the age of 72 and was diagnosed with PD 1 year later. Initially, he did not take any treatment, but then came back to the clinic with slowness, clumsiness, and a rest tremor in his right hand. On examination, he showed an amimic face, hypophonia, and dysarthria. He had a resting tremor (1/4) in the right hand as well as rigidity in the neck (3/4 neck stiffness), right arm (2/4), and left arm (1/4). He had a moderate global akinesia (3/4) with axial disturbance. Reflexes were normal. He was diagnosed with PD—score 2 according to the Hoehn and Yahr (H&Y) scale. This condition was treated with carbidopa/levodopa (75/300 mg/d) with improvement, and a year later cabergoline (2 mg/d) and rasagiline (1 mg/d) were administered. Later, cabergoline was replaced with rotigotine (8 mg/d). At the age of 78, he was autonomous in daily life, but a year later, his postural instability increased with more clumsiness when turning in bed and hypophonia. At the age of 80, he had an important gait disturbance with freezing but not falls, oscillations, and moderate dyskinesias without cognitive impairment. He died at the age of 86 of respiratory infection.
He had a family history of PD with a paternal aunt with a diagnosis of PD, a father with a possible resting tremor in the elderly, a brother who died without PD at the age of 77, a healthy sister (87 years old), a brother with cognitive impairment (86 years old), a daughter with PD (58 years old; figure, A; patient A2), and a daughter without PD (53 years old).
Family A: Patient A2.
This patient was first seen at 49 years of age with a 6-month evolution of bradykinesia in her right hand affecting her daily activities, including writing, moving, dressing, and so on. She also noticed in writing notes that her letters were smaller. She reported trouble sleeping because of symptoms compatible with REM sleep behavior disorder (RBD), associated with anxiety precipitated by a family conflict and restless legs syndrome (RLS) in the last 2 years. On examination, she showed mild amimia as well as neck and right arm rigidity. She had a slight decrease in arm movements in her walk, micrography, and bilateral babinski signs with mild hyperreflexia. Brain MRI study was normal, and DATSCAN showed bilateral striatal hypocaptation, mainly on the left side. She was diagnosed with PD, and initially began treatment with rasagiline (1 mg/d) and later with carbidopa-levodopa (75/300 mg/d). In the following years, she has been presenting with increasing oscillations and generalized dyskinesias that have progressed to severe oscillations, difficulty in walking with some degree of ataxia, and severe instability with falls over the last year. Her cognitive state remains normal.
Family B: Patient B1.
This patient was examined for the first time at the age of 70, when he presented with tremor in his right hand of 6 months of duration. At examination, he showed rest tremor and akinesia with mild rigidity in his right hand. He was diagnosed with PD—score 1 according to the H&Y scale—and was treated with carbidopa-levodopa (75/300 mg daily) and selegiline with good response. In the following 3 years, his symptoms progressed to bilateral and he developed dyskinesias 6 years after levodopa initiation.
He was autonomous in daily life until the age of 83, and he could go outside without assistance. He subsequently had postural instability, hypophonia, dysarthria, and dysexecutive mild cognitive impairment, but never became demented. At the age of 93, he suffered from lateral bulbar infarction related to embolism because of atrial fibrillation, with minor sequels. Two months later, he had a fall with a chest contusion and fracture of the 10th rib. During his hospital admission, he had acute delirium, dyspnea, and finally died.
He had a family history of PD. His mother, who died at the age of 75, suffered from PD and rest tremor since the age of 70. Her brother died of pancreatic cancer at the age of 79. This brother had 4 children: a son who was diagnosed with PD at the age of 51 (he is now 59 years old) as well as 2 other sons and 1 daughter who died without neurodegenerative disease. The patient (patient B1) had 3 children: a son who died of sepsis at the age of 53, a 68-year-old healthy daughter, and a 63-year-old daughter with postural tremor (figure, A; patient B2).
Family B: Patient B2.
This patient was first examined at the age of 57 when she presented with tremor in the hands of 2 years of evolution. She feared having fPD related to her father's PD. She had postural tremor of small amplitude and frequency 8 Hz in the hands and was diagnosed with enhanced physiologic tremor that aggravated by anxiety. At the age of 61, she came back to the clinic because of her tremor. She had shaking hands without cephalic postural tremor, but neither rigidity nor slowness was observed. Her tremor increased with nervousness. She reported to have symptoms suggestive of RLS. No constipation, hyposmia, dizziness, or symptoms suggestive of RBD were observed. The levels of thyroid hormones and other analytic studies were normal.
Genetic results.
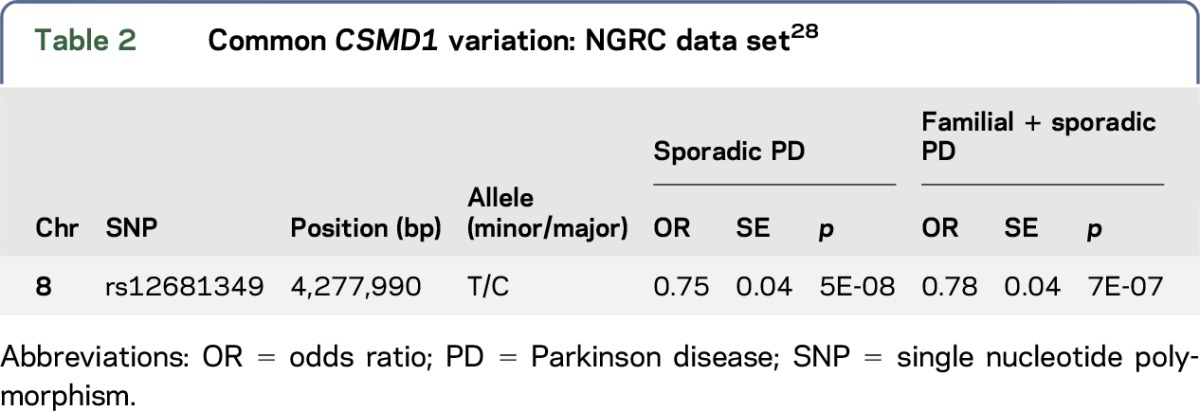
WES approaches were performed in 3 different family members (cases A1, A2, and B1) belonging to 2 PD families (families A and B) and 1 isolated familial case. Between 96.38% (case A2) and 93.56% (case A1) of the target exome at 20-fold coverage or higher was captured for all sequenced samples. This led to the identification of 979 coding genetic variations for case A1, 1,560 for case A2, 1,132 for case B1, and 907 for the isolated familial case. After filtering and by including only novel or with very low frequency genomic variation (1E-04–1E-06), 26 SNVs (12 novel and 14 with low frequency) were found to be shared by the 2 affected members of family A. We then searched if any of the genes, where the shared SNVs were located, were also mutated in family B and the isolated case, and identified 2 novel heterozygous mutations, not previously reported in public databases, in the CSMD1 gene (MIM# 608397) in both affected families A and B. No novel genetic variation was found to be shared between the 2 families (A and B) and the isolated fPD case. A G-to-A transition (c.5885G>A) resulting in p.Arg1962His amino acid substitution was identified in both affected members from family A, while a G-to-A transition (c.8959G>A) resulting in p.Gly2987Arg amino acid substitution was identified in the only family member of family B subject to WES (table 1). Analysis of this second mutation (p.Gly2987Arg) in additional family members revealed that this mutation segregated with the disease status (figure, A and B). We also found that both mutated CSMD1 amino acids are highly conserved across different species (figure, B) as well as CSMD2 and CSMD3 proteins (data not shown) and that both mutations are located in different complement control protein (CCP) domains of the translated CSMD1 protein (figure, C). The CCP domains, containing approximately 60 amino acid residues, have been identified in several proteins of the complement system that is part of the innate immune system. The CSMD1 gene is also found to be weakly expressed in most tissues, except in the brain, where it is expressed at an intermediate level in the cerebellum, substantia nigra, hippocampus, and fetal brain.27 Both novel CSMD1 mutations were predicted highly pathogenic by various computational methods. We then screened both CSMD1 mutations in 372 control chromosomes, including 188 ethnicity-matched control chromosomes, and did not identify any additional mutation carrier. No mutation carrier was identified in 115 DNA samples of Spanish patients with PD tested through Sanger sequencing. However, a genome-wide significant association between a CSMD1 nucleotide variation (rs12681349; intron 2) and PD has been recently reported using the NeuroGenetics Research Consortium (NGRC) data set, which includes 435 fPD, 1,565 sporadic PD, and 1,986 control cases (table 2).28 This association remained highly significant when including all PD (familial and sporadic) and sporadic PD cases. And although it was also observed when considering only fPD, this was not statistically significant, probably because of power limitations.
Table 1.
CSMD1 mutations in familial PD
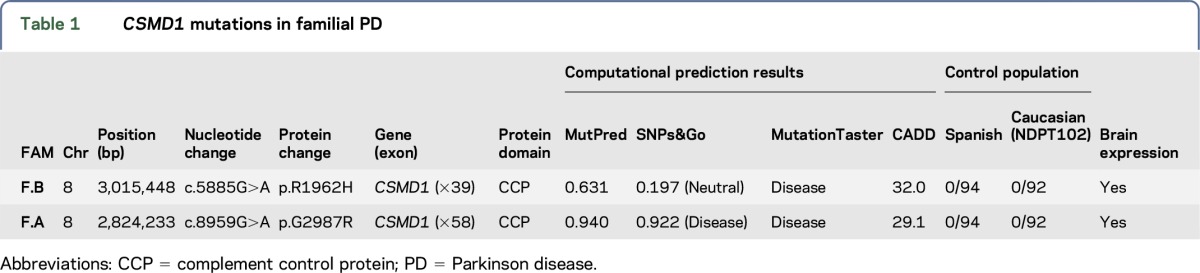
Table 2.
Common CSMD1 variation: NGRC data set28
DISCUSSION
We here described the identification of a novel gene (CSMD1) to be mutated in unrelated Basque families with LOPD in which known PD genes were previously excluded.18 Although patients from both families mainly presented with classical symptoms of PD, the disease course was observed with variable age at onset, ranging from 49 to 72 years, and variable phenotypic heterogeneity, with 2 patients presenting with resting tremor, akinesia, and rigidity, and later on postural instability (patients B1 and A1), 1 patient (patient B2) presenting with tremor of the hands that aggravated with anxiety and RLS, and the youngest patient with severe bradykinesia affecting her daily activities, RBD, and RLS, as well as atypical symptoms such as bilateral Babinski signs and ataxic gait (patient A2). We do not know whether the patient B2, currently diagnosed with postural tremor, will manifest PD in the near future, but we believe that she might develop PD at a more advanced age as her father did. Despite the advance age of some of the patients, only 1 patient presented with dysexecutive mild cognitive impairment (patient B1). However, we cannot discard that the other 2 reported mutation carriers, who are still relatively young, might develop some kind of cognitive dysfunction at an advanced age.
Because of the same ethnicity and geographical region of the examined patients, we first searched for novel and rare genetic variations shared between all affected individuals, and although we did not find any mutation to be shared by all examined patients, we found the same gene, CSMD1, to be mutated in both families. Both CSMD1 mutations segregated with disease status, were not previously reported in public databases, were absent in a large number of neurologically normal individuals, were highly conserved among other orthologous, and were predicted to be pathogenic by various computational methods. This, along with previously reported CSMD1 association with the risk of PD,28 led us to believe in a possible role of CSMD1 genetic variability in the pathogenesis of PD. The CSMD1 gene contains 70 coding exons and encodes for a large protein (3,564 aa) that contains multiple CUB and Sushi, also known as CCP, domains. It is primarily synthesized in the developing CNS and epithelial tissues, and its encoding protein is known to act as a regulator of complement activation and inflammation in the developing CNS.29 Complement activation is essential for synaptic pruning and plasticity and has recently been implicated in several brain-related disorders and functions, including schizophrenia, Alzheimer disease (AD), immediate episodic memory, and information processing.30–32 In particular, based on mice models of AD, it can be said that both the microglia and the component pathway might act as early mediators of hippocampal synapse loss and dysfunction before plaque formation and neuroinflammation.32 In addition, copy number variations within the CSMD1 locus have been reported in patients with AD vs controls,33 further supporting a role of the CSMD1 genetic variability and function in the pathogenesis of AD.
Therefore, taking into consideration the previous associations of CSMD1 and the HLA region with the risk of PD and the identification of disease-segregating CSMD1 mutations in familial LOPD, we hypothesize that CSMD1 genetic variability might also contribute to PD pathogenesis through mechanisms implicated in immune-related synaptic dysfunction. Although it will be important to address these pathologic functions, this study may highlight the complement pathway as an important therapeutic target in PD as it has been suggested in AD.
ACKNOWLEDGMENT
The authors thank the families, their relatives, and other participants for their contribution to this study. They also thank Vladimir Makarov for his contribution to data processing.
GLOSSARY
- AD
Alzheimer disease
- CCP
complement control protein
- fPD
familial Parkinson disease
- H&Y
Hoehn and Yahr
- INDEL
insertions/deletions
- LOPD
late-onset PD
- PD
Parkinson disease
- RBD
REM sleep behavior disorder
- RLS
restless legs syndrome
- SNV
single nucleotide variant
- WES
whole-exome sequencing
NOTE ADDED IN PROOF
The authors have added a citation for an article that was published online after acceptance of this manuscript.34
AUTHOR CONTRIBUTIONS
Dr. Martí-Massó and Dr. Paisán-Ruiz: study concept and design. Dr. Ruiz-Martínez, Dr. Azcona, Dr. Bergareche, and Dr. Paisán-Ruiz: acquisition of data. Dr. Ruiz-Martínez, Dr. Azcona, Dr. Bergareche, and Dr. Paisán-Ruiz: analysis and interpretation of data. Dr. Ruiz-Martínez, Dr. Bergareche, Dr. Martí-Massó, and Dr. Paisán-Ruiz: critical revision of the manuscript for important intellectual content. Dr. Martí-Massó and Dr. Paisán-Ruiz: study supervision.
STUDY FUNDING
This work was supported by the National Institute of Neurological Disorders and Stroke of the NIH (NINDS; R01NS079388; C.P.-R.).
DISCLOSURE
J. Ruiz-Martínez, L. Azcona, J. Bergareche, and J. Martí-Massó report no disclosures. C. Paisán-Ruiz serves on the editorial board of BMC Neurology and receives funding from the NINDS (R01NS079388) and the American Parkinson's Disease Association (APDA). Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Halliday G, Lees A, Stern M. Milestones in Parkinson's disease–clinical and pathologic features. Mov Disord 2011;26:1015–1021. [DOI] [PubMed] [Google Scholar]
- 2.Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet 2009;373:2055–2066. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Lewis P, Revesz T, Lees A, Paisan-Ruiz C. The genetics of Parkinson's syndromes: a critical review. Curr Opin Genet Dev 2009;19:254–265. [DOI] [PubMed] [Google Scholar]
- 4.Kalinderi K, Bostantjopoulou S, Fidani L. The genetic background of Parkinson's disease: current progress and future prospects. Acta Neurol Scand 2016;134:314–326. [DOI] [PubMed] [Google Scholar]
- 5.Hamza TH, Chen H, Hill-Burns EM, et al. . Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson's disease modifier gene via interaction with coffee. PLoS Genet 2011;7:e1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paisan-Ruiz C, Jain S, Evans EW, et al. . Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 2004;44:595–600. [DOI] [PubMed] [Google Scholar]
- 7.Polymeropoulos MH, Lavedan C, Leroy E, et al. . Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997;276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 8.Zimprich A, Benet-Pages A, Struhal W, et al. . A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011;89:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nalls MA, Pankratz N, Lill CM, et al. . Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 2014;46:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paisan-Ruiz C. LRRK2 gene variation and its contribution to Parkinson disease. Hum Mutat 2009;30:1153–1160. [DOI] [PubMed] [Google Scholar]
- 11.Paisan-Ruiz C, Lewis PA, Singleton AB. LRRK2: cause, risk, and mechanism. J Parkinsons Dis 2013;3:85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamza TH, Zabetian CP, Tenesa A, et al. . Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet 2010;42:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.International Parkinson Disease Genomics C, Nalls MA, Plagnol V, Hernandez DG, et al. . Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 2011;377:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pihlstrom L, Axelsson G, Bjornara KA, et al. . Supportive evidence for 11 loci from genome-wide association studies in Parkinson's disease. Neurobiol Aging 2013;34:1708.e7–1708.e13. [DOI] [PubMed] [Google Scholar]
- 15.Sidransky E, Nalls MA, Aasly JO, et al. . Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foo JN, Tan LC, Irwan ID, et al. . Genome-wide association study of Parkinson's disease in East Asians. Hum Mol Genet 2017;26:226–232. [DOI] [PubMed] [Google Scholar]
- 17.Marti-Masso JF, Bergareche A, Makarov V, et al. . The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and parkinsonism. J Mol Med (Berl) 2013;91:1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorostidi A, Marti-Masso JF, Bergareche A, Rodriguez-Oroz MC, Lopez de Munain A, Ruiz-Martinez J. Genetic mutation analysis of Parkinson's disease patients using multigene next-generation sequencing panels. Mol Diagn Ther 2016;20:481–491. [DOI] [PubMed] [Google Scholar]
- 19.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol 1999;56:33–39. [DOI] [PubMed] [Google Scholar]
- 20.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruiz-Martinez J, Krebs CE, Makarov V, Gorostidi A, Marti-Masso JF, Paisan-Ruiz C. GIGYF2 mutation in late-onset Parkinson's disease with cognitive impairment. J Hum Genet 2015;60:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DePristo MA, Banks E, Poplin R, et al. . A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makarov V, O'Grady T, Cai G, Lihm J, Buxbaum JD, Yoon S. AnnTools: a comprehensive and versatile annotation toolkit for genomic variants. Bioinformatics 2011;28:724–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Exome Variant Server. NHLBI Exome Sequencing Project (ESP). Seattle: Available at: http://evsgswashingtonedu/EVS/. Accessed July 20, 2017. [Google Scholar]
- 26.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagase T, Kikuno R, Ohara O. Prediction of the coding sequences of unidentified human genes. XXI. The complete sequences of 60 new cDNA clones from brain which code for large proteins. DNA Res 2001;8:179–187. [DOI] [PubMed] [Google Scholar]
- 28.Hill-Burns EM, Wissemann WT, Hamza TH, Factor SA, Zabetian CP, Payami H. Identification of a novel Parkinson's disease locus via stratified genome-wide association study. BMC Genomics 2014;15:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraus DM, Elliott GS, Chute H, et al. . CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J Immunol 2006;176:4419–4430. [DOI] [PubMed] [Google Scholar]
- 30.Athanasiu L, Giddaluru S, Fernandes C, et al. . A genetic association study of CSMD1 and CSMD2 with cognitive function. Brain Behav Immun 2016;61:209–216. [DOI] [PubMed] [Google Scholar]
- 31.Havik B, Le Hellard S, Rietschel M, et al. . The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol Psychiatry 2011;70:35–42. [DOI] [PubMed] [Google Scholar]
- 32.Hong S, Beja-Glasser VF, Nfonoyim BM, et al. . Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016;352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swaminathan S, Kim S, Shen L, et al. . Genomic copy number analysis in Alzheimer's disease and mild cognitive impairment: an ADNI Study. Int J Alzheimers Dis 2011;2011:729478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vilas D, Fernández-Santiago R, Sanchez-Rodriguez E, et al. . A novel p.Glu298Lys mutation in the ACMSD gene in sporadic Parkinson's disease. J Parkinsons Dis Epub 2017 Jun 30. [DOI] [PubMed] [Google Scholar]