

Multi-targeted nanocarrier
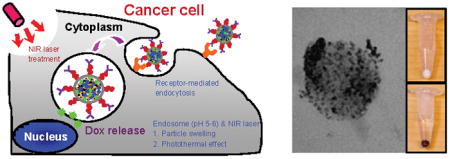
Multi-targeted drug delivery vehicles encapsulating doxorubicin within a pH-sensitive matrix, embedded with gold nanoparticles, and decorated on the surface with Herceptin®-polyethylene glycol conjugates were synthesized for enhanced specific binding and tumoricidal efficacy in HER2 overexpressing SK-BR-3 cells. We demonstrated that multiple modes of targeting were complementary and have potential for use as a cancer therapy.
Keywords: pH-sensitive nanoparticles, photothermal therapy, Au nanoparticles, drug delivery, cancer nanotechnology
Breast cancer is the second leading cause of cancer related deaths in women, surpassed only by lung cancer.[1] Current clinical therapies target estrogen receptor (ER) and human epidermal growth factor receptor-2 (HER2) to reduce cancer cell proliferation. These methods are often used in conjunction with surgery, chemotherapy, and/or radiation in efforts to eradicate the disease. However, 25% of patients face tumor recurrence and resistance within 5 years after treatment.[2] Ideally, the initial treatment would supply a robust, broad spectrum therapy to eliminate all cancer cells while minimizing damage to healthy tissue.
Tumor targeting strategies have the potential to improve the therapeutic efficacy of chemotherapeutic agents relative to systemic approaches. To date, targeted therapeutics are engineered using receptor-mediated or stimuli-responsive methods. Molecules that recognize and/or inhibit receptor function – alone, pegylated, or tethered to a cargo – improved targeting and reduced cancer cell proliferation and/or migration (e.g., folate,[3] anti-CXCR4,[4] anti-HER2).[5] Stimuli-sensitive therapeutics tailored delivery to the tumor site via activation by a physiological change or an external source, minimizing off-target effects. We previously synthesized a pH-sensitive drug delivery vehicle that triggered the release of paclitaxel in acidic tumor environments.[6] Thermal ablation of tumors, resulting from localized heating of near infrared (NIR) light activated carbon nanotubes[7] or gold particles,[8] were effective in the treatment of skin,[9] breast,[10] liver,[11] and ovarian cancers.[12] Individually, each targeted therapy provided a substantial advantage over nontargeted methods.
Integrating two targeting methods in one vehicle has improved tumoricidal efficacy relative to each approach used independently. A drawback of merging two targeting approaches is additional synthesis and purification steps that often result in low encapsulation of drug. Doxorubicin (Dox) encapsulation can reduce the overall efficacy of the chemotherapeutic relative to systemic administration.[13] Dox, an effective and widely used chemotherapeutic agent due to its hydrophilicity and high toxicity, suffers from low drug loading and fast release rates when encapsulated within polymeric nanoparticles. In addition, the simultaneous or step-wise delivery of heat and drug may drastically affect the therapeutic result.[14] A tunable drug delivery strategy engineered to provide the greatest therapeutic impact would be desirable.
In this report, we synthesized a single drug carrier that targets HER2, triggers Dox release, and thermally ablates breast cancer cells. The particles encapsulate Dox within a pH-sensitive matrix that is embedded with Au nanoparticles and decorated with polyethylene glycol (PEG)-HER2 conjugates within five steps (Figure 1). Dithiolated dimethylaminoethyl methacrylate (dT-DMAEMA), the pH-responsive monomer, was synthesized by reversible addition-fragmentation chain-transfer (RAFT) polymerization and characterized by nuclear magnetic resonance (NMR) (Figure S1, supporting information). Particles encapsulating Dox were prepared via an oil in water emulsification. Tuning the ratio of dT-DMAEMA/HEMA and buffer pH regulated the swelling properties of the matrix. We have previously shown that DMAEMA/HEMA matrices are sensitive to incremental changes in pH (>0.2 pH units).[6, 15] The pH-responsive matrix effectively acts as a logic gate, entrapping Dox within the matrix until the network is opened.
Figure 1.
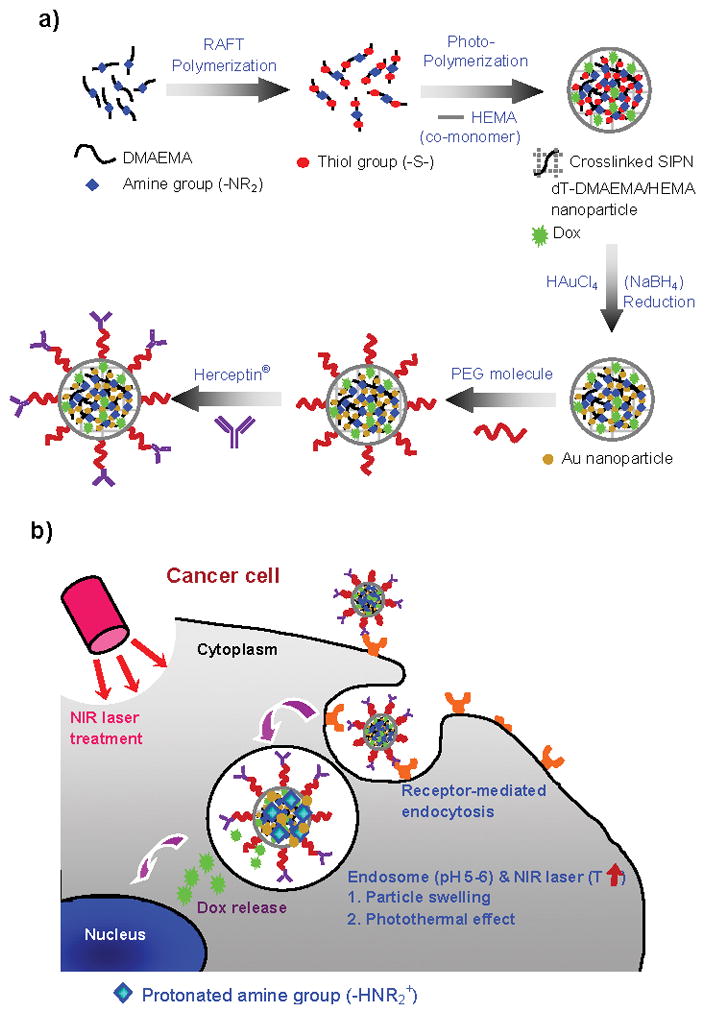
Schematic diagrams of a) Herceptin®-conjugated, Dox encapsulating, and pH-sensitive dT-DMAEMA/HEMA (HPG-Dox-30D70H) nanoparticle synthesis and b) cancer therapy.
Subsequently, gold (Au) nanoparticles are homogeneously formed throughout the pH-responsive matrix. Thiol groups in dT-DMAEMA have affinity for itself and both colloidal Au and Au ions. dT-DMAEMA/HEMA nanoparticles were immersed in an aqueous potassium tetrachloroaurate (KAuCl4) solution and followed by reduction in a sodium borohydride (NaBH4) solution.[16] As shown in Figure 2a, Au nanoparticles were homogeneously distributed within Dox encapsulating dT-DMAEMA/HEMA nanoparticles (30/70, mol/mol). This was also observed visually; white particles became brown after Au synthesis (Figure 2a–i and 2a–ii).
Figure 2.
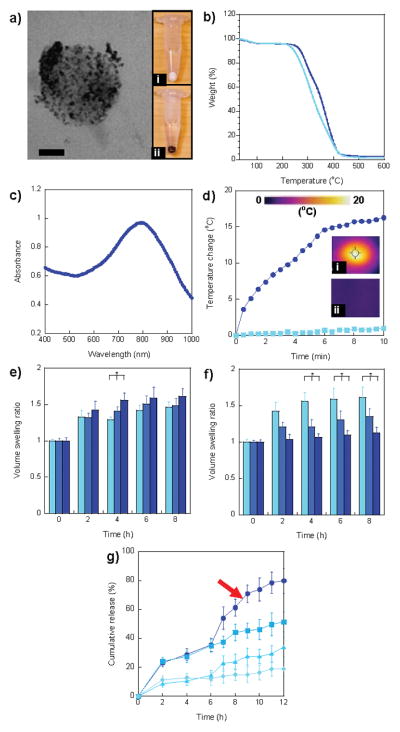
Characterization of multi-targeted nanoparticles. a) TEM image of HPG-Dox-30D70H nanoparticles. Au nanoparticles were homogeneously distributed in nanoparticles. The images of nanoparticle pellets after centrifugation of (i) Dox-30D70H (white) and (ii) HPG-Dox-30D70H (dark brown). Scale bar = 50 nm. b) TGA curves of HPG-Dox-10D90H (light blue) and HPG-Dox-30D70H (dark blue). c) UV-vis spectrum of HPG-Dox-30D70H nanoparticles exhibited a peak at 795 nm. d) Temperature change of HPG-Dox-30D70H in distilled water (5 mg/mL) and nanoparticle-free distilled water after NIR laser irradiation for 10 min. Images were taken using a NIR thermal camera for (i) HPG-Dox-30D70H in distilled water and (ii) only distilled water at 10 min NIR laser irradiation. e) Volume swelling ratios of HPG-Dox-10D90H (light blue), HPG-Dox-20D80H (blue), and HPG-Dox-30D70H (dark blue) in pH 5.5 20mM phosphate buffer at 0, 2, 4, 6, and 8 h. f) Volume swelling ratios of HPG-Dox-30D70H in pH 5.5 (light blue), 6.5 (blue), and 7.4 (dark blue) 20 mM phosphate buffers at 0, 2, 4, 6, and 8 h. g) Cumulative Dox release from HPG-Dox-30D70H (5 mg/mL) in pH 5.5 (square, dark blue curve) and 7.4 (diamond, light blue curve) 20 mM phosphate buffers over 12 h. To evaluate the photothermal effect induced by NIR irradiation, HPG-Dox-30D70H was incubated at pH 5.5 (circle) and 7.4 (triangle) in 20 mM phosphate buffer and then irradiated under a NIR laser for 6 h. The error is the standard deviation from the mean, where n = 3. * is P < 0.05.
Particles were functionalized on the exterior with a difunctional polyethylene glycol (SH-PEG-COOH, 10 μmol/10 mg particles), having both thiol and carboxylic acid groups at each end. Introduction of a thiol group allowed the noncovalent attachment of PEG to Au and dT-DMAEMA. Thiol-functionalized PEGs are useful for triggering the release of PEG within the glutathione rich cytoplasmic environment.[17] After separating unanchored PEG from free PEG in solution by dialysis, Herceptin® was conjugated via a carbodiimide reaction (9 nmol/10 mg particles). Herceptin® has been shown clinically to target and reduce proliferation in HER2+ breast tumors.[18] Herceptin®-PEG labeled, Gold embedded, Dox encapsulating, pH-responsive (30 dT-DMAEMA/70 HEMA, mol/mol) (HPG-Dox-30D70H) nanoparticles were designed to target HER2+ breast cancer cells, trigger the release of Dox, and induce hyperthermia upon activation with near infrared (NIR) laser irradiation.
The size and morphology of all nanoparticles were examined by transmission electron microscopy (TEM) (Figure 2a) and dynamic light scattering (Table S1, supporting information). Examination of the nanoparticles revealed a uniform and smooth surface morphology as shown in Figure S2a (supporting information). The average diameter of HPG-Dox-30D70H nanoparticles was 186.4 ± 18.6 nm (Figure S2b, supporting information) with a negative surface charge of −7.3 ± 2.1 mV. The size and charge of HPG-Dox-30D70H are suitable for systemic delivery; these nanocarrier traits have resulted in the enhanced permeability and retention (EPR).[19] The negative charge of HPG-Dox-30D70H is caused by unreacted PEG-COOH. The amine groups in DMAEMA become protonated as the pH decreases. As the dT-DMAEMA content increased from 10 to 30%, the zeta potential rose from −18.9 to −7.3 mV in parallel. Nanoparticle aggregation in serum free medium was not observed during cell viability and targeting experiments.
The Au content of HPG-Dox-10D90H (10 dT-DMAEMA/90 HEMA, mol/mol) and HPG-Dox-30D70H was characterized by thermogravimetric analysis (TGA), via heating from 20 to 600°C under continuous argon flow (Figure 2b). TGA analysis of Au embedded, pH-sensitive nanoparticles revealed the content of Au nanoparticles was 1.45 ± 0.10% and 2.71 ± 0.22% (w/w) for HPG-Dox-10D90H and HPG-Dox-30D70H, respectively. Au density increased with dT-DMAEMA content. Our previous report demonstrated that under similar reaction conditions the in situ synthesis of Au colloids was dictated by initial thiol content.[16b] The encapsulation of Dox had no effect on the ability to synthesize Au colloids within dT-DMAEMA/HEMA matrices. The UV-vis spectrum of HPG-Dox-30D70H nanoparticles indicated absorbance in the near infrared range with a peak occurring at 795 nm (Figure 2c).
To demonstrate the potential of HPG-Dox-30D70H for photothermal cancer therapy, HPG-Dox-30D70H nanoparticles were exposed under NIR laser irradiation at 810 nm with 5 W/cm2 for 10 min. Changes in temperature were measured and imaged by a thermal camera over the duration of the experiment (Figure 2d–i, 2d–ii). Representative thermal images taken at different time points of NIR exposure for HPG-Dox-30D70H nanoparticles (5 mg/mL) show the temperature distribution within each well (Figure S3). The HPG-Dox-30D70H solution resulted in a 15.3°C increase compared to distilled water, which increased by almost 1°C. During NIR laser irradiation, particle size was not affected by changes in temperature.
In addition to local heating, the nanocarriers were designed to swell upon changes in pH, triggering the release of Dox. Volumetric swelling was measured as a function of the molar ratio of dT-DMAEMA to HEMA and the pH of the swelling medium (Figure 2e and 2f, respectively). The volume swelling ratio is defined as the nanoparticle diameter after swelling divided by the original diameter.[20] Figure 2e depicts the swelling ratio for molar ratios of 10/90, 20/80, and 30/70 dT-DMAEMA/HEMA (mol/mol) as a function of time. The swelling ratio of dT-DMAEMA/HEMA nanoparticles increased with the dT-DMAEMA content due to the greater number of protonated amine groups. After 4 h at pH 5.5, the swelling ratio of HPG-Dox-10D90H and HPG-Dox-30D70H increased from 1.29 ± 0.04 and 1.56 ± 0.09, respectively. To investigate the pH sensitivity of nanoparticles, the swelling ratio of HPG-Dox-30D70H was measured as a function of pH (Figure 2f). The swelling ratio increased from 1.13 ± 0.07 at pH 7.4 to 1.61 ± 0.14 at pH 5.5. The nanoparticle formulation may be tuned to control pH-induced swelling.
Dox was encapsulated during particle synthesis. The encapsulation efficiencies of Dox in HP-Dox-30D70H (without gold) and HPG-Dox-30D70H nanoparticles were 95.2 ± 3.1% and 72.3 ± 6.7%. The reduction in Dox loading was a result of multiple washing steps after Au synthesis and antibody conjugation. Dox encapsulation is high within liposome formulation (greater than 90%)[21]; polymeric nanoparticles have failed to show similar results.[22]
To confirm that pH-induced swelling triggered Dox, the release of Dox was measured as a function of time at pH 5.5 and 7.4 (Figure 2g). Dox release was significantly enhanced under acidic conditions. After 8 h, 44.05 ± 5.79% of encapsulated Dox was released at pH 5.5 compared to 15.01 ± 3.80% at pH 7.4. In comparison, other pH-sensitive nanocarriers reported 25% Dox release after 24 h at pH 5.5[23] or exhibited a significant burst release (50% of loaded Dox).[13b] Our pH-sensitive nanocarriers exhibited a slow, controlled release of Dox at pH 7.4 and then a rapid release upon a decrease in pH.
We also investigated the ability of HPG-Dox-30D70H nanoparticles to release Dox by NIR laser irradiation. HPG-Dox-30D70H nanoparticles were allowed to release Dox in pH 5.5 buffer for 6 h and then heated (810 nm, 5 W/cm2, 6 h). As depicted in Figure 2g, Dox release was enhanced at pH 5.5 after laser irradiation compared to the nonirradiated control. The temperature of the well increased from 37.7 to 48.9°C after 5 min NIR irradiation which significantly increased Dox release (Figure S3, supporting information). Approximately 30% of the initial dose was delivered via a pH change; subsequent irradiation delivered an additional 50% of the entrapped drug. At pH 7.4, only 10% of the initial dose was available after 6 h, which increased to 30% of the entrapped drug after laser irradiation. Coupling pH-triggered release and localized heating into one carrier yielded enhanced Dox delivery. This synergistic delivery approach provided tunable release of Dox within the tumor microenvironment.
The HPG-Dox-30D70H nanoparticles were conjugated with Herceptin® for targeting HER2+ breast cancer cells. The number of Herceptin® molecules on the nanoparticle surface was determined by a DC protein assay (Table S2, supporting information).
Approximately, 116 and 117 Herceptin® molecules were conjugated on HPG-30D70H and HPG-Dox-30D70H nanoparticles, respectively. The binding affinity of HPG-Dox-30D70H to a breast cancer cell line with low HER2 expression, MCF-7, and a HER2 overexpressing breast cancer cell line, SK-BR-3, were measured using flow cytometry (Figure 3). As a control, immunoglobulin G-PEG labeled, gold embedded, Dox encapsulating, pH-responsive (30 dT-DMAEMA/70 HEMA, mol/mol) (IPG-Dox-30D70H) nanoparticles were prepared. As expected, HPG-Dox-30D70H nanoparticles exhibited a 5.4-fold enhancement in binding SK-BR-3 cells relative to MCF-7 cells. These results confirmed previous reports claiming that Herceptin® may be used to distinguish between cell lines with low and high HER2 expression.[24] Our HPG-Dox-30D70H nanocarriers exhibited similar results compared to prior reports of HER2 targeting.[22]
Figure 3.
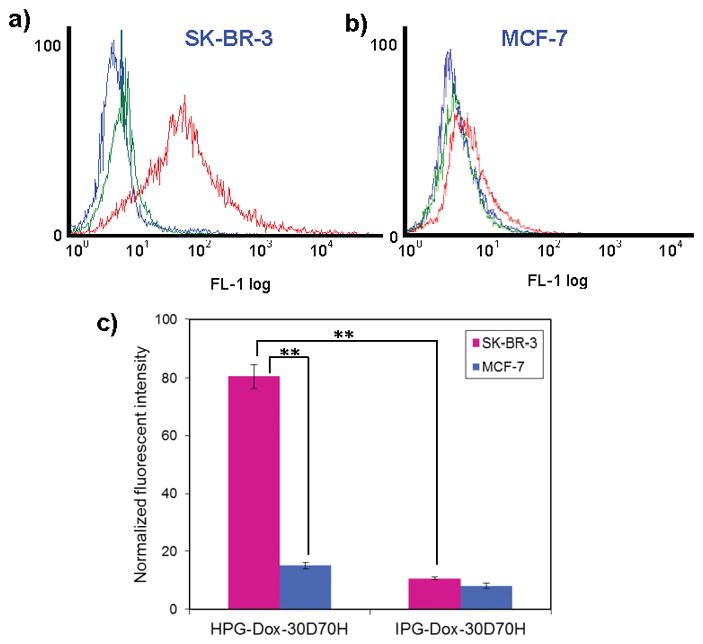
Flow cytometry analysis of a) SK-BR-3 and b) MCF-7 cells treated with HPG-Dox-30D70H or IPG-Dox-30D70H. Blue, green, and red curves represent non-treated, IPG-Dox-30D70H treated, and HPG-Dox-30D70H treated samples, respectively. c) Normalized fluorescent intensities using flow cytometry. The error is the standard deviation from the mean, where n = 3. ** is P < 0.001.
To investigate the therapeutic efficacy of HPG-Dox-30D70H, SK-BR-3 and MCF-7 cells were treated with HPG-Dox-30D70H with and without laser irradiation (at 0.26 μM Dox equivalent). This dose was 10-fold lower than the half maximal inhibitory concentration (IC50) for free Dox (Figure S4, supporting information). Nontargeting IPG-Dox-30D70H was used as a control. Cell viability was measured quantitatively using a fluorescent plate reader and qualitatively via microscopy. SK-BR-3 cells treated with HPG-30D70H (without Dox) and HPG-Dox-30D70H without laser irradiation at pH 7.4 exhibited 77.31 ± 4.13% and 51.81 ± 5.39% cell viability, respectively. After NIR laser irradiation, SK-BR-3 cell viability significantly decreased to 31.23 ± 7.68% and 14.49 ± 3.60% for HPG-30D70H and HPG-Dox-30D70H, respectively. HPG-Dox-30D70H (Figure 4a–b) showed an enhanced tumoricidal effect compared to the antibody labeled nanoparticle without Dox, HPG-30D70H (Figure 4c–d). When laser irradiation was added, the tumoricidal effect was enhanced by both increased temperature and Dox release. SK-BR-3 cells, treated with nanoparticles that coupled receptor targeting, chemotherapy, and thermal ablation, resulted in a maximal 14% cell survival. Given the low DOX concentration and mild irradiation conditions, this level of toxicity is significantly more efficacious than use of systemic Dox or combinations of Herceptin®/Dox, Herceptin®/NIR, or Dox/NIR.
Figure 4.
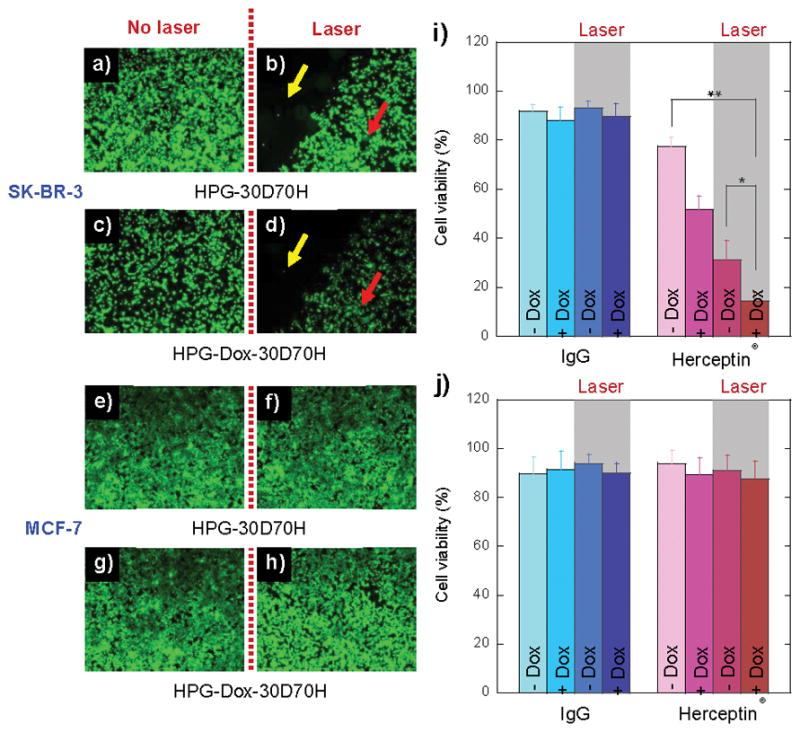
Fluorescent microscopy images of SK-BR-3 (a–d) and MCF-7 (e–h) cells treated with HPG-30D70H and HPG-Dox-30D70H with or without NIR laser illumination. Multi-targeted nanocarriers were conjugated with Herceptin® (a–h) or IgG (Figure S3c–l, supporting information). Yellow and red arrows indicate the NIR laser treated and non-treated areas, respectively. Cell viabilities of SK-BR-3 (i) and MCF-7 (j) cells were determined by a resazurin-based toxicology assay. Cells were treated with nanocarriers conjugated with either Herceptin® or IgG. The error is the standard deviation from the mean, where n = 3. * is p < 0.05 and ** is P < 0.01.
Conversely, MCF-7 cells did not show a significant difference between HPG-30D70H and HPG-Dox-30D70H in the absence of NIR laser irradiation. After NIR laser exposure, the cell viability of MCF-7 cells treated with HPG-30D70H (Figure 4e–f) and HPG-Dox-30D70H (Figure 4g–h) were not significantly different. This is attributed to the lack of targeting, low dose, and reduced Dox release.
IPG-30D70H and IPG-Dox-30D70H were used as controls for cell toxicity experiments. As shown in graphs Figure 4i–j and fluorescent images in Figure S5 (supporting information), the tumoricidal effect was hindered due to non-specific targeting. The nanocarriers without Dox were not cytotoxic (approximately 91–93% cell viability for both cell lines with and without Au-synthesis). The viability results were similar to poly(lactic-co-glyoclide) (PLGA) nanoparticles (nearly 93% for both cell lines), which are widely-used for systemic drug delivery (Figure S6, supporting information).
Other “multifunctional” vehicles have incorporated two modalities, either Dox/NIR,[25] Dox/targeting,[26] or targeting/NIR.[22] These papers reported significantly better results than the uni-dimensional control: either Dox, NIR, or targeting moiety alone. Ideally, Dox delivery would be localized, to avoid off-target effects, and released quickly. Dox encapsulating PLGA nanoparticles exhibit slow release.[27] Stimuli-triggered delivery could be beneficial. However, previous reports of pH-sensitivity exhibited either slow release or an uncontrolled burst release.[28] The vehicles presented here are different; they have a high encapsulation efficiency and can trigger Dox release within hours given a small pH change.
Functionalization of particles with an antibody or peptide is a conventional approach for targeting. Nanocarriers that couple both a targeting moiety and either Au nanoparticles[22] or Dox[26] on the surface, show a significant decline in targeting ability. We chose Herceptin® because it was currently used in the clinic. Herceptin® functionalized nanoparticles (HPG-30D/70H), without Dox or laser irradiation, resulted in 77% cell viability. Herceptin® alone did not result in a significant tumoricidal effect. Targeting with the addition of encapsulated Dox has shown to be useful; however, we have shown that some cancer cell types are resistant to this type of therapy and require alternate methods to be employed.[29]
Laser irradiation is a powerful technique that can achieve penetration depths of up to 50 mm.[30] HPG-30D/70H nanoparticles exhibited 30% cell viability after laser irradiation. Cancer cells were reported to be most vulnerable to hyperthermia and chemotherapeutics above 43°C.[14] Here, we used mild irradiation conditions (810 nm, 10 min, 5 W/cm2) to achieve a 15°C temperature increase whereas other reports required increased laser power to achieve similar results (10 min, 15 W/cm2;[22] 3 min, 32 W/cm2;[31] 7 min, 80 W/cm2).[32]
Our multi-targeted nanoparticles have gold nanoparticles embedded homogeneously throughout the matrix. Many drug delivery vehicles that induce local heating via NIR irradiation use gold surface coatings.[22] Instead, our method employed polymers as a template for Au colloids. We have demonstrated that the NIR thermal effect on Au nanoparticles is similar to Au coatings (Fig. 2d). Additionally, our templating method is suitable for manufacturing and scale-up.
The fusion of three targeting modalities exhibited a significant benefit relative to each method alone. The multi-targeted drug delivery platform resulted in 14% cell viability whereas the equivalent dosage of free Dox resulted in 81% cell viability. Addition of Herceptin® coupled with pH-triggered Dox or NIR ablation increased toxicity over free Dox but did not attain the levels of combining all three methods.
In conclusion, we have synthesized a unique multi-targeted vehicle that couples three modalities: targeting, triggered release and thermal ablation. HPG-Dox-30D70H vehicles had a high loading efficiency and exhibited pH-responsive Dox release, which was enhanced by laser irradiation. Enhanced targeting relative to IgG labeled nanocarriers was achieved by conjugating Herceptin®-PEG conjugates to the surface. Critical to translation, these particles are relatively simple to prepare with high reproducibility. Our acquired toxicity suggested that multi-targeted delivery has great potential as a cancer therapy. HPG-Dox-30D70H is the first carrier to integrate targeting, stimuli-responsive drug delivery, and thermal ablation for use in breast cancer research.
Supplementary Material
Footnotes
The authors would like to thank Prof. David Mooney and Dr. Praveen Arany for use of the NIR laser device and Dariela Almeda for FACS analysis. We gratefully acknowledge financial support from the Kavli Institute for Bionano Science and Technology at Harvard University. This work was supported by the NIH (1DP2CA174495) and NSF (DMR-0820484). This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the National Science Foundation under NSF award no. ECS-0335765. CNS is part of the Faculty of Arts and Sciences at Harvard University.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Prof. Jin-Oh You, Department of Industrial Chemical Engineering, Chungbuk National University, Cheongju, 361-763 (Republic of Korea)
Dr. Peng Guo, Department of Surgery, Harvard Medical School, Boston, MA 02115 (USA). Vascular Biology Program, Children’s Hospital Boston, Boston, MA 02115 (USA). Department of Biomedical Engineering, The City College of New York, New York, NY 10031 (USA)
Prof. Debra T. Auguste, Department of Surgery, Harvard Medical School, Boston, MA 02115 (USA). Vascular Biology Program, Children’s Hospital Boston, Boston, MA 02115 (USA). Department of Biomedical Engineering, The City College of New York, New York, NY 10031 (USA)
References
- 1.Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, Franssen E, Pritchard KI, Slingerland JM. Nat Med. 1997;3:227. doi: 10.1038/nm0297-227. [DOI] [PubMed] [Google Scholar]
- 2. [Accessed on September 27, 2012]; http://www.umm.edu/patiented/articles/how_serious_breast_cancer_000006_6.htm.
- 3.Poon Z, Chen S, Engler AC, Lee HI, Atas E, von Maltzahn G, Bhatia SN, Hammond PT. Angew Chem Int Ed. 2010;49:7266. doi: 10.1002/anie.201003445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ottaiano A, di Palma A, Napolitano M, Pisano C, Pignata S, Tatangelo F, Botti G, Acquaviva AM, Castello G, Ascierto PA, Iaffaioli RV, Scala S. Cancer Immunol Immunother. 2005;54:781. doi: 10.1007/s00262-004-0636-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Cancer Res. 1998;58:2825. [PubMed] [Google Scholar]
- 6.You JO, Auguste DT. Biomaterials. 2008;29:1950. doi: 10.1016/j.biomaterials.2007.12.041. [DOI] [PubMed] [Google Scholar]
- 7.a) Moon HK, Lee SH, Choi HC. ACS Nano. 2009;3:3707. doi: 10.1021/nn900904h. [DOI] [PubMed] [Google Scholar]; b) Fisher JW, Sarkar S, Buchanan CF, Szot CS, Whitney J, Hatcher HC, Torti SV, Rylander CG, Rylander MN. Cancer Res. 2010;70:9855. doi: 10.1158/0008-5472.CAN-10-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Huang XH, Jain PK, El-Sayed IH, El-Sayed MA. Lasers Med Sci. 2008;23:217. doi: 10.1007/s10103-007-0470-x. [DOI] [PubMed] [Google Scholar]; b) Zharov VP, Mercer KE, Galitovskaya EN, Smeltzer MS. Biophys J. 2006;90:619. doi: 10.1529/biophysj.105.061895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan H, Fales AM, Vo-Dinh T. J Am Chem Soc. 2012;134:11358. doi: 10.1021/ja304180y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci USA. 2003;100:13549. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen JY, Wang DL, Xi JF, Au L, Siekkinen A, Warsen A, Li ZY, Zhang H, Xia YN, Li XD. Nano Lett. 2007;7:1318. doi: 10.1021/nl070345g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu SY, Liang ZS, Gao F, Luo SF, Lu GQ. J Mater Sci - Mater Med. 2010;21:665. doi: 10.1007/s10856-009-3895-x. [DOI] [PubMed] [Google Scholar]
- 12.Chen WX, Bardhan R, Bartels M, Perez-Torres C, Pautler RG, Halas NJ, Joshi A. Mol Cancer Ther. 2010;9:1028. doi: 10.1158/1535-7163.MCT-09-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Lee SM, Park H, Choi JW, Park YN, Yun CO, Yoo KH. Angew Chem Int Ed. 2011;50:7581. doi: 10.1002/anie.201101783. [DOI] [PubMed] [Google Scholar]; b) Du JZ, Sun TM, Song WJ, Wu J, Wang J. Angew Chem Int Ed. 2010;49:3621. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]
- 14.Park JH, von Maltzahn G, Ong LL, Centrone A, Hatton TA, Ruoslahti E, Bhatia SN, Sailor MJ. Adv Mater. 2010;22:880. doi: 10.1002/adma.200902895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) You JO, Auguste DT. Nano Lett. 2009;9:4467. doi: 10.1021/nl902789s. [DOI] [PubMed] [Google Scholar]; b) You JO, Auguste DT. Biomaterials. 2010;31:6859. doi: 10.1016/j.biomaterials.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 16.a) You YZ, Manickam DS, Zhou QH, Oupicky D. J Controlled Release. 2007;122:217. doi: 10.1016/j.jconrel.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) You JO, Auguste DT. Langmuir. 2010;26:4607. doi: 10.1021/la100294p. [DOI] [PubMed] [Google Scholar]
- 17.Hong R, Han G, Fernández JM, Kim BJ, Forbes NS, Rotello VM. J Am Chem Soc. 2006;128:1078. doi: 10.1021/ja056726i. [DOI] [PubMed] [Google Scholar]
- 18.a) Baselga J. Eur J Cancer. 2001;37:S18. [PubMed] [Google Scholar]; b) Miles DW. Breast Cancer Res. 2001;3:380. doi: 10.1186/bcr326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torchilin VP. Pharm Res. 2007;24:1. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 20.Caldorera-Moore M, Kang MK, Moore Z, Singh V, Sreenivasan SV, Shi L, Huang R, Roy K. Sofr Matter. 2011;7:2879. [Google Scholar]
- 21.Dos Santos N, Cox KA, McKenzie CA, van Baarda F, Gallagher RC, Karlsson G, Edwards K, Mayer LD, Allen C, Bally MB. BBA-Biomembranes. 2004;1661:47. doi: 10.1016/j.bbamem.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Lee J, Kang J, Oh SJ, Ko HJ, Son JH, Lee K, Suh JS, Huh YM, Haam S. Adv Mater. 2009;21:4339. doi: 10.1002/adma.200900334. [DOI] [PubMed] [Google Scholar]
- 23.Lee CH, Cheng SH, Huang IP, Souris JS, Yang CS, Mou CY, Lo LW. Angew Chem Int Edit. 2010;49:8214. doi: 10.1002/anie.201002639. [DOI] [PubMed] [Google Scholar]
- 24.Wartlick H, Michaelis K, Balthasar S, Strebhardt K, Kreuter J, Langer K. J Drug Target. 2004;12:461. doi: 10.1080/10611860400010697. [DOI] [PubMed] [Google Scholar]
- 25.Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ. Proc Natl Acad Sci USA. 2009;107:981. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi M, Ho K, Keating A, Shoichet MS. Adv Funct Mater. 2009;19:1689. [Google Scholar]
- 27.a) Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. Eur J Pharm Sci. 2009;37:141. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Betancourt T, Brown B, Brannon-Peppas L. Nanomedicine. 2007;2:219. doi: 10.2217/17435889.2.2.219. [DOI] [PubMed] [Google Scholar]
- 28.a) Hruby M, Konak C, Ulbrich K. J Control Release. 2005;103:137. doi: 10.1016/j.jconrel.2004.11.017. [DOI] [PubMed] [Google Scholar]; b) Gao ZG, Lee DH, Kim DI, Bae YH. J Drug Target. 2005;13:391. doi: 10.1080/10611860500376741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo P, You JO, Yang J, Moses MA, Auguste DT. Biomaterials. 2012;33:8104. doi: 10.1016/j.biomaterials.2012.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esnouf A, Wright PA, Moore JC, Ahmed S. Acupunct Electrother Res. 2007;32:81–86. doi: 10.3727/036012907815844165. [DOI] [PubMed] [Google Scholar]
- 31.You J, Zhang R, Zhang G, Zhong M, Liu Y, Van Pelt CS, Liang D, Wei W, Sood AK, Li C. J Controlled Release. 2012;158:319. doi: 10.1016/j.jconrel.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loo C, Lowery A, Halas N, West J, Drezek R. Nano Lett. 2005;5:709. doi: 10.1021/nl050127s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.