

Abstract
Background and Purpose
In the present study, we reviewed the outcome of ketogenic diet (KD) use for the management of epilepsy in children with tuberous sclerosis complex (TSC).
Methods
A total of 12 children with intractable epilepsy associated with TSC who were treated with KD at our hospital between March 1, 2008 and February 28, 2015 were retrospectively enrolled.
Results
The mean age at the time of KD initiation was 73.1 ± 38.0 months. Patients were medically refractory to a mean of 4.8 ± 1.7 antiepileptic drugs. Nine patients (75.0%) had a history of infantile spasms. At 3 months after KD initiation, 10 patients (83.3%) had > 50% seizure reduction. Moreover, 7 patients (58.3%) exhibited qualitative improvements in cognition and behavior after KD initiation, as reported by caregivers/parents. The mean duration of dietary therapy was 14.8 ± 12.8 months. Half of the patients in this study eventually underwent epilepsy surgery due to persistent seizures or seizure relapse.
Conclusion
KD is an important non-pharmacological treatment option for patients with intractable epilepsy associated with TSC. KD may improve cognition and behavior in addition to reducing seizure frequency.
Keywords: Ketogenic diet, Epilepsy, Tuberous sclerosis complex, Children
Introduction
Tuberous sclerosis complex (TSC) is an inherited multisystem disorder that is typically caused by mutations in TSC1 or TSC2, affecting 1 in every 6,000 or 10,000 persons, respectively.1–3 TSC is characterized by pleomorphic features involving several organ systems and hallmark central nervous system (CNS) findings including cortical tubers, subependymal nodules (SEN), and subependymal giant cell astrocytoma (SEGA).1,2,4,5 Neurologic involvement in TSC is associated with disease-related disability such as epilepsy, behavioral disorders, and cognitive deficits,4–8 with epilepsy being the most common, reported in 75% to 90% of patients.6,9 Moreover, 30% to 50% of patients with TSC are diagnosed with infantile spasms (IS), an epileptic encephalopathy syndrome, in their 1st year of life.10–13 TSC-associated neuropsychiatric disorder is another major pathological feature in patients, which affects clinical outcome as well as quality of life.5,7
Several treatment options are available to optimize neurodevelopmental function and manage epilepsy in patients with TSC, including antiepileptic drugs (AEDs), ketogenic diet (KD), epilepsy surgery (resective surgery or corpus callosotomy), vagus nerve stimulation, and mammalian target of rapamycin (mTOR) inhibitor therapy.6,9,12,14,15 In this study, we reviewed use of a KD in children with TSC and drug-resistant epilepsy and evaluated its effects on seizure burden and cognitive as well as behavioral outcomes.
Methods
We performed a retrospective chart review of all patients seen for TSC at the pediatric neurology department of Severance Children’s Hospital between March 1, 2008 and February 28, 2015. Patients were included if they had clinically definite TSC, had been on a KD for > 3 months, had seizures intractable to at least 2 AEDs, and were available for formal neuropsychological (NP) evaluation and parental global assessment for diet outcome. We excluded patients who had been on the low glycemic index diet, those with poor compliance for KD maintenance < 3 months, and those whose parents had difficulty counting seizures to determine seizure frequency. Finally, 12 patients were enrolled.
A KD (3:1 or 4:1 ratio of fat to carbohydrate and protein) or a modified Atkin’s diet was started without fasting. Patient data including gender, age at last follow up, brain magnetic resonance imaging (MRI) findings, age at seizure onset, number of AEDs used, history of infantile spasms, developmental status, epilepsy classification at KD initiation, age at KD initiation, lead-time to KD initiation, peak effect of KD, duration of KD, seizure outcome after KD, and parental global assessment of overall KD outcome were collected by chart review. Minor adjustments of AEDs were done during KD maintenance in some cases. Seizure outcomes were assessed at 3 months, 6 months, and 12 months after starting the KD, and were classified as follows: seizure free, > 90% reduction, 50% to 90% reduction, and < 50% reduction in seizures. The parental global assessment of overall KD outcome regarded the patient’s overall condition, including seizure burden as well as cognitive and behavioral features. Parental assessments were obtained at clinic visits, and were classified as much improved, somewhat improved, no interval change, and worsened.
NP evaluations were available in all 12 patients. As per formal NP evaluations, patients with an intelligence quotient (IQ) < 70 (or sometimes a Developmental quotient [DQ] or social quotient [SQ] in cases where an IQ was not obtained) were considered to be cognitively impaired.
The institutional review board of the Severance Hospital approved this study.
Results
Patient demographic and clinical characteristics
Of 156 children with TSC who were seen at our hospital during the study period, 133 (85.3%) were treated for epilepsy with multiple treatment modalities such as AEDs, KD, and surgery. KD was initiated in 18 patients. 6 patients were excluded due to loss to follow up or early discontinuation of the KD within 3 months. Ultimately, 12 of these patients were included in our analysis.
The 12 patients enrolled in this study were 5 boys and 7 girls with a mean age of 73.1 ± 38.0 months (range, 26.4–144.2 months). All 12 patients of had cortical tubers and SEN, and 3 patients (25.0%) had SEGA on brain MRI. Ten patients (83.3%) had experienced seizures in the 1st year of life and the mean age at seizure onset was 9.6 ± 12.5 months (range, 1.0–47.4 months). Patients were treated with a mean of 4.8 ± 1.7 AEDs (range, 2–7) for seizure control. Nine patients (75.0%) had a history of IS and all of them (9 patients with IS) failed to control spasms with vigabatrin, which is an effective in the treatment of TSC children with IS. Upon KD initiation, 6 patients (50%) had IS, 1 patient had Lennox-Gastaut Syndrome (LGS), and 5 (41.7%) patients had focal epilepsy. The mean age at KD initiation was 23.2 ± 21.7 months (range, 2.9–76.5 months) and the lead-time to KD initiation (i.e., the time interval from seizure onset to KD initiation) was 13.7 ± 13.5 months (range, 1.5–38.0 months). The mean duration of KD maintenance was 14.8 ± 12.8 months (range, 3.3–43.6 months). Seven patients (58.3%) had cognitive impairment at the time of KD initiation (Table 1, 2).
Table 1.
Demographics of patients
| Parameters | Value |
|---|---|
| Gender, n (%) | |
| Male | 5 (41.7) |
| Female | 7 (58.3) |
| Mean age at last follow up, mean ± SD (months) | 73.1 ± 38.0 (range, 26.4–144.2) |
| Brain MRI findings, n (%) | |
| Cortical tubers | 12 (100.0) |
| SEN | 12 (100.0) |
| SEGA | 3 (25.0) |
| Seizure onset in the 1st year of life, n (%) | 10 (83.3) |
| Seizure onset age, mean ± SD (months) | 9.6 ± 12.5 (range, 1.0–47.4) |
| No. of tried AEDs, mean ± SD (months) | 4.8 ± 1.7 (range, 2–7) |
| History of infantile spasms, n (%) | 9 (75.0) |
| Developmental status at KD initiation, n (%) | |
| IQ, DQ, or SQ 70 | 5 (41.7) |
| IQ, DQ, or SQ < 70 | 7 (58.3) |
SD, standard deviation; MRI, magnetic resonance imaging; SEN, subependymal nodules; SEGA, subependymal giant cell astrocytoma; No., Number; AEDs, anti-epileptic drugs;IQ, intelligence quotient; DQ, developmental quotient; SQ, social quotient.
Table 2.
Clinical characteristics and seizure outcomes related to KD
| Parameters | Value |
|---|---|
| Mean age at initiation of KD, mean ± SD (months) | 23.2 ± 21.7 (range, 2.9–76.5) |
| Lead-time to KD, mean ± SD (months) (i.e the time interval from seizure onset to KD initiation) | 13.7 ± 13.5 (range, 1.5–38.0) |
| Mean duration of KD, mean ± SD (months) | 14.8 ± 12.8 (range, 3.3–43.6) |
| Epilepsy classification at KD, n (%) | |
| IS | 6 (50.0) |
| LGS | 1 (8.3) |
| Focal epilepsy | 5 (41.7) |
| Seizure free after 3 months of KD, n (%) | 5 (41.7) |
| Seizure reduction more than 50% after 3 months of KD, n (%) | 10 (83.3) |
| Epilepsy surgery after KD, n (%) | 6 (50.0) |
KD, ketogenic diet; SD, standard deviation; IS, infantile spasms; LGS, Lennox-Gastaut syndrome.
Seizure outcomes
After 3 months of the KD, 4 patients (33.3%) were seizure free and 10 patients (83.3%) achieved a > 50% reduction in seizures. Two patients discontinued the KD after 3 months due to seizure aggravation. Among 10 patients who continued the KD for 6 months, 6 (60.0%) were seizure free and 8 (80%) achieved a > 50% reduction in seizures. Reasons for KD discontinuation after 6 months were limited effectiveness in 2 patients (50.0%), general weakness and weight loss in 1 patient (25.0%), and parental will in 1 patient (25.0%). Half of the initial 12 patients maintained the dietary therapy for more than 12 months. Of these, 2 (33.3%) were seizure free, 2 achieved a 90% reduction in seizures, and 2 had a < 50% reduction in seizures after 12 months of the KD (Table 2, 3).
Table 3.
Detailed profiles of all patients: tuberous sclerosis complex patients with epilepsy treated by KD
| No. of patients | Age at KD initiation (months) | Epilepsy classification at KD initiation | Lead time to KD (months) | Ratio of KD | Duration of KD (months) | Peak effect of KD |
|---|---|---|---|---|---|---|
| 1 | 2.9 | IS | 1.8 | 3:1 → 4:1 | 3.3 | No interval change |
| 2 | 4.5 | IS | 1.5 | 4:1 | 3.9 | 90% Reduction for 3 months → aggravation |
| 3 | 76.5 | Focal epilepsy | 29.1 | MAD → 4:1 | 6.1 | No interval change |
| 4 | 6.0 | IS | 3.7 | 3:1 → 4:1 | 7.6 | 50% Reduction for 6 months |
| 5 | 3.4 | Focal epilepsy | 38.0 | 4:1 | 7.8 | No interval change |
| 6 | 15.0 | Focal epilepsy | 10.7 | 4:1 | 10.3 | Seizure free for 6 months → recur |
| 7 | 4.0 | IS | 2.3 | 3:1 → 4:1 → 2:1 | 12.2 | Seizure free for 3 months → recur |
| 8 | 8.0 | IS | 6.1 | 4:1 | 12.5 | 90% Reduction for 12 months → aggravation |
| 9 | 3.9 | IS | 4.5 | 3:1 → MAD | 12.5 | Seizure free for 9 months → recur |
| 10 | 6.9 | Focal epilepsy | 13.3 | 4:1 | 20.9 | 75% Reduction for 20 months → aggravation |
| 11 | 7.0 | Focal epilepsy | 17.0 | 4:1 → MAD | 36.5 | Seizure free for 8 months → recur |
| 12 | 9.0 | LGS | 35.8 | 4:1 → 3:1 | 43.6 | Seizure free over 60 months |
KD, ketogenic diet; No., number; IS, infantile spasms; LGS, Lennox-Gastaut syndrome; MAD, modified Atkin’s diet.
Of the 12 patients included in our analysis, 6 patients underwent epilepsy surgery after trying the KD; 3 of these patients (50.0%) experienced no effect on KD, 2 of them (33.3%) were seizure free with KD for initial several months, but recurred, and 1 of them (16.7%) had only partial effect by KD (75% reduction of seizure frequency for less than 3 months). 5 of above mentioned 6 patients (83.3%) were seizure free after surgery and 1 patient still experienced weekly seizures. Therefore, 8 of the 12 original patients included in our analysis achieved seizure free status at last follow up; 1 (12.5%) was seizure free with KD maintenance, 5 (62.5%) were seizure free after surgery, and 2 (25.0%) were seizure free after finding an appropriate AED regimen, even after KD discontinuation.
Cognitive and behavioral features
Efficacy of the KD was mainly assessed by seizure outcome; however, the global outcome by parental assessment included cognitive and behavioral features in addition to seizure burden. Four patients (33.3%) were “much improved,” 5 patients (41.7%) were “somewhat improved,” 2 patients (16.7%) showed “no interval change,” and 1 patient was (8.3%) “worsened” after 3 months of the KD (Fig. 1).
Figure 1.
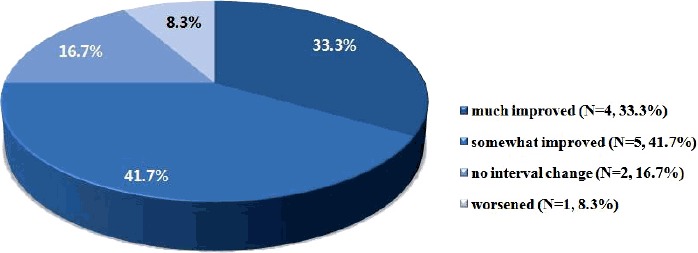
Global outcome by parental assessment for ketogenic diet on seizure, cognition and behavior.
Discussion
The present results suggest that KD maintenance is useful for the management of intractable seizures in patients with TSC. KD maintenance not only had beneficial effects on seizure frequency, but also produced overall improvements in cognition or behavior as rated by patient’s caretakers/parents. One-third of patients were seizure free and 83.3% patients showed a > 50% reduction in seizure frequency after 3 months of the KD. After 12 months of the KD, 4 of 6 patients (66.7%) achieved a > 50% reduction in seizures. Furthermore, 75.0% patients who maintained the KD for at least 3 months were reported to have overall improvement (much or somewhat improved) as per parental assessments. Given that cognitive and behavioral problems are main features of CNS involvement in TSC,5,8,16 and that 58.3% of patients had cognitive impairment defined by an IQ, SQ, or DQ of < 70 prior to dietary therapy in our study, it is possible that the KD has multifaceted utility for addressing CNS symptoms in TSC.
Few previous studies have reported the efficacy of a KD for the treatment of epilepsy associated with TSC.17,18 Our finding of a 50% to 90% reduction in seizure frequency at 6 months after diet initiation is consistent with that a previous study by Kossoff et al.17 Another study indicated that low glycemic index dietary therapy also produced good responses with minimal side effects in patients with epilepsy related to TSC.19
Patients with TSC are at high risk for early-onset seizures including focal seizures and IS.9 Approximately 30% of infants with TSC present with epileptic encephalopathy, which has a poor prognosis.6 In our study, 83.3% patients developed seizures in the 1st year of life and 75.0% of patients had a history of IS. Indeed, half of our study population started KD to treat IS. Future studies should examine whether the KD has particular usefulness in a subset of patients with a history of IS or active IS.
Neuropsychological deficits of TSC are associated with abnormalities in the mTOR signaling pathway, which has a crucial role in brain development. Previous studies have identified the ability of a KD to decrease mTOR activation in animal models, providing a biological basis for the mechanism of action for dietary treatment.3,20 Yet, the exact mechanism of KD-mediated benefits in TSC remains unknown and warrants further research.
This study had several limitations. First, we enrolled a small number of patients and lacked precise data about seizure types, electroencephalogram findings, and genetic studies. Second, considering the retrospective nature of the study, data may have been incomplete despite our efforts to be thorough in chart/record collection and review.
In conclusion, KD therapy is an important treatment option in cases of medically intractable epilepsy related to TSC. Considering the efficacy of the KD for seizure reduction and neuropsychological improvement in our study, additional studies should explore the therapeutic value and mechanism of a KD in TSC.
Acknowledgements
This study was supported by the Soonchunhyang University Research Fund.
Footnotes
Conflicts of Interest
The authors have no financial conflicts of interest.
References
- 1.Kassiri J, Snyder TJ, Bhargava R, Wheatley BM, Sinclair DB. Cortical tubers, cognition, and epilepsy in tuberous sclerosis. Pediatr Neurol. 2011;44:328–32. doi: 10.1016/j.pediatrneurol.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Krueger DA. Management of CNS-related Disease Manifestations in Patients With Tuberous Sclerosis Complex. Curr Treat Options Neurol. 2013;15:618–33. doi: 10.1007/s11940-013-0249-2. [DOI] [PubMed] [Google Scholar]
- 3.Jülich K, Sahin M. Mechanism-based treatment in tuberous sclerosis complex. Pediatr Neurol. 2014;50:290–6. doi: 10.1016/j.pediatrneurol.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krueger DA, Northrup H International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49:255–65. doi: 10.1016/j.pediatrneurol.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015;14:733–45. doi: 10.1016/S1474-4422(15)00069-1. [DOI] [PubMed] [Google Scholar]
- 6.Saxena A, Sampson JR. Epilepsy in Tuberous Sclerosis: Phenotypes, Mechanisms, and Treatments. Semin Neurol. 2015;35:269–76. doi: 10.1055/s-0035-1552616. [DOI] [PubMed] [Google Scholar]
- 7.de Vries PJ, Whittemore VH, Leclezio L, et al. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. 2015;52:25–35. doi: 10.1016/j.pediatrneurol.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaroff CM, Devinsky O, Miles D, Barr WB. Cognitive and behavioral correlates of tuberous sclerosis complex. J Child Neurol. 2004;19:847–52. doi: 10.1177/08830738040190110101. [DOI] [PubMed] [Google Scholar]
- 9.Curatolo P, Jóźwiak S, Nabbout R. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16:582–6. doi: 10.1016/j.ejpn.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Thiele EA. Managing and understanding epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51( Suppl 1):90–1. doi: 10.1111/j.1528-1167.2009.02458.x. [DOI] [PubMed] [Google Scholar]
- 11.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–41. doi: 10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moavero R, Cerminara C, Curatolo P. Epilepsy secondary to tuberous sclerosis: lessons learned and current challenges. Childs Nerv Syst. 2010;26:1495–504. doi: 10.1007/s00381-010-1128-8. [DOI] [PubMed] [Google Scholar]
- 13.Ohtsuka Y, Ohmori I, Oka E. Long-term follow-up of childhood epilepsy associated with tuberous sclerosis. Epilepsia. 1998;39:1158–63. doi: 10.1111/j.1528-1157.1998.tb01306.x. [DOI] [PubMed] [Google Scholar]
- 14.Curatolo P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol. 2015;52:281–9. doi: 10.1016/j.pediatrneurol.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 15.Holmes GL, Stafstrom CE. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48:617–30. doi: 10.1111/j.1528-1167.2007.01035.x. [DOI] [PubMed] [Google Scholar]
- 16.Cusmai R, Moavero R, Bombardieri R, Vigevano F, Curatolo P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav. 2011;22:735–9. doi: 10.1016/j.yebeh.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 17.Kossoff EH, Thiele EA, Pfeifer HH, McGrogan JR, Freeman JM. Tuberous sclerosis complex and the ketogenic diet. Epilepsia. 2005;46:1684–6. doi: 10.1111/j.1528-1167.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- 18.Coppola G, Klepper J, Ammendola E, et al. The effects of the ketogenic diet in refractory partial seizures with reference to tuberous sclerosis. Eur J Paediatr Neurol. 2006;10:148–51. doi: 10.1016/j.ejpn.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Larson AM, Pfeifer HH, Thiele EA. Low glycemic index treatment for epilepsy in tuberous sclerosis complex. Epilepsy Res. 2012;99:180–2. doi: 10.1016/j.eplepsyres.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 20.McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:e7–11. doi: 10.1111/j.1528-1167.2011.02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]