

Abstract
Mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome is a rare autosomal dominant disorder caused by heterozygous POLD1 mutations. To date, 13 patients affected by POLD1 mutation-caused MDPL have been described. We report a clinically undiagnosed 11-year-old male who noted joint contractures at 6 years of age. Targeted exome sequencing identified a known POLD1 mutation [NM_002691.3:c.1812_1814del, p.(Ser605del)] that diagnosed him as the first Japanese/East Asian MDPL case.
Mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL, MIM#615381) syndrome is a rare autosomal dominant systemic disorder resulting from heterozygous mutations in POLD1 (MIM#174761).1–3 MDPL is clinically characterized by prominent loss of subcutaneous fat, characteristic facial appearance, metabolic abnormalities involving insulin resistance and diabetes mellitus, and sensorineural deafness occurring late in the first or second decades of life. To date, 18 patients affected by this syndrome have been described, including 1 Indian, 2 Hispanic and 15 Caucasians.1,2,4–6 Among the 13 POLD1 mutation-caused MDPL cases, all have been caused by one of two different POLD1 mutations: an in-frame deletion (Ser605del, 11 cases) and a missense mutation (R507C, 2 cases).2,4–6 In most of these cases, the mutation occurred de novo.2,4–6 Although these POLD1 mutations appear to occur in genic hotspots regardless of race/ethnicity, no East Asian cases (including Japanese) have been reported.
We herein report the first Japanese/East Asian case of MDPL in an 11-year-old male with characteristics of MDPL. We used targeted exome sequencing (TES) as a genome-first approach in a clinically undiagnosed Japanese patient and determined that he was carrying a known heterozygous POLD1 mutation.
The patient was an 11-year-old, first-born male child of healthy, nonconsanguineous Japanese parents with unremarkable family history (Figure 1a). He was born through normal vaginal delivery at full term with birth weight of 2.692 kg (−0.8 s.d.), body length of 46.6 cm (−1.1 s.d.) and occipitofrontal circumference of 33.2 cm (−0.1 s.d.). His early developmental milestones were normal, but his parents noticed poor height and weight gain when he was 3 years of age. At 6 years of age, school teachers noticed that he was unable to either perform kicking motions while swimming or sit on his heels, and an orthopedist pointed out the presence of joint contractures. At 7 years of age, he was referred to a pediatric endocrinology department due to his short stature. He presented with prominent eyes, beaked nose, mandibular hypoplasia, crowded teeth, small mouth and testicular hypoplasia. He was also diagnosed with moderate sensorineural bilateral hearing loss and he started to wear hearing aids. Growth hormone (GH) levels in a GH stimulation test were observed to be normal. Standard karyotyping using peripheral blood revealed no abnormalities (46,XY). No results consistent with known metabolic syndromes were obtained from metabolic surveys, including serum amino acids and urine organic acids. At 9 years of age, tight skin around his cheeks, hepatic steatosis and mild liver dysfunction were noted. At the age of 11, due to his short stature (weight; 24.6 kg (−1.7 s.d.), height; 125.2 cm (−2.9 s.d.) and body mass index; 15.7), facial features, deafness, primary hypogonadism, joint contractures and thin arms and legs with a wide trunk, the patient visited a division of clinical genetics to consider any genetic diseases (with informed consent from his parents). Although the patient’s clinical features were retrospectively consistent with the recently proposed clinical spectrum of MDPL caused by POLD1 mutations (Table 1),1,2,4–6 the patient remained undiagnosed. As a result, TES was considered using a panel of multiple potential disease-causing genes.
Figure 1.
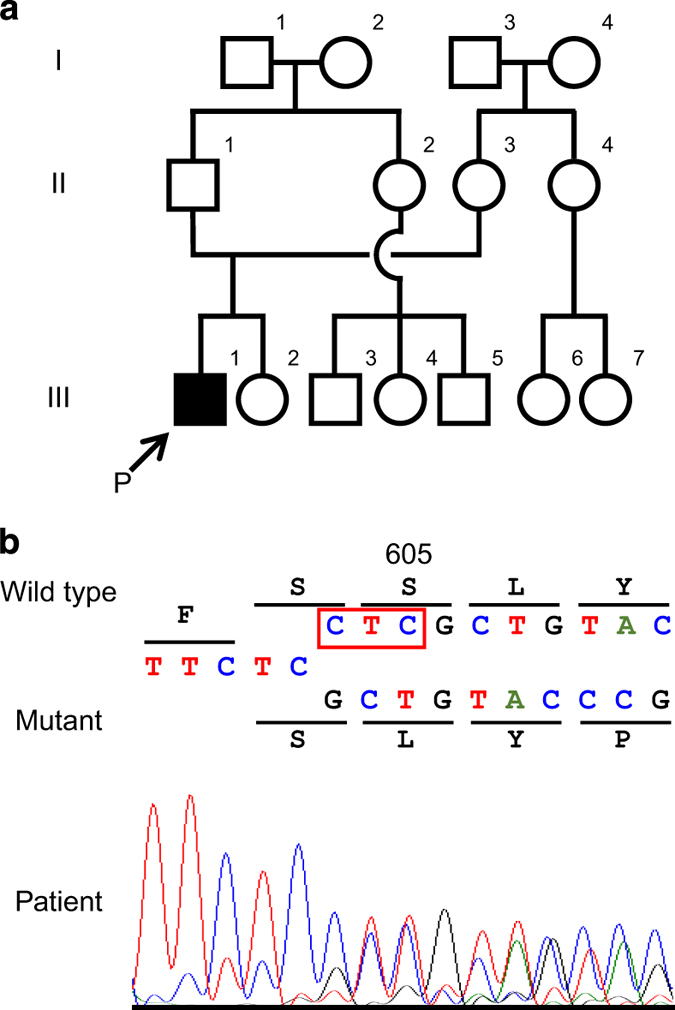
(a) Family pedigree; arrow shows the proband (P). (b) Partial sequence chromatograms around codon 605 on exon 15 of POLD1 in the patient. The red box denotes the deleted bases. The DNA and corresponding amino acid sequences of the wild-type and mutant POLD1 alleles are also shown.
Table 1. Clinical characteristics of the POLD1 mutation-caused MDPL patient presented here compared to previously described subjects.
| Clinical features | Study patient | Previous studiesa (n=13) |
|---|---|---|
| Age (years; range, median) | 11 | 10–62, 25 |
| Sex | Male | 6 males, 7 females |
| Birth weight (kg; range, mean) | 2.692 | 2.4–4.2, 3.23 (n=9) |
| Height (cm) | 125.2 | — |
| Weight (kg) | 24.58 | — |
| BMI (kg/m2; range, mean) | 15.7 | 13.8–26.8, 17.5 |
|
Metabolic profile | ||
| Diabetes mellitus | N | 5/13 |
| Hepatic steatosis | Y | 4/6 |
| ALT (U/l) | 52 (5–40) | Abnormal LFT 5/8 |
| Total cholesterol (mg/dl) | 141 (130–220) | High 8/10 |
| Triglycerides (mg/dl) | 125 (35–150) | High 9/11 |
| Leptin (ng/ml) | 10.9 | 4.4–8.2, 5.6 (n=4) |
|
Morphology | ||
| Short stature | Y | 8/13 |
| Tight skin around cheeks and small nasal bones | Y | 13/13 |
| Mandibular underdevelopment | Y | 12/13 |
| Dental overcrowding/irregular teeth | Y | 10/13 |
| Telangiectasia | N | 9/13 |
| Thin arms and legs with wide trunk | Y | 13/13 |
| High pitched voice | Y | 9/12 |
| Hearing impairment | Y | 10/13 |
|
Musculoskeletal | ||
| Joint contractures | Y | 5/13 |
| Muscle wasting | Y | 11/13 |
| Kyphosis/scoliosis | N | 4/5 |
| Hypogonadism | Y | 4/5 males |
| Abnormal cognitive function | N | 1/12 |
Abbreviations: ALT, alanine aminotransferase; BMI, body mass index; LFT, liver function test; MDPL, mandibular hypoplasia, deafness, progeroid features and lipodystrophy; N, no; Y, yes.
Previously reported cases with POLD1 mutation-caused MDPL.
After informed consent was obtained from the parents, molecular diagnosis was performed using genomic DNA extracted from the patient’s blood sample. The study was approved by the ethics committees of Tokushima University. To screen known disease-associated genes for molecular diagnosis, we used a TruSight One Sequencing Panel (Illumina, San Diego, CA, USA) with a MiSeq sequencer (Illumina), followed by our pipeline for next generation sequencing (NGS) data analysis as previously described,7,8 with a minor modification due to a software update specific for a bioinformatics pipeline.8 To identify presumably pathogenic single-nucleotide variants, we excluded sequence variants with low-allele frequencies, that is, >0.01 included in the 1000 Genomes Project database (http://www.1000genomes.org), National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS), Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB) and integrative Japanese Genome Variation Database (https://ijgvd.megabank.tohoku.ac.jp). Copy-number variations analysis using TES data was also performed as described elsewhere.8,9 These analyses detected an in-frame heterozygous deletion in exon 15 of POLD1, NM_002691.3(POLD1_v001):c.1812_1814del, affecting the polymerase-active site, NM_002691.3(POLD1_i001):p.(Ser605del), which was confirmed by Sanger sequencing (Figure 1b). This mutation has been shown to cause most cases of MDPL.2,6 No other variants or gross deletions were detected in the coding regions of other progeroid-related genes (data not shown). As a result of this molecular diagnosis and the re-evaluation of the affected patient’s clinical features, together with the clinical spectrum of patients harboring POLD1 mutations (Table 1),1,2,4–6 the patient was diagnosed with MDPL caused by a known frameshift deletion in POLD1. Because parental DNA was not available, we were unable to determine if the mutation occurred de novo.
To the best of our knowledge, the patient described herein is the 19th MDPL and the 14th POLD1 mutation-caused MDPL case reported worldwide. Notably, this patient is the first Japanese or East Asian case with MDPL, which is caused by the most common POLD1 in-frame deletion mutation (c.1812_1814del). Our case supports the hypothesis that POLD1 mutations causing MDPL, at least this in-frame deletion mutation, commonly occur at hotspots irrespective of race/ethnicity. The CTCCT motif occurs within two CTC triples, one of which is deleted in most MDPL cases, and corresponds to the complement of the mirror image of the ‘deletion hotspot consensus sequences’ TG(A/G) (A/G) (G/T) (A/C).10,11 In addition, (A/T)GGAG is one of the specific native DNA sequences known to arrest DNA synthesis by DNA polymerase α.12 Therefore, the arrest of DNA synthesis at this DNA polymerase pause site may increase the possibility that a slipped mispairing was mediated by direct repeats and/or secondary structure formation promoted by symmetric elements and might cause the commonly observed c.1812_1814del mutation in POLD1 in MDPL patients.5,11
Although clinical characteristics of the patient, including his phenotype, history of the disease and metabolic profile showed features fully and retrospectively compliant with the diagnosis of MDPL (Table 1),1,2,4–6 clinical diagnosis could not be evoked because of the rarity of this disease. Indeed, reported MDPL cases are clinically and/or genetically diagnosed at a relatively higher age (median age >20 years).1,2,4–6 By facilitating differential diagnosis of this syndrome and related diseases in a cost-effective manner, molecular diagnosis by a genome-first approach may be crucially important for providing appropriate therapeutic options and optimized health care in patients with unclassified segmental progeroid syndromes.5 In addition, correct diagnosis through this approach also can be useful in establishing recurrence risks and for providing appropriate genetic counseling to the family.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
We thank the patient and his family for their participation in this study. Parts of this work were performed at the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University. This work was supported by Japan Society for the Promotion of Science (16K15618, 15K19620) and Japan Agency for Medical Research and Development (16kk0205012h001, 16ek0109151h002).
Footnotes
The authors declare no conflict of interest.
References
- Shastry S, Simha V, Godbole K, Sbraccia P, Melancon S, Yajnik CS et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab 2010; 95: E192–E197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet 2013; 45: 947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas E, Golemis EA, Arora S. POLD1: central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016; 590: 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosini C, Martinelli S, Ceccarini G, Magno S, Barone I, Basolo A et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism 2014; 63: 1385–1389. [DOI] [PubMed] [Google Scholar]
- Reinier F, Zoledziewska M, Hanna D, Smith JD, Valentini M, Zara I et al. Mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome in the context of inherited lipodystrophies. Metabolism 2015; 64: 1530–1540. [DOI] [PubMed] [Google Scholar]
- Lessel D, Hisama FM, Szakszon K, Saha B, Sanjuanelo AB, Salbert BA et al. POLD1 germline mutations in patients initially diagnosed with Werner syndrome. Hum Mutat 2015; 36: 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Naruto T, Kohmoto T, Komori T, Imoto I. A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum Genome Var 2014; 1: 14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Nakagawa R, Naruto T, Kohmoto T, Suga K, Goji A et al. A novel missense mutation of COL5A2 in a patient with Ehlers-Danlos syndrome. Hum Genome Var 2016; 3: 16030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Hayabuchi Y, Ono A, Naruto T, Horikawa H, Kohmoto T et al. Detection of 1p36 deletion by clinical exome-first diagnostic approach. Hum Genome Var 2016; 3: 16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Cooper DN. Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum Genet 1991; 86: 425–441. [DOI] [PubMed] [Google Scholar]
- Ball EV, Stenson PD, Abeysinghe SS, Krawczak M, Cooper DN, Chuzhanova NA. Microdeletions and microinsertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat 2005; 26: 205–213. [DOI] [PubMed] [Google Scholar]
- Weaver DT, DePamphilis ML. Specific sequences in native DNA that arrest synthesis by DNA polymerase alpha. J Biol Chem 1982; 257: 2075–2086. [PubMed] [Google Scholar]
Data Citations
- Imoto Issei.HGV Database. 2017. 10.6084/m9.figshare.hgv.1393. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Imoto Issei.HGV Database. 2017. 10.6084/m9.figshare.hgv.1393. [DOI]