

Abstract
Background
Aberrant activation of the intracellular tyrosine kinase Src has been implicated as a mechanism of acquired chemotherapy resistance in metastatic colorectal cancer (mCRC). Here, the oral tyrosine kinase Src inhibitor, dasatinib, was investigated in combination with FOLFOX and cetuximab.
Methods
We performed a phase IB/II study of 77 patients with previously-treated mCRC. Primary objectives were to determine the MTD, dose-limiting-toxicities, pharmacodynamics, and efficacy. Using a 3+3 design, patients received FOLFOX6 with cetuximab and escalating doses of dasatinib(100, 150, 200mg daily), followed by a 12 patient expansion cohort at 150mg. Phase II studies evaluated FOLFOX plus dasatinib 100mg in KRAS c12/13mut patients, or in combination with cetuximab if KRAS c12/13WT. FAK and paxillin were utilized as surrogate blood biomarkers of Src inhibition, and paired biopsies of liver metastases were obtained in patients in the expansion cohort.
Results
In Phase IB, the dose-limiting-toxicities were grade 3/4 fatigue(20%), and neutropenia(23%). In Phase II, grade 3/4 fatigue(23%) and pleural effusions(11%) were present. Response rates were 20%(6/30) in the Phase IB escalation and expansion cohort, and 13%(3/24) and 0%(0/23) in the KRAS c12/13WT and mutant cohorts of Phase II, respectively. Median PFS was 4.6, 2.3, and 2.3 months, respectively. There was no evidence of Src inhibition based on surrogate blood biomarkers or paired tumor biopsies.
Conclusions
The combination of dasatinib plus FOLFOX with or without cetuximab showed only modest clinical activity in refractory CRC. This appears to be primarily due to a failure to fully inhibit Src at the achievable doses of dasatinib.
Keywords: Dasatinib, Src inhibitor, Colorectal cancer, Phase I/II
BACKGROUND
Colorectal cancer (CRC) remains one of the most common cancers worldwide, and the second and third leading cause of death in men and women in the United States, respectively.(1) For patients with surgically unresectable disease, the expected 5-year relative survival is less than 15%.(2) Despite significant advances in treatment of metastatic colorectal cancer (mCRC) with currently available regimens, response rates for those that progress after first line treatment are only 4–17%.(3,4)
Src family kinases have been implicated in drug resistance in mCRC. Cellular stimuli induce conformational changes that increase Src kinase activity via dephosphorylation of residue Y530 and autophosphorylation of residue Y418.(5) Src activation results in a variety of downstream signaling pathways involved in survival, angiogenesis, proliferation, and migration.(6–9) Src can potentiate the effects of receptor tyrosine kinases as well as directly activate PI3 kinase pathway that results in reduction of apoptosis signaling and increased cellular survival. Further, signaling through the RAS/RAF/MAPK results in increased proliferation and numerous other pro-proliferation phenotypes. By additionally interacting with cytoskeletal components including focal adhesion kinase (FAK), paxillin, and E-cadherin, among others, Src is a key regulator of tumor cell adhesion, migration, and invasiveness.(10) Src expression and activity have been shown to be higher in primary colon tumors compared to benign polyps, and in metastatic compared to non-metastatic colon cancer, highlighting its role in colorectal progression.(11–15)
Colon cancer cell lines with defective Src kinases are particularly sensitive to oxaliplatin-induced apoptosis.(16) Metastatic CRC patients who received neoadjuvant oxaliplatin demonstrated higher levels of Src pathway signaling in hepatic metastases, a finding associated with poorer relapse-free survival.(17)
Dasatinib is an orally bioavailable, potent, multi-targeted kinase inhibitor and an effective therapeutic agent for the treatment of several malignancies.(18) Although relatively specific for the Src and Abl family kinases, dasatinib possesses broad-spectrum inhibition of kinases including Kit, PDGFR, EphA receptors and many others.(19) Several preclinical studies have demonstrated the ability of Src inhibitors like dasatinib to overcome chemoresistance as well as resistance to targeted agents, such as the EGFR monoclonal antibody cetuximab.(20–23) Other preclinical studies suggest that the combination of dasatinib and oxaliplatin has additive, and possibly synergistic activity. CRC tumors treated with the combination of dasatinib and oxaliplatin demonstrate markedly decreased microvessel activity and increased oxidative stress.(9,24) In a mouse model of colorectal liver metastases, 28-day treatment with oxaliplatin resulted in chronic Src activation, and the combination of dasatinib plus oxaliplatin resulted in significantly smaller tumors compared to single agent treatment with oxaliplatin alone, corresponding with reduced proliferation and angiogenesis.(25)
We thus conducted a phase IB/II study combining the Src inhibitor dasatinib with a modified FOLFOX regimen with or without cetuximab in patients with previously treated mCRC, to evaluate its efficacy as well as its toxicity profile in this patient population.
PATIENTS AND METHODS
Eligibility Criteria
Eligible patients were required to have histologically or cytologically confirmed colorectal adenocarcinoma with metastatic disease documented on diagnostic imaging studies, mass-spectroscopy genotyping confirmed KRAS mutation status (exon 1, codon 12, 13), evaluable or measurable disease by Response Evaluation Guide for Solid Tumors (RECIST) version 1.0, and Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0 or 1. Patients also had to be ≥ 18 years of age with adequate hematopoietic, hepatic, and kidney function and a life expectancy ≥ 3 months. Each patient must have had previously progressed, either clinically or radiographically, on systemic therapy for mCRC, with no limit on the number of prior regimens. Patients in the Phase II cohort must have progressed on fluorouracil (5-FU) or capecitabine and oxaliplatin if KRAS mutant, and either cetuximab or panitumumab if KRAS wild type. Key exclusion criteria included recent (within 4 weeks of the first infusion of study drugs on this study) or planned participation in another experimental therapeutic drug study; systemic chemotherapy, radiotherapy, or major surgery within 21 days prior to the first infusion of study drugs; radiographic evidence of pleural effusions in the last 30 days prior to enrollment; known brain metastases; known dihydropyrimidine dehydrogenase deficiency; long QT syndrome; history of clinically significant ventricular arrhythmias; concurrent severe and/or uncontrolled medical conditions including uncontrolled high blood pressure (≥140/90), unstable angina or stable angina markedly limiting ordinary physical activity, New York Heart Association (NYHA) ≥ grade 2 congestive heart failure, myocardial infarction within 6 months of study enrollment, history of stroke within 6 months of study enrollment, unstable symptomatic arrhythmia requiring medication, clinically significant peripheral vascular disease, uncontrolled diabetes and serious active or uncontrolled infection. The trial was conducted in accordance with the Declaration of Helsinki. The protocol (ClinicalTrials.gov identifier: NCT00501410) was approved by the Institutional Review Board at U.T. MD Anderson Cancer Center, and written informed consent was obtained for all patients before performing study-related procedures.
For the Phase II cohort, patients were categorized as KRAS G12 and G13 mutant or wild type based on mass spectroscopy genotyping for all RAS mutations in tumor tissue.
Drug Administration and Study Design
We conducted a single-institution, open-label, investigator-initiated phase IB/II study in refractory mCRC patients treated with modified FOLFOX6, cetuximab, and dasatinib, with dasatinib dose escalation by cohort. The primary objectives of the Phase IB portion of the study were to determine the maximum tolerated dose (MTD) and dose limiting toxicity (DLT) of the combination of dasatinib, cetuximab and modified FOLFOX6 in adult patients with mCRC and to determine if biological activity of the combination regimen on c-Src activity occurred at the MTD in the expansion cohort. The secondary objectives were to demonstrate the feasibility of peripheral blood biomarkers of Src inhibition, to determine the safety profile and tolerability of the regimen, and to document its antitumor effects.
The primary objective of the Phase II arm was to determine the response-rate distribution of dasatinib and FOLFOX with or without cetuximab. The secondary objectives were determination of time to treatment failure (TTF) and ratio of current TTF to TTF of immediate prior regimen, determination of overall survival (OS) on this regimen, and evaluation of its safety profile and tolerability.
A Bayesian design was utilized to assess efficacy, with a target response rate of interest of greater than 10%. Continuous reassessment was utilized for futility monitoring. The sample size ensured that the 95% confidence interval (CI) would have a width at most of 0.22 under the assumption of a 10% response rate. Operating characteristics can be found in Supplementary Materials and Supplemental Table 1.
Treatment
The regimen consisted of cetuximab (400 mg/m2 week 1 followed by weekly doses of 250 mg/m2), oxaliplatin (85 mg/m2 q2weeks), bolus 5-FU (400 mg/m2 q2weeks) and leucovorin (400 mg/m2 q2weeks), followed by a 46-hour infusion of 5-FU (2400 mg/m2 q2weeks). Dasatinib was dosed orally daily in cohorts of 100 mg/d, 150 mg/d, and 200 mg/d, administered without interruption. Dasatinib dose escalation was performed using a standard “3+3” design. Dose reductions were required for all grade 3 or 4 toxicities attributed to study medications. The symptom-specific dose adjustment table can be found in Supplemental Table 2. The dose reduction algorithm for all drugs can be found in Supplemental Table 3. Treatment was continued until disease progression, unacceptable toxicities, or withdrawal of consent. Adverse event grading was performed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 3.0. Appropriate radiographic images were collected for all patients enrolled at baseline and at all post-baseline evaluations by the same radiological method, in order to assess response. All radiological tests that demonstrated tumor at baseline were repeated after every 4 cycles and at discontinuation of study treatment. For the Phase II arm, mass-spectroscopy genotyping for somatic gene mutations was performed on resected tumor tissue prior to any treatment with cetuximab. DNA extracted from formalin-fixed paraffin-embedded resected tumor tissue was analyzed with Sequenom MassArray technology (Sequenom, Inc, San Diego, CA) using the protocol developed in one of our institutional core facilities.(26) Patients deemed to be KRAS c12/13 wild type received the MTD from the Phase I arm, while KRAS mutant patients received the same, without cetuximab.
Correlative studies
Previous studies have demonstrated that peripheral blood mononuclear cells can provide information on Src activity.(27) To demonstrate the feasibility of examining Src phosphorylation and phosphorylation of selected Src targets, peripheral blood mononuclear cells from normal subjects were treated ex-vivo with dasatinib 300 nM or a low dose of hydrogen peroxide as a positive control. The Src substrates, FAK and paxillin, were utilized for this study as they are expressed in detectable levels in mononuclear cells. Assay optimization demonstrated that protease and phosphatase inhibition were required to maintain signal. As shown in Supplemental Figure 1, ex-vivo treatment with dasatinib significantly inhibited phosphorylation of FAK at tyrosine 861. Similarly, phosphorylation of FAK at tyrosine 118 on paxillin was reduced. Treatment with hydrogen peroxide led to a modest increase, as expected (Supplemental Figure 1).
Further, we sought to correlate peripheral blood mononuclear cell findings with pSrc levels in paired core liver biopsies obtained in the 12 patients in the expansion cohort. pFAK as categorized by standardized immunohistochemical grading on a scale of 0 to + 4 was compared to baseline levels by the Wilcoxon Matched-Pairs Signed-Ranks test. Serum and peripheral blood mononuclear cells were collected at baseline, day 8 of the second and fourth cycle, and optionally at the time of study treatment discontinuation. Paired liver biopsies were obtained prior to therapy (within 4 weeks of initiation of therapy), and between day 8 and day 14 of cycle 2 or 3. See Supplemental Information for further details on methods.
RESULTS
Safety and Efficacy
Thirty patients were enrolled in the Phase IB portion of the study. Eighteen patients were enrolled in the dose escalation cohort and 12 patients in the expansion cohort. Baseline characteristics can be found in Table 1. Notable toxicities included Grade 2 or 3 thrombocytopenia in 27%, (representing platelet counts < 75,000), neutropenia (23% Grade 3 or 4) and fatigue (20% Grade 3 and 4) (Table 2). Other toxicities such as diarrhea, nausea, vomiting and mucositis were consistent with prior experience with the non-investigational components. Pleural effusions were commonly seen in this patient population. These pleural effusions appeared to be dose related and were present predominantly in patients with pre-existing effusions or metastatic disease to the lung. These were not dose limiting and rarely required intervention with thoracentesis.
Table 1.
Baseline characteristics
| Phase IB | Phase II | |
|---|---|---|
| N = 30 | N = 47 | |
| Patient and Disease Characteristics |
Number of Patients (%) | Number of Patients (%) |
| Age, years | ||
| Median, Range | 54, (34–76) | 58, (26–74) |
|
| ||
| Female sex | 19 (63) | 18 (38) |
|
| ||
| ECOG Performance Status | ||
| 0 | 13 (43) | 23 (49) |
| 1 | 17 (57) | 24 (51) |
|
| ||
| Location of tumor | ||
| Liver | 29 (97) | 30 (64) |
| Lung | 14 (47) | 12 (26) |
| Peritoneum | 10 (33) | 10 (21) |
| Intact primary tumor | 7 (23) | 24 (51) |
| Non-regional lymph nodes | 6 (20) | 2 (4) |
| Omentum | 0 | 4 (9) |
| Mesentery | 0 | 2 (4) |
|
| ||
| Prior Treatment* | ||
| Median | 4 | 3 |
| Range | 2–7 | 1–10 |
|
| ||
| KRAS c12/13 Mutation Status | ||
| Wild type | 24 (51) | |
| Mutant | 23 (49) | |
|
| ||
| Prior EGFR therapy | ||
| Treatment | 24 (80) | 24 (51) |
| Documented Refractory | 23 (77) | 24 (100) |
|
| ||
| Prior Oxaliplatin therapy | ||
| Treatment | 29 (97) | 47 (100) |
| Documented Refractory | 21 (70) | 46 (97) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor.
Prior chemotherapy, immunotherapy, hormonal, biologic, or small molecule targeted therapy regimens.
Table 2.
Summary of Adverse Events While Receiving Treatment, Irrespective of Causality
| Adverse Event | Phase IB | Phase II | ||
|---|---|---|---|---|
| N = 30 | N = 47 | |||
| Number of patients (%) | Number of patients (%) | |||
| All Grades | Grade ≥ 3 | All Grades | Grade ≥ 3 | |
| Fatigue | 19 (63) | 6 (20) | 17 (36) | 11 (23) |
|
| ||||
| Anemia | 10 (33) | 4 (13) | 1 (2) | 1 (2) |
|
| ||||
| Nausea/vomiting | 9 (30) | 1 (3) | 3 (6) | 2 (4) |
|
| ||||
| Thrombocytopenia | 8 (26) | 1(3) | 5 (11) | 0 |
|
| ||||
| Neutropenia | 7 (23) | 7 (23) | 6 (13) | 3 (6) |
|
| ||||
| Diarrhea | 5 (17) | 2 (7) | 7 (15) | 3 (6) |
|
| ||||
| Pleural Effusion | 0 | 0 | 7 (9) | 5 (11) |
|
| ||||
| Peripheral neuropathy | 0 | 0 | 5 (11) | 0 |
|
| ||||
| Acute renal failure | 0 | 0 | 4 (9) | 0 |
|
| ||||
| Anorexia | 0 | 0 | 3 (6) | 2 (4) |
|
| ||||
| Mucositis | 0 | 0 | 3(6) | 1 (2) |
|
| ||||
| Dyspnea | 0 | 0 | 1 (2) | 1 (2) |
|
| ||||
| Bleeding | 0 | 0 | 1 (2) | 1 (2) |
|
| ||||
| Hypokalemia | 0 | 0 | 1 (2) | 1 (2) |
|
| ||||
| Hemorrhagic shock (death) | 0 | 0 | 1 (2) | 1 (2) |
|
| ||||
| Deep vein thrombosis | 0 | 0 | 1 (2) | 0 |
|
| ||||
| Rash | 0 | 0 | 1 (2) | 0 |
|
| ||||
| Cardiomyopathy | 0 | 0 | 1 (2) | 0 |
No maximum tolerated dose was identified during the dose limiting toxicity period. A dasatinib dose of 200 mg was cleared based on MTD criteria after two week assessment. However, due to high rates of dasatinib dose reduction after two weeks, 150 mg was utilized for the expansion cohort.
Of the 30 Phase IB patients treated at all dose levels, including in the expansion cohort, 25 patients had measurable disease. The remaining 5 patients either had early toxicities that precluded assessment within the first two months or had disease that was not measurable by standard radiologic criteria. Of the 30 patients, 47% had their disease controlled after one restaging scan. Six of these 30 patients had a reduction in the size of their measurable tumors by > 30%, representing a response rate of 20% (Figure 1A). Of these 6 patients, 5 were previously refractory to oxaliplatin. One had received prior oxaliplatin but this was discontinued secondary to neuropathy. Four of the 6 patients had received prior EGFR based therapy (3 with cetuximab and 1 with panitumumab), and all had failed to respond or subsequently progressed on this treatment. One of these responding patients was treated at the 100 mg dose of dasatinib and 5 received a 150 mg dose. There were no responses in the 200 mg cohort. Dose reductions were eventually required in 4 of the 6 patients due to myelosuppression and/or fatigue. In addition to a dasatinib dose reduction in all four patients, 1 patient also had a 5-FU and oxaliplatin dose reduction, and 1 other patient had a cetuximab dose reduction.
Figure 1.
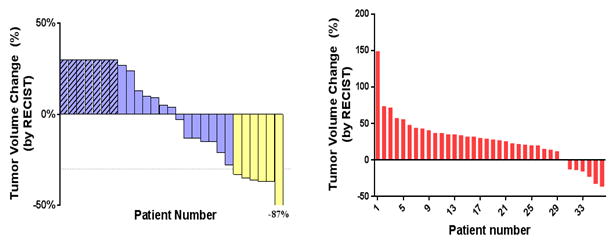
Clinical efficacy of the FOLFOX + cetuximab + dasatinib treatment. A) Waterfall plot of best response for the regimen in the Phase IB study demonstrates 6 patients with a RECIST response. An additional 7 patients had tumor regression that did not meet RECIST criteria for response. Patients with progressive disease noted by new lesions are listed as +30% growth. B) Waterfall plot of patients in the Phase II study treated with dasatinib, FOLFOX, with or without cetuximab, measuring the maximum change from baseline in the sum of the longest diameter for target lesions.
Of the 6 responding patients, 5 had a wild type KRAS oncogene, while the KRAS status was unknown for 1 patient. The median progression free survival for this cohort was 4.6 months and 23% of the patients were free of progression and continued on treatment at 6 months (Figure 2A).
Figure 2.
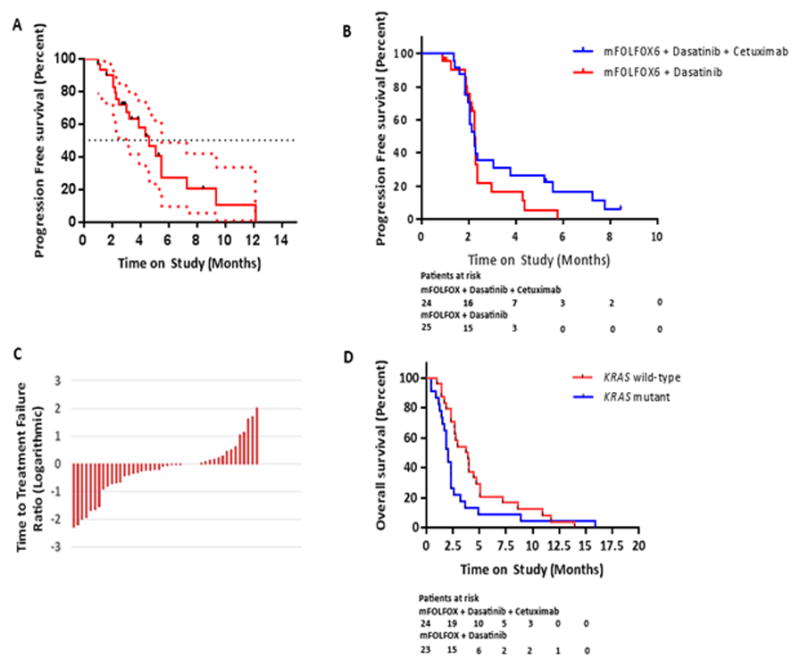
A) Progression free survival of patients treated on the FOLFOX, cetuximab and dasatinib study, with 95% confidence intervals shown. Median PFS was 4.6 months (95% confidence interval of 3.0 to 5.5 months). B) Time to treatment failure on study drugs. All censored patients in both treatment arms were assumed to have had an event at the date of censoring. C) The ratio of time to treatment failure on study regimen to time to treatment failure on prior regimen, assessed logarithmically in all patients. D) Kaplan-Meier curve of overall survival calculated as time from trial initiation to time of death, stratified by tumor KRAS mutational status.
Twenty-four patients with KRAS c12/13 wild type and 23 patients with KRAS c12/13 mutant CRC participated in the Phase II portion of the study. Baseline characteristics of these patients can be found in Table 1. The most common toxicities of any grade while on treatment were fatigue (36%), diarrhea (15%), and neutropenia (13%). Overall, 21 of 47 patients experienced at least one grade 3 AE while on study. The most common grade 3 toxicities were fatigue and pleural effusions. There was one grade 4 episode of neutropenia and one grade 5 episode of hemorrhagic shock (Table 2). There was one toxicity related death due to hemorrhagic shock. Sixteen patients (34%) had AEs leading to dose interruption across all cycles. The most common causes of dose interruption included pleural effusion (n =3) and fatigue (n=3).
Forty of the 47 patients in this trial had post-baseline scans. Fourteen of forty-seven patients (30%; 95% confidence interval (CI), 0.17 to 0.45) experienced an overall response. There were no CRs. At the first restaging interval, three patients (6%; 95% CI, 0.1 to 0.18), all KRAS wild type, and all who had been refractory to an oxaliplatin based regimen and cetuximab previously, had a confirmed PR, which was durable for a median of 5.2 months (range, 1.6 to 8.5 months). Eleven patients (23%; 95% CI, 0.12 to 0.38) had stable disease; 6 of these patients were KRAS wild type. Thirty-six patients (77%; 95% CI, 0.63 to 0.87) discontinued their regimen due to progressive disease after a median of 4 cycles of therapy (range, 1 to 16 cycles).
The median TTF on the immediate prior regimen to the study was 3.8 months in KRAS c12/13 wild type patients (range, 0 to 14.0 months), and 2.0 months in KRAS mutant patients (range, 0 to 16.0 months). The median TTF on our study regimen was 2.3 months in the KRAS wild type cohort (range, 1.4 to 8.5 months), and 2.3 months in the KRAS mutant cohort (range, 0.9 to 5.8 months) (Figure 2B). The median ratio of TTF on study regimen to TTF on prior regimen was 0.9 for all patients (range, 0.1 to 5.5), and 0.8 (range, 0.1 to 7.6) and 1.0 (range, 0.1 to 5.5) in the KRAS wild type and mutant patients, respectively (Figure 2C). The median overall survival was 6.7 months (range, 1.0 to 28.4 months) in all patients, 7.7 months in KRAS wild type patients (range, 2.0 to 22.6 months), and 5.9 months (range, 1.0 to 28.4 months) in KRAS mutant patients (Figure 2D).
Peripheral blood mononuclear cell pharmacodynamics
Peripheral blood mononuclear cell samples at baseline and cycle 2, day 8 were available for 29 of the 30 patients. These lysates were initially analyzed by Western Blot for both phospho-FAK(861) and phospho-paxillin(118). However, it became clear that phosphorylation of FAK was not maintained during processing as there was no detectable phosphorylation band that was evident in baseline nor cycle 2, day 8 samples. Conversely, paxillin maintained a phosphorylation signal on Western Blotting and was used as the standard for effects of dasatinib in subsequent studies.
As shown in Figure 3, there was a significant increase in phospho-paxillin in cycle 2, day 8 of therapy. When compared to baseline, an almost 4-fold increase was observed by densitometry (P=0.004). There was no discernible correlation between the dose of dasatinib utilized and phospho-paxillin, although there were limited samples available from the patients remaining on the 200 mg dose. Likewise, there was not a correlation between phosphorylation and response to therapy or time on therapy (Figure 3).
Figure 3.
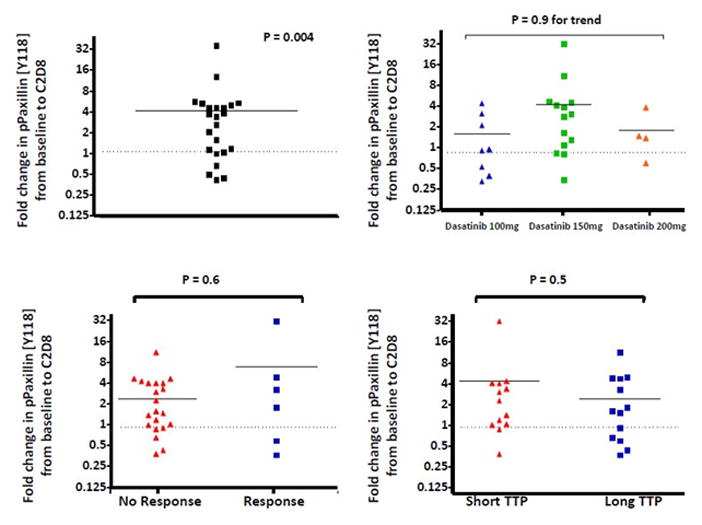
Quantification of pPaxillin [Y118] from PBMCs of patients treated on the FOLFOX + cetuximab + dasatinib clinical trial. Samples were collected at baseline and after 3 weeks of therapy (at day 8 of cycle 2). A) pPaxillin [Y118] was increased after 3 weeks on treatment. # paired t-test for fold change by densitometry. B) No difference is seen in the induction of pPaxillin by cohort. C) There is no correlation between change in pPaxillin and response to therapy, nor time to progression (TTP) (Figure D). Short and long TTP was defined as less than 4 months and greater than or equal to 4 months, respectively (approximately the median TTP).
Pharmacodynamics in Tumor Tissue
Of the 12 patients in the expansion cohort, all consented for paired liver biopsies. These biopsies were obtained by interventional radiology as per Phase IB Methods. There were no complications noted for these 24 procedures. Adequate and viable tissue was available for all 24 patients. An average of 4 core biopsies was obtained from each patient with a range of 2 to 6 at each time point. A minimum of 4 biopsies was obtained in 22 of the 24 procedures. Frozen sections of the tumor tissue were stained for phospho-Src with tyrosine 418 and total Src. The results from viable sections as assessed from adjacent H&E slides were assessed qualitatively. The staining of these tumors was assessed semi-quantitatively and the results are shown in Figure 4. There was no change in phospho-Src [Y418] staining after 3 weeks of combination therapy (P = 0.3 by the Wilcoxon signed rank test). However, there was a statistically significant increase in total Src after treatment (P = 0.049). As there were only 2 responders in this cohort there was a limited ability to detect correlations between response and modulation. The second biopsy for one of the responders only contained acellular fibrosis without evidence of viable tumor in the region sampled, and so could not be assessed for Src substrates.
Figure 4.
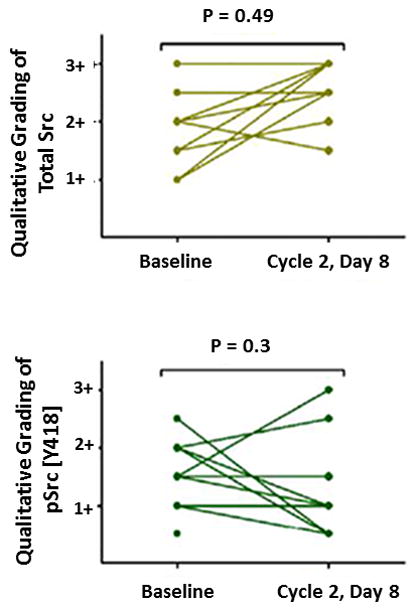
In patients with evaluable paired biopsies in the expansion cohort semi-quantitative grading of total Src and phospho-Src [Y418] staining was performed. Each line represents an individual patient. There was a statistically significant increase in total Src staining after 3 weeks of treatment (P = 0.049). There was no change in phospho-Src staining (P = 0.3).
As the second biopsy was obtained at cycle 2, day 8, the liver biopsy samples are correlated with peripheral blood mononuclear cells in Supplemental Figure 2 in an attempt to evaluate the surrogacy of the peripheral mononuclear cell signal. This analysis is exploratory given the limited sample size in the biopsy cohort. There was no significant correlation of Src activity in the peripheral blood mononuclear cells (measured by fold change in phospho-Paxillin) and increased Src or phospho-Src expression in the liver biopsy.
DISCUSSION
Aberrant activation of the intracellular tyrosine kinase Src has been implicated as a mechanism of acquired chemotherapy resistance in mCRC. Preclinical studies have shown synergistic effects of Src inhibition with both oxaliplatin and cetuximab. Based on these results, we conducted a Phase IB/II study of the oral tyrosine kinase Src inhibitor, dasatinib, in combination with FOLFOX and cetuximab in patients with heavily pretreated mCRC.
The Phase IB portion of our study determined a Phase II dose of dasatinib that could be used safely in combination. Correlative studies were performed which included evaluation of peripheral mononuclear cells and paired liver biopsies from the expansion cohort. The enrolled population in the Phase IB portion was heavily pretreated with a median of 4 prior lines of therapy. Ninety-seven percent of the patients had been exposed to prior oxaliplatin and 80% had been exposed to prior EGFR therapy. Response rate in this population was 20% in the evaluable subset. In patients who were previously refractory to oxaliplatin there was a 25% response rate. Similarly, in patients who were refractory to EGFR based therapy the response rate was 20%. The median progression-free survival of 4.6 months was better than would be expected in a similar cohort of patients treated with regorafenib (1.9 months) or TAS-102 (2.0 months).(28,29)
The toxicities associated with this regimen included fatigue and myelosuppression. The dose of 150 mg daily of dasatinib was chosen for further study based on the higher rates of myelosuppression seen with the 200 mg dose. Even at this reduced dose there were no patients who were able to maintain a 150 mg dose beyond 3 months. The most common reason for dose reduction was myelosuppression and fatigue. The degree of fatigue was also greater than would be expected from this chemotherapy regimen alone in this patient population.(30) However, a recent phase I study demonstrated that dasatinib 70 mg daily could be safely combined with capecitabine, oxaliplatin and bevacizumab—a dose of dasatinib that was lower than anticipated.(31) Thus, the intensity of this chemotherapy regimen in a heavily refractory population should not be discounted, and lower doses of dasatinib may be considered for further studies in this setting.
The pharmacodynamic studies incorporated in the Phase IB portion of the study demonstrated that treatment with the dasatinib-containing regimen failed to inhibit Src, and instead demonstrated a paradoxical increase in Src activation. This may, in part, be due to increased Src activity after oxaliplatin therapy, as shown in our tissue analysis after staining for phospho-Src. Murine studies have suggested that treatment with dasatinib at a higher dose is sufficient to inhibit Src activation after oxaliplatin.(25) However, in our study, the analysis of the peripheral blood mononuclear cells demonstrated increased phosphorylation in the Src substrate of paxillin. This increase was robust and was statistically significant, suggesting that dasatinib was unable to inhibit Src activity in the presence of concurrent chemotherapy in this surrogate tissue. Similarly, paired biopsies failed to demonstrate phospho-Src inhibition. This is likely due to the robust activation of Src after oxaliplatin alone due to induction of reactive oxygen species, as we have previously described.(25) If confirmed in further testing, this may impact how Src inhibitors are used in combination therapy with oxaliplatin.
In the Phase II arm we evaluated the role of KRAS mutation status on response to treatment and time to disease progression. There was a 30% overall response rate, which was restricted to the KRAS wild type arm. Six patients who were refractory to an oxaliplatin based regimen and cetuximab previously, had a confirmed PR, which was durable for a median of 5.2 months. There was no meaningful activity in the KRAS mutant arm. The median overall survival was 6.7 months in all patients, and slightly better in KRAS wild type patients.
There are several possible explanations for the response rates seen with this regimen. First, previous prospective, randomized phase II and III trials in advanced CRC have shown that intensified, repeated short courses of FOLFOX, including oxaliplatin reintroduction after progression, result in significantly improved response rates, progression free survival, and overall survival.(32–37) In particular, these studies have shown a PFS of approximately 3 months(33,34), a partial response rate of 33%(37), a stable disease rate ranging from 38% to 42.7%(33,37), a median OS ranging from 9.9 to 22.1 months(34,36), and an independent significant positive impact on OS (hazard ratio = 0.56, P = 0.009).(35)
Similarly, the efficacy of cetuximab rechallenge is an area of active research and may provide a further explanation for the response rates seen with this regimen. Circulating tumor DNA profiles of CRC patients that had acquired resistance to cetuximab has revealed that upon antibody withdrawal KRAS clones decay, whereas the population regains drug sensitivity.(38,39) The results from both the FOLFOX and cetuximab studies indicate that the CRC genome adapts dynamically to intermittent drug schedules and provide an explanation for the efficacy of rechallenge therapies.
Further, it is well established that Src family kinases and the EGFR cooperate during tumorigenesis and cause resistance to cetuximab. This has prompted several preclinical studies that have demonstrated the ability of Src inhibitors to overcome chemoresistance as well as resistance to targeted agents such as cetuximab.(20–23) Also, it may be that partial Src inhibition is sufficient to produce a modest clinical benefit in a subset of patients.
Finally, the clinical activity observed in the current study may have occurred via alternative dasatinib targets. Li et al. identified nearly 40 distinct kinase targets of dasatinib, of which the platelet-derived growth factor receptor (PDGFR) and c-KIT were found to correlate with clinical activity in NSCLC cell lines.(40) Copy number gain in ephrin receptors, Abl, or SFK are factors that affect NSCLC’s sensitivity to dasatinib in vitro.(41) Future studies of dasatinib in CRC could incorporate the measurement of gene copy numbers and mutational status for all dasatinib targets.
Taken together, our findings suggest that in subsequent work the study design would need to be altered to separate out the effects of the above mentioned possible contributions to drug response, prior to clinical use. If the responses are cetuximab mediated, one could use circulating tumor DNA to better predict patients that may respond to such treatment.
Our study had several limitations. Our sample size was small and thus our study was underpowered to observe small improvements in PFS or OS. Further, our attempts to expand upon the molecular analysis of our paraffin embedded tumor tissue samples by deep sequencing techniques were unsuccessful due to DNA degradation. Finally, the use of tumor tissue to study acquired resistance to anti-EGFR antibodies has several limitations. First, the availability of post-treatment tumor tissue is not universal. Second, if post-treatment tumor tissue is available, sampling bias may confound interpretation, precluding assessment of genetic heterogeneity. Also, acquired mutations are known to be present in low allele frequencies, necessitating high sensitivity assays.
In summary, our Phase II study showed only modest benefit of FOLFOX plus cetuximab plus dasatinib in this heavily pretreated patient population. This appears to be primarily due to a failure to fully inhibit Src at the achievable doses of dasatinib. It is unclear if we may have seen greater clinical activity if we were able to fully inhibit Src in this study, but given the requirement that enrolling patients have documented disease progression on cetuximab, acquired resistant KRAS mutant clones may have been present, limiting future strategies to reverse EGFR resistance. Additional early stage clinical trials are needed to further clarify the best approaches to targeting resistance to cetuximab, and the role of Src tyrosine kinase inhibitors in mCRC.
Supplementary Material
Statement of Significance.
The combination of dasatinib plus FOLFOX with or without cetuximab did not show meaningful clinical activity in refractory colorectal cancer due to failure to fully inhibit Src.
Translational Relevance.
Our Phase II study showed minimal benefit to the addition of dasatinib to the widely used regimen of FOLFOX plus cetuximab in metastatic colorectal cancer. This appears to be primarily due to a failure to fully inhibit Src at the achievable doses of dasatinib. It is unclear if we may have seen some clinical activity if we were able to fully inhibit Src in this study, but given the requirement that enrolling patients have documented disease progression on cetuximab, acquired resistant KRAS mutant clones may have been present, limiting future strategies to reverse EGFR resistance. Additional early stage clinical trials are needed to further clarify the best approaches to targeting resistance to cetuximab, and the role of Src tyrosine kinase inhibitors in mCRC.
Acknowledgments
Financial Support:
Investigator-initiated study supported by BMS
Footnotes
Conflicts of Interest:
S.K. has received remuneration as a scientific advisory board member of BMS
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104–17. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 3.Rothenberg ML, Cox JV, DeVore RF, Hainsworth JD, Pazdur R, Rivkin SE, et al. A multicenter, phase II trial of weekly irinotecan (CPT-11) in patients with previously treated colorectal carcinoma. Cancer. 1999;85(4):786–95. [PubMed] [Google Scholar]
- 4.Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22(2):229–37. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 5.Roskoski R., Jr Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331(1):1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Elfiky A, Han M, Chen C, Saif MW. The role of Src in colon cancer and its therapeutic implications. Clin Colorectal Cancer. 2014;13(1):5–13. doi: 10.1016/j.clcc.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–8. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 8.Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24(19):3110–20. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- 9.Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335(1):249–59. doi: 10.1007/s00441-008-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12(5):1398–401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- 11.Aligayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer. 2002;94(2):344–51. doi: 10.1002/cncr.10221. [DOI] [PubMed] [Google Scholar]
- 12.Bolen JB, Veillette A, Schwartz AM, DeSeau V, Rosen N. Activation of pp60c-src protein kinase activity in human colon carcinoma. Proc Natl Acad Sci U S A. 1987;84(8):2251–5. doi: 10.1073/pnas.84.8.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cartwright CA, Kamps MP, Meisler AI, Pipas JM, Eckhart W. pp60c-src activation in human colon carcinoma. J Clin Invest. 1989;83(6):2025–33. doi: 10.1172/JCI114113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91(1):53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Termuhlen PM, Curley SA, Talamonti MS, Saboorian MH, Gallick GE. Site-specific differences in pp60c-src activity in human colorectal metastases. J Surg Res. 1993;54(4):293–8. doi: 10.1006/jsre.1993.1046. [DOI] [PubMed] [Google Scholar]
- 16.Griffiths GJ, Koh MY, Brunton VG, Cawthorne C, Reeves NA, Greaves M, et al. Expression of kinase-defective mutants of c-Src in human metastatic colon cancer cells decreases Bcl-xL and increases oxaliplatin- and Fas-induced apoptosis. J Biol Chem. 2004;279(44):46113–21. doi: 10.1074/jbc.M408550200. [DOI] [PubMed] [Google Scholar]
- 17.Kopetz S, Morris VK, Parikh N, Overman MJ, Jiang ZQ, Maru D, et al. Src activity is modulated by oxaliplatin and correlates with outcomes after hepatectomy for metastatic colorectal cancer. BMC Cancer. 2014;14:660. doi: 10.1186/1471-2407-14-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587–95. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 19.Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk Lymphoma. 2008;49(4):615–9. doi: 10.1080/10428190801896103. [DOI] [PubMed] [Google Scholar]
- 20.Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67(17):8240–7. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 21.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res. 2004;10(7):2307–18. doi: 10.1158/1078-0432.ccr-1183-3. [DOI] [PubMed] [Google Scholar]
- 22.Pengetnze Y, Steed M, Roby KF, Terranova PF, Taylor CC. Src tyrosine kinase promotes survival and resistance to chemotherapeutics in a mouse ovarian cancer cell line. Biochem Biophys Res Commun. 2003;309(2):377–83. doi: 10.1016/j.bbrc.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 23.George JA, Chen T, Taylor CC. SRC tyrosine kinase and multidrug resistance protein-1 inhibitions act independently but cooperatively to restore paclitaxel sensitivity to paclitaxel-resistant ovarian cancer cells. Cancer Res. 2005;65(22):10381–8. doi: 10.1158/0008-5472.CAN-05-1822. [DOI] [PubMed] [Google Scholar]
- 24.Kim YM, Kim YM, Lee YM, Kim HS, Kim JD, Choi Y, et al. TNF-related activation-induced cytokine (TRANCE) induces angiogenesis through the activation of Src and phospholipase C (PLC) in human endothelial cells. J Biol Chem. 2002;277(9):6799–805. doi: 10.1074/jbc.M109434200. [DOI] [PubMed] [Google Scholar]
- 25.Kopetz S, Lesslie DP, Dallas NA, Park SI, Johnson M, Parikh NU, et al. Synergistic activity of the SRC family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer Res. 2009;69(9):3842–9. doi: 10.1158/0008-5472.CAN-08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serrels A, Macpherson IR, Evans TR, Lee FY, Clark EA, Sansom OJ, et al. Identification of potential biomarkers for measuring inhibition of Src kinase activity in colon cancer cells following treatment with dasatinib. Mol Cancer Ther. 2006;5(12):3014–22. doi: 10.1158/1535-7163.MCT-06-0382. [DOI] [PubMed] [Google Scholar]
- 28.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–12. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 29.Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372(20):1909–19. doi: 10.1056/NEJMoa1414325. [DOI] [PubMed] [Google Scholar]
- 30.Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25(12):1539–44. doi: 10.1200/JCO.2006.09.6305. [DOI] [PubMed] [Google Scholar]
- 31.Strickler JH, McCall S, Nixon AB, Brady JC, Pang H, Rushing C, et al. Phase I study of dasatinib in combination with capecitabine, oxaliplatin and bevacizumab followed by an expanded cohort in previously untreated metastatic colorectal cancer. Invest New Drugs. 2014;32(2):330–9. doi: 10.1007/s10637-013-0042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maindrault-Goebel F, Tournigand C, Andre T, Carola E, Mabro M, Artru P, et al. Oxaliplatin reintroduction in patients previously treated with leucovorin, fluorouracil and oxaliplatin for metastatic colorectal cancer. Ann Oncol. 2004;15(8):1210–4. doi: 10.1093/annonc/mdh305. [DOI] [PubMed] [Google Scholar]
- 33.Tournigand C, Cervantes A, Figer A, Lledo G, Flesch M, Buyse M, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer--a GERCOR study. J Clin Oncol. 2006;24(3):394–400. doi: 10.1200/JCO.2005.03.0106. [DOI] [PubMed] [Google Scholar]
- 34.Suenaga M, Mizunuma N, Matsusaka S, Shinozaki E, Ozaka M, Ogura M, et al. Phase II study of reintroduction of oxaliplatin for advanced colorectal cancer in patients previously treated with oxaliplatin and irinotecan: RE-OPEN study. Drug Des Devel Ther. 2015;9:3099–108. doi: 10.2147/DDDT.S85567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Gramont A, Buyse M, Abrahantes JC, Burzykowski T, Quinaux E, Cervantes A, et al. Reintroduction of oxaliplatin is associated with improved survival in advanced colorectal cancer. J Clin Oncol. 2007;25(22):3224–9. doi: 10.1200/JCO.2006.10.4380. [DOI] [PubMed] [Google Scholar]
- 36.Andre T, Tournigand C, Mineur L, Fellague-Chebra R, Flesch M, Mabro M, et al. Phase II study of an optimized 5-fluorouracil-oxaliplatin strategy (OPTIMOX2) with celecoxib in metastatic colorectal cancer: a GERCOR study. Ann Oncol. 2007;18(1):77–81. doi: 10.1093/annonc/mdl336. [DOI] [PubMed] [Google Scholar]
- 37.Adams RA, Meade AM, Seymour MT, Wilson RH, Madi A, Fisher D, et al. Intermittent versus continuous oxaliplatin and fluoropyrimidine combination chemotherapy for first-line treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet Oncol. 2011;12(7):642–53. doi: 10.1016/S1470-2045(11)70102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):827. doi: 10.1038/nm0715-827b. [DOI] [PubMed] [Google Scholar]
- 39.Morelli MP, Overman MJ, Dasari A, Kazmi SM, Mazard T, Vilar E, et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann Oncol. 2015;26(4):731–6. doi: 10.1093/annonc/mdv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6(4):291–9. doi: 10.1038/nchembio.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119(6):1727–40. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.