

Abstract
Primary tumors are often heterogeneous, composed of therapy-sensitive and emerging therapy-resistant cancer cells. Interestingly, treatment of therapy-sensitive tumors in heterogeneous tumor microenvironments results in apoptosis of therapy-resistant tumors. In this study, we identify a Par-4 amino-terminal fragment (PAF) that is released by diverse therapy-sensitive cancer cells following therapy-induced caspase cleavage of the tumor suppressor Par-4 protein. PAF caused apoptosis in cancer cells resistant to therapy and inhibited tumor growth. A VASA segment of Par-4 mediated its binding and degradation by the ubiquitin ligase Fbxo45, resulting in loss of Par-4 pro-apoptotic function. Conversely, PAF, which contains this VASA segment, competitively bound to Fbxo45 and rescued Par-4-mediated induction of cancer cell-specific apoptosis. Collectively, our findings identify a molecular decoy naturally generated during apoptosis that inhibits a ubiquitin ligase to overcome therapy resistance in tumors.
Keywords: Par-4, Fbxo45, Apoptosis
Introduction
Most tumors are phenotypically and functionally heterogeneous owing to their multi-clonal nature, genetic instability and tumor cell plasticity (1,2). Primary tumors are usually responsive to the standard-of-care therapy aimed largely at reducing tumor bulk, and therapy-resistant tumor cells emerge mainly at distant metastatic sites, and less frequently at the primary site, within a few years after the primary tumor has regressed (3). These observations indicate that a dynamic relationship exists between the treatment of primary tumors and emergence of resistant clones. To understand the mechanisms of tumor resistance to therapy, we are studying the relationship between therapy-sensitive and therapy-resistant tumor cells. Our studies indicated that therapy-resistant cancer cells undergo apoptosis if the therapy-sensitive cancer cells in a heterogeneous tumor microenvironment are induced to undergo apoptosis. This paracrine effect of the therapy-sensitive tumor cells undergoing apoptosis was attributed to a therapy-induced, caspase-cleavage fragment of the tumor suppressor prostate apoptosis response-4 (Par-4) protein.
Par-4 is a leucine zipper domain containing protein that is ubiquitously expressed in diverse cell types and tissues (4,5). Par-4 induces apoptosis specifically in cancer cells but not in normal cells by intracellular and extracellular mechanisms (6). Par-4-knockout mice develop spontaneous, as well as inducible tumors (7), and corollarily, transgenic mice overexpressing Par-4 are resistant to the growth of spontaneous or oncogene-inducible tumors (8). Consistent with its tumor suppressor function, a number of laboratories have reported that Par-4 is inactivated, down-regulated, or mutated in different types of cancers (9–11).
Par-4 protein is secreted in response to various endoplasmic stress-producing agents or small molecule secretagogues and induces apoptosis specifically in cancer cells leading to growth inhibition of tumors (12–14). Secreted Par-4 binds, via its SAC domain, to its receptor GRP78 on the surface of cancer cells and induces apoptosis by a FADD/caspase 8/caspase 3 dependent pathway (15). Normal cells, on the other hand, are resistant to apoptosis by extracellular Par-4 as they express low to undetectable levels of basal or inducible cell surface GRP78 receptors (15).
Several reports have indicated that cells undergoing apoptosis can release factors that induce paracrine apoptosis of neighboring cells in the microenvironment; however, the precise factor(s) has been elusive (16). We tested the hypothesis that treatment of therapy-sensitive cancer cells with apoptosis-inducing therapeutics may induce paracrine apoptosis in neighboring therapy-resistant cancer cells grown in heterogeneous microenvironments. Rather unexpectedly, we identified an amino-terminal fragment of Par-4 that is naturally produced and released from therapy-sensitive cancer cells following caspase-dependent cleavage of Par-4. This Par-4 fragment specifically enters cancer cells, including those that are resistant to therapy, and induces apoptosis.
Materials and Methods
Cells
Human prostate cancer cells DU145, PC-3, LNCaP, lung cancer cells A549 and H460, breast cancer cells MCF7, and primary human lung fibroblast cells HEL were from ATCC, MD. Human colon cancer cells KM20 were from Mark Evers (University of Kentucky). Parent A549 and A549TR cells were from Bruce Zetter (Boston Children’s Hospital, MA) (17). Mel1617 and Mel1617BR melanoma cells were from Meenhard Herlyn (Wistar Institute, PA)(18). Beas2B and arsenic-transformed Beas2B cells were from Xianglin Shi (University of Kentucky). C4-2B cells were from Leland Chung (Ceder Sinai Medical Center, LA). Normal human prostate stromal cells PrS and prostate epithelial cells PrE were from Lonza Inc. (Allendale, NJ). KP7B cells were from Tyler Jacks (MIT, MA). PC-3/dnFADD or PC-3/FLIP cells were previously described (15). Par-4+/+ and Par-4−/− MEFs were from wild type and Par-4-null C57BL/6 mouse embryos. All the cell lines were obtained between 2011 and 2012 and authenticated by us in August 2012, and again in December 2016, via STR profiling by Genetica DNA Laboratories (Burlington, NC). Mycoplasma contamination in the cell cultures was ruled out by using the MycoAlert™ Mycoplasma Detection Kit (from Lonza). The length of time between thawing and use of the cell lines in the described experiments was two to four weeks.
Recombinant Proteins
Purified recombinant thioredoxin (TRX), TRX-Par-4 and TRX-PAF were procured from ProMab Biotechnologies Inc, as described previously (15). Purified His-PAF or His-TRX was prepared using the baculoviral protein expression system (GenScript, NJ).
Reagents
Paclitaxel injection vials were obtained from the Markey Cancer Center pharmacy for research use only. Caspase inhibitors z-VAD-fmk, z-DEVD, z-IETD-fmk, z-LEHD-fmk were from BioVision Inc. Recombinant human TRAIL was from Calbiochem. BFA was procured from Sigma. PLX4720 was from Plexxikon Inc., CA.
Plasmid Constructs
Plasmids for expression of vector pCB6+, pCB6+/GFP, non GFP tagged pCB6+/Par-4, Par-4-GFP, and pcDNA3.1-nV5 Par-4/3A mutant have been described previously (8, 19). The expression constructs for PAF, PAF/3A mutant and deletion mutants (132-340aa; or 30-131aa, 60-131aa, and 90-131aa tagged with GFP at the C-terminus) were cloned into the pCB6+ vector backbone following PCR amplification of the cDNA using pCB6+/Par-4 or pcDNA3.1-nV5 Par-4-3A constructs, respectively, as templates. The pCB6+ vector was digested using the restriction enzyme EcoR1, and the cDNA was then ligated into the vector. Fidelity of the sequence was confirmed by nucleotide sequencing. The shRNA for Par-4 was constructed using the pLKO.1 vector and stable cell lines were generated by Sunil Nooti in our laboratory. The 16aa fragment containing wild type VASA domain (ELNNNL) or VASA mutant (ALANAL) was synthesized by GenScript, NJ.
Antibodies
Antibodies for Par-4 N-terminal (R334); GRP78 (N20), and Col1A1 (H-197), were from Santa Cruz Biotechnology, Inc. Par-4 C-terminal antibody (P5367) was from Sigma-Aldrich, USA. Fbxo45 antibody (ab136614) was from Abcam or was custom prepared as previously described (19). Active caspase 3 antibody (Asp175) (5A1E) was from Cell Signaling. The β-actin antibody was from Sigma-Aldrich.
Co-immunoprecipitation and Western blot analysis
Cells were lysed using RIPA (Radio Immuno Precipitation Assay) buffer and Complete Protease Inhibitor cocktail (Roche) on ice. The lysates were centrifuged at 13000 rpm for 5 minutes at 4°C and the supernatant was used for the pull-down. The protein concentration was determined by Bradford assay using a cuvette based spectrophotometer. The proteins were immunoprecipitated using 1 mg of input and 2 μg antibody and 25 μl bed volume of magnetic protein A/G beads from ThermoFisher Scientific. The proteins were then eluted and resolved using SDS-PAGE. The Western blot analysis was carried out as described previously (10).
Apoptosis and Cell Proliferation Assays
Cancer cell killing by PAF was measured in blinded experiments using CellTiter 96® AQueous MTS Reagent (Promega). The cells were seeded in 96- well plates and after 72 h of treatment, the MTS assay was performed as per the manufacturer’s instructions. The absorbance was measured at 490nm using a microplate reader (Spectra MR, DYNEX Technologies). Apoptotic cells were identified by immunocytochemical (ICC) analysis for active caspase-3, and apoptotic nuclei were revealed by 4, 6-diamidino-2-phenylindole (DAPI) staining as previously described (12). A total of three independent and blinded experiments were performed; and approximately 500 cells were scored in each experiment for apoptosis under a fluorescent microscope. Data shown represent mean of three independent experiments ± Standard Deviation (SD).
Animal experiments
Male athymic mice (nu/nu; from Jackson Laboratories at 6 weeks of age) were injected s.c. with 1.5×106 cells of either A549 or A549TR/GFP cells into both the flanks. In the group with mixed tumors, 1.5×106 cells of each cell type (A549 and A549TR/GFP) were mixed and injected into the flanks. Following tumor formation, the animals were treated with Paclitaxel (12 mg/kg) or injectable saline as vehicle control. The animal procedures were performed blinded. The tumors were measured using Vernier calipers every 3 days for period of 24 days. The tumors were excised and subjected to GFP immunohistochemical analysis or TUNEL staining to evaluate apoptosis.
Lewis Lung Carcinoma (LLC1) cells (from ATCC, MD; 0.4 × 106 cells) were injected into the athymic (nu/nu) mice through the tail vein, and 5 h later, the animals received 250 μg of protein via the i.v. route. The animal procedures were performed as blinded experiments. The tumors were allowed to develop for a period of 18 days, and then the animals were humanely killed and the lungs were stained with India ink. The tumors nodules were counted with the aid of a dissection microscope. All animal procedures were performed with University of Kentucky IACUC approval.
Statistical analysis
All in vivo and cell death experiments were blinded, and the cell death experiments were performed independently at least three different times, to verify the reproducibility of the findings. A mean of at least 3 experiments ± Standard Deviation (S.D.) was calculated. Statistical analyses were carried out with Graphpad Prism or Microsoft Excel software, and P values were calculated using the Student t test.
Results
Tumor cell heterogeneity permits cancer therapeutics to overcome therapy resistance
Paclitaxel, representing the taxane family of drugs, is commonly used as a standard of care therapy for a broad range of cancers (20,21). To test the effect of Paclitaxel on taxane-resistant cancer cells grown within the microenvironment of taxane-sensitive cancer cells, we treated co-cultures of RFP-tagged Paclitaxel-sensitive lung cancer cells A549 and GFP-tagged Paclitaxel-resistant A549TR cells with Paclitaxel. Treatment of the co-cultures with Paclitaxel resulted in apoptosis of the A549/RFP cells, as well as A549TR/GFP cells (Figure 1A). On the other hand, when grown separately, A549/RFP cells were susceptible to Paclitaxel induced apoptosis but the A549TR/GFP cells were resistant to Paclitaxel (Figure 1A). The conditioned medium (CM) from Paclitaxel-treated A549/RFP cells induced apoptosis in A549TR/GFP (Supplementary Figure S1A), implying that apoptosis was induced by a factor released by A549/RFP cells.
Figure 1. Paclitaxel treatment of heterogeneous cultures induces apoptosis in both sensitive and resistant cells.
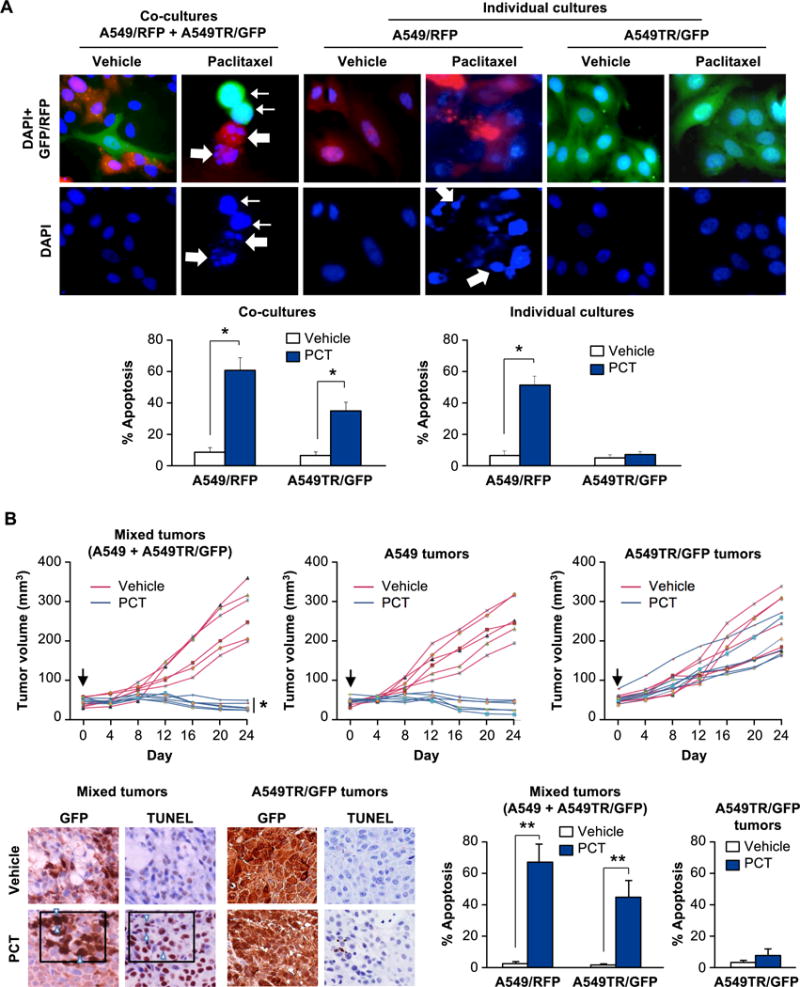
(A) Paclitaxel induces apoptosis in Paclitaxel-resistant cells A549TR/GFP co-cultured with Paclitaxel-sensitive cells A549/RFP. Cells were grown separately (as individual cultures, upper middle and right panels) or co-cultured as a 1:1 mixture (1 × 106 each) (upper left panel), and treated with Paclitaxel (PCT, 25 nM) or vehicle for 24 h. The cells were then stained with DAPI to reveal their nuclei (upper panels). Apoptotic cells were quantified (lower panels) as indicated in Supplemental Materials and Methods section. Three independent experiments were carried out, and the results in the graphs represent mean ± SD from three independent experiments. Asterisk (*) indicates statistical significance (P < 0.001) based on Student’s t test. A549/RFP cells (thick arrows) and A549TR/GFP cells (thin arrows) underwent apoptosis when treated with Paclitaxel in co-cultures.
(B) Paclitaxel induces apoptosis in Paclitaxel-resistant cells within tumors containing Paclitaxel-sensitive cells. A549 cells or A549TR/GFP cells were injected separately (upper middle and upper right panels) or co-injected as a 1:1 mixture (1.5 × 106 cells of each) (upper left panel) into the flanks of nude mice. When the tumors had grown to a volume of approximately 50 mm3 (Day 0, black arrow), the mice were injected i.p. with Paclitaxel (PCT) or vehicle. Six mice were used for each treatment group and tumor volumes for each mouse over a 24-day period are shown. Asterisk (*) indicates that the mixed tumors treated with PCT were significantly smaller in volume (P < 0.025 by Student’s t test) compared to A549TR/GFP tumors treated with PCT.
Sections of the mixed tumors or A549TR-tumors were scored for GFP expression or apoptosis by TUNEL assays (lower left panels). Data show percentage of GFP-positive cells that were also TUNEL-positive in three different tumors, and mean ± SD values are presented. Arrowheads indicate representative GFP-positive cells that are also TUNEL-positive. Asterisks (**) indicate statistical significance (P < 0.001) based on Student’s t test.
To determine the in vivo significance of these observations in a heterogeneous tumor microenvironment, we injected a mixture of A549 and A549TR/GFP cells into the flanks of nude mice. As control, mice were injected with either A549 cells or A549TR/GFP cells. Interestingly, Paclitaxel treatment caused remarkable inhibition of mixed tumor-cell xenografts containing A549 and A549TR/GFP cells (Figure 1B). By contrast, xenografts of A549TR/GFP cells injected separately in the flanks of mice were resistant to Paclitaxel (Figure 1B). As expected, A549-derived xenografts were sensitive to Paclitaxel (Figure 1B). TUNEL assays confirmed significant apoptosis with Paclitaxel in the A549TR/GFP cells within the tumors arising from co-injection of A549 and A549TR/GFP cells (Figure 1B). Together, these findings indicate paracrine apoptosis in Paclitaxel-resistant lung cancer cells and tumor shrinkage produced by a soluble factor, which was released by Paclitaxel-sensitive lung cancer cells undergoing apoptosis in response to Paclitaxel.
We next performed an unbiased screen of potential proteins that are secreted from A549 cells and may induce apoptosis in Paclitaxel-resistant lung cancer cells. A549 cells were treated with Paclitaxel and the CM was applied to the A549TR cells. The CM was incubated with antibodies against potential candidate proteins that are known to be secreted and induce apoptosis. Rather surprisingly, although the A549 or A549TR cells are resistant to Par-4, the Par-4 antibody caused ~70% decrease in apoptosis by the CM, relative to the control IgG (Figure S1B). By contrast, the maspin antibody caused ~30% decrease in apoptosis by the CM and antibodies for TRAIL, IGFBP3, GDF15 did not inhibit apoptosis by the CM (Figure S1B). These findings implicated a Par-4-like factor in paracrine apoptosis of Paclitaxel-resistant A549TR cells.
A Par-4-like candidate factor is involved in paracrine apoptosis of diverse therapy-resistant cancer cells
To identify the Par-4-like factor that produced apoptosis in therapy-resistant cells, we treated A549 lung cancer cells with Paclitaxel and applied the CM to A549TR cells in the presence of a control IgG antibody, Par-4 N-terminal antibody, or Par-4 C-terminal antibody. Interestingly, apoptosis by the CM was neutralized by the N-terminal antibody but not by the C-terminal antibody or the control antibody (Figure 2A, lower left panel). Although residual PCT is expected to be carried over in the CM, A549TR cells are resistant to PCT (Figure 2A, lower right panel). Apoptosis of A549TR cells treated with the CM from A549 cells is caused by Par-4, as the apoptotic activity of the CM is neutralized by the Par-4 antibody (Figure 2A, lower left panel). Western blot analysis of the CM from lung cancer cells that were treated with Paclitaxel (Figures 2A and S2B), TRAIL or doxorubicin (Figure S2A), which induce apoptosis, resolved 3 distinct bands: (1) full-length Par-4 at ~40 kDa detected by both the N- and C-terminal Par-4 antibodies, (2) a 25 kDa band detected by the C-terminal antibody, and (3) a 15 kDa band detected by the N-terminal antibody. Because the N-terminal antibody, which neutralized the apoptotic action of the CM, detected full length Par-4 and the 15 kDa band, and because A549 cells or A549TR cells were resistant to the action of full-length Par-4 (12,22), the 15 kDa band was a prime candidate for the paracrine apoptotic effect of Paclitaxel in A549TR cells.
Figure 2. The 15 kDa amino-terminal fragment of Par-4 is involved in paracrine apoptosis of therapy-resistant cancer cells.
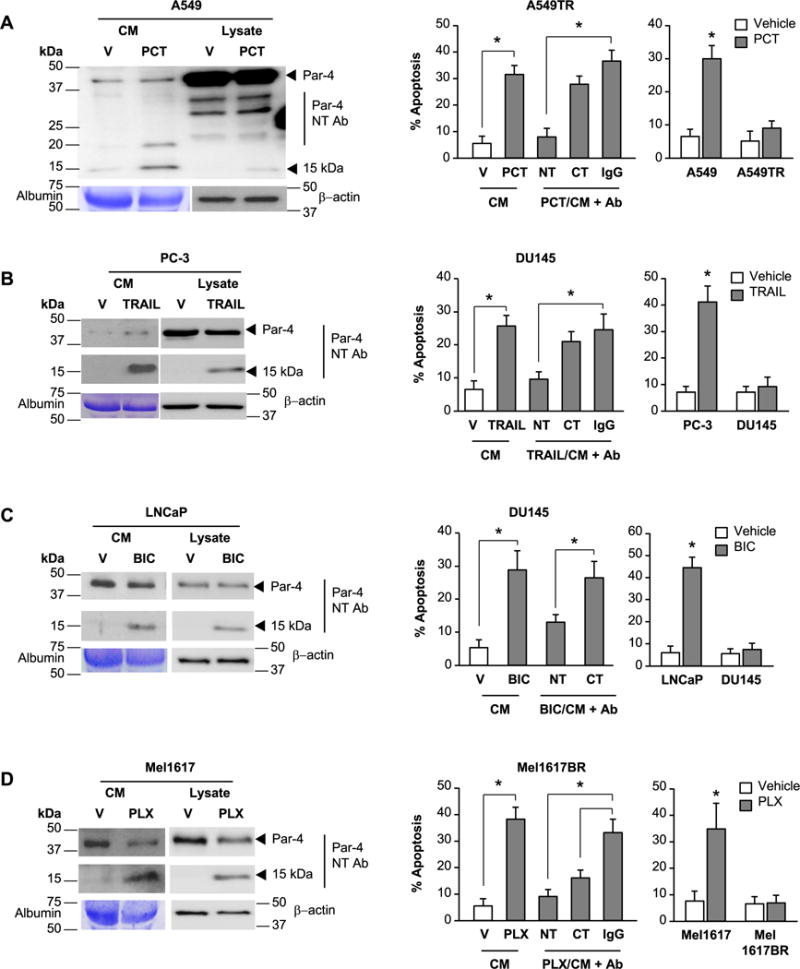
(A) Paracrine apoptosis of Paclitaxel-resistant A549TR cells. A549 cells were treated for 24 h with vehicle (V) or Paclitaxel (PCT, 25 nM). The CM from the vehicle- or PCT-treated cells was then pre-incubated with Par-4 N-terminal (NT) or C-terminal (CT) antibody (Ab) or control IgG, and then applied to the PCT-resistant A549TR cells for 24 h (middle panel). As controls, the A549 or A549TR cells were directly treated with vehicle (V) or PCT for 24 h (right panel).
(B) Paracrine apoptosis of TRAIL-resistant DU145 cells. PC-3 cells were treated for 24 h with vehicle (V) or TRAIL (100 ng/ml). The CM from the vehicle- or TRAIL-treated cells was pre-incubated with Par-4 (NT) or (CT) antibody or control IgG, and then applied to the TRAIL-resistant DU145 cells for 24 h (middle panel). As controls, PC-3 and DU145 cells were directly treated with vehicle (V) or TRAIL (100 ng/ml) for 24 h (right panel).
(C) Paracrine apoptosis of bicalutamide-resistant DU145 cells. LNCaP cells were treated for 24 h with vehicle (V) or bicalutamide (BIC, 70 μM). The CM from the vehicle- or BIC-treated cells was pre-incubated with Par-4 (NT) or (CT) antibody or control IgG, and then applied to the BIC-resistant DU145 cells for 24 h (middle panel). As controls, LNCaP and DU145 cells were directly treated with vehicle (V) or BIC (70 μg/ml) for 24 h (right panel).
(D) Paracrine apoptosis of PLX4720-resistant Mel1617BR cells. Mel1617 cells were treated for 24 h with vehicle (V) or PLX4720 (PLX, 2.5 μM). The CM from the vehicle- or PLX4720-treated Mel1617 cells was pre-incubated with Par-4 (NT) or (CT) antibody or control IgG, and then applied to the PLX472–resistant Mel1617BR cells for 24 h (middle panel). As controls, Mel1617 and Mel1617BR cells were directly treated with vehicle (V) or PLX4720 (2.5 μM) (right panel).
(A–D) Western blot analysis was performed on the whole-cell lysates and CM with the indicated antibodies (left panels). Uncut gels are shown in Supplementary Figure S2 A–D. The cells were scored for apoptosis by ICC for active caspase 3 (middle and right panels). Three independent experiments were carried out, and the apoptosis data represent mean ± SD from three independent experiments. Asterisk (*) indicates statistical significance (P < 0.001) based on Student’s t test.
We further validated the concept using diverse therapy-resistant cell culture models. TRAIL-sensitive PC-3 cells were treated with TRAIL, and the CM was applied to TRAIL-resistant DU145 cells (Figures 2B and S2B). Similarly, LNCaP cells, which are sensitive to bicalutamide, were treated with bicalutamide and the CM was applied to bicalutamide-resistant DU145 cells (Figures 2C and S2B). We also treated MEL cells Mel1617, which are sensitive to the mutant BRAF inhibitor PLX4720, and the CM was applied to Mel1617BR melanoma cells, which are resistant to PLX4720 (Figures 2D and S2B). Interestingly, the CM from the TRAIL, bicalutamide, or PLX4720-treated therapy-sensitive cancer cells produced apoptosis in the corresponding therapy-resistant cells (Figures 2 B–D). Importantly, the apoptotic activity in the CM from the TRAIL-treated or bicalutamide-treated therapy-sensitive cancer cells was neutralized by the N-terminal Par-4 antibody but not the C-terminal Par-4 antibody, and the 15 kDa band detected by the Par-4 N-terminal antibody was present in the CM (Figures 2B, 2C and S2B). The 15 kDa band was also present in the CM from the PLX4720 treated therapy-sensitive cancer cells, and the apoptotic activity of the CM was neutralized by the N-terminal Par-4 antibody, as well as by the C-terminal Par-4 antibody (Figures 2D and S2B). These findings suggested that the 15 kDa band represented a candidate factor released by therapy-sensitive cancer cells that may effectively induce apoptosis in therapy-resistant cancer cells.
Identification of PAF
Since TRAIL, Paclitaxel, bicalutamide or PLX4720 induced apoptosis through the activation of intracellular caspases (20,23–25), we considered the possibility that the 15 kDa and 25 kDa bands represented N- and C-terminal caspase-cleavage fragments, respectively, of Par-4. An in silico search of caspase cleavage sites within Par-4 identified a potential cleavage site at the D131 position (D131EEEPD). During the course of these studies, several groups identified D131 as a target for caspase-3 and focused on the role of the 25 kDa fragment, which contains the SAC effector domain of Par-4, but did not specifically identify the 15 kDa fragment (26–28). Pretreatment of H460 cells with TRAIL in the presence of cycloheximide, which is known to inhibit de novo protein biosynthesis, did not inhibit production of the 25 kDa and 15 kDa bands (Figure S3A), indicating that production of these fragments was not dependent on new gene expression events. We also transfected H460 cells with Par-4 that was tagged with GFP at the C-terminus (Par-4 CT-GFP), which is known to activate caspase-dependent apoptosis (29). Despite fusion of GFP at the C-terminus, Par-4-CT-GFP produced the 15 kDa band, indicating that the 15 kDa fragment was not a result of alternate splicing or antibody cross-reaction with other proteins (Figure S3B). On the other hand, pretreatment of cells with the pan-caspase inhibitor zVAD-fmk inhibited the generation of the 25 kDa and 15 kDa fragments, confirming that these fragments were produced by caspase-dependent cleavage of Par-4 (Figure 3A). As expected, ectopic expression of full length Par-4, which produces apoptosis in H460 cells, produced the 25 kDa and 15 kDa fragments, and the products of the 1-131aa or 132-340aa deletion mutant constructs of Par-4 co-migrated with the 15 kDa or 25 kDa band, respectively (Figure 3A). By contrast, overexpression of Par-4 D131A mutant, which is expected to be resistant to caspase activity, failed to produce the Par-4 cleavage fragments (Figure 3A). Moreover, TRAIL, which activated caspase-3 (25,30), further induced the production of the 25 kDa and 15 kDa bands in TRAIL-sensitive cells transfected with the Par-4 expression construct but not with the D131A mutant of Par-4, relative to vector (Figure S4). Together, these findings indicated that the 15 kDa band was produced by caspase cleavage of Par-4, and that the D131 residue was critical for caspase induced cleavage of Par-4. We designated this 1-131aa/15 kDa, fragment as PAF (Par-4 Amino-terminal Fragment).
Figure 3. Discovery and validation of PAF.
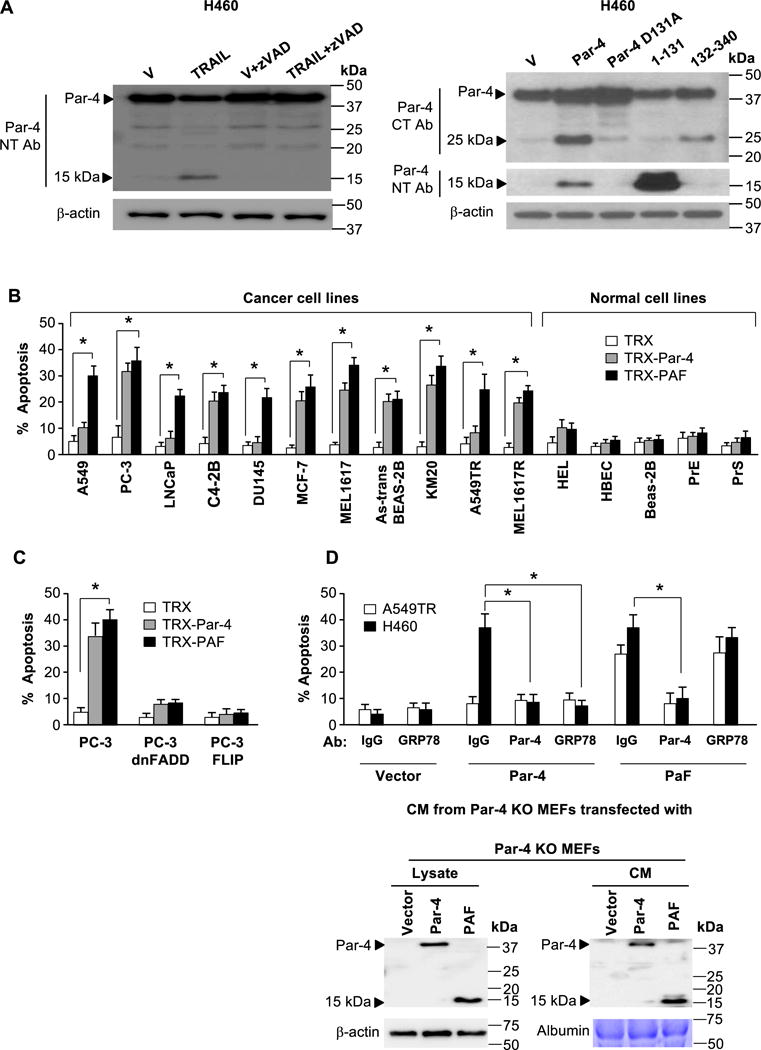
(A) The 25 kDa and 15 kDa fragments are produced by caspase-dependent cleavage of Par-4 at its D131 residue. Cells were treated with vehicle or TRAIL (100 ng/ml) in the presence or absence of zVAD-fmk (left panel), or transfected with the indicated expression constructs or vector control (right panel). Whole cell lysates were then subjected to Western blot analysis.
(B) Recombinant PAF induces apoptosis specifically in cancer cells. A panel of cancer or normal cell lines was treated with recombinant TRX, TRX-Par-4 or TRX-PAF (100 nM each) for 24 h and scored for apoptosis.
(C) PAF induced apoptosis is dependent on FADD and caspase 8. PC-3 cells expressing dnFADD or FLIP, or parent PC-3 cells were treated with recombinant TRX, TRX-Par-4 or TRX-PAF (100 nM each) for 24 h and scored for apoptosis.
(D) PAF induced apoptosis is independent of cell surface GRP78. Par-4-null MEF cells were transfected with vector, Par-4 or PAF and expression of Par-4 or PAF in the CM was confirmed by Western blot analysis (lower panel). The CM was preincubated with antibody (Ab) for Par-4 (NT), GRP78 or control IgG, then applied to A549TR and H460 cells for 24 h to score for apoptosis (upper panel).
(B–D) The cells were scored for apoptosis by ICC for active caspase 3. The apoptosis data represent mean ± SD from three independent experiments. Asterisk (*) indicates statistical significance (P < 0.001) based on Student’s t test.
PAF induces apoptosis selectively in cancer cells
To interrogate whether PAF was sufficient to induce apoptosis, we transfected 3T3 fibroblasts with vector, human Par-4 or PAF expression constructs, and the CM from the cells was transferred to diverse normal or cancer cell lines. The CM from PAF transfected cells induced apoptosis in diverse cancer types, regardless of whether they were sensitive or resistant to apoptosis by the CM from Par-4 transfected cells, but not in normal cells (Figure S5). To further confirm our findings on the cancer-specific apoptotic potential of PAF, we used recombinant PAF. As seen in Figure 3B, recombinant PAF induced apoptosis in Par-4-resistant cancer cells, as well as Par-4-sensitive cancer cells, but not in normal cells. Together, these findings indicate that PAF induces apoptosis specifically in cancer cells, including those that are resistant to apoptosis by exogenous full length Par-4.
Mechanism of PAF induced apoptosis
As Par-4 induces apoptosis via the caspase 8/caspase 3-dependent pathway (15), we sought to determine whether PAF utilized a similar or distinct apoptotic mechanism. We transfected MEFs with vector, Par-4 or PAF expression constructs, and the CM was applied to A549 cells in combination with various caspase inhibitors. Apoptosis by PAF was blocked by the pan-caspase inhibitor (zVAD-fmk), caspase 8 inhibitor (zIETD-fmk), and caspase 3 inhibitor (zDEVD-fmk), and relatively weakly by the caspase 9 inhibitor (zLEHD-fmk) (Figure S6).
The caspase 8/caspase 3 pathway requires functional FADD for activation of caspase 8 activation, which then directly activates effector caspase 3 or the mitochondrial/caspase 9 pathway through cleavage of t-BID (31–33). To confirm the requirement of the caspase 8/caspase 3 pathway in the action of PAF, we treated PC-3 parent cells or PC-3 transfectants expressing dominant negative (dn) FADD (to block FADD activation) or FLIP (to inhibit caspase 8 activation) with recombinant PAF, Par-4, or TRX control protein. PAF induced apoptosis in PC-3 cells with an intact extrinsic apoptotic pathway but not in PC-3 cells expressing dnFADD or FLIP (Figure 3C). These findings indicated that FADD/caspase 8/caspase 3 pathway was essential for apoptosis by PAF.
Extracellular Par-4 is known to induce apoptosis by binding to GRP78 at the cell surface (15). PAF is able to induce apoptosis in Par-4 resistant cell types such as A549 or DU145, which display low levels of cell surface GRP78 (15), suggesting that the action of PAF may be independent of GRP78. To directly verify the cell surface GRP78-independent action of PAF, we transfected Par-4-null MEFs with vector, Par-4 and PAF and the CM was applied to A549TR and H460 cells in the presence of antibodies to Par-4 and GRP78. The Par-4 antibody inhibited apoptosis by both PAF and Par-4 (Figure 3D). The antibody to GRP78 was unable to inhibit apoptosis in PAF treated cells but apoptosis in Par-4 treated cells was significantly reduced (Figure 3D), thereby confirming that PAF induced apoptosis independent of cell surface GRP78.
The VASA domain within PAF is essential for apoptosis by Par-4 or PAF
To identify the domains of PAF that are critical for apoptosis of cancer cells, we generated deletion mutants of PAF. MEFs were transfected with these mutants and the CM was tested for apoptosis in A549 and PC-3 cells. These experiments indicated that the most active region of PAF was localized between amino acids 60 and 90 (Figure S7 A–C).
A screen for molecular signatures of potentially functional domains within the 60-90aa residues of PAF revealed a VASA segment (ELNNNL) that is important for Par-4-binding to the SPRY domain of the E3 ubiquitin ligases, Fbxo45, SPSB1, SPSB2, and SPSB4 (19,34). Our recent studies indicated that Fbxo45 causes rapid degradation of Par-4 by binding to the VASA segment (19). We therefore examined whether the VASA segment within PAF was essential for apoptosis of cancer cells. H460 cells were transfected with expression constructs for Par-4, Par-4 with an ALANAL mutation introduced within its VASA segment (Par-4/3A), and vector for control. Western blot analysis of the CM from the H460 transfectants indicated cleavage of both wild type Par-4 and Par-4/3A mutant to generate the 15 kDa PAF fragment or PAF-mutant fragment, respectively (Figure 4A). The CM from Par-4 transfectants but not from Par-4/3A mutant transfectants induced apoptosis in the Par-4- and/or therapy-resistant A549 and A549TR cells (Figure 4A). As expected, Par-4-sensitive PC-3 and Mel1617BR cells were sensitive to both Par-4 and Par-4/3A mutant in the CM (Figure 4A).
Figure 4. The VASA segment of PAF is essential for apoptosis.
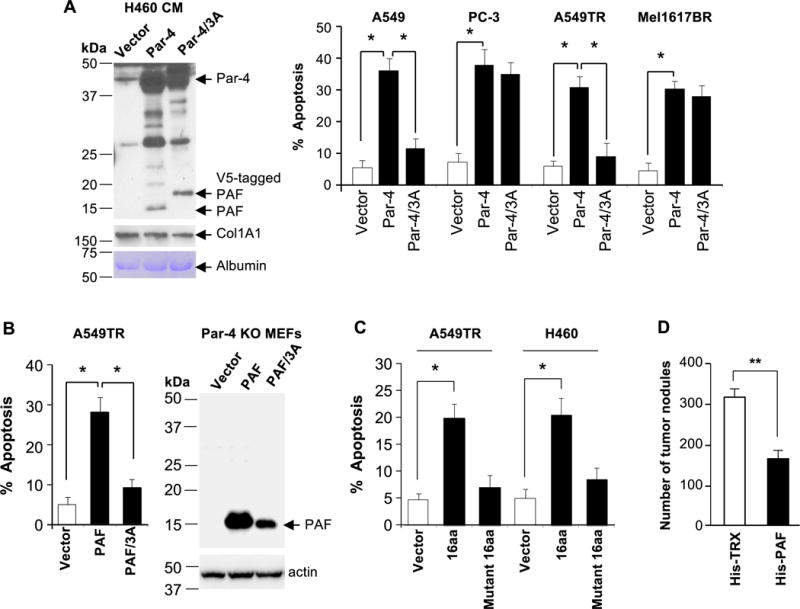
(A) The VASA domain is necessary for Par-4-induced apoptosis in Par-4-resistant cancer cells. H460 cells were transfected with expression constructs for Par-4, Par-4/ALANAL mutant (Par-4/3A), or vector for control for 24 h. The CM from the transfectants was confirmed for generation of the PAF fragment (left panel) and then transferred to cancer cells for 24 h. The cells were scored for apoptosis (right panel).
(B) The VASA domain is necessary for PAF induced apoptosis. Par-4 null MEFs were transfected with expression constructs for PAF, PAF/ALANAL (PAF/3A) mutant or vector and expression of the constructs was confirmed by Western blot analysis (right panel). The CM from the transfectants was then applied to A549TR cells for 24 hours to score for apoptosis (left panel).
(C) A short peptide containing the VASA domain induces apoptosis. Cancer cells were treated for 24 h with a 16aa synthetic peptide containing the VASA segment (ELNNNL), a 16 amino acid VASA/3A mutant (ALANA) peptide, or vehicle control. The cells were scored for apoptosis.
(A–C) The apoptosis data represent mean ± SD from three independent experiments. Asterisk (*) indicates statistical significance (P < 0.001) based on Student’s t test.
(D) PAF inhibits tumor growth in vivo. Immunocompromised mice were injected with LLC1 cells, and then with His-PAF or His-TRX protein (250 μg). Six mice were used for each treatment group. After 18 days, the animals were sacrificed, the lungs were injected with India ink and the tumor nodules were counted. Asterisks (**) indicates statistical significance (P < 0.02) based on Student’s t test.
We also generated a PAF/3A-mutant containing an ELNNNL to ALANAL mutation and transfected Par-4-null MEF cells with this mutant or with PAF expression construct or vector for control. The CM from PAF transfectants but not from PAF/3A mutant transfectants induced apoptosis in A549TR cells (Figure 4B). To determine whether the VASA segment was sufficient to induce apoptosis in therapy-sensitive and therapy-resistant cells, we used a 16aa synthetic peptide containing Par-4 residues 68 to 73 or a 16aa VASA mutant containing the ALANAL sequence. The 16aa peptide but not the mutant peptide induced apoptosis in both therapy-sensitive and therapy-resistant cancer cells (Figure 4C). Collectively, these findings indicate that the pro-apoptotic activity of PAF was dependent on its VASA segment.
Finally, to test the efficacy of PAF in tumor growth inhibition, immunocompromised mice were injected intravenously with LLC-1 cells followed by injection of the His-PAF or His-TRX control protein. His-PAF induced significant inhibition of lung tumor growth compared to control protein (Figure 4D). These results indicate that PAF is sufficient to cause tumor growth inhibition in vivo.
Extracellular PAF selectively enters cancer cells and competes with Par-4 binding to Fbxo45
To determine whether intracellular Par-4 function was essential for apoptosis by PAF, Par-4-null MEFs were transfected with PAF or PAF-ALANA mutant, Par-4, or vector for control. The CM from the transfectants was applied to the A549 cells with either shRNA knockdown levels of Par-4 or control shRNA. Cells with basal levels of Par-4 but not knockdown levels of Par-4 showed apoptosis in response to PAF; and mutant PAF failed to induce apoptosis (Figure 5A). These findings indicate that intracellular Par-4 is essential for apoptosis by extracellular PAF.
Figure 5. Mechanism of apoptosis by PAF.
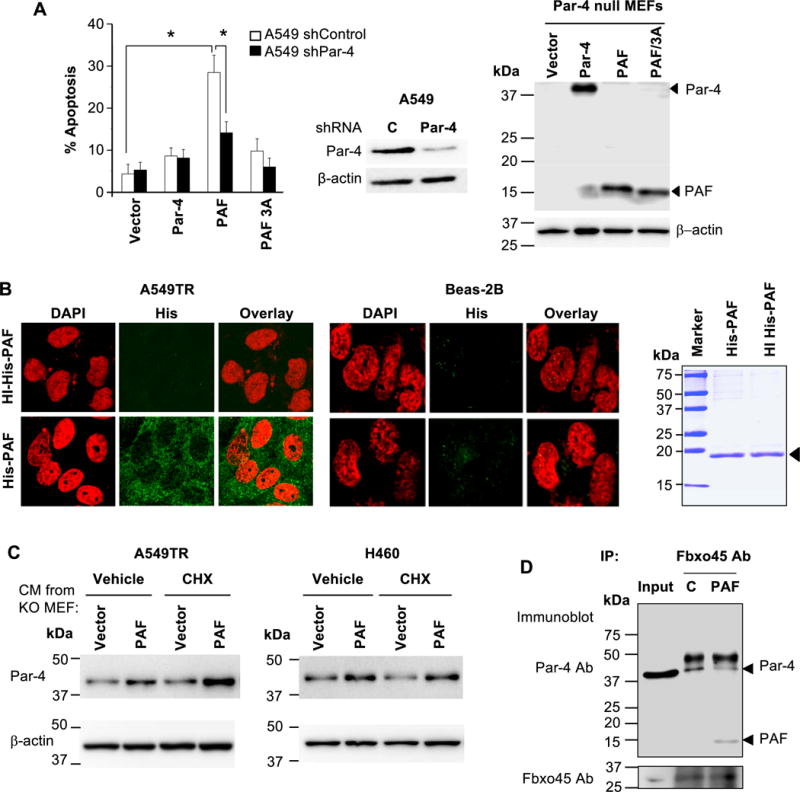
(A) Intracellular Par-4 function is essential for apoptosis by extracellular PAF. Par-4 null MEFs were transfected with expression constructs for Par-4, PAF, PAF/3A mutant or vector and expression of the constructs was confirmed by Western blot analysis (right panel). The CM was applied to A549 cells that were stably expressing control shRNA or Par-4 shRNA. Par-4 knockdown was confirmed by Western blot analysis (middle panel). The cells were scored for apoptosis by ICC for active caspase 3 after 24 hours of treatment (left panel). The apoptosis data represent mean ± SD from three independent experiments. Asterisk (*) indicates statistical significance (P < 0.001) based on Student’s t test.
(B) Extracellular PAF selectively enters cancer cells, not normal cells. Cells were treated with His-tagged PAF (100 nM) or heat-inactivated (HI) PAF (100 nM) as control, and the intracellular presence of the His-tagged protein was examined by ICC and confocal microscopy (left panels). Image magnification = 40×. PAF was analyzed by SDS-PAGE and Coomassie blue staining (right panel).
(C) Extracellular PAF stabilizes intracellular Par-4. CM from Par-4-knock out (KO) MEFs transfected with PAF or vector (as indicated above in Panel A), was transferred to A549TR or H460 cells for 5 h in the presence of cycloheximide (10 μg/ml) or vehicle control. Cell lysates were examined by Western blot analysis for stabilization of intracellular Par-4 by PAF from the CM.
(D) Extracellular PAF binds to Fbxo45 and prevents Par-4 binding to Fbxo45. A549TR cells were treated with purified His-PAF or heat-inactivated His-PAF (C) and the lysates were subjected to co-immunoprecipitation with Fbxo45 antibody and Western blot analysis for Par-4 and PAF with the Par-4 N-terminal antibody. Input lysate was used as control.
We then examined whether extracellular PAF can translocate into the cells to utilize intracellular Par-4 function. Cells were treated with His-tagged PAF and intracellular presence of the His-tagged peptide was examined by ICC and confocal microscopy. His-PAF was detected in the cytoplasm in A549TR and H460 cells but not in Beas-2B and MEF cells (Figure 5B and Figure S8). By contrast, heat-inactivated His-PAF control peptide failed to translocate into the cells. These observations indicate that extracellular PAF selectively enters the cancer cells to execute its effect intracellularly.
As intracellular Par-4 is ubiquitinated and degraded following binding to Fbxo45 (19), we examined whether extracellular PAF stabilizes intracellular Par-4. Par-4-null MEFs were transfected with PAF or vector for control, and the CM was transferred to A549TR or H460 cells for 5 h in the presence of cycloheximide, which prevents de novo protein synthesis. Treatment with CM containing extracellular PAF but not CM containing PAF/3A caused stabilization of intracellular Par-4 levels (Figure 5C).
As intracellular Par-4 is degraded by ubiquitin ligase Fbxo45, yet PAF stabilizes intracellular Par-4, we examined whether PAF competes with binding of Par-4 to Fbxo45. A549TR cells were treated with His-PAF or heat-inactivated His-PAF and the lysates were subjected to co-immunoprecipitation with Fbxo45 antibody. As seen in Figure 5D, extracellular His-PAF, but not heat-inactivated His-PAF, reduced binding of Par-4 to Fbxo45. Collectively these results indicate that application of PAF extracellularly facilitates the stabilization of Par-4 by preventing degradation of Par-4.
Discussion
Heterogeneity of tumors poses a significant challenge to cancer therapy due to the existence of drug resistant cell populations. Our present study indicates that treatment of therapy-sensitive cancer cells with chemotherapeutic agents leads to apoptosis of therapy-resistant cancer cells in culture. Importantly, growth of therapy-resistant tumor cells was also inhibited by therapy that was administered to xenografts containing a heterogeneous population of both therapy-sensitive and therapy-resistant tumor cells. By contrast, xenografts containing only therapy-resistant tumors were not inhibited by the same therapeutic regimen. These observations supported the release of a factor(s) from dying therapy-sensitive tumor cells to induce apoptosis in therapy-resistant tumor cells, leading to tumor growth inhibition. Although previous studies indicate that dying cells release tumor cell-inhibitory factors, the precise factor(s) released during the process has been elusive (16). We identified this factor as PAF, a previously unidentified 15 kDa caspase-cleavage fragment of the tumor suppressor Par-4. PAF is produced by cleavage of Par-4 at the D131 residue and is preferentially shed into the tumor cell microenvironment for paracrine apoptosis of both therapy-sensitive and therapy-resistant tumor cells, including those that are resistant to secreted Par-4. We noted that PAF protein inhibits the growth of tumors in mice. Most importantly, we identified the VASA domain of Par-4 that resides within PAF, as an essential domain for the apoptotic action of PAF. Intracellular Par-4 is typically sequestered and degraded by the ubiquitin ligase Fbxo45 (19). Our studies indicated that PAF translocates selectively into cancer cells and serves as a decoy by binding, via its VASA segment, to Fbxo45, and thereby releases intracellular Par-4 for apoptosis of cancer cells. Thus, cancer therapeutics that target sensitive populations of tumor cells exhibit a built-in mechanism for targeting therapy-resistant cells via cleavage of Par-4 and release of PAF. However, as tumor cells ultimately develop resistance to therapy, our ongoing studies are directed toward understanding the parameters that may contribute to resistance to PAF-mediated apoptosis, in an effort to harness PAF to overcome resistance to therapy.
PAF extends the target range of Par-4 to therapy-resistant cancer cells
Tumor heterogeneity is a roadblock in the treatment of cancer. However, as sensitive cells within the population are expected to release diverse factors when treated with cancer therapeutics, we hypothesized that such factors may perturb the resistant population. Rather unexpectedly, we identified PAF, a fragment of Par-4 shed by therapy-sensitive cancer cells undergoing apoptosis that can kill both therapy-sensitive and therapy-resistant cancer cells. The target range of PAF is not limited to any single type of cancer. We noted that lung cancer cells resistant to taxanes, prostate cancer cells resistant to TRAIL or bicalutamide, and melanoma cells resistant to the mutant-BRAF-inhibitor PLX4720 were sensitive to apoptosis by PAF. Interestingly, when Par-4 is intact, it does not induce cell death in most of the therapy-resistant lung or prostate cancer cells. In fact, the VASA domain within the PAF residues in intact Par-4 compromises the stability of Par-4 by serving as a binding motif for ubiquitin ligases such as Fbxo45 that degrade Par-4 (19). Our studies indicate that when Par-4 is naturally cleaved by caspase action to produce PAF, or when PAF is provided exogenously to cancer cells, it can enter cancer cells then bind to Fbxo45 and stabilize intracellular Par-4. The finding that PAF induces apoptosis, as judged by caspase 3 ICC and DAPI positivity, was corroborated in an independent automated assay for cell death (Figure S9 A–D). We further showed that PAF competes with the binding of ubiquitin ligase Fbxo45 to Par-4, thereby allowing Par-4 to stay physically and functionally intact and induce apoptosis of the cancer cells, regardless of whether they are sensitive or resistant to therapy.
The VASA domain of PAF interacts with ubiquitin ligase complexes
Our present studies indicated that the VASA domain located within PAF is important for competitive binding of PAF to Fbxo45, in order to release Par-4 for apoptosis. Fbxo45 binds, via its SPRY module, to the VASA domain of Par-4 (19). The SPRY domain of several other proteins, such as the SSB family proteins (SSB-1, -2, and -4), binds to the VASA module of Par-4 (34). Like many SOCS box-containing proteins, the SSB family proteins could function as receptors for substrate proteins that are ubiquitinated by cullin-based ubiquitin ligases (35, 36); however, their precise role in Par-4 regulation in cancer cells has not been fully delineated. The findings of the present study raise the possibility that the PAF domain, acting as a decoy via its VASA sequence, may modulate the interaction of other members of the family of SPRY domain containing proteins with their substrates.
PAF induces apoptosis selectively in cancer cells
It is particularly noteworthy that PAF induces apoptosis exclusively in cancer cells but not in normal cells. This finding is consistent with the finding that PAF selectively enters cancer cells, not normal cells. Accordingly, in the presence of PAF, binding of ubiquitin ligases to intracellular Par-4 is blocked in cancer cells, so that Par-4 induces apoptosis via activation of FADD/caspase 8/caspase 3. As cellular entry is essential for apoptosis of the cancer cells, the precise mechanism conferring selective cellular entry to PAF in cancer verses normal cells is currently under investigation in the laboratory.
PAF action in therapy-resistant tumors
Our study suggests that cancer-therapeutics that can induce a pro-apoptotic response in therapy-sensitive cancer cells may alter the tumor microenvironment, thereby making it conducive to apoptosis of therapy-resistant tumor cells. However, we noted that only about 50–60% of the cells in culture undergo apoptosis with PAF. This finding implies that PAF is unable to achieve its full potential. Escape from PAF-mediated apoptosis may be attributed to several factors that constitute the complex and heterogeneous tumor microenvironment. A recent study has indicated the emergence of therapy-resistant clones within heterogeneous tumors owing to activation of cell survival kinases such as Akt1 (37). As Akt1 is known to bind to Par-4 and inhibit Par-4 inducible apoptosis (10), emerging clones with elevated Akt1 activity may be resistant to PAF-induced apoptosis.
In summary, we identified a caspase-cleavage fragment of Par-4, designated as PAF, which is naturally produced and shed into the microenvironment by diverse cancer cells undergoing apoptosis in response to chemotherapeutic agents. PAF kills both therapy-sensitive and therapy-resistant cancer cells, causing growth inhibition of heterogeneous tumors containing therapy-resistant tumor cells. The action of PAF was attributed to its ability to translocate selectively into cancer cells and bind, via its VASA segment, to ubiquitin ligases such as Fbxo45 that contain the SPRY domain. Thus, PAF acts as a molecular decoy to rescue Par-4 from ubiquitin ligase-dependent degradation, so that Par-4 can induce apoptosis of cancer cells. As PAF selectively induces apoptosis in cancer cells but not in normal cells, this function of PAF can be harnessed to overcome therapeutic resistance in tumors.
Supplementary Material
Acknowledgments
We thank Jason Brandon for participating in the mouse studies, and the Markey Biospecimen and Tissue Procurement Shared Resource Facility for immuno-histochemical analysis and TUNEL of mouse tumors.
Financial Support: V.M. Rangnekar was recipient of NIH grants R01CA187273, R01CA165469, and R21CA179283.
References
- 1.Hanahan D, Weinberg Robert A. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 3.Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nature Reviews Clinical Oncology. 2015;13:335–47. doi: 10.1038/nrclinonc.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sells SF, Wood DP, Jr, Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA, et al. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1994;5:457–66. [PubMed] [Google Scholar]
- 5.El-Guendy N, Rangnekar VM. Apoptosis by Par-4 in cancer and neurodegenerative diseases. Exp Cell Res. 2003;283:51–66. doi: 10.1016/s0014-4827(02)00016-2. [DOI] [PubMed] [Google Scholar]
- 6.Shrestha-Bhattarai T, Rangnekar VM. Cancer-selective apoptotic effects of extracellular and intracellular Par-4. Oncogene. 2010;29:3873–80. doi: 10.1038/onc.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.García-Cao I, Duran A, Collado M, Carrascosa MJ, Martín-Caballero J, Flores JM, et al. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep. 2005;6:577–83. doi: 10.1038/sj.embor.7400421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Y, Burikhanov R, Qiu S, Lele SM, Jennings CD, Bondada S, et al. Cancer resistance in transgenic mice expressing the SAC module of Par-4. Cancer Research. 2007;67:9276–85. doi: 10.1158/0008-5472.CAN-07-2124. [DOI] [PubMed] [Google Scholar]
- 9.Cook J, Krishnan S, Ananth S, Sells SF, Shi Y, Walther MM, et al. Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene. 1999;18:1205–08. doi: 10.1038/sj.onc.1202416. [DOI] [PubMed] [Google Scholar]
- 10.Goswami A, Burikhanov R, de Thonel A, Fujita N, Goswami M, Zhao Y, et al. Binding and phosphorylation of Par-4 by Akt Is essential for cancer cell survival. Molecular Cell. 2005;20:33–44. doi: 10.1016/j.molcel.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I, et al. Inactivation of the candidate tumor suppressor Par-4 in endometrial cancer. Cancer Research. 2007;67:1927–34. doi: 10.1158/0008-5472.CAN-06-2687. [DOI] [PubMed] [Google Scholar]
- 12.Burikhanov R, Sviripa VM, Hebbar N, Zhang W, Layton WJ, Hamza A, et al. Arylquins target vimentin to trigger Par-4 secretion for tumor cell apoptosis. Nature Chemical Biology. 2014;10:924–26. doi: 10.1038/nchembio.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burikhanov R, Shrestha-Bhattarai T, Hebbar N, Qiu S, Zhao Y, Zambetti Gerard P, et al. Paracrine apoptotic effect of p53 mediated by tumor suppressor Par-4. Cell Reports. 2014;6:271–77. doi: 10.1016/j.celrep.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Burikhanov R, Brandon J, Qiu S, Shelton BJ, Spear B, et al. Systemic Par-4 inhibits non-autochthonous tumor growth. Cancer Biology & Therapy. 2011;12:152–57. doi: 10.4161/cbt.12.2.15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377–88. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou W, Zhang Q, Yan Z, Chen R, Zeh HJ, III, Kang R, et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4:e966. doi: 10.1038/cddis.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel N, Chatterjee SK, Vrbanac V, Chung I, Mu CJ, Olsen RR, et al. Rescue of paclitaxel sensitivity by repression of Prohibitin1 in drug-resistant cancer cells. Proceedings of the National Academy of Sciences. 2010;107:2503–08. doi: 10.1073/pnas.0910649107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Sahasrabuddhe AA, Szankasi P, Chung F, Basrur V, Rangnekar VM, et al. Fbxo45-mediated degradation of the tumor-suppressor Par-4 regulates cancer cell survival. Cell Death Differ. 2014;21:1535–45. doi: 10.1038/cdd.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weigel TL, Lotze MT, Kim PK, Amoscato AA, Luketich JD, Odoux C. Paclitaxel-induced apoptosis in non–small cell lung cancer cell lines is associated with increased caspase-3 activity. The Journal of Thoracic and Cardiovascular Surgery. 2000;119:795–803. doi: 10.1016/S0022-5223(00)70016-X. [DOI] [PubMed] [Google Scholar]
- 21.Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S, et al. Resistance to Taxol in lung cancer cells associated with increased microtubule dynamics. Proceedings of the National Academy of Sciences. 2001;98:11737–42. doi: 10.1073/pnas.191388598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burikhanov R, Shrestha-Bhattarai T, Qiu S, Shukla N, Hebbar N, Lele SM, et al. Novel mechanism of apoptosis resistance in cancer mediated by extracellular PAR-4. Cancer Research. 2012;73:1011–19. doi: 10.1158/0008-5472.CAN-12-3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proceedings of the National Academy of Sciences. 2008;105:3041–46. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee ECY, Zhan P, Schallhom R, Packman K, Tenniswood M. Antiandrogen-induced cell death in LNCaP human prostate cancer cells. Cell Death Differ. 2003;10:761–71. doi: 10.1038/sj.cdd.4401228. [DOI] [PubMed] [Google Scholar]
- 25.Schneider P, Thome M, Burns K, Bodmer J-L, Hofmann K, Kataoka T, et al. TRAIL Receptors 1 (DR4) and 2 (DR5) Signal FADD-dependent apoptosis and activate NF-κB. Immunity. 1997;7:831–36. doi: 10.1016/s1074-7613(00)80401-x. [DOI] [PubMed] [Google Scholar]
- 26.Chaudhry P, Singh M, Parent S, Asselin E. Prostate Apoptosis Response 4 (Par-4), a novel substrate of caspase-3 during apoptosis activation. Molecular and Cellular Biology. 2011;32:826–39. doi: 10.1128/MCB.06321-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Thonel A, Hazoumé A, Kochin V, Isoniemi K, Jego G, Fourmaux E, et al. Regulation of the proapoptotic functions of prostate apoptosis response-4 (Par-4) by casein kinase 2 in prostate cancer cells. Cell Death Dis. 2014;5:e1016. doi: 10.1038/cddis.2013.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Treude F, Kappes F, Fahrenkamp D, Müller-Newen G, Dajas-Bailador F, Krämer OH, et al. Caspase-8-mediated PAR-4 cleavage is required for TNFα-induced apoptosis. Oncotarget. 2014;5:2988–98. doi: 10.18632/oncotarget.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Guendy N, Zhao Y, Gurumurthy S, Burikhanov R, Rangnekar VM. Identification of a unique core domain of Par-4 sufficient for selective apoptosis Induction in cancer cells. Molecular and Cellular Biology. 2003;23:5516–25. doi: 10.1128/MCB.23.16.5516-5525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122–33. doi: 10.1038/sj.onc.1204282. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Zhu H, Xu C-j, Yuan J. Cleavage of BID by Caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 32.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, Mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 33.Muzio M. An induced proximity model for caspase-8 activation. Journal of Biological Chemistry. 1998;273:2926–30. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 34.Filippakopoulos P, Low A, Sharpe TD, Uppenberg J, Yao S, Kuang Z, et al. Structural Basis for Par-4 recognition by the SPRY domain- and SOCS Box-containing proteins SPSB1, SPSB2, and SPSB4. Journal of Molecular Biology. 2010;401:389–402. doi: 10.1016/j.jmb.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo J-S, Suh H-Y, Park S-Y, Oh B-H. Structural basis for protein recognition by B30. 2/SPRY Domains Molecular Cell. 2006;24:967–76. doi: 10.1016/j.molcel.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HME, Hilton DJ. The SOCS box: a tale of destruction and degradation. Trends in Biochemical Sciences. 2002;27:235–41. doi: 10.1016/s0968-0004(02)02085-6. [DOI] [PubMed] [Google Scholar]
- 37.Spagnolo F, Ghiorzo P, Queirolo P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget. 2014;5:10206–21. doi: 10.18632/oncotarget.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.