

Abstract
Purpose of the Review
To review the evidence that the Alzheimer peptide β-amyloid (Aβ) interacts with the blood coagulation system and influences the pathophysiology of the disease.
Recent findings
That Aβ can interact with fibrinogen and blood coagulation Factor XII and trigger ischemia and inflammation.
Summary
Aβ interacts with fibrinogen and FXII. These interactions can lead to increased clotting, abnormal clot formation, persistent fibrin deposition, and generation of pro-inflammatory molecules. These events can damage neurons and could contribute to the cognitive decline in Alzheimer’s disease patients.
Keywords: Alzheimer’s disease, β-amyloid peptide, fibrinogen, Factor XII
Introduction
Alzheimer’s disease (AD) leads to cognitive impairment and is eventually fatal. The cognitive decline is associated with extensive neuronal degeneration. The most well-known pathological features of AD are extracellular Aβ plaques, intracellular tau tangles, neuroinflammation, and neuronal loss. Less discussed is that AD is often associated with cerebrovascular abnormalities [1]. Recently, the vasculature of 2083 AD postmortem samples were analyzed, and the conclusion was “concurrent vascular disease strongly correlates with cognitive dysfunction” in AD [2]. The symptoms of AD and cerebrovascular pathology could be independent co-morbidities, with both being increased in aging populations. However, it is also possible that there is a mechanistic link between AD and vascular pathology.
If AD and vascular pathology are connected, what are the mechanistic links?
While the mechanisms behind neuronal dysfunction and cognitive decline in AD are heterogeneous and complex, abundant evidence points to accumulation of Aβ as one cause of AD. Aβ is generated when amyloid-β precursor protein (APP) is cleaved by β- and γ-secretases. Although the primary physiological function of APP is still unclear, it can participate in neurogenesis, synapse formation, and cell adhesion [3, 4]. Once liberated from APP on the cell surface, Aβ is released into the extracellular space and can form aggregation-prone oligomers, protofibrils, and fibrils in the brain parenchyma. Normally, the generation of Aβ in the brain is balanced by its clearance, and its accumulation in AD is thought to be the result of progressively impaired or overwhelmed clearance mechanisms. As Aβ is cleared from the brain, it can access extracellular fluids like cerebrospinal fluid (CSF) and blood [5], which would bring it in contact with plasma proteins. Since much of the Aβ produced in the brain is cleared through the cerebral vasculature, Aβ levels are higher in CNS venous blood compared to arterial blood [6] with some studies showing that elevated plasma Aβ levels are an early event in AD development [7, 8]. During clearance, Aβ can also accumulate around blood vessels as cerebral amyloid angiopathy (CAA).
A possible role of Aβ-fibrinogen interaction in AD
AD patients show increased fibrin deposition in the brain, including areas associated with cell death [9–13], suggesting that fibrin accumulation may contribute to neurodegeneration in AD. This idea is supported by work in AD mice. Similar to AD patients, AD mice have increased fibrin deposition in their brains, with fibrin co-localizing with dystrophic neurites [9, 10, 14]. Pharmacological or genetic reduction of circulating fibrinogen in AD mice reduces fibrin deposition in the brain and improves their cognitive performance [10], indicating that fibrin deposition participates in the development and/or progression of AD in this model. Furthermore, fibrinogen-derived peptides are also increased in the plasma and CSF of AD patients [15–17], indicating that fibrinogen or fibrinogen-derived peptides could be potential biomarkers for diagnosis in some AD patients.
The inflammatory properties of fibrin together with its potential role in CNS signal transduction provide a possible mechanistic link between fibrin deposition in the brain, neuroinflammation, and neuronal dysfunction [18, 19]. In addition to its pathological effects in the brain parenchyma, persistent fibrin within blood vessel walls and in the vessel lumen can also contribute to AD pathology. Fibrin clots can occlude capillaries and restrict blood flow, leading to microinfarcts and damage to downstream cells.
In AD, fibrin(ogen) deposition may be precipitated and/or exacerbated by its interaction with Aβ [20–22], which has been investigated using electron microscopy, X-ray crystallography, and other in vitro techniques. Aβ specifically interacts with fibrinogen with a Kd of 26.3 ± 6.7 nM and also binds to pre-formed fibrin fibrils [20, 21]. Aβ-fibrin(ogen) binding is mediated by the central region of Aβ and by two regions on fibrin(ogen), the C-terminus of the β-chain and the αC region of the α-chain [20–22]. Binding of Aβ to fibrinogen has functional consequences: it induces a structural change in the C-terminal region of the fibrinogen β-chain (β384–393) [22], precipitates fibrinogen oligomerization [20], and results in the formation of fibrin with increased resistance to fibrinolysis [10, 21]. Aβ delays fibrinolysis via two mechanisms: 1) by inducing a tighter fibrin network composed of thinner fibers; and 2) by inhibiting the plasmin(ogen)-fibrin interaction [21], likely through steric interference with plasminogen’s binding sites on fibrin Aα residues 148–160 and on the αC region of fibrin, which are in close spatial proximity to the β-chain and αC binding sites of Aβ, respectively. Another consequence of Aβ binding to the αC region of fibrinogen and blocking plasmin-mediated cleavage at this site is the generation of increased levels of a plasmin-resistant fibrin degradation fragment [22], which if found in AD patients, could serve as a possible marker of fibrin(ogen)-related AD pathology.
In the brain parenchyma, extravasated fibrin(ogen) could come in contact with high concentrations of soluble Aβ released from neurons and with soluble Aβ surrounding Aβ plaques. In the vessel wall, fibrinogen may interact with increased levels of soluble Aβ near CAA deposits [23, 24]. Thus, the interaction between Aβ and fibrinogen in the brain parenchyma and/or vessels may result in the formation of abnormal, persistent fibrin and in its deposition along cerebrovascular walls. Over time, this could lead to microinfarcts with subsequent hemorrhage, inflammation and blood-brain barrier disruption, all pathologies commonly observed in AD.
Aβ has been shown to interact with numerous other circulating components including erythrocytes [25, 26], tissue plasminogen activator [27], apolipoprotein E [28], apolipoprotein J [29], α2-macroglobulin [30], albumin [31], and members of the classical and alternative complement cascades [32, 33]. These interactions between Aβ and mediators of coagulation and inflammation could predispose blood to clotting. Indeed, there is evidence that circulating Aβ can elicit a clinically meaningful prothrombotic state, since plasma Aβ levels are correlated with infarctions as determined by MRI [34]. This prothrombotic state might be manifest in areas most sensitive to circulatory deficits, such as the hippocampus [35], providing one mechanism by which circulating Aβ could contribute to initiation of neurodegeneration in that region. At the same time, these interactions could contribute to the development of a systemic proinflammatory state. The idea that peripheral inflammation may contribute to neuropathology in AD is supported by studies demonstrating that peripheral markers of inflammation increase the risk of AD, that incidence of systemic infections increase the risk of dementia, and that cognitive impairment in AD is exacerbated during and after systemic infection [36–38]. Thus, activation and/or modulation of the delicately balanced coagulation and inflammatory systems by Aβ could lead to minor but chronic and pathological occlusion and inflammation, both of which could contribute to the neuronal death observed in AD.
The FXII contact system is significant in AD pathophysiology in humans and mouse models
Aβ also binds to and activates coagulation factor XII (FXII), which can then initiate the contact system [39–43], a proteolytic cascade that leads both to clot formation through the intrinsic coagulation pathway and also to inflammation via bradykinin release after high molecular weight kininogen (HK) cleavage. Aβ-mediated activation of FXII could be one mechanism behind the increased contact system activation observed in the plasma and CSF of AD patients and mouse models [39, 40]
The contact system can initiate vascular pathology and inflammation, both of which are implicated in AD. Thus, this system could contribute to these pathologies in AD. Connections between FXII and AD have been reported: Aβ plaques contain FXII [44], the AD brain parenchyma exhibits higher plasma kallikrein activity [45], and AD patients have increased HK cleavage in their CSF [46]. In addition, AD patients and mice have higher plasma levels of activated FXII (FXIIa) and increased HK cleavage compared to controls [39].
To determine if FXII is causally related to AD, plasma FXII was depleted in AD mice using antisense oligonucleotide (ASO)-mediated messenger RNA knockdown. This depletion inhibited HK cleavage in plasma and reduced neuroinflammation, fibrin(ogen) deposition, and neuronal degeneration in the brain. The improved brain pathology was accompanied by better cognitive function [47]. These results provide a mechanistic link between Aβ and the contact system with resulting neuroinflammation, neuronal degeneration, and cognitive impairment.
There is no effective treatment for AD. However, a link between FXII activation and the pathogenesis of AD provides a possible novel approach to treatment. The contact system is an attractive target for therapy [48]. Humans deficient in FXII and mice with knockout of the FXII, FXI, or HK gene all have normal hemostasis. Deficiencies in the contact system protect mice from thrombogenic challenges such as clotting after arterial injury and experimental cerebral ischemia [49, 50].
Since preventing FXII activation attenuates AD pathology in a mouse model [47], therapies designed to block the contact system might slow disease progression while not affecting normal hemostasis. In addition, this approach would block bradykinin release from HK and thereby reduce inflammation in this disease. Thus, the contact system might represent new targets to suppress both thrombotic and inflammatory contributions to AD progression. Positive results might be able to be applied to AD patients rapidly. For example, a small molecule inhibitor of PK, ecallantide, is currently approved for treatment of hereditary angioedema [51]. An antibody inhibitor of PK [52] has also been developed, which is slated for a phase 3 trial and possible FDA approval by 2018. Some of these reagents might be useful for the treatment of AD in the future.
Conclusion
The data reviewed here suggest that the interaction of Aβ with fibrin(ogen) can lead to increased fibrin deposition in cerebral blood vessels, and that these accumulated fibrin deposits may disrupt cerebral blood flow and induce microinfarcts, inflammation, and BBB damage, all of which are aspects of cerebrovascular dysfunction observed in AD. At the same time, Aβ’s ability to activate FXII may contribute to increased fibrin generation through the intrinsic coagulation pathway as well as to increased inflammation and vascular permeability through bradykinin release from HK. These possible roles of Aβ in thrombosis, fibrinolysis, and inflammation via its interaction with fibrinogen and FXII are summarized in Figure 1.
Figure 1.
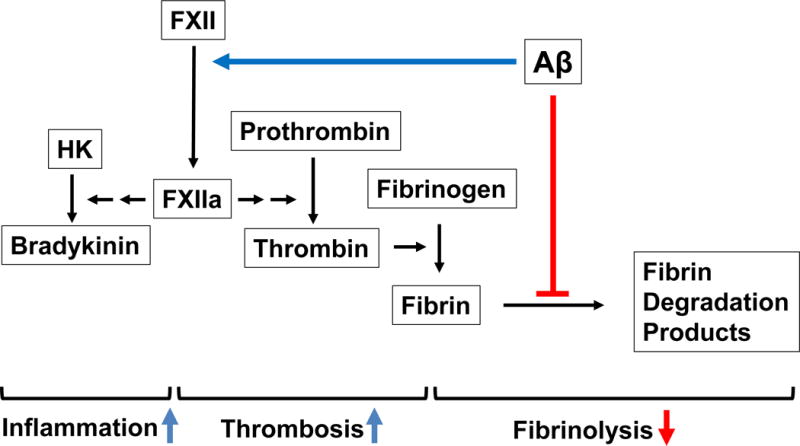
Proposed model for the role of Aβ in thrombosis, fibrinolysis, and inflammation via its interaction with fibrinogen and FXII. FXII activation by Aβ may induce inflammation through bradykinin activation and increase fibrin generation through the intrinsic coagulation pathway. Furthermore, the interaction of Aβ with fibrin(ogen) can decrease fibrinolysis via abnormal fibrin clot formation as well as by inhibiting the plasmin(ogen)-fibrin interaction. Together, these processes may increase the level of fibrin and inflammation in the cerebral blood vessels, which could induce or enhance the cerebrovascular dysfunction observed in AD patients.
Combination therapy has been indispensable in making therapeutic breakthroughs in other complex diseases like cancer and AIDS. AD is an extremely complex disease with many probable pathogenic mechanisms, and treatment will likely involve combination therapy targeting various aspects of the pathological network [53]. The interactions of Aβ with fibrinogen and FXII described here are possible components of this network. If these mechanisms are involved in AD pathophysiology, targeted therapy could be designed for AD patients identified as having circulatory abnormalities stemming from Aβ’s interaction with these blood components. Given the heterogeneity of this disease, targeting a combination of different pathogenic pathways in different individuals will likely be a valuable approach to treatment.
Key Points.
Aβ can interact with fibrinogen leading to structurally abnormal, persistent blood clots
Aβ can interact with FXII leading to increased clotting and release of the pro-inflammatory peptide bradykinin
The interaction of Aβ with fibrinogen and/or FXII could participate in Alzheimer’s disease pathophysiology
Acknowledgments
We thank the members of the Strickland laboratory for their help. H.J.A., Z.C., E.H.N., and S.S. were supported by NIH grant NS050537; Cure Alzheimer’s Fund; Rudin Family Foundation; Mellam Family Foundation; Louis Herlands; John A. Herrmann Jr.; Mary & James G. Wallach Foundation. D.Z. is an employee and shareholder of Regeneron Pharmaceuticals, Inc.
References
- 1.Humpel C. Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease? Exp Gerontol. 2011;46(4):225–32. doi: 10.1016/j.exger.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serrano-Pozo A, et al. Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J Neuropathol Exp Neurol. 2013;72(12):1182–92. doi: 10.1097/NEN.0000000000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Priller C, et al. Synapse formation and function is modulated by the amyloid precursor protein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26(27):7212–21. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westmark CJ, et al. Novel roles of amyloid-beta precursor protein metabolites in fragile × syndrome and autism. Molecular psychiatry. 2016;21(10):1333–41. doi: 10.1038/mp.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarasoff-Conway JM, et al. Clearance systems in the brain-implications for Alzheimer disease. Nature reviews. Neurology. 2015;11(8):457–70. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts KF, et al. Amyloid-beta efflux from the central nervous system into the plasma. Ann Neurol. 2014;76(6):837–44. doi: 10.1002/ana.24270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pesini P, et al. Reliable Measurements of the beta-Amyloid Pool in Blood Could Help in the Early Diagnosis of AD. Int J Alzheimers Dis. 2012;2012:604141. doi: 10.1155/2012/604141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schupf N, et al. Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2008;105(37):14052–7. doi: 10.1073/pnas.0805902105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cortes-Canteli M, et al. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36(2):608–17. doi: 10.1016/j.neurobiolaging.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortes-Canteli M, et al. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66(5):695–709. doi: 10.1016/j.neuron.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hultman K, et al. Plasmin deficiency leads to fibrin accumulation and a compromised inflammatory response in the mouse brain. J Thromb Haemost. 2014;12(5):701–12. doi: 10.1111/jth.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narayan PJ, et al. Assessing fibrinogen extravasation into Alzheimer’s disease brain using high-content screening of brain tissue microarrays. J Neurosci Methods. 2015;247:41–9. doi: 10.1016/j.jneumeth.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Viggars AP, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci Lett. 2011;505(1):25–30. doi: 10.1016/j.neulet.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 14.Klohs J, et al. Contrast-enhanced magnetic resonance microangiography reveals remodeling of the cerebral microvasculature in transgenic ArcAbeta mice. J Neurosci. 2012;32(5):1705–13. doi: 10.1523/JNEUROSCI.5626-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noguchi M, et al. Roles of serum fibrinogen alpha chain-derived peptides in Alzheimer’s disease. International journal of geriatric psychiatry. 2014;29(8):808–18. doi: 10.1002/gps.4047. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura Y, et al. Plasma protein profiling for potential biomarkers in the early diagnosis of Alzheimer’s disease. Neurological research. 2017;39(3):231–238. doi: 10.1080/01616412.2017.1281195. [DOI] [PubMed] [Google Scholar]
- 17.Vafadar-Isfahani B, et al. Identification of SPARC-like 1 protein as part of a biomarker panel for Alzheimer’s disease in cerebrospinal fluid. J Alzheimers Dis. 2012;28(3):625–36. doi: 10.3233/JAD-2011-111505. [DOI] [PubMed] [Google Scholar]
- 18.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34(1):43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 19.Ryu JK, Davalos D, Akassoglou K. Fibrinogen signal transduction in the nervous system. J Thromb Haemost. 2009;7(Suppl 1):151–4. doi: 10.1111/j.1538-7836.2009.03438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn HJ, et al. Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A. 2010;107(50):21812–7. doi: 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamolodchikov D, Strickland S. Aβ delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood. 2012;119:3342–3351. doi: 10.1182/blood-2011-11-389668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamolodchikov D, et al. Biochemical and structural analysis of the interaction between beta-amyloid and fibrinogen. Blood. 2016;128(8):1144–51. doi: 10.1182/blood-2016-03-705228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki N, et al. High tissue content of soluble beta 1–40 is linked to cerebral amyloid angiopathy. Am J Pathol. 1994;145(2):452–60. [PMC free article] [PubMed] [Google Scholar]
- 24.Shinkai Y, et al. Amyloid beta-proteins 1–40 and 1–42 (43) in the soluble fraction of extra- and intracranial blood vessels. Annals of neurology. 1995;38(3):421–8. doi: 10.1002/ana.410380312. [DOI] [PubMed] [Google Scholar]
- 25.Mattson MP, et al. Abeta25–35 induces rapid lysis of red blood cells: contrast with Abeta1–42 and examination of underlying mechanisms. Brain Res. 1997;771(1):147–53. doi: 10.1016/s0006-8993(97)00824-x. [DOI] [PubMed] [Google Scholar]
- 26.Kuo YM, et al. Amyloid-beta peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem Biophys Res Commun. 2000;268(3):750–6. doi: 10.1006/bbrc.2000.2222. [DOI] [PubMed] [Google Scholar]
- 27.Kingston IB, Castro MJ, Anderson S. In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid beta-peptide analogues. Nat Med. 1995;1(2):138–42. doi: 10.1038/nm0295-138. [DOI] [PubMed] [Google Scholar]
- 28.Strittmatter WJ, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(17):8098–102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghiso J, et al. The cerebrospinal-fluid soluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J. 1993;293(Pt 1):27–30. doi: 10.1042/bj2930027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du Y, et al. alpha2-Macroglobulin as a beta-amyloid peptide-binding plasma protein. J Neurochem. 1997;69(1):299–305. [PubMed] [Google Scholar]
- 31.Biere AL, et al. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271(51):32916–22. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 32.Rogers J, et al. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89(21):10016–20. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradt BM, WP Kolb, NR Cooper. Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J Exp Med. 1998;188(3):431–8. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toledo JB, et al. Factors affecting Abeta plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011;122(4):401–13. doi: 10.1007/s00401-011-0861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Crit Rev Neurobiol. 1991;6(3):149–82. [PubMed] [Google Scholar]
- 36.Holmes C, et al. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74(6):788–9. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelhart MJ, et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61(5):668–72. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- 38.Dunn N, et al. Association between dementia and infectious disease: evidence from a case-control study. Alzheimer Dis Assoc Disord. 2005;19(2):91–4. doi: 10.1097/01.wad.0000165511.52746.1f. [DOI] [PubMed] [Google Scholar]
- 39.Zamolodchikov D, et al. Activation of the factor XII-driven contact system in Alzheimer’s disease patient and mouse model plasma. Proc Natl Acad Sci U S A. 2015;112(13):4068–73. doi: 10.1073/pnas.1423764112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergamaschini L, et al. Activation of the contact system in cerebrospinal fluid of patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12(2):102–8. doi: 10.1097/00002093-199806000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Maas C, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118(9):3208–18. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zamolodchikov D, Renne T, Strickland S. The Alzheimer’s disease peptide Abeta promotes thrombin generation through activation of coagulation factor XII. J Thromb Haemost. 2015 doi: 10.1111/jth.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, et al. A plasma proteolysis pathway comprising blood coagulation proteases. Oncotarget. 2016;7(27):40919–40938. doi: 10.18632/oncotarget.7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasuhara O, Walker DG, McGeer PL. Hageman factor and its binding sites are present in senile plaques of Alzheimer’s disease. Brain Res. 1994;654(2):234–40. doi: 10.1016/0006-8993(94)90484-7. [DOI] [PubMed] [Google Scholar]
- 45.Ashby EL, Love S, Kehoe PG. Assessment of activation of the plasma kallikrein-kinin system in frontal and temporal cortex in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2012;33(7):1345–55. doi: 10.1016/j.neurobiolaging.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 46.Bergamaschini L, et al. The region 1–11 of Alzheimer amyloid-beta is critical for activation of contact-kinin system. Neurobiol Aging. 2001;22(1):63–9. doi: 10.1016/s0197-4580(00)00174-3. [DOI] [PubMed] [Google Scholar]
- 47.Chen ZL, et al. Depletion of coagulation factor XII ameliorates brain pathology and cognitive impairment in Alzheimer’s disease mice. Blood. 2017 doi: 10.1182/blood-2016-11-753202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renne T, et al. In vivo roles of factor XII. Blood. 2012;120(22):4296–303. doi: 10.1182/blood-2012-07-292094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Renne T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol. 2012;34(1):31–41. doi: 10.1007/s00281-011-0288-2. [DOI] [PubMed] [Google Scholar]
- 50.Merkulov S, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111(3):1274–81. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheffer AL, et al. Outcomes after ecallantide treatment of laryngeal hereditary angioedema attacks. Ann Allergy Asthma Immunol. 2013;110(3):184–188. e2. doi: 10.1016/j.anai.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Chyung Y, et al. A phase 1 study investigating DX-2930 in healthy subjects. Ann Allergy Asthma Immunol. 2014;113(4):460–6. e2. doi: 10.1016/j.anai.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 53.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen ’. Nat Neurosci. 2015;18(6):800–6. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]