

Myelodysplastic syndromes (MDS) are poorly understood and rare hematologic malignancies in children. In recent years several germline mutations have been implicated in a subset of familial cases of MDS, other myeloid neoplasms, or bone marrow failure syndromes, including mutations in GATA2, ETV6, and DDX41 (1, 2). However, for some families, such as those first reported nearly 30 years ago (3, 4), who present with MDS and monosomy 7 (OMIM 252270), the predisposing genetic alteration has remained elusive. Here we report our findings on a family without historical suggestions of MDS/AML predisposition but with three siblings with monosomy 7 and MDS.
A 4-year-old male (proband/SJ015856/Sibling 1) was found to have severe neutropenia, macrocytosis, and thrombocytopenia (ANC=400/mm3, MCV=101.7fL, platelets=111×109/L). Bone marrow evaluation revealed hypocellularity with trilineage dysplasia and 1% blasts (Figure 1A). His 3-year-old sister (SJ015855/Sibling 2) also had low peripheral cell counts and bone marrow dysplasia, while his youngest brother (14-months-old/SJ018228/Sibling 3) had normal peripheral counts and a hypocellular bone marrow with megakaryocytic dysplasia. Cytogenetic analysis was significant for monosomy 7 in all siblings (Figure 2 & Supplemental Figure 1) and thus all 3 were diagnosed with refractory cytopenia of childhood. All three siblings had a transient thrombocytopenic phase at birth requiring a platelet transfusion, but had otherwise normal developmental histories and stature-for-age (at or above the 75th percentile). The proband was born with severe hypospadias and a bifid scrotum requiring multiple corrective surgeries without any bleeding complications. A karyotype during the neonatal period demonstrated a normal male karyotype. No other congenital abnormalities were noted in the siblings. Chromosome breakage studies for Fanconi Anemia were negative. The two eldest siblings have been treated with matched, unrelated donor BM transplants while the youngest sibling remains asymptomatic with normal peripheral blood counts and is monitored annually.
Figure 1. Diagnostic work up of 3 siblings with MDS and monosomy 7.
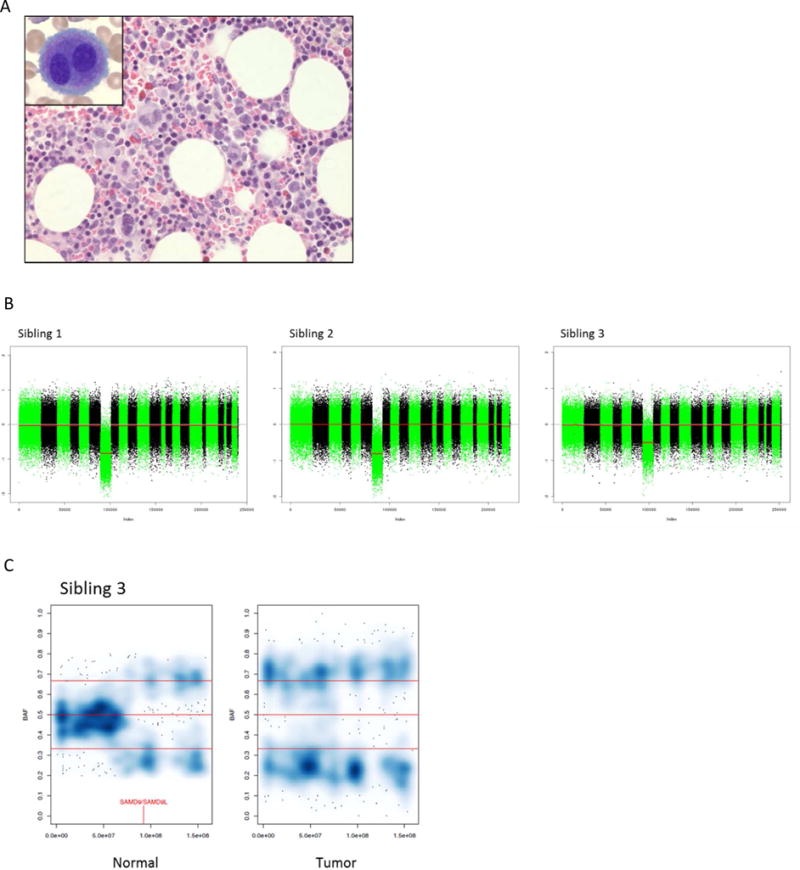
A.) Bone marrow core biopsy and aspirate (inset) showing dysplastic megakaryocytes in Sibling 1. B.) Copy number analysis demonstrating monosomy 7 in myeloid cells all three siblings. C.) Smoothed color density plots of chromosome 7 B-allele frequency (BAF) of lymphocyte (normal) and myeloid (tumor) DNA in Sibling 3 showing a subclonal population with allelic imbalance, despite the lack of a copy number change, consistent with CN-LOH in the lymphocytes. Darker blue signifies a higher data point (SNP alternative allele frequencies) density. Red vertical line indicates genomic location of SAMD9 and SAMD9L.
Figure 2. Sequencing validation and biologic characterization of the SAMD9 p.E1136Q mutation.
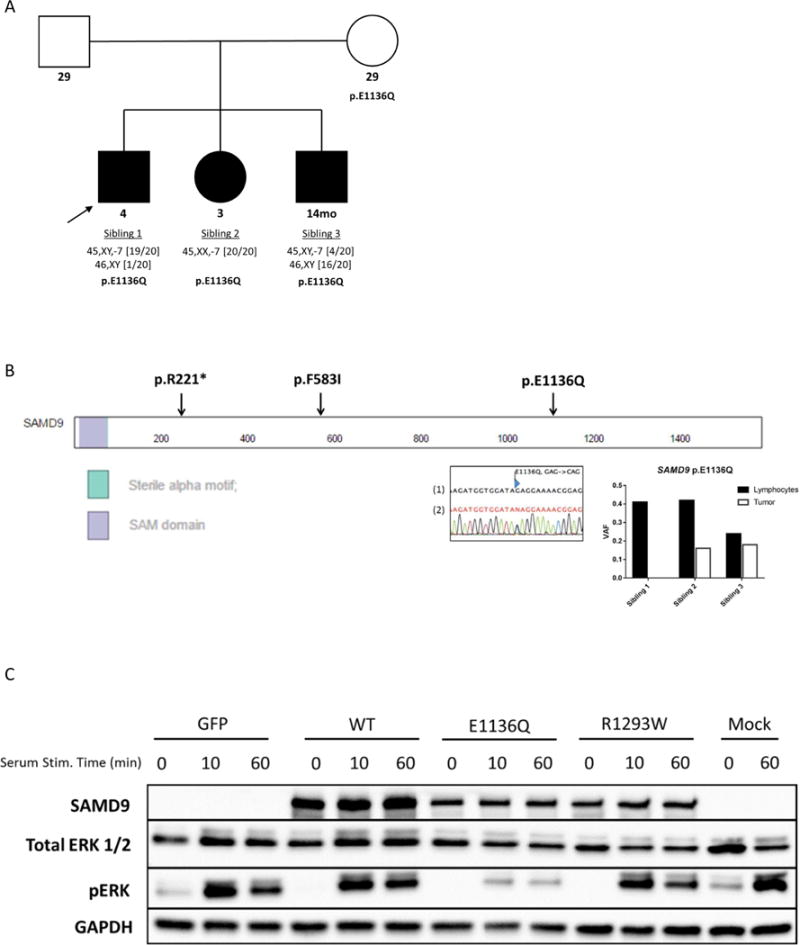
A.) Family pedigree identifying those with a SAMD9 mutation and their monosomy 7 status (filled: monosomy 7; open: untested karyotype). Ages at diagnosis are below pedigree symbols, bracketed numbers adjacent to karyotype below indicate fraction of metaphases affected. B.) Locations of mutations within the SAMD9 protein, a Sanger sequencing electropherogram (1-Reference, 2-Reverse primer) confirming the SAMD9 p.E1136Q mutation in the mother, and tumor/normal variant allele frequency (VAF) values for the p.E1136Q mutation. C.) Western blot (representative of multiple biological replicates) showing less ERK phosphorylation in 293T cells transiently expressing the mutant GFP-SAMD9 protein when compared to the wild-type at 0, 10, and 60 minutes following serum stimulation. The R1293W mutation was identified in some patients with MIRAGE syndrome (8). GFP indicates the empty GFP-fusion vector. The following antibodies were used: SAMD9: abcam (ab180575), From Cell Signaling Technologies: Total ERK (4695S), Phos-ERK (9101S), and GAPDH (2118S).
Bone marrow samples from the 3 siblings were banked for research after informed consent was obtained. All studies were approved by the Institutional Review Board at St. Jude Children’s Research Hospital. Cryopreserved marrow samples were flow-sorted into lymphocyte (source of “germline” DNA), as an alternate source of germline DNA was not available (5), and bulk myeloid populations. WES was performed on paired tumor/normal samples from the three siblings. WGS was also performed on the paired samples from Sibling 2 and only the normal samples from Siblings 1 and 3. Genomic data have been deposited in the European Genome-Phenome Archive (EGAS00001002202).
The loss of chromosome 7 was confirmed in the myeloid cells of all three siblings using WES data (6), with a clear subclonal loss in Sibling 3 (Figure 1B). No copy number loss was observed in the lymphocyte samples; however, a copy-number neutral loss of heterozygosity (CN-LOH) event was noted at 7q (chr7:78Mb–159Mb, q21.11–q36.3) in Sibling 3 (Figure 1C). A total of 19 non-synonymous shared high-confidence germline variants were present when intersecting the WES and WGS data from all three siblings, 10 of which were predicted to functionally impact the gene product by either SIFT or Polyphen2 (Supplemental Figure 2 & Supplemental Table 1). These analyses pointed to a heterozygous missense mutation (c.3406G>C) in SAMD9 (NM_017654), resulting in a p.E1136Q mutation (NP_010124; CADD score:19.7), as a potential causal lesion for MDS and monosomy 7 in this family (7). This mutation affects an evolutionarily conserved position and is not present in the ExAC or in-house databases. Sanger sequencing of genomic DNA isolated from the peripheral blood of the parents observed this mutation in the clinically asymptomatic mother, confirming this as an inherited variant (Figure 2A & B). Furthermore, a CLIA-approved laboratory confirmed the presence of the SAMD9 p.E1136Q in a buccal swab sample obtained from the proband. Similar SAMD9 missense mutations have recently been reported in patients with MIRAGE syndrome, a multisystem disorder associated with MDS and monosomy 7 (8).
Although present in the patients’ lymphocytes, the mutant allele was less common in the myeloid cell fraction (see Figure 2B), suggesting a preferential loss of the copy of chromosome 7 that harbors the SAMD9 variant. A similar decrease in VAF was reported in children with MIRAGE syndrome, presumably through a mechanism referred to as “adaptation by aneuploidy,” to deal with the growth-restrictive properties of SAMD9 mutant proteins (8). Similar to those mutations present in MIRAGE syndrome, the p.E1136Q mutation results in decreased induction of ERK phosphorylation (Figure 2C) after serum stimulation. Additional sibling-specific alterations involving SAMD9 (Supplemental Figure 3 & 4) were also observed in the lymphocyte fractions: a p.R221* (c.661C>T) in Sibling 1, p.F583I (c.1747C>A) in Sibling 2, and the aforementioned CN-LOH event in Sibling 3. The p.R221* and p.F583I were confirmed to be in cis with the p.E1136Q (Supplemental Figure 3C). These additional mutations are likely acquired rather than germline variants. Unlike the germline SAMD9 p.E1136Q, the p.R221* mutation was not present in DNA isolated from the buccal swab sample from Sibling 1. Further, the p.F583I in Sibling 2 and the CN-LOH event in Sibling 3 are subclonal events in the lymphocyte fraction. Sibling 2 also harbored a somatic mutation in the myeloid cells in ETV6 (NM_001987;p.R369W) at a codon previously observed in MDS (9). No somatic mutations recurrently reported in MDS were observed in the other two siblings.
We believe this to be the first report of SAMD9 mutations in isolated familial MDS. Although the in vitro impact on ERK phosphorylation appears similar to that reported in MIRAGE syndrome, clearly the p.E1136Q results in a milder phenotype as these siblings lack the other clinical manifestations of that syndrome. In addition, this family demonstrates a maternal pattern of inheritance yet the mother has no documented history of cytopenias. The reason for this variable penetrance is unclear, but could be explained by monoallelic gene expression, which has been observed in some families with MDS and GATA2 mutations (10). Of note, mutations in the related gene SAMD9L have recently been reported in Ataxia-Pancytopenia Syndrome (11, 12) and in some families with MDS (13), and loss of SAMD9L can result in myelodysplasia in mice (14). Tesi and colleagues described a phenomenon of somatic revertant mosaicism in which additional SAMD9L mutations could relieve the deleterious impact of the initial germline mutation (13), which may explain the additional SAMD9 variants we observed in these siblings. Collectively, these findings establish SAMD9/SAMD9L mutations as a new class of germline lesions with variable clinical phenotypes, including familial MDS. It is likely that the MDS observed in these cases is not due to the SAMD9/SAMD9L mutations per se. Rather, the MDS likely is a direct consequence of haploinsufficiency of multiple genes on chromosome 7 (15) and this genetic loss may represent a cellular adaptation to these deleterious germline SAMD9/SAMD9L mutations. Future studies to explore the hematopoietic impact of these mutations and to determine their recurrency in larger cohorts of families with MDS are clearly warranted.
Supplementary Material
Acknowledgments
We thank the family in this study and for the clinical staff involved in their care and management. The authors thank Tina Motroni for BM histology images and the Hartwell Center at SJCRH for providing sequencing assistance. This work was funded in part by the American Lebanese and Syrian Associated Charities (ALSAC) of SJCRH and the US National Institutes of Health (K08 HL116605-J.M.K.). J.M.K. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund.
Footnotes
Conflict of Interest Statement
The authors have nothing to disclose.
References
- 1.Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–97. doi: 10.1182/blood-2015-09-669937. quiz 518. [DOI] [PubMed] [Google Scholar]
- 2.Babushok DV, Bessler M, Olson TS. Genetic predisposition to myelodysplastic syndrome and acute myeloid leukemia in children and young adults. Leuk Lymphoma. 2016;57(3):520–36. doi: 10.3109/10428194.2015.1115041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shannon KM, Turhan AG, Chang SS, Bowcock AM, Rogers PC, Carroll WL, et al. Familial bone marrow monosomy 7. Evidence that the predisposing locus is not on the long arm of chromosome 7. J Clin Invest. 1989;84(3):984–9. doi: 10.1172/JCI114262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll WL, Morgan R, Glader BE. Childhood bone marrow monosomy 7 syndrome: a familial disorder? J Pediatr. 1985;107(4):578–80. doi: 10.1016/s0022-3476(85)80027-5. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937–41. doi: 10.1038/ng.2698. [DOI] [PubMed] [Google Scholar]
- 6.Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48(12):1551–6. doi: 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48(7):792–7. doi: 10.1038/ng.3569. [DOI] [PubMed] [Google Scholar]
- 9.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–27. doi: 10.1182/blood-2013-08-518886. quiz 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al Seraihi A, Rio-Machin A, Tawana K, et al., editors. Variable Penetrance Is Linked with Monoallelic Gene Expression in Inherited GATA2-Mutated MDS/AML. American Society of Hematology 58th Annual Meeting and Exposition; San Diego, CA. 2016. [Google Scholar]
- 11.Chen DH, Below JE, Shimamura A, Keel SB, Matsushita M, Wolff J, et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet. 2016;98(6):1146–58. doi: 10.1016/j.ajhg.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li FP, Hecht F, Kaiser-McCaw B, Baranko PV, Potter NU. Ataxia-pancytopenia: syndrome of cerebellar ataxia, hypoplastic anemia, monosomy 7, and acute myelogenous leukemia. Cancer Genet Cytogenet. 1981;4(3):189–96. doi: 10.1016/0165-4608(81)90013-3. [DOI] [PubMed] [Google Scholar]
- 13.Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SC, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS and neurological symptoms. Blood. 2017 doi: 10.1182/blood-2016-10-743302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A, et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell. 2013;24(3):305–17. doi: 10.1016/j.ccr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Wong JC, Weinfurtner KM, Alzamora Mdel P, Kogan SC, Burgess MR, Zhang Y, et al. Functional evidence implicating chromosome 7q22 haploinsufficiency in myelodysplastic syndrome pathogenesis. Elife. 2015;4 doi: 10.7554/eLife.07839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.