

Abstract
Clear cell renal cell carcinoma (ccRCC) is characterized by BAP1 and PBRM1 mutation, which are associated with tumors of different grade and prognosis. However, whether BAP1 and PBRM1 loss causes ccRCC and determines tumor grade is unclear. We conditionally targeted Bap1 and Pbrm1 (with Vhl) in the mouse using several Cre drivers. Sglt2 and Villin proximal convoluted tubule drivers failed to cause tumorigenesis, challenging the conventional notion of ccRCC origins. In contrast, targeting with Pax8, a developmental lineage-specific transcription factor, led to ccRCC of different grade. Bap1-deficient tumors were high grade and showed greater mTORC1 activation than Pbrm1-deficient tumors, which exhibited longer latency. Disrupting one allele of the mTORC1 negative regulator, Tsc1, in Pbrm1-deficient kidneys triggered higher grade ccRCC. This study establishes Bap1 and Pbrm1 as lineage-specific drivers of ccRCC and histological grade, implicates mTORC1 as a tumor grade rheostat, and suggests that ccRCC may arise from Bowman capsule cells.
INTRODUCTION
Kidney cancer is one of the top 10 most common cancers in the U.S. with ~60,000 new cases diagnosed annually (1). Most cases of renal cell carcinoma (RCC) are sporadic (>95%), and over 70% are clear cell renal cell carcinoma (ccRCC) (2). Inactivation of the von Hippel-Lindau (VHL) gene by either mutation or methylation is observed in over 80% of ccRCC (3–6). Germline loss-of-function mutations in VHL lead to VHL syndrome and a high incidence of ccRCC (7). The VHL gene, which encodes an E3 ubiquitin ligase that negatively regulates hypoxia inducible factor (HIF) α subunits, is a typical two-hit tumor suppressor gene, in which both alleles are inactivated for tumor development. Typically, the first allele is inactivated by an intragenic mutation, and the second allele is lost as part of a deletion event resulting in loss of heterozygosity (LOH) (8), often involving the whole chromosome 3p arm where the VHL gene is located. Intragenic mutation in one copy of VHL followed by 3p LOH are early events in ccRCC tumorigenesis, in fact the only known “truncal” events (9, 10).
Recurrent mutations in other tumor suppressor genes have also been identified in ccRCC with varying frequencies. Mutations in the Polybromo-1 (PBRM1) gene, also known as BRG1-associated factor 180 (BAF180), a component of the SWI/SNF-B (PBAF) chromatin-remodeling complex, are observed in roughly 50% of ccRCC (11, 12). BRCA1 associated protein-1 (BAP1), a deubiquitinating enzyme of the ubiquitin carboxyl-terminal hydrolase (UCH) family, and Set domain-containing 2 (SETD2), a histone methyltransferase that is specific for lysine 36 of histone H3, are found to be mutated in ~10 to 15% of ccRCC (13–15). Like VHL, all three of these genes are located on chromosome 3p, within a 50 Mb region, and one copy is deleted along with VHL when 3p is lost (16).
Remarkably, PBRM1 mutation tends to anticorrelate with BAP1 mutation in ccRCC, and tumors with BAP1 versus PBRM1 mutation exhibit distinct biology with markedly different outcomes (4, 17). Tumors with BAP1 mutation are higher grade and more aggressive compared with PBRM1-deficient tumors, and patients have significantly shorter survival rates (13, 17). In addition, whereas BAP1-deficient tumors exhibit higher levels of mammalian target of rapamycin complex 1 (mTORC1) activation, levels are reduced in PBRM1-deficient tumors (13, 17). These differences led to the first molecular genetic classification of ccRCC (4, 13), however, whether the difference in tumor grade and patient survival is actually driven by BAP1 and PBRM1 loss is not known.
An important limitation of the field for many years has been the lack of a genetically engineered mouse model (GEMM) reproducing genetic events in human ccRCC. Despite the discovery of the VHL gene in 1993 (18), VHL targeting failed to generate such a model. The discovery that BAP1 and PBRM1, two other frequently mutated ccRCC tumor suppressor genes, reside on 3p, together with the finding that this region is lost in the majority of ccRCCs, led us to hypothesize that RCC tumorigenesis may require the simultaneous loss of VHL and BAP1 (or PBRM1) (16). Surprisingly, while VHL heterozygous patients are predisposed to developing ccRCC, Vhl heterozygous mice do not develop ccRCC (19–23). A possible explanation for this may be that in the mouse, Vhl is located on chromosome 6 while Pbrm1 and Bap1 are on chromosome 14, and thus LOH in the Vhl region in chromosome 6 would still leave two copies of Pbrm1 and Bap1 intact. We previously modeled genetic events in human RCC by targeting Vhl and Bap1 in nephron progenitor cells (NPCs). However, Six2-Cre;VhlF/F;Bap1F/F mice died soon after birth. Nevertheless, Six2-Cre;VhlF/F;Bap1F/+ mice developed ccRCC and the kidneys bore resemblance to kidneys of patients with VHL syndrome (24). Unfortunately, however, the mice died from renal failure at around 8 months of age (as did Six2-Cre;VhlF/F mice), and these tumors remained uniformly small. Moreover, a GEMM of Pbrm1-deficient ccRCC has not been reported. Additionally, choosing an appropriate Cre driver is confounded by limited knowledge regarding the cell type of origin. Based on previous IHC evidence, it has been proposed that ccRCC arises from the epithelium of proximal convoluted tubule cells (25, 26). Subsequent gene expression analyses from expression data of normal tissue microdissection from various regions of the nephron (27, 28) supported this (29, 30): ccRCC resembles the glomerulus and proximal convoluted tubule leading to the notion that it arises from the proximal nephron area (29).
The paired box 8 (PAX8) gene encodes a transcription factor that plays a critical role in kidney development (31, 32). PAX8 is expressed in the mesonephros, metanephros, nephric duct, and ureteric bud (33, 34). PAX8 is also a sensitive and specific diagnostic marker for renal tumors routinely used in clinical practice (35–37). Here, we report a novel RCC GEMM based on Pax8-Cre deletion of Vhl together with either Bap1 or Pbrm1 (34), and show that Bap1 and Pbrm1 are not only drivers of RCC, but also determinants of tumor grade.
RESULTS
Human BAP1- and PBRM1-Deficient Tumors Express PAX8
To optimally model ccRCC in the mouse, we sought to identify a driver that could be used to broadly target genes implicated in RCC pathogenesis. Previously, we used the Six2-Cre driver, which is expressed broadly in nephron progenitor cells. While Six2-Cre;VhlF/F;Bap1F/F mice died shortly after birth, Six2-Cre;VhlF/F;Bap1F/+ mice survived and developed ccRCCs. However, the mice died at around 8 months of age due to kidney failure, and ccRCCs were uniformly small (24). We searched for other potential Cre drivers with more limited expression. PAX8, a known nephric lineage transcription factor, is activated later in development than Six2, and was particularly attractive as it is routinely used in clinical practice for diagnosis of primary and metastatic RCC (36, 37). Immunohistochemical (IHC) staining for PAX8 yields a robust nuclear signal in normal tubular epithelial cells, and in both primary and metastatic ccRCC (Fig. 1A and not shown). Tissue microarray (TMA) analyses of 123 molecularly annotated UT Southwestern ccRCCs showed positive staining for PAX8 in normal renal tubules and 97% (119/123) of ccRCCs. Importantly, nearly all BAP1- and PBRM1-deficient ccRCCs expressed PAX8 (Fig. 1B and C). In normal human kidney, PAX8 is expressed in most epithelial cells of proximal and distal renal tubules, loops of Henle, collecting ducts, and the parietal epithelial cells of Bowman capsule. Consistent with prior reports, and as shown in Fig. 1A, PAX8 staining is stronger in the parietal cells of Bowman capsule, distal tubules, and collecting ducts compared to the proximal tubules, in which weak cytoplasmic staining is also seen. A similar Pax8 pattern of expression was seen in normal adult mouse kidney (Fig. 1A).
Figure 1.
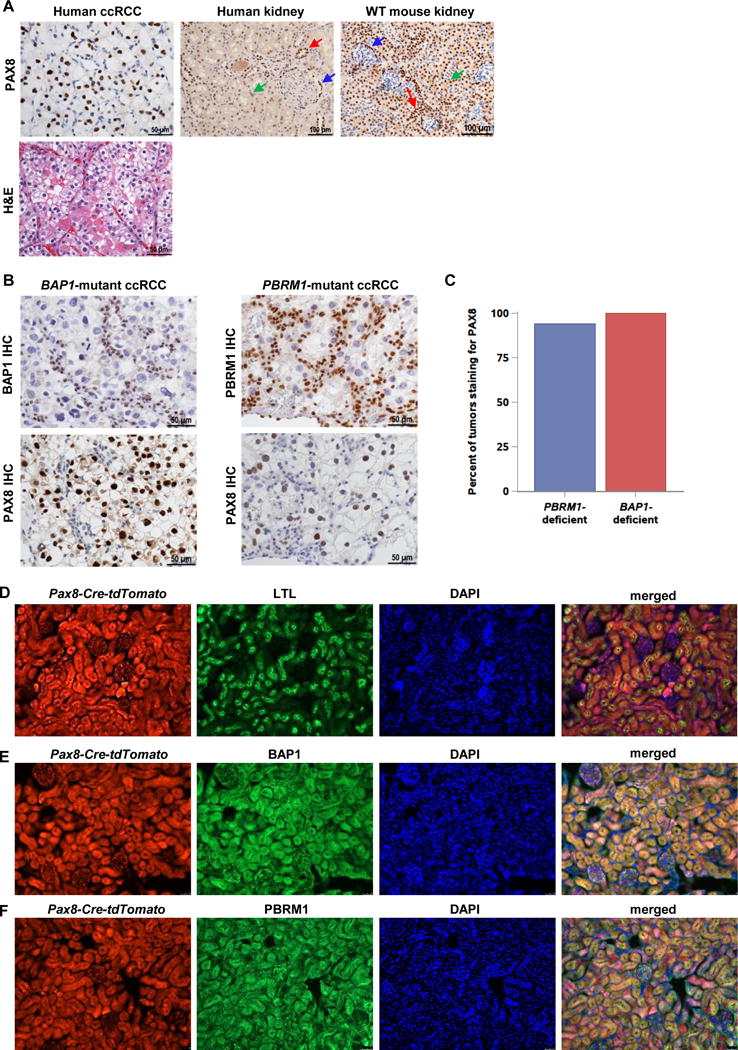
PAX8 is a marker of ccRCC and drives effective recombination of Bap1 and Pbrm1 in the mouse kidney. A, Representative PAX8 immunohistochemistry (IHC) of human ccRCC (along with hematoxylin and eosin (H&E) micrograph), normal human kidney, and wild-type (WT) mouse kidney. (Blue arrow, Bowman capsule; green arrow, proximal tubules; red arrow, distal tubules and collecting ducts) B, Representative images of PAX8 IHC in BAP1-mutant and PBRM1-mutant human RCC tissue microarray samples. Note that nuclei of tumor cells, which are larger and pleomorphic (but not those of stromal cells, which serve as internal positive controls) are BAP1 (or PBRM1) deficient. Conversely, tumor nuclei (larger), but not nuclei of stromal cells (smaller) are PAX8 positive. C, Quantification of PAX8 staining in PBRM1- and BAP1-mutant ccRCC samples from patient TMA (63/67 PBRM1-deficient and 19/19 BAP1-deficient ccRCCs expressed PAX8). D, Lineage tracing experiments of kidney sections from 3-month-old mice showing Pax8-lineage cells (Pax8-Cre-tdTomato; red), Lotus tetragonolobus lectin (LTL, a proximal tubule marker; green), DAPI (nuclei; blue), and a merged image. Co-staining shows that Pax8-lineage cells contribute to proximal tubules. (Magnification = 200×) E, Representative immunofluorescence images of kidney sections from 3-month-old Pax8-Cre-tdTomato;Bap1lacZ/+ gene trap mice showing lineage tracing (Pax8-Cre-tdTomato; red), Bap1 expression (β-gal; green), DAPI (nuclei; blue), and a merged image. (Magnification = 200×) F, Representative immunofluorescence images of kidney sections from 3-month-old Pax8-Cre-tdTomato;Pbrm1lacZ/+ gene trap mice showing lineage tracing of Pax8-expressing cells (Pax8-Cre-tdTomato; red), Pbrm1 expression (β-gal; green), DAPI (nuclei; blue), and merged image. (Magnification = 200×).
Pax8 Lineage Cells Express Bap1 and Pbrm1
To explore Pax8 as a driver, we evaluated the overlap between the Pax8 lineage and cells expressing Bap1 and Pbrm1. We crossed Pax8-Cre mice with a Rosa26-CAG-loxP-stop-loxP-tdTomato reporter line to generate Pax8-Cre-tdT mice. Expression of Pax8-Cre recombinase in these mice resulted in indelible labeling of these cells and their progeny with tdTomato (tdT) fluorescence (Fig. 1D). Lineage tracing experiments showed that glomerular and renal tubular cells were broadly derived from Pax8-Cre-expressing cells (Fig. 1D). Co-immunostaining with Lotus tetragonolobus lectin (LTL), a proximal tubule marker, revealed that Pax8-lineage cells included proximal tubules (Fig. 1D).
Next, we determined the overlap between Bap1- and Pbrm1-expressing cells and Pax8-lineage cells. We crossed Pax8-Cre-tdT mice with transgenic Bap1-LacZ and Pbrm1-LacZ mice harboring a β-galactosidase gene trap in the respective loci. As assessed by β-galactosidase immunostaining, Bap1 was widely expressed in the kidney, and we observed that most Bap1-expressing cells were labeled with tdTomato indicating that a Pax8 driver would disrupt Bap1 expression in most Bap1-expressing cells. (Fig. 1E). Similar results were observed for Pbrm1 expressing cells (Fig. 1F). These data show that Pax8 is a suitable driver to disrupt normal Bap1 and Pbrm1 function in the kidney.
Loss of Bap1 in Pax8-Lineage Cells Leads to High-Grade ccRCC
Mouse cohorts harboring Pax8-Cre-driven deletion of Vhl and Bap1 were generated. As shown by PCR genotyping analysis, recombination of LoxP sites occurred primarily in the kidney (Supplementary Fig. S1A). As further confirmation, western blot analyses demonstrated that Vhl and Bap1 protein levels were greatly reduced in kidneys of Pax8-Cre;VhlF/F;Bap1F/F mice (Fig. 2A). As established by the evaluation of downstream targets, deletion of both Vhl and Bap1 resulted in activation of the corresponding effector pathways (Fig. 2B). Deletion of Vhl leads to Hif-2 and Hif-1 activation and increased expression of target genes, including CyclinD1, Pai-1, Vegf, Glut1, Igfbp3, Pgk1, and Tgfa (Fig. 2B). A HIF target gene overexpressed in ccRCC is carbonic anhydrase IX (CAIX) (38, 39), which was also markedly upregulated (Fig. 2A). BAP1 deubiquitinates H2A (lysine 119 in both human and mouse) (40–42) and the level of ubiquitinated histone H2A protein (Ub-H2A) was dramatically increased (Fig. 2A). These data demonstrate that deletion of Vhl and Bap1 with a Pax8 driver leads to activation of expected downstream pathways.
Figure 2.
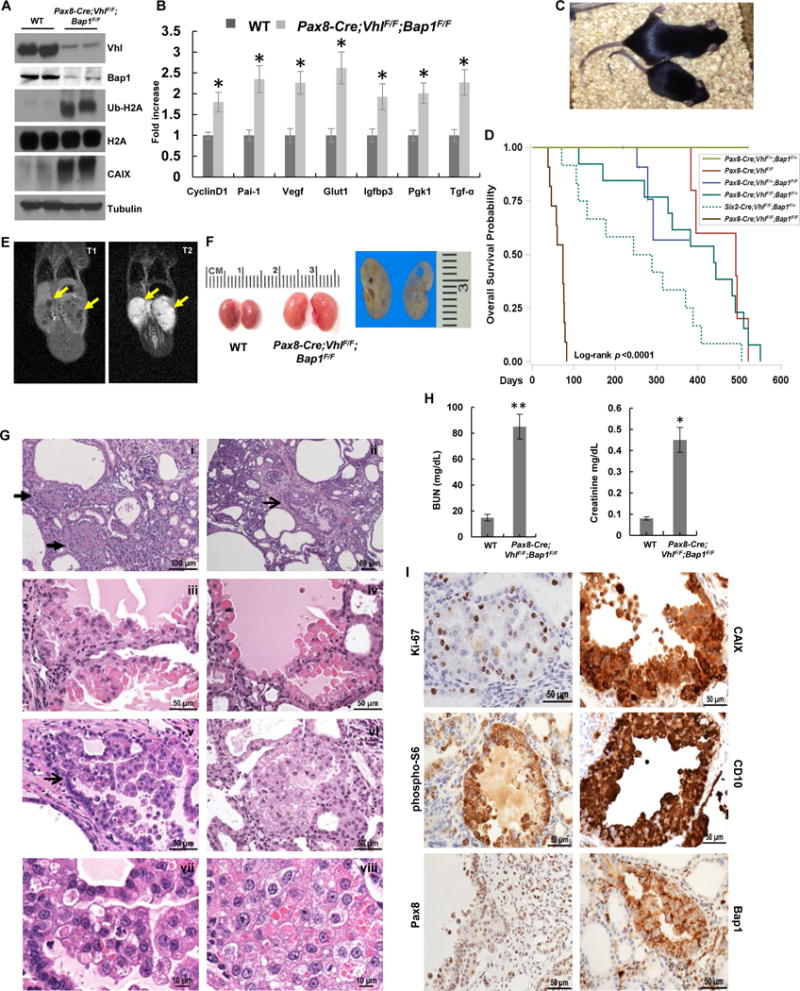
RCC development and premature death of Pax8-Cre;VhlF/F;Bap1F/F mice. A, Western blot analysis of the indicated proteins in Pax8-Cre;VhlF/F;Bap1F/F kidneys and controls. B, qRT–PCR for the indicated Hif target genes in kidneys of Pax8-Cre;VhlF/F;Bap1F/F mice and controls (n=3 independent mice; PCR done in triplicate). C, Pax8-Cre;VhlF/F;Bap1F/F mice are smaller and runted compared to littermate controls. D, Survival curve of Pax8-Cre;VhlF/F;Bap1F/F mice (n=12) compared to other genotype configurations (Pax8-Cre;VhlF/+;Bap1F/+, n=20; Pax8-Cre;VhlF/F, n=5; Pax8-Cre;VhlF/+;Bap1F/F, n=5; Pax8-Cre;VhlF/F;Bap1F/+, n=13). Six2-Cre;VhlF/F;Bap1F/+ (n=12) are shown as a reference (24). E, Coronal T1-weighted (left panel) and T2-weighted (right panel) images of a moribund Pax8-Cre;VhlF/F;Bap1F/F mouse (77 days old) with clearly visible focal renal lesions with marked hyperintensity on T2-weighted images (yellow arrows). F, Representative macroscopic images of kidneys from Pax8-Cre;VhlF/F;Bap1F/F mutant and control mice showing multiple cystic lesions. G, Representative kidney sections from Pax8-Cre;VhlF/F;Bap1F/F mice showing (i-ii) cysts and RCCs: (iii-v) cystic lesions with atypical proliferative lining; (vi) solid lesion with focal cytoplasmic clearing; (vii-viii) neoplastic cells with pleomorphism, anaplasia, and mitosis. Arrows: (i) solid lesions; (ii, v) cystic lesions. H, Plasma BUN and Creatinine measurements in Pax8-Cre;VhlF/F;Bap1F/F mice and controls (n=3 independent mice for each group). I, IHC showing increased expression of Ki-67, CAIX, phospho-S6, and CD10 in RCC in the tumors of Pax8-Cre;VhlF/F;Bap1F/F mice. Retained Pax8 expression, and loss of nuclear Bap1 also shown. *, P < 0.05; **, P < 0.01. (WT, age-matched VhlF/F;Bap1F/F mice without Cre; for survival curve, D, n=12, while n=16 for mice analyzed for histology.)
Pax8-Cre;VhlF/F;Bap1F/F mice were born at expected Mendelian ratios, and lived longer than Six2-Cre;VhlF/F;Bap1F/F mice, which die shortly after birth (24). However, pups were small and sickly, and died at around 3 months of age (Fig. 2C and D). MRI scans on moribund mice showed small kidney lesions (Fig. 2E). On gross examination, the kidneys were enlarged and pale with macroscopic cortical cystic lesions that varied in size from 0.1 to 1.25 mm (Fig. 2F). Histologically, there was architectural distortion with multiple large cysts and dilatation of the tubules, although the glomeruli appeared normal. Cystic changes, which were seen in all 16 mice (2–4 months of age) (Table 1), were more pronounced after age 2.5 months (Fig. 2G). Blood urea nitrogen (BUN) and creatinine were significantly increased, indicating kidney dysfunction as the probable cause of death (Fig. 2H).
Table 1.
List of genotypes and associated histological features.
| Genotype | Age | Architecture | Simple cysts, tubular & Bowman space dilatation | Preneoplastic end Neoplastic lesions | ||||
|---|---|---|---|---|---|---|---|---|
| Cystic ccRCCs | Solid ccRCC | |||||||
| Atypical cysts | High grade cystic solid ccRCC | Low grade solid-nested ccRCC | High grade solid-nested ccRCC | Cytoplasmic clearing | ||||
| Pax8-Cre;VhlF/F | 16–18 months (n=10) | Normal | rare | neg | neg | neg | neg | neg |
| Pax8-Cre; VhlF/+;Bap1 F/+ | 11–18 months (n=9) | Normal | nag | neg | neg | neg | neg | neg |
| Pax8-Cre; VhlF/+;Bap1 F/F | 2–9 months (n=2) | Normal | rare | neg | neg | neg | neg | neg |
| Pax8-Cre; VhlF/F;Bap1 F/+ | 6–11 months (n=6) | Normal | rare | neg | neg | neg | neg | neg |
| 11–20 months (n=8) | Distorted | present (++) | present (+++) | present (+++) | neg | neg | present (++) | |
| Pax8-Cre; VhlF/F;Bap1 F/F | 2–4 months (n=16) | Distorted | present (+++) | present (+++) | present (+) | neg | neg | present (+) |
| Pax8-Cre; Pbrm1F/F | 17–18 month (n=5) | Normal | neg | neg | neg | neg | neg | neg |
| Pax8-Cre; VhlF/+;Pbrm1F/F | 16–28 months (n=23) | Normal | rare | neg | neg | neg | neg | neg |
| Pax8-Cre; VhlF/+;Pbrm1F/+ | 14–18 month (n=11) | Normal | rare | neg | neg | neg | neg | neg |
| Pax8-Cre; VhlF/F;Pbrm1F/F | 4–7 months (n=6) | Normal | rare | neg | neg | neg | neg | neg |
| 9–13 months (n=7) | Distorted | present (+) | present (+) | rare | present (++) | neg | present (+) | |
| 14–19 months (n=13) | Distorted | present (+) | present (+) | present (+) | present (+++) | neg | present (++) | |
| Pax8-Cre;VhlF/F;Pbrm1F/F;Tsc1F/+ | 12–19 months (n=10) | Distorted | present (+) | present (+) | rare | present (+++) | present (+++) | present (++) |
| Sglt2-Cre;VhlF/F;BaplF/F | 20–32 months (n≥14) | Normal | neg | neg | neg | neg | neg | neg |
| Villin-Cre;VhlF/F,Bap1F/F | 12–30 months (n≥14) | Normal | rare | neg | neg | neg | neg | neg |
| Sglt2-Cre;VhlF/F ;Pbrm1F/F | 20–27 months (n≥14) | Normal | neg | neg | neg | neg | neg | neg |
| Villin-Cre;VhlF/Fr;Pbrm1F/F | 20–26 months (n≥14) | Normal | neg | neg | neg | neg | neg | neg |
Even though the mice died by 3 months, by that time small RCCs and atypical cystic lesions were observed in all mice. The number and size of lesions increased with the age of the mice. Akin to the kidneys from patients with VHL syndrome, we observed a spectrum of premalignant and malignant tumors. Throughout the cortex, numerous simple cysts lined by a single layer of plump columnar epithelial cells with varying degree of cytoplasmic clearing and nuclear atypia were seen (Fig. 2Gi,ii). Some of the cysts showed varying degree of epithelial proliferation from focal papillation to prominent intracystic growth (Fig. 2Giii–v). Solid tumor masses were infrequent, but also observed (Fig. 2Gvi). The cytologic characteristics of the cells across these lesions were similar and most consistent with neoplastic cells displaying varying degree of nuclear enlargement, pleomorphism, hyperchromasia, nuclear membrane irregularities, and prominent nucleoli. Mitotic figures and apoptotic bodies were frequently identifiable (Fig. 2Gvii–viii). The cell size was larger than that of normal renal tubular epithelium and the cytoplasm varied from eosinophilic to clear and had prominent intracytoplasmic eosinophilic inclusions (Fig. 2Gvi–viii). The morphologic features were indistinguishable from those observed in the Six2-Cre;VhlF/F;Bap1F/+ mice reported previously (24), suggesting that the cell of origin is retained in the narrower Pax8 lineage.
Increased expression of the cell proliferation marker, Ki-67, was seen in atypical cysts and neoplastic nodules (Fig. 2I). CAIX, a HIF target and marker of ccRCC, along with CD10, another routinely used ccRCC marker, exhibited the classic membranous (and cytoplasmic) staining pattern in cysts and neoplastic nodules in the mutants (Fig. 2I). Pax8 staining was consistently positive in neoplastic cells in the lesions of Pax8-Cre;VhlF/F;Bap1F/F mutants (Fig. 2I).
Pax8-Cre;VhlF/F;Bap1F/+ mice survived much longer, with median survival of around 14.5 months (Fig. 2D). Renal tumors developed in these mice (Fig. 3A) and were observed at around 11 months of age (Table 1). Small atypical cysts could be seen starting at around 10 months of age (no lesions were seen in mice less than 7.5 months old). While the spectrum of cysts and tumors was similar to the Pax8-Cre;VhlF/F;Bap1F/F mice, these lesions were both more numerous and larger in size. The tumors consisted of expansile cystic masses (0.7–2.4 mm in size) with epithelial proliferation and occasional central cavitation, and included in some cases serous fluid (Fig. 3Bi,ii,iv). Scattered solid cystic masses, occasionally exhibiting a nested pattern and vascular network characteristic of human ccRCC, were observed (Fig. 3Bii,iii,vi,vii). Cytologic features were similar to Pax8-Cre;VhlF/F;Bap1F/F cells, but were more pronounced including pleomorphism, nucleolar prominence, atypia, and mitosis (Fig. 3B). Focal lymphovascular invasion was also identified (Fig. 3Bv). As observed in human ccRCC, these tumors accumulate neutral lipids and glycogen (Fig. 3C). Similar to the Pax8-Cre;VhlF/F;Bap1F/F mice and as observed in human ccRCC, the expression of Ki-67, CAIX, CD10, and vimentin was increased (Fig. 3D). Bap1 protein expression by IHC was reduced across lesions and completely absent in approximately 30% (Fig. 3D). As expected, Hif-2 and Hif-1 target genes were up-regulated (Fig. 3E and F). Finally, BUN and creatinine levels were also significantly elevated in moribund Pax8-Cre;VhlF/F;Bap1F/+mice likely explaining their death (Fig. 3G).
Figure 3.
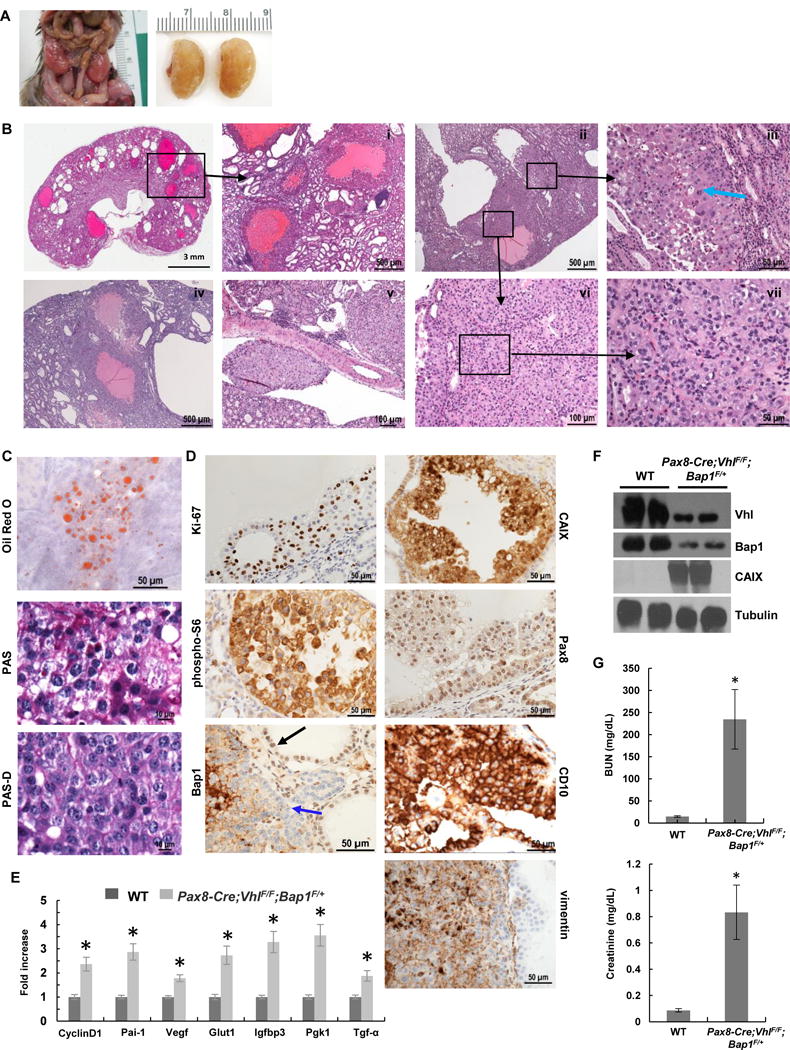
Pax8-Cre;VhlF/F;Bap1F/+ mice develop high grade ccRCC. A, Representative images from Pax8-Cre;VhlF/F;Bap1F/+ mutant mice showing enlarged nodular kidneys. B, H&E staining of kidney sections from Pax8-Cre;VhlF/F;Bap1F/+ mice (n=14) showing (i) cystic and solid ccRCCs, (i,iv) multiple large cystic tumors with marked proliferative lining, (iii,vi,vii) solid tumor with thin arborizing vascular network, (iii,vii) neoplastic cells with marked pleomorphism, anaplasia, prominent nucleoli, and mitosis (blue arrow indicates atypical mitosis), and (v) lymphovascular spread. C. Oil Red O staining for triglycerides and neutral lipids, and Periodic acid-Schiff staining without (PAS) and with (PAS-D) diastase highlight the abundant lipid and glycogen in neoplastic cells. D, IHC for Ki-67, CAIX, phospho-S6, Pax8, Bap1 (blue arrow, tumor cells; black arrow, retained Bap1 staining), CD10, and vimentin in RCCs. Complete loss of nuclear Bap1 expression in Pax8-Cre;VhlF/F;Bap1F/+ mice was seen in 30% (21/69 lesions (n=8)), however, no appreciable differences in morphology, size, or grade across lesions were seen. E, qRT–PCR for the indicated Hif target genes in kidneys of Pax8-Cre;VhlF/F;Bap1F/+ mice and controls (n=3 independent mice; PCR done in triplicate). F, Western blot analysis of the indicated proteins in Pax8-Cre;VhlF/F;Bap1F/+ mice and controls. G, Plasma BUN and Creatinine measurements in Pax8-Cre;VhlF/F;Bap1F/+ mice and controls (n=3 independent mice for each group). *, P < 0.05. (WT, age-matched VhlF/F;Bap1F/+ mice without Cre.)
As a reference, Pax8-Cre;VhlF/F mice survived 16–18 months and histologically, the kidneys appeared normal with occasional manifestations of chronic interstitial inflammation, mild tubular dilatation or rare simple cysts in older mice. However, no proliferative lesions were observed (Supplementary Fig. S1B), despite evidence of functional Vhl inactivation (Supplementary Fig. S1B). These data are consistent with the notion that loss of Vhl is insufficient for tumorigenesis.
We previously showed increased staining of phosphorylated S6 ribosomal protein [p-S6 (Ser240/244)], a marker of mammalian target of rapamycin complex 1 (mTORC1) activation, in BAP1-deficient human ccRCC (13). Provocatively, this was also observed in Vhl;Bap1-deficient neoplastic cells (Fig. 2I and 3D), but was not seen in Pax8-Cre;VhlF/F mice (Supplementary Fig. S1B).
Overall, these data show that the lesions that arise in kidney of Pax8-Cre;VhlF/F;Bap1F/F and Pax8-Cre;VhlF/F;Bap1F/+ mice mimic the morphological and immunohistochemical characteristics of the corresponding tumors in humans, including increased phospho-S6 staining.
Pbrm1 Loss Leads to Low Grade ccRCC
We deleted Pbrm1 using the same Pax8-Cre driver (Supplementary Fig. S2A). As demonstrated by western blot analysis, Vhl and Pbrm1 protein levels were greatly reduced in kidneys of Vhl;Pbrm1-deficient mice (Fig. 4A).
Figure 4.
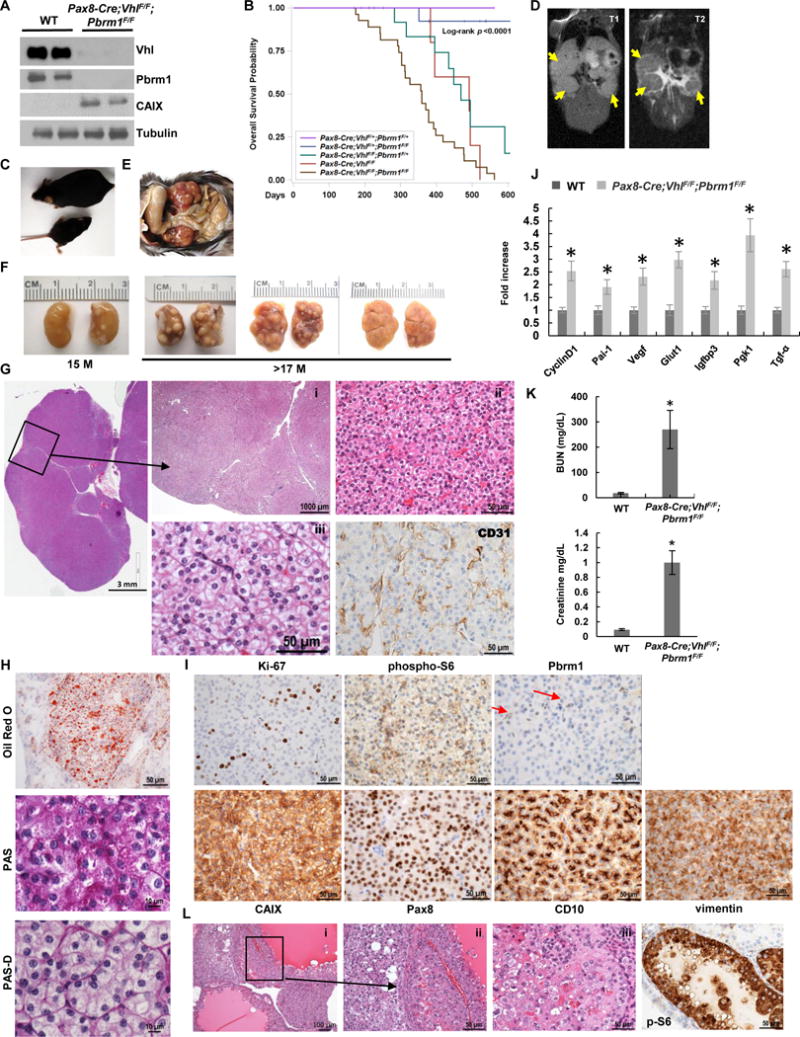
Pax8-Cre;VhlF/F;Pbrm1F/F mice develop ccRCC of lower grade. A, Western blot analysis of the indicated proteins in Pax8-Cre;VhlF/F;Pbrm1F/F kidneys and controls. B, Survival curve of Pax8-Cre;VhlF/F;Pbrm1F/F mice (n=27) compared to Pax8-Cre;VhlF/+;Pbrm1F/+ controls (n=20), and other genotype configurations (Pax8-Cre;VhlF/F;Pbrm1F/+, n=15; Pax8-Cre;Pbrm1F/F, n=5; Pax8-Cre;VhlF/+;Pbrm1F/F, n=17). C, Pax8-Cre;VhlF/F;Pbrm1F/F mice are smaller and runted compared to littermate controls. D, Coronal T1-weighted (left) and T2-weighted (right) images of a Pax8-Cre;VhlF/F;Pbrm1F/F mouse with clearly visible homogeneous tumors with intermediate-to-low signal intensity on T2-weighted images (yellow arrows). E, Kidneys in situ in Pax8-Cre;VhlF/F;Pbrm1F/F mutant mouse. F, Representative macroscopic images of kidneys from Pax8-Cre;VhlF/F;Pbrm1F/F mutant mice at 15 and >17 months of age showing large solid tumors studding the entire cortex. G, Representative H&E microphotographs of kidney sections from Pax8-Cre;VhlF/F;Pbrm1F/F mice showing large ccRCCs completely replacing the kidney. (i) Multiple large solid tumors with pushing borders; (ii) solid tumor with thin arborizing vascular network, monomorphic neoplastic cells with minimal atypia, inconspicuous nucleoli, and mitosis; (iii) prominent cytoplasmic clearing with morphology indistinguishable from low grade human ccRCC. CD31 IHC highlights vasculature. H. Oil Red O staining for triglycerides and neutral lipids, and Periodic acid-Schiff staining without (PAS) and with (PAS-D) diastase highlighting the abundant lipid and moderate glycogen in the neoplastic cells. I, IHC of Ki-67, phospho-S6 (weak and focal), Pbrm1 (lost in tumor nuclei, but retained in normal endothelial cells (red arrows)), CAIX, Pax8, CD10, and vimentin in RCCs of Pax8-Cre;VhlF/F;Pbrm1F/F mice. J, qRT–PCR for the indicated Hif target genes in the kidneys of Pax8-Cre;VhlF/F;Pbrm1F/F mice and controls (n=3 independent mice; PCR done in triplicate). K, Plasma BUN and Creatinine measurements in Pax8-Cre;VhlF/F;Pbrm1F/F mice and controls (n=3 independent mice for each group). L, H&E (i-iii) and phospho-S6 (p-S6) IHC of rare higher grade cystic tumors with pleomorphism and atypia (seen in 4.3% (11/253) of the lesions in 20% of the mice (n=5/26) as well as prominent phospho-S6 resembling those observed in Bap1-targeted kidneys.*, P < 0.05. (WT, age-matched VhlF/F;Pbrm1F/F mice without Cre.)
Median survival of Pax8-Cre;VhlF/F;Pbrm1F/+ mice was around 15 months (Fig. 4B), and the kidneys looked similar to wild-type mice (not shown). Unlike the Pax8-Cre;VhlF/F;Bap1F/+mice, no lesions developed in the kidneys of Pax8-Cre;VhlF/F;Pbrm1F/+ mice (Supplementary Fig. S2B). The kidneys also appeared normal histologically, with occasional manifestations of inflammation or cysts in older mice. Similarly, Pax8-Cre;VhlF/+;Pbrm1F/F mice looked indistinguishable from wild-type mice, surviving for >24 months (16–28 months) (Supplementary Fig. S2B and Fig. 4B).
In contrast, Pax8-Cre;VhlF/F;Pbrm1F/F mice were born at expected Mendelian ratios, but were runty and smaller over the course of their lifetime compared to littermate controls (Fig. 4C). Median survival was ~12 months (Fig. 4B). MRI analyses revealed the presence of extensive large bilateral tumors (Fig. 4D). However, despite their large size, tumors appeared quite homogeneous with low signal intensity on T2-weighted images, and lacked central necrosis.
Grossly, the kidneys of moribund mice were enlarged and firm, with nodular capsular surface. The cut surface showed numerous variably sized tan masses (Fig. 4E and F). Histologically, the renal architecture of young Pax8-Cre;VhlF/F;Pbrm1F/F mice looked normal until about 7 months of age. Microscopic tumors could be detected in mice at around 9 months of age (Fig. 4G, Table 1). These tumorlets ranged in number from 1 to 26 and were observed in all but one mouse (6/7) analyzed between the ages of 9–13 months. However, these tumors were uniformly small. The background kidney also showed small, cystically-dilated tubules, dilated Bowman space, simple cysts, and rare scattered atypical cysts. All mice older than 13 months of age showed multiple tumors (100%), with tumor size increasing with the age of the mice, and tumor nodules almost completely replacing the entire kidney after 16 months.
These solid tumors were very different from those seen in Vhl;Bap1-floxed mice. They consisted of solid, circumscribed masses (0.5–12 mm in size) with pushing borders (Fig. 4Gi). Cells were arranged in small nested architecture with a delicate interconnected vascular network (highlighted by CD31 IHC), a pattern prototypic of low grade human ccRCC (Fig. 4Gi,iii). Cells had a moderate amount of cytoplasm varying from predominantly (or exclusively) eosinophilic to moderately clear (Fig. 4Gi,iii). Nuclei were monomorphic and without prominent nucleoli (Fig. 4Giii). Cystic dilatation of tubules and glomerular capsule was observed to varying degrees in the background.
Tumors showed accumulation of lipid and glycogen (Fig. 4H), and an immunohistochemical staining pattern similar to low grade human ccRCC, as depicted by positive membranous CAIX, CD10, moderate Ki-67, and strong vimentin expression (Fig. 4I). As expected, Pax8 expression was observed in Vhl-Pbrm1-deficient lesions (Fig. 4I), while Pbrm1 showed loss of nuclear reactivity in the lesions and most of the normal tubules (Fig. 4I). Notably, in contrast to the RCC arising in Bap1-floxed kidneys, these solid tumors exhibited weak to largely negative phospho-S6 expression (Fig. 4I). As expected, there was increased expression of Hif target genes (Fig. 4A, I, and J).
A significant increase in BUN and creatinine levels was observed, suggesting that these mice had impaired kidney function (Fig. 4K). However, in contrast to Vhl;Bap1-deficient kidneys, there were less cystic changes of the renal parenchyma, with relatively preserved background renal parenchyma, therefore suggesting that the cause of renal failure may be tumor replacement.
We generated a cell line from one tumor, which was confirmed to be Vhl and Pbrm1 deficient by PCR (Supplementary Fig. S2C–E). Immunoblotting confirmed the loss of Vhl and Pbrm1 proteins (Supplementary Fig. S2F). Consistent with their origin, these cells showed significant induction of both Hif-1α and Hif-2α, as well as the Hif target, CAIX, akin to what is often observed in human ccRCC (Supplementary Fig. S2F).
mTORC1 Activation in Rare High-Grade Pbrm1-Deficient ccRCC
Spontaneous renal tumor development in Pax8-Cre;VhlF/F;Pbrm1F/F mice, but not other genetic configurations, indicates that Pax8-Cre-driven inactivation of both alleles of Vhl and Pbrm1 is required for ccRCC tumorigenesis. The ccRCCs observed in Pax8-Cre;VhlF/F;Pbrm1F/F kidneys are reminiscent of VHL;PBRM1-deficient human tumors in their morphology and immunohistochemical features. In addition to the solid, low grade tumors that were predominantly observed, rare, high grade cystic tumors similar to those seen in the Vhl;Bap1-floxed mice were found in 20% of the mice (Fig. 4L). We observed 11 high grade cystic Bap1-like tumors and 242 low grade solid tumors. Notably, the cystic, high grade tumors showed significantly stronger phospho-S6 expression (Fig. 4L) similar to tumors in Pax8-Cre;VhlF/F;Bap1F/F mice suggesting that mTORC1 may contribute to higher grade.
Activation of mTORC1 Induces High Grade Pbrm1-Deficient ccRCC Development
To assess the role of mTORC1 in ccRCC tumor grade, we disrupted a single allele of the mTORC1 negative regulator, Tsc1. Tsc1 forms a complex with Tsc2 that functions as a GAP towards the obligatory activator of mTORC1 Rheb. Reduced Tsc1/Tsc2 complexes increase Rheb-GTP leading to mTORC1 activation (43, 44). Interestingly, loss of one copy of Tsc1 in Pax8-Cre;VhlF/F;Pbrm1F/F;Tsc1F/+ mice led to the development of higher grade tumors in fifty percent of the mice analyzed (Fig. 5A) (5/10 mice). While the majority of ccRCC were still of low grade (12 high grade vs. 112 low grade), the high grade ccRCC were solid and their morphology suggested that they arose from the lower grade Pax8-Cre;VhlF/F;Pbrm1F/F tumors. These higher grade ccRCC had increased nuclear pleomorphism, large nuclei, prominent nucleoli, more abundant cytoplasm with focal prominent clearing, and tumor necrosis (Fig. 5B). In contrast, no high grade solid tumors were observed among the 242 solid tumors evaluated in Pax8-Cre;VhlF/F;Pbrm1F/F mice. Consistent with our hypothesis, strong phospho-S6 was seen in the high grade tumors while low grade tumors in the same mice had weak (or absent) phospho-S6 expression (Fig. 5C). Overall, these data suggest that loss of one Tsc1 allele promotes the progression of lower grade Pbrm1-deficient tumors, into higher grade ccRCC, albeit at low frequency. In addition, in Pax8-Cre;VhlF/F;Pbrm1F/F;Tsc1F/+ mice tumor development appeared to be accelerated by ~3 months. Finally, while TSC1 mutations are infrequent in ccRCC, an analysis of COSMIC identifies several human ccRCC with mutations in VHL, PBRM1 and TSC1, establishing a proof-of-principle for this gene combination (Supplementary Fig. S2G).
Figure 5.
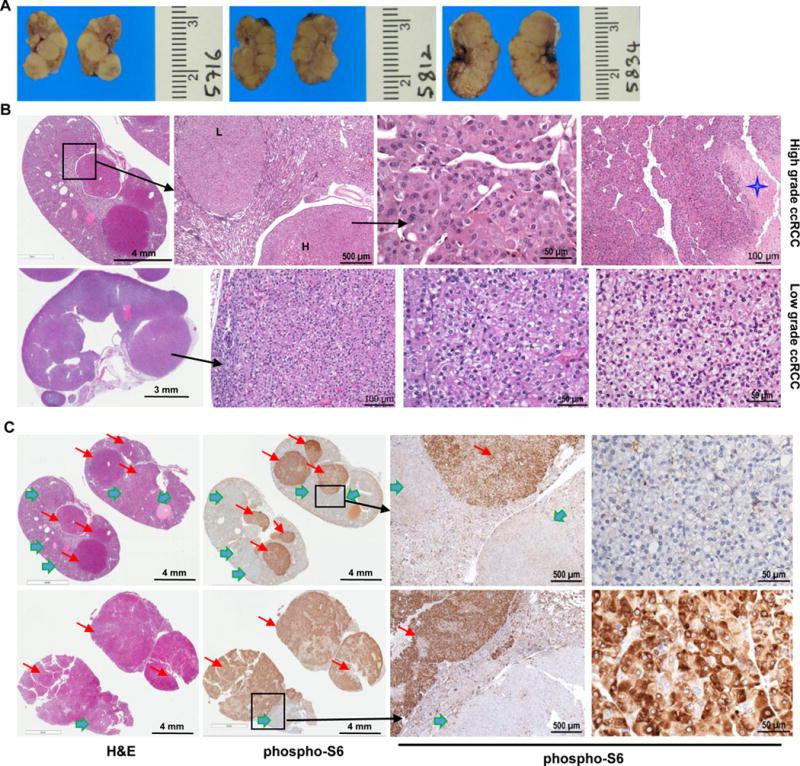
Targeting one allele of Tsc1 increases high grade tumors and accelerates tumorigenesis in Pax8-Cre;VhlF/F;Pbrm1F/F mice. A, Representative macroscopic images of bisected kidneys from Pax8-Cre;VhlF/F;Pbrm1F/F;Tsc1F/+ mutant mice at 13 months of age. B, Representative H&E microphotographs of kidney sections from Pax8-Cre;VhlF/F;Pbrm1F/F;Tsc1F/+ mice (n=10) showing numerous large solid RCCs of both high grade (upper panel; star highlights necrosis; L=low grade; H=high grade) and low grade (lower panel) almost completely replacing the normal kidney. C, Representative kidney sections with H&E and corresponding phospho-S6 IHC showing weak to negative phospho-S6 expression in low grade tumors (green arrows) and high expression in high grade tumors (red arrows).
Bap1 and Pbrm1 Targeting To Proximal Convoluted Tubules Fails to Give Rise to RCC
Careful histological analyses in the Pax8-Cre;VhlF/F;Pbrm1F/F kidneys in particular, but also scattered in other genotypes, showed alterations of the lining parietal epithelial cells of Bowman capsule suggesting that these cells may give rise to ccRCC. These alterations were of different magnitude and could be arranged in what appeared to be a series of progressive steps with increased abnormalities (Fig. 6A). The lining parietal epithelial cells showed nuclear enlargement and variable cytoplasmic clearing (Fig. 6Ai–ii). Some of these dilated formerly Bowman capsule structures showed only a minute rudimentary glomerular tuft, suggesting that they are the source of cysts (Fig. 6Aiii). Occasionally, parietal epithelial cell proliferation was observed (Fig. 6Aiv–vi; arrow points to entrapped glomerular tuft). Interestingly, similar features were also observed in uninvolved renal parenchyma adjacent to human ccRCC (Fig. 6Bi–vi). These data suggest that the parietal epithelial cell of the Bowman capsule, rather than the proximal convoluted tubule cell, may be the likely cell of origin of these tumors.
Figure 6.
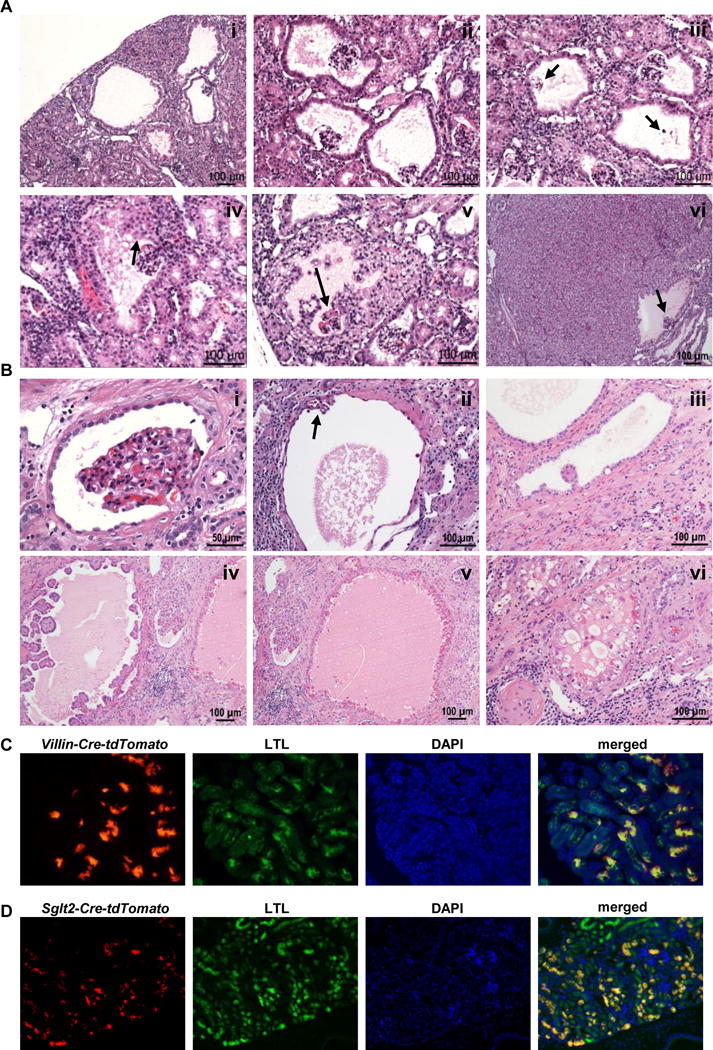
Proximal-tubule-specific deletion of Vhl;Bap1 or Vhl;Pbrm1 is not sufficient to drive tumor development. A, (i-vi) Representative H&E microphotographs from Pax8-Cre;VhlF/F;Pbrm1F/F lesions supporting the notion that RCC originates in Bowman capsule. (i-ii) multiple dilated Bowman spaces; (iii) dilated Bowman spaces with vanishing glomerular tufts (arrows) seemingly giving rise to cysts; (iv) dilated Bowman space with varying degree of parietal epithelial cell proliferation (arrow); (v) dilated Bowman space with extensive parietal epithelial cell proliferation and entrapped glomerular tuft (arrow); (vi) large solid low grade RCC with preserved dilated Bowman space and entrapped glomerular tuft (arrow). B, Representative H&E microphotographs from human kidneys adjacent to ccRCC showing similar features to those observed in our GEMMs, and further supporting the notion that RCC originates in Bowman capsule. C, Immunofluorescence images of kidney sections showing staining for Villin-lineage cells (Villin-Cre-tdTomato; red) or D, Sglt2-lineage cells (Sglt2-Cre-tdTomato; red), together with LTL (green; a proximal tubule marker), DAPI (nuclei; blue), and merged showing that the Villin and Sglt2 lineages contribute to proximal tubules.
To provide further insight, we targeted Vhl together with either Bap1 or Pbrm1 using Cre-drivers expressed in proximal tubules, and not in Bowman capsule: Villin-Cre and Sglt2-Cre (34, 45–47). As shown in Fig. 6C and D, Villin- and Sglt2-lineage cells are mainly distributed in proximal tubules. Interestingly, murine cohorts harboring Villin-Cre- or Sglt2-Cre-driven deletion of both alleles of Vhl together with both alleles of either Bap1 or Pbrm1 (n ≥ 14 for each genotype) failed to develop tumors even at an old age (24 to 30 months) (Supplementary Fig. S3A). One caveat is that significantly fewer cells are targeted by Villin and Sglt2 drivers than with Pax8 (Fig. 6C and D). However, substantial recombination rates were observed (Supplementary Fig. S3B and C) and the recombination events were associated with loss of Bap1 and Pbrm1 (Supplementary Fig. S3A) and activation of downstream Vhl targets such as CAIX (Supplementary Fig. S3A). While we cannot exclude that failure to develop tumors with Villin and Sglt2 drivers results from a reduced number of targeted cells, taken together, our data suggest that Villin- and Sglt2-lineage cells are not transformed by the combined loss of Vhl and either Bap1 or Pbrm1 and that these cells are not the likely source for ccRCC. Finally, an examination of Villin (VIL1) and SGLT2 (SLC5A2) in ccRCCs from TCGA (and a UTSW cohort) reveals expression levels that are comparable or below those found in normal kidneys (Supplementary Fig. S3D).
DISCUSSION
Genomic analyses of ccRCC by us and others identified frequent mutation of the PBRM1 and BAP1 genes in 50% and 15% of tumors, respectively (11, 13). However, whether loss of BAP1 and PBRM1 causes ccRCC is not well understood. Germline mutations in BAP1 and PBRM1 have been associated with ccRCC familial syndromes, but these mutations are rare and BAP1 germline mutations are primarily associated with other tumor types such as melanoma and mesothelioma (48–51). We previously showed that combined targeting of Bap1 and Vhl in the mouse kidney using a Six2 driver results in ccRCC development, indicating that BAP1 and VHL cooperate in tumor development (24). However, even in the conditional setting, Bap1 loss caused perinatal lethality and mice with loss of Vhl and one copy of Bap1 in the kidneys survived only for approximately 8 months (24). In addition, ccRCC were small. Here, we used Pax8, a more restricted lineage-specific transcription factor than Six2 (52), and a commonly used RCC clinical diagnostic marker. We show that there is substantial overlap between Bap1 expression and the Pax8 lineage, and that by targeting Bap1 in this compartment, the survival of mice can be extended allowing for greater time to observe tumor development. Tumors developing in these mice are indistinguishable from those developing in the Six2 lineage, but they are more advanced and larger. These data also support that the narrower Pax8 lineage contains the cell of origin of Bap1-deficient ccRCC.
We previously reported that BAP1 and PBRM1 mutations in ccRCC tend to anticorrelate (13, 16). One potential explanation is that BAP1- and PBRM1-deficient tumors arise from different cell types. Here, we show that targeting Pbrm1 (along with Vhl) to the Pax8 lineage also causes ccRCC. These data show that the Pax8 lineage contains cells capable of transformation by the loss of both Bap1 and Pbrm1. In contrast, similar gene targeting of the Sglt2 and Villin lineages did not lead to ccRCC. However, effective gene targeting with both Sglt2 and Villin drivers in proximal convoluted cells was demonstrated by lineage tracing experiments, PCR, and IHC analysis showing protein loss and downstream effector pathway activation. These data suggest that gene targeting in proximal tubules is not sufficient to give rise to tumors challenging the notion that ccRCC arises from this compartment. One caveat is that not all proximal convoluted tubule cells were targeted by Sglt2 and Villin. Our morphological analyses suggest that ccRCC arises from parietal cells of the Bowman capsule, which are derived from the Pax8 lineage but not the Sglt2 and Villin lineages. Similar observations in kidneys from patients with ccRCC suggest that they are relevant to humans. However, whether it is the same cell or different cells that lead to Bap1- and Pbrm1-deficient tumors remains to be determined.
We observed a spectrum of cystic and solid lesions in both the Bap1- and Pbrm1-deficient kidneys that recapitulated the morphology and biologic behavior of human ccRCC. Our mouse models reproduce the features of patient tumors, including CD10, vimentin, CAIX and Pax8 expression. In humans, BAP1-deficient tumors tend to be of high grade and PBRM1-deficient tumors of low grade. In addition, BAP1-deficient tumors are associated with significantly worse patient survival than PBRM1-deficient tumors (17, 30, 53–56). Whether BAP1 and PBRM1 are responsible for these differences, however, is unclear. Here we show that, as in humans, Bap1-deficient ccRCCs in the mouse are different than Pbrm1-deficient tumors, and that they are of high grade. In contrast, Pbrm1-deficient tumors arise later and tend to be homogeneous and of low grade. The grade of Bap1-deficient ccRCC corresponds to Fuhrman nuclear grade 3 versus Fuhrman nuclear grade 1 or 2 for Pbrm1-deficient ccRCC. Overall, our data show that BAP1 and PBRM1 are determinants of tumor grade and likely the drivers of the differences in patient survival.
Interestingly, a minority of Pbrm1-deficient tumors (~4%) were similar to those observed following Bap1 loss and of high grade. Unlike lower grade tumors, these tumors displayed strong phospho-S6 staining indicative of mTORC1 activation. This led us to hypothesize that mTORC1 may be implicated in tumor grade. To test the role of mTORC1, we inactivated one allele of the mTORC1 negative regulator Tsc1. Targeting Tsc1 in kidneys along with Vhl and Pbrm1 led to the development of tumors that were similar to those observed in kidneys with intact Tsc1, but of higher grade. These tumors exhibited focal pleomorphism, large nuclei, prominent nucleoli, more abundant cytoplasm with prominent clearing, and tumor necrosis. In contrast, no high grade solid tumors were observed in Vhl and Pbrm1 deficient kidneys. As expected, strong mTORC1 activation was observed in the higher grade, but not lower grade tumors. Overall, these data suggest that inactivation of one Tsc1 allele promotes the progression of lower grade Pbrm1-deficient tumors to high grade. These data show that mTORC1 is a driver of the transformation of PBRM1-deficient tumors from low to high grade. Interestingly, while somatic mutations in TSC1 are infrequent in human ccRCC, several human ccRCC with mutations in VHL, PBRM1 and TSC1 have been reported. It is worth noting, in this context, that human BAP1-deficient ccRCC, which are of high grade, also tend to exhibit higher levels of mTORC1 activity (13). These data suggest that mTORC1 plays a broad role in tumor grade determination. There is a biological basis for this observation as tumor grade is heavily influenced by nuclear and nucleolar size, which are regulated by mTORC1 (57–59).
While this manuscript was under review, a manuscript was published reporting the generation of mice with targeted disruption of Vhl and Pbrm1 in the mouse kidney (60). There are several important differences between the studies. First, Nargund et al., targeted Vhl and Pbrm1 by using the traditional Ksp-Cre driver, and the phenotype was dominated by the development of large cysts sometimes taking up half of the kidney (60). In contrast, cysts are infrequently observed in mice with targeted loss of Vhl and Pbrm1 using a Pax8 driver, and the mice survive significantly longer. While the role of VHL and PBRM1 in human polycystic kidney disease is unclear, the Ksp-Cre mice represent an excellent model with progressive cystic development leading to renal failure and eventually the death of the mice by 1 year of age. Second, while the authors were able to detect occasional tumors, they were only found in 30% of the mice and tumors were uniformly small. In contrast, in our model, tumors were observed with 100% penetrance, were often multiple, and reached sizes as large as one third of the kidney. Third, the small size of the tumors in the study by Nargund precluded studies of tumor progression, which required transplantation in immunocompromised mice (60). Fourth, detection of levels of mTORC1 activation higher than the normal kidney led the authors to conclude that mTORC1 is a dominant feature of Pbrm1-deficient tumors. In contrast, our extensive analysis of tumors arising from loss of Bap1 and Pbrm1 and through Tsc1 targeting, led us to the conclusion that mTORC1 activity characterizes higher grade tumors. This is also consistent with the role of mTORC1 in the regulation of cell and nuclear size. In addition, by targeting both Bap1 and Pbrm1 in the same compartment, we could draw conclusions about their role in driving tumor grade, which are otherwise not possible. Finally, our careful histological studies point to the parietal cell of the Bowman capsule as a cell of origin for ccRCC. Overall, our data implicate BAP1 and PBRM1 loss as drivers not only of ccRCC, but also of tumor grade, and establish mTORC1 as a nuclear grade rheostat.
METHODS
Phenotype Validation and Histopathology
Kidneys were removed and fixed in 10% (vol/vol) neutral-buffered formalin for ∼24 h, and then routinely processed, embedded in paraffin, sectioned at 5 μm, and stained with H&E. Stained sections were evaluated by a board-certified pathologist (P.K.). H&E sections of kidneys from mice that were found dead and showed significant autolysis precluding accurate morphologic assessment were not included in the morphologic analysis. The number of tumors were counted on a single representative H&E slide from each mouse kidney, which might underestimate the numbers owing to unidentified small tumors.
BUN and Creatinine Measurements
Blood was collected before mice were sacrificed, maintained at room temperature (RT) for 30 min before centrifugation, and serum was obtained by spinning down samples at 3,000 rpm for 10 min. Serum samples were kept in the -20 °C freezer until submission to the mouse metabolic phenotyping core for measurement of BUN and Creatinine levels by VITROS MicroSlideTM technology (University of Texas Southwestern Medical Center).
Immunostaining
For immunofluorescence, kidneys were fixed in 4% (wt/vol) PFA, embedded in OCT compound, kept in -80 °C freezer, and sectioned on a cryostat. Frozen sections were washed with PBS, treated with 0.2% (vol/vol) Triton X-100, blocked with 5% (wt/vol) BSA/PBS for 1 hour at RT, incubated with a primary antibody to anti–β-galactosidase (ab9361; Abcam), 1:1000 dilution, at 4° C overnight, and detected by secondary antibodies with Cy2 (Jackson ImmunoResearch Laboratories). Sections counterstained with LTL were incubated with biotinylated Lotus tetragonolobus agglutinin (FL-1321; Vector Laboratories) at RT for 30 min before washing. Nuclei were stained by DAPI (Thermo Fisher Scientific Inc.). For immunohistochemistry, paraffin-embedded tissue was sectioned at 4 μm and staining was performed as described (13, 24). Immunohistochemistry for Bap1, Pbrm1, CD10, CD31, phospho-S6 ribosomal protein (Ser240/244), Ki-67, CAIX, and Pax8 were performed using an AutostainerLink 48 (Dako, Agilent Technologies). Briefly, kidneys were formalin-fixed, paraffin-embedded tissue was cut in 3- to 4-μm sections and air-dried overnight. The sections were deparaffinized, rehydrated, and subjected to heat-induced epitope retrieval using low pH target retrieval solution for 15 minutes (for CD31, phospho-S6 and Ki-67 IHC) and high pH for target retrieval solution (for CD10, Bap1, Pbrm1, CAIX, Pax8, and vimentin) (Envision FLEX Target Retrieval Solution, High and Low pH; Dako). Sections were incubated with primary antibodies from: Cell Signaling: phospho-S6 ribosomal protein − phospho-S6 (Ser240/244), 1:100 dilution, #5364; Ki-67, 1:100 dilution, #12202; vimentin, 1:100 dilution, #5741; Thermo Fisher Scientific: CAIX, 1:400 dilution; PA1-16592; Proteintech: Pax8, 1:200 dilution, 10336-1-AP; Bethyl Laboratories: Pbrm1, 1:2000 dilution, A301-591A; Dako, Agilent Technologies: CD10, 1:40, M7308; CD31, 1:50 dilution, M0823. Bap1 antibody provided by Genentech, 1:1000 dilution. For signal detection, the Envision FLEX System (Dako, Agilent Technologies) was used according to the manufacturer’s protocols for all antibodies except for Ki-67 for which avidin/biotin detection system (Vector Laboratories) was used. Slides were developed using 3,3′-diaminobenzidine chromogen and counterstained with hematoxylin. Appropriate positive and negative controls were used for each run of immunostaining.
Special Stains
Representative kidneys were embedded in OCT media and 5 μm thick cryosections were cut. Oil Red O stains to evaluate neutral lipids and triglycerides were performed according to manufacturer’s instructions (Poly Scientific R&D Corp.). Periodic acid-Schiff staining without (PAS) and with (PAS-D) diastase were performed on paraffin-embedded sections in accordance with the manufacturer’s instructions (Poly Scientific R&D Corp.).
Patient RCC Tissue
All RCC patient samples were from the UTSW tissue bank. PAX8 IHC staining analysis on patient tissue was performed by double-blind experiment. The study was performed in accordance with the protocol approved by the Institutional Review Board of the University of Texas Southwestern Medical Center. Approval numbers: 012011-190 and STU-22013-052.
Mice
Bap1tm2a(EUCOMM)hmgu, Pbrm1tm1a(EUCOMM)Wtsi ES cells were purchased from European Conditional Mouse Mutagenesis Program and injected into the cavity of Day 3.5 blastocysts from C57Bl/6N mice at the University of Texas Southwestern Medical Center Transgenic Core. Male chimeras were mated with C57BL/6NTac female mice to generate germline transmission. Mice heterozygous for the Bap1 or Pbrm1 null allele (Bap1/Pbrm1 lacZ/+) were used for β-galactosidase staining to determine Bap1/Pbrm1 expression. Pbrm1-LacZ/+ mice were mated with FLP mice (Jackson Laboratory) to introduce FRT recombinase reactivity for excision of the LacZ element to generate floxed Pbrm1 mice. Floxed VhlF/F mice were kindly provided by Dr. Volker H. Haase (Vanderbilt University Medical Center, Nashville, TN) (19). Bap1F/F mice were kindly provided by Dr. Vishva M. Dixit (Genentech, San Francisco, CA) (61). Sglt2-Cre mice were kindly provided by Dr. Michel Tauc (University of Nice-Sophia Antipolis, France) (47). Villin-Cre mice (Jackson Laboratory; JAX 004586) were kindly provided by Dr. Joshua Mendell (University of Texas Southwestern Medical Center). Rosa26-CAG-loxP-stop-loxP-tdTomato mice were from the Jackson Laboratory. Mouse protocols were approved by the Institutional Animal Care and Use Committee (APN#2015-100932) at the University of Texas Southwestern Medical Center.
Cell Culture and Transfection
The tumor cell line was generated as described in Tran et al. (62). Briefly, tumor tissue was dissected from the kidney in cold PBS, placed in MEM with Earle’s salts, 2 mM glutamine, 50 μg/ml of gentamicin [Life Technologies], 1% Pen/Strep, and 1% amphotericin B [Life Technologies], and minced into small pieces. Subsequently, 15 ml of tissue digestion medium (transport medium with 0.1 mg/ml collagenase [Sigma], 0.1 mg/ml hyaluronidase [Sigma], and 20 μg/ml DNase I [Sigma; D5025]) was added. Dissociated cells were plated in primary RCC cell medium (MEM with Earle’s salts, 2 mM glutamine, 10 ng/ml EGF (Life Technologies), 1% MEM nonessential amino acid (CellGro), 0.4 μg/ml hydrocortisone (Sigma), 1% Pen/Strep, and 10% FBS) and cells were maintained in 37°C incubator with 5% CO2. Cell medium was changed every 3 days and cells were split 1:2–3 until confluent. Cells were not tested for mycoplasma.
HEK293T cells [ATCC, negative for mycoplasma on August 17, 2016, using MycoAlert™ Mycoplasma Detection Kit (Lonza, LT07–118)] were grown in DMEM with 1% Pen/Strep, and 10% FBS and maintained at 37°C with 5% CO2. Plasmids for mouse Hif-1α and Hif-2α (Addgene #44028 and #44027 respectively; laboratory plasmid database #966 and #967, respectively) were transfected using Lipofectamine® 2000 (Thermo Fisher Scientific) into HEK293T cells. After 48 hours, transfected cells were harvested as positive controls for expression of mouse Hif-1α and Hif-2α.
Genotyping
Genotyping primer sequences available upon request.
Quantitative PCR
For qRT-PCR, frozen tissue was homogenized, and RNA was extracted as previously described (63). cDNA was synthesized from 1 to 4 μg of total RNA (RNA was isolated from 3 independent mice) using random primers (Invitrogen) and Moloney MLV reverse transcriptase (Invitrogen). PCR was performed in triplicate. Primer sequences available upon request.
Gene Expression Analysis of VIL1 and SLC5A2
Expression data for the TCGA KIRC patients were downloaded and combined with a UTSW cohort (64). Quantile normalization was carried out on log-transformed gene expression matrix. Two-tailed t test was used to test for differences in gene expression values on the log scale.
Western Blotting
Tissues were homogenized in lysis buffer (50 mM Tris-HCl pH 7.4, 250 mM NaCl, 0.5% (vol/vol) Igepal with complete protease and phosphatase inhibitors [protease inhibitors: 0.1 μm aprotonin (USB), 0.02 mM leupeptin (USB), 0.01 mM pepstatin (USB), 0.5 mM PMSF (Sigma); phosphatase inhibitors: 2 mM imidazole (Sigma), 1.15 mM sodium molybdate (Sigma), 1 mM sodium orthovanadate (Sigma), 5 nM microcystin (Calbiochem)]. Western blotting was performed with the following antibodies from: Cell Signaling Technology: BAP1 (#13271); Bethyl Laboratories: PBRM1 (A301-591A), Hif-1α (A300-286A); Novus Biologicals: CAIX (AF2344), Hif-2α (AF2997); Santa Cruz Biotechnology: VHL (sc-1534); Millipore: H2A (07-146), Ubiquityl-Histone H2A (05-678); Sigma: Tubulin (T5168); Thermo Fisher Scientific: HRP-conjugated goat anti-mouse IgG (#31430) and HRP-conjugated goat anti-rabbit IgG (#31460); Novus Biologicals: Donkey anti-Goat IgG Secondary Antibody (HAF109).
MRI Imaging
All imaging was performed on a 1T Desktop MR scanner (M2 Compact, Aspect Imaging, Israel), using a mouse volume coil. The general T1-weighted and T2-weighted imaging were performed with a spin echo (SE) (TR/TE = 326/13 ms) and a fast spin echo (FSE) (TR/TE = 2500/80 ms) sequence respectively (prone position).
Supplementary Material
Significance.
Determinants of tumor grade and aggressiveness across cancer types are poorly understood. Using ccRCC as a model, we show that BAP1 and PBRM1 loss drive tumor grade. Furthermore, we show that the conversion from low grade to high grade can be accelerated by activation of mTORC1.
Acknowledgments
The authors wish to thank the patients who generously provided tissues and participated in our studies. We thank Dr. Joshua Mendell for the Villin-Cre mice. We thank members of the Brugarolas Lab for assistance and helpful discussions.
Grant Support
This work was supported by Cancer Prevention and Research Institute of Texas grant RP130603 (J. Brugarolas) and NIH Grants R01CA175754 (J. Brugarolas), R01CA154475 (I. Pedrosa), and P50CA196516 (J. Brugarolas, P. Kapur, I. Pedrosa, T. Wang). The authors would like to acknowledge the assistance of the UT Southwestern Small Animal Imaging Resource, which is supported in part through an NCI Cancer Center Support Grant, 1P30 CA142543.
Footnotes
Disclosure of Potential Conflicts of Interest: J. Brugarolas is on the Scientific Advisory Board of Bethyl Laboratories.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Baldewijns MM, van Vlodrop IJ, Schouten LJ, Soetekouw PM, de Bruine AP, van Engeland M. Genetics and epigenetics of renal cell cancer. Biochimica et Biophysica Acta. 2008;1785(2):133–55. doi: 10.1016/j.bbcan.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nature Genetics. 1994;7(1):85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 4.Brugarolas J. Molecular genetics of clear-cell renal cell carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32(18):1968–76. doi: 10.1200/JCO.2012.45.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, Toro JR, Karami S, Hung R, Gerard GF, Linehan WM, Merino M, Zbar B, Boffetta P, Brennan P, Rothman N, Chow WH, Waldman FM, Moore LE. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clinical Cancer Research : an official journal of the American Association for Cancer Research. 2008;14(15):4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nature Reviews Cancer. 2002;2(9):673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 8.Gatto F, Nookaew I, Nielsen J. Chromosome 3p loss of heterozygosity is associated with a unique metabolic network in clear cell renal carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(9):E866–75. doi: 10.1073/pnas.1319196111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, Rabinowitz A, Spencer-Dene B, Gulati S, Bates PA, Stamp G, Pickering L, Gore M, Nicol DL, Hazell S, Futreal PA, Stewart A, Swanton C. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature Genetics. 2014;46(3):225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England Journal of Medicine. 2012;366(10):883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, Bignell G, Butler A, Cho J, Dalgliesh GL, Galappaththige D, Greenman C, Hardy C, Jia M, Latimer C, Lau KW, Marshall J, McLaren S, Menzies A, Mudie L, Stebbings L, Largaespada DA, Wessels LF, Richard S, Kahnoski RJ, Anema J, Tuveson DA, Perez-Mancera PA, Mustonen V, Fischer A, Adams DJ, Rust A, Chan-on W, Subimerb C, Dykema K, Furge K, Campbell PJ, Teh BT, Stratton MR, Futreal PA. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469(7331):539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teh BT, Varela I, Tarpey P, Raine K, Huang D, Ong CK, Furge KA, Campbell PJ, Stratton MR, Futreal PA. Identification of mutations of the SWI/SNF complex gene PBRM1 by exome sequencing in renal carcinoma. Journal of Clinical Oncology : official journal of the American Society of Clinical Oncology. 2011;29(15_suppl):4571. [Google Scholar]
- 13.Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, Kinch L, Hambuch T, Jain S, Lotan Y, Margulis V, Sagalowsky AI, Summerour PB, Kabbani W, Wong SW, Grishin N, Laurent M, Xie XJ, Haudenschild CD, Ross MT, Bentley DR, Kapur P, Brugarolas J. BAP1 loss defines a new class of renal cell carcinoma. Nature Genetics. 2012;44(7):751–9. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O’Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brugarolas J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer Journal. 2013;19(4):324–32. doi: 10.1097/PPO.0b013e3182a102d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pena-Llopis S, Christie A, Xie XJ, Brugarolas J. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Research. 2013;73(14):4173–9. doi: 10.1158/0008-5472.CAN-13-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavia-Jimenez A, Rathmell WK, Xie XJ, Brugarolas J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. The Lancet Oncology. 2013;14(2):159–67. doi: 10.1016/S1470-2045(12)70584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 19.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(4):1583–8. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, Emmert-Buck MR, Westphal H, Klausner RD, Linehan WM. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9102–7. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, Moch H, Krek W. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. The EMBO Journal. 2008;27(12):1747–57. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer research. 2006;66(5):2576–83. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death and Differentiation. 2008;15(4):650–9. doi: 10.1038/sj.cdd.4402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang SS, Gu YF, Wolff N, Stefanius K, Christie A, Dey A, Hammer RE, Xie XJ, Rakheja D, Pedrosa I, Carroll T, McKay RM, Kapur P, Brugarolas J. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(46):16538–43. doi: 10.1073/pnas.1414789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace AC, Nairn RC. Renal tubular antigens in kidney tumors. Cancer. 1972;29(4):977–81. doi: 10.1002/1097-0142(197204)29:4<977::aid-cncr2820290444>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida SO, Imam A, Olson CA, Taylor CR. Proximal renal tubular surface membrane antigens identified in primary and metastatic renal cell carcinomas. Archives of pathology & laboratory medicine. 1986;110(9):825–32. [PubMed] [Google Scholar]
- 27.Cheval L, Pierrat F, Rajerison R, Piquemal D, Doucet A. Of mice and men: divergence of gene expression patterns in kidney. PLoS One. 2012;7(10):e46876. doi: 10.1371/journal.pone.0046876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, Hacker KE, Bhanot G, Gordenin DA, Chu A, Gunaratne PH, Biehl M, Seth S, Kaipparettu BA, Bristow CA, Donehower LA, Wallen EM, Smith AB, Tickoo SK, Tamboli P, Reuter V, Schmidt LS, Hsieh JJ, Choueiri TK, Hakimi AA, Cancer Genome Atlas Research N. Chin L, Meyerson M, Kucherlapati R, Park WY, Robertson AG, Laird PW, Henske EP, Kwiatkowski DJ, Park PJ, Morgan M, Shuch B, Muzny D, Wheeler DA, Linehan WM, Gibbs RA, Rathmell WK, Creighton CJ. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26(3):319–30. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen F, Zhang Y, Senbabaoglu Y, Ciriello G, Yang L, Reznik E, Shuch B, Micevic G, De Velasco G, Shinbrot E, Noble MS, Lu Y, Covington KR, Xi L, Drummond JA, Muzny D, Kang H, Lee J, Tamboli P, Reuter V, Shelley CS, Kaipparettu BA, Bottaro DP, Godwin AK, Gibbs RA, Getz G, Kucherlapati R, Park PJ, Sander C, Henske EP, Zhou JH, Kwiatkowski DJ, Ho TH, Choueiri TK, Hsieh JJ, Akbani R, Mills GB, Hakimi AA, Wheeler DA, Creighton CJ. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep. 2016;14(10):2476–89. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narlis M, Grote D, Gaitan Y, Boualia SK, Bouchard M. Pax2 and pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. Journal of the American Society of Nephrology : JASN. 2007;18(4):1121–9. doi: 10.1681/ASN.2006070739. [DOI] [PubMed] [Google Scholar]
- 32.Carroll TJ, Vize PD. Synergism between Pax-8 and lim-1 in embryonic kidney development. Developmental Biology. 1999;214(1):46–59. doi: 10.1006/dbio.1999.9414. [DOI] [PubMed] [Google Scholar]
- 33.Bouchard M, Souabni A, Mandler M, Neubuser A, Busslinger M. Nephric lineage specification by Pax2 and Pax8. Genes & Development. 2002;16(22):2958–70. doi: 10.1101/gad.240102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouchard M, Souabni A, Busslinger M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis. 2004;38(3):105–9. doi: 10.1002/gene.20008. [DOI] [PubMed] [Google Scholar]
- 35.Tong GX, Yu WM, Beaubier NT, Weeden EM, Hamele-Bena D, Mansukhani MM, O’Toole KM. Expression of PAX8 in normal and neoplastic renal tissues: an immunohistochemical study. Modern Pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2009;22(9):1218–27. doi: 10.1038/modpathol.2009.88. [DOI] [PubMed] [Google Scholar]
- 36.Barr ML, Jilaveanu LB, Camp RL, Adeniran AJ, Kluger HM, Shuch B. PAX-8 expression in renal tumours and distant sites: a useful marker of primary and metastatic renal cell carcinoma? Journal of Clinical Pathology. 2015;68(1):12–7. doi: 10.1136/jclinpath-2014-202259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozcan A, de la Roza G, Ro JY, Shen SS, Truong LD. PAX2 and PAX8 expression in primary and metastatic renal tumors: a comprehensive comparison. Archives of Pathology & Laboratory Medicine. 2012;136(12):1541–51. doi: 10.5858/arpa.2012-0072-OA. [DOI] [PubMed] [Google Scholar]
- 38.Genega EM, Ghebremichael M, Najarian R, Fu Y, Wang Y, Argani P, Grisanzio C, Signoretti S. Carbonic anhydrase IX expression in renal neoplasms: correlation with tumor type and grade. Am J Clin Pathol. 2010;134(6):873–9. doi: 10.1309/AJCPPPR57HNJMSLZ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Research. 2000;60(24):7075–83. [PubMed] [Google Scholar]
- 40.Daou S, Hammond-Martel I, Mashtalir N, Barbour H, Gagnon J, Iannantuono NV, Nkwe NS, Motorina A, Pak H, Yu H, Wurtele H, Milot E, Mallette FA, Carbone M, Affar el B. The BAP1/ASXL2 Histone H2A Deubiquitinase Complex Regulates Cell Proliferation and Is Disrupted in Cancer. The Journal of Biological Chemistry. 2015;290(48):28643–63. doi: 10.1074/jbc.M115.661553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun. 2016;7:10292. doi: 10.1038/ncomms10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11(5):525–34. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 44.Wilson C, Idziaszczyk S, Parry L, Guy C, Griffiths DF, Lazda E, Bayne RA, Smith AJ, Sampson JR, Cheadle JP. A mouse model of tuberous sclerosis 1 showing background specific early post-natal mortality and metastatic renal cell carcinoma. Hum Mol Genet. 2005;14(13):1839–50. doi: 10.1093/hmg/ddi190. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3(2):169–81. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. The Journal of Biological Chemistry. 2002;277(36):33275–83. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 47.Rubera I, Poujeol C, Bertin G, Hasseine L, Counillon L, Poujeol P, Tauc M. Specific Cre/Lox recombination in the mouse proximal tubule. Journal of the American Society of Nephrology : JASN. 2004;15(8):2050–6. doi: 10.1097/01.ASN.0000133023.89251.01. [DOI] [PubMed] [Google Scholar]
- 48.Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d’Enghien C, Richaudeau B, Renaudin X, Sellers J, Nicolas A, Sastre-Garau X, Desjardins L, Gyapay G, Raynal V, Sinilnikova OM, Andrieu N, Manie E, de Pauw A, Gesta P, Bonadona V, Maugard CM, Penet C, Avril MF, Barillot E, Cabaret O, Delattre O, Richard S, Caron O, Benfodda M, Hu HH, Soufir N, Bressac-de Paillerets B, Stoppa-Lyonnet D, Stern MH. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet. 2013;92(6):974–80. doi: 10.1016/j.ajhg.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A, Vocke CD, Ricketts CJ, Peterson J, Middelton L, Kinch L, Grishin N, Merino MJ, Metwalli AR, Xing C, Xie XJ, Dahia PL, Eng C, Linehan WM, Brugarolas J. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res. 2013;11(9):1061–71. doi: 10.1158/1541-7786.MCR-13-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benusiglio PR, Couve S, Gilbert-Dussardier B, Deveaux S, Le Jeune H, Da Costa M, Fromont G, Memeteau F, Yacoub M, Coupier I, Leroux D, Mejean A, Escudier B, Giraud S, Gimenez-Roqueplo AP, Blondel C, Frouin E, Teh BT, Ferlicot S, Bressac-de Paillerets B, Richard S, Gad S. A germline mutation in PBRM1 predisposes to renal cell carcinoma. J Med Genet. 2015;52(6):426–30. doi: 10.1136/jmedgenet-2014-102912. [DOI] [PubMed] [Google Scholar]
- 51.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nature reviews Cancer. 2013;13(3):153–9. doi: 10.1038/nrc3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brodbeck S, Englert C. Genetic determination of nephrogenesis: the Pax/Eya/Six gene network. Pediatr Nephrol. 2004;19(3):249–55. doi: 10.1007/s00467-003-1374-z. [DOI] [PubMed] [Google Scholar]
- 53.Gossage L, Murtaza M, Slatter AF, Lichtenstein CP, Warren A, Haynes B, Marass F, Roberts I, Shanahan SJ, Claas A, Dunham A, May AP, Rosenfeld N, Forshew T, Eisen T. Clinical and pathological impact of VHL, PBRM1, BAP1, SETD2, KDM6A, and JARID1c in clear cell renal cell carcinoma. Genes Chromosomes Cancer. 2014;53(1):38–51. doi: 10.1002/gcc.22116. [DOI] [PubMed] [Google Scholar]
- 54.Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, Reuter VE, Russo P, Cheng EH, Sander C, Motzer RJ, Hsieh JJ, cc RCCCGARNi Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clinical Cancer Research : an official journal of the American Association for Cancer Research. 2013;19(12):3259–67. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joseph RW, Kapur P, Serie DJ, Parasramka M, Ho TH, Cheville JC, Frenkel E, Parker AS, Brugarolas J. Clear Cell Renal Cell Carcinoma Subtypes Identified by BAP1 and PBRM1 Expression. J Urol. 2016;195(1):180–7. doi: 10.1016/j.juro.2015.07.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, Nagata Y, Yoshida K, Kon A, Suzuki Y, Chiba K, Tanaka H, Niida A, Fujimoto A, Tsunoda T, Morikawa T, Maeda D, Kume H, Sugano S, Fukayama M, Aburatani H, Sanada M, Miyano S, Homma Y, Ogawa S. Integrated molecular analysis of clear-cell renal cell carcinoma. Nature Genetics. 2013;45(8):860–7. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes & Development. 2000;14(21):2712–24. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsang CK, Bertram PG, Ai W, Drenan R, Zheng XF. Chromatin-mediated regulation of nucleolar structure and RNA Pol I localization by TOR. The EMBO Journal. 2003;22(22):6045–56. doi: 10.1093/emboj/cdg578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982;6(7):655–63. doi: 10.1097/00000478-198210000-00007. [DOI] [PubMed] [Google Scholar]
- 60.Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, Aras O, Han S, Oyama T, Takeda S, Ray CE, Dong Z, Berge M, Hakimi AA, Monette S, Lekaye CL, Koutcher JA, Leslie CS, Creighton CJ, Weinhold N, Lee W, Tickoo SK, Wang Z, Cheng EH, Hsieh JJ. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep. 2017;18(12):2893–906. doi: 10.1016/j.celrep.2017.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, Wu J, Phu L, Katavolos P, LaFave LM, Abdel-Wahab O, Modrusan Z, Seshagiri S, Dong K, Lin Z, Balazs M, Suriben R, Newton K, Hymowitz S, Garcia-Manero G, Martin F, Levine RL, Dixit VM. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337(6101):1541–6. doi: 10.1126/science.1221711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tran TA, Leong HS, Pavia-Jimenez A, Fedyshyn S, Yang J, Kucejova B, Sivanand S, Spence P, Xie XJ, Pena-Llopis S, Power N, Brugarolas J. Fibroblast Growth Factor Receptor-Dependent and -Independent Paracrine Signaling by Sunitinib-Resistant Renal Cell Carcinoma. Mol Cell Biol. 2016;36(13):1836–55. doi: 10.1128/MCB.00189-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pena-Llopis S, Brugarolas J. Simultaneous isolation of high-quality DNA, RNA, miRNA and proteins from tissues for genomic applications. Nat Protoc. 2013;8(11):2240–55. doi: 10.1038/nprot.2013.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, Zhang N, Toffessi-Tcheuyap V, Nguyen TT, Pahuja KB, Chen YJ, Saleem S, Chaudhuri S, Heldens S, Jackson M, Pena-Llopis S, Guillory J, Toy K, Ha C, Harris CJ, Holloman E, Hill HM, Stinson J, Rivers CS, Janakiraman V, Wang W, Kinch LN, Grishin NV, Haverty PM, Chow B, Gehring JS, Reeder J, Pau G, Wu TD, Margulis V, Lotan Y, Sagalowsky A, Pedrosa I, de Sauvage FJ, Brugarolas J, Seshagiri S. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nature Genetics. 2015;47(1):13–21. doi: 10.1038/ng.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.