

Abstract
With an estimated global population of cancer survivors exceeding 32 million and growing, there is a heightened awareness of the long-term toxicities resulting from cancer treatments and their impact on quality of life. Unexplained heterogeneity in the persistence and development of toxicities, as well as an incomplete understanding of their mechanisms have generated a growing need for the identification of predictive pharmacogenomic markers. Early studies addressing this need used a candidate gene approach; however, over the last decade, unbiased and comprehensive genome-wide association studies (GWAS) have provided markers of phenotypic risk and potential targets to explore the mechanistic and regulatory pathways of biological functions associated with chemotherapeutic toxicity. In this review, we provide the current status of GWAS of chemotherapeutic toxicities with an emphasis on examining the ancestral diversity of the representative cohorts within these studies. Persistent calls to incorporate both ancestrally diverse and/or admixed populations into genomic efforts resulted in a recent rise in the number of studies utilizing cohorts of East Asian descent; however, few pharmacogenomic studies to date include cohorts of African, Indigenous American, Southwest Asian, and admixed populations. Through comprehensively evaluating sample size, composition by ancestry, genome-wide significant variants, and population-specific minor allele frequencies as reported by HapMap/dbSNP using NCBI PubMed, and the NHGRI-EBI GWAS Catalog, we illustrate allele frequencies and effect sizes tend to vary among individuals of differing ancestries. In an era of Personalized Medicine, the lack of diversity in genome-wide studies of anticancer agent toxicity may contribute to the health disparity gap.
Keywords: GWAS, toxicity, anticancer agent, ethnic differences
Introduction
Over 1.5 million new cancer cases are diagnosed in the United States every year with overall five-year survival rates approaching two thirds of all diagnoses (1). Patients diagnosed at less than twenty years of age exhibit even higher five-year survival rates at roughly 80% (1). The total number of cancer survivors world-wide has increased over 33% between 2002 and 2013, from an estimated 24.2 million survivors to over 32 million (2–4). The growing population of survivors has led to a growing awareness of the debilitating long-term toxicities of chemotherapy. Toxicities often drastically alter a patient’s quality of life and exhibit comorbidities, including cardiovascular and endocrine-related diseases (5,6). Toxicities can be life-long, and their permanence can have a dramatic impact on a patient’s physical and psychological wellbeing. In a study of 1,713 diverse childhood cancer survivors, 48–65% displayed impaired pulmonary functions, hearing loss, endocrine dysfunction, cardiac disorders, and neurocognitive impairments at a median of 25 years after diagnosis (7–9). Neurocognitive disorders and hearing loss, although not typically life threatening, can be particularly disruptive to quality of life. For instance, cisplatin-related hearing loss in children impedes speech and language development with irreversible effects (10). Inadequate understanding of underlying mechanisms and inter-patient variability represent major obstacles facing clinical actionability. In the era of genome-wide association studies (GWAS), pharmacogenomic studies aim to address variability in drug response and/or toxicity and suggest plausible mechanisms for phenotypic variation. Genetic predictors of adverse effects allow for the alteration of regimens and doses according to the patient’s genetic susceptibilities, providing a personalized approach to mitigating toxicities. Besides moving medicine into a more preventative paradigm, they also reveal toxicological etiology (11–13). In this review, we address some lessons learned from GWAS of adverse effects of chemotherapy, focusing on findings pertaining to pharmacoethnicity. By comprehensively evaluating the literature, we reveal stark disparities in population representation despite numerous calls to include more diverse cohorts (14–16). These disparities could contribute to widening gaps in health outcomes.
GWAS of Anticancer Agent Toxicity
The basic principle of genetic studies is to statistically associate genetic variants to a particular phenotype. Early studies exploring the genetic contribution to chemotherapeutic toxicities relied heavily upon candidate gene approaches, associating polymorphisms in genes encoding known drug metabolizing enzymes, DNA repair pathways, receptors, and transporters (17–23). Though candidate gene studies successfully identified clinically applicable variants associated with chemotherapeutic toxicities, they lack the ability to identify risk loci outside of already well studied pathways (24). The advent of GWAS allowed researchers to detect novel associations while avoiding variant ascertainment bias, with a downside of an incurred burden of multiple hypothesis testing (i.e. >1 million independent tests). Thus, statistical significance can only be achieved with considerable sample and effect sizes, and false positives are likely without the careful accounting of confounding variables and stringent criteria. Confounders such as inter-population differences in toxicity, differing minor allele frequencies (MAF), and haplotype/linkage disequilibrium (LD) structures all compromise the accuracy of estimates across populations (25). Informative variants in one group may therefore not be useful in another. Ancestral heterogeneity can be accounted for with various methods, including sample exclusion. However, the inclusion of ancestrally diverse participants in GWAS is necessary to gain broader insights into genetic architectures and to capitalize on genomic variation.
Ancestral differences in prevalence of chemotherapeutic toxicities
Inter-population differences in incidence and severity of adverse reactions to chemotherapeutic treatment have been observed (26). Bevacizumab, an anti-angiogenic monoclonal antibody targeting vascular endothelial growth factor A (VEGFA), has several documented population discrepancies in toxicities. Among 4,308 lung cancer patients, East Asian populations were found more likely to develop bevacizumab-induced thromboembolism (RR = 3.65) and severe bleeding (RR = 2.17), and were less susceptible to proteinuria (RR = 0.43) compared to others (27). Two studies have shown increased susceptibility of African Americans to bevacizumab-related hypertension; one showing a 1.6-fold increase in bevacizumab-induced hypertension (14), and a second showing bevacizumab-induced exacerbation of pre-existing hypertension disproportionately affecting African Americans (28). The former study included a GWAS of bevacizumab-induced hypertension which excluded the African American sample during quality control to avoid confounding population substructure (29).
Cancer patients are at a 4–7 fold increase for venous thromboembolism (VTE) compared to the general populace (30,31), making it a leading cause of mortality. VTE can manifest as a direct result of cancer via the aberrant activation of pro-coagulatory pathways (32,33), or due to chemotherapy-mediated complications (34,35). Among 1,295 acute lymphocytic leukemia and acute myeloid leukemia patients, African Americans were more likely to develop VTE (33.3% vs. 20.3%; P = 0.04) (36). Increased rates of VTE in African Americans has also been reported in the general population (37).
Peripheral neuropathy is the most common non-hematologic toxicity associated with chemotherapy (38). One study showed African Americans were roughly twice as likely as European Americans to have dose reductions due to taxane-induced peripheral neuropathy (39). Some rare variants in the Charcot-Marie-Tooth gene SET binding factor 2 (SBF2) increase the frequency of paclitaxel-induced peripheral neuropathy in African Americans (23). Conversely, patients of European descent are more likely to develop vincristine-induced neurotoxicity than African Americans, partially due to allelic differences in the gene coding cytochrome P450 enzyme 3A5 (40,41).
Higher rates of toxicities have been observed in East Asian populations compared to European and North American populations that frequently lead to dose-limiting restrictions (42,43). Some rates of toxicity remain higher in East Asians despite dose titration (44). Rates of neutropenia among patients treated with cisplatin, docetaxel, and 5-FU remained twice as high (19% vs. 8%) in Japanese individuals, despite an 80% dose reduction compared to US counterparts, with no significant change in overall response or survival (45,46). A retrospective study of breast cancer from five international centers found East Asian participants to be twice as likely to experience hematological toxicities from equivalent FEC100 (fluorouracil, epirubicin, cyclophosphamide) treatment compared to patients of European and African ancestry (32% vs. 16% and 10% respectively; P < 0.05) (47).
Cisplatin-induced ototoxicity affects up to 80% of treated adults with severe/profound hearing loss in 18%, and over 60% of children (10,48). Hearing loss rates as high as 77% have been observed in an adult Japanese cohort (49), and a single South African study reported rates of over 55% (50) suggesting the prevalence of drug-induced ototoxicity may not be uniform across ancestry, although different study methodologies make comparisons less straightforward.
Higher rates of therapeutic toxicities could be attributed to pharmacokinetic mechanisms such as metabolism and/or, clearance as well as pharmacodynamics, and genetic studies often obscure the distinction. A hypothetical example would be a membrane drug transporter expressed in both the renal epithelium and neuronal tissue. In the kidneys, it might excrete the drug into the collecting duct and be considered to have pharmacokinetic functions. In the neuron, it might mitigate neurotoxicity by lowering intracellular drug concentration and therefore be considered to play a pharmacodynamic role. Accounting for variability in drug pharmacokinetics is paramount to provide accurate estimates of drug exposure, an increasingly prominent necessity in pharmacogenomic studies (51). Large GWAS of anticancer agent toxicities in diverse, well-characterized cohorts could resolve many ambiguities and partition trait heritability to specific chromosomal regions and biological pathways (11,51). Several GWAS of chemotherapeutic toxicities have been conducted in recent years, and we have queried them using databases to make inferences about pharmacoethnic differences.
Literature Query
We utilized two publically available databases to assess total peer-reviewed pharmacogenomic anticancer agent toxicity GWAS: MEDLINE (PubMed) and the GWAS Catalog, a continuously curated database of all published English-language GWAS under the partnership of the European Bioinformatics Institute and the US National Human Genome Research Institute [https://www.ebi.ac.uk/gwas/home]. A diagram of the query is presented in Figure 1. Animal and cell based studies, review articles, and candidate gene studies were excluded in subsequent filtering. All three searches were performed on March 3, 2017. We additionally queried minor allele frequencies (MAF) of important SNPs based on HapMap via dbSNP at the National Center for Biotechnology Information [www.ncbi.nlm.nih.gov/projects/SNP/]. We used Yoruban (YRI), Han Chinese (HCB), and European American (CEU) populations as representatives of African, East Asian, and European populations respectively.
Figure 1. Literature search of all current pharmacogenomic anticancer chemotherapeutic induced toxicity GWAS.
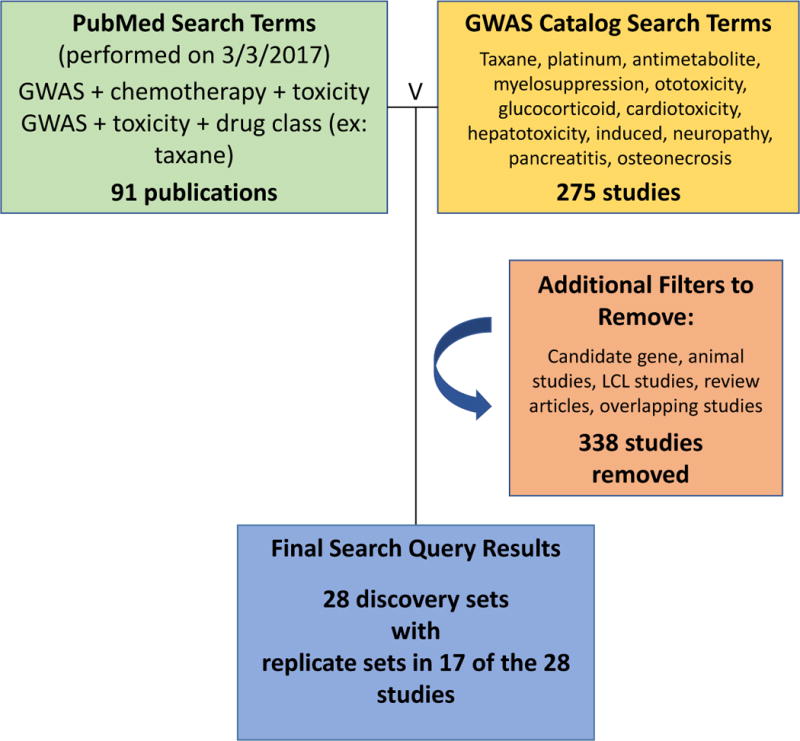
Filters were designed to maximize initial results using PubMed [www.ncbi.nlm.nih.gov/pubmed] and the NHGRI-EBI GWAS Catalog [www.ebi.ac.uk/gwas/], followed by removing all candidate gene studies, studies of non-anticancer drugs, animal models, lymphoblastoid cell-line based GWAS and review articles. Numbers in blue boxes indicate initial query results; −338 indicates the studies that did not pass filtering criteria. The final result was 28 non-overlapping discovery studies with 17 studies including at least one replication set. Note: Search terms that yielded no results were excluded.
Results
In Table 1 we list 28 GWAS of anticancer agent that were performed, 17 of which included at least one replication set (22,29,39,52–77). Of the discovery studies, 22 were ancestrally homogeneous. Any number of non-specific ancestral descriptors in a single manuscript including white and black are used. Often, the term African is used in reference to admixed individuals from North America and the term Asian is used without population or region specificity. These observations are consistent with a study performed by Panofsky and Bliss that found ambiguity in ethnic descriptors among geneticists including the tendency to use both racially based terms and geographic descriptors of populations (78).
Table 1. List of Genome Wide Association Studies of Chemotherapeutic Toxicity.
Query Results by Study
| Drug | Toxicity | Study Population (ndiscovery){nby population}[nreplicate]* | Reference |
|---|---|---|---|
| methotrexate | myelosuppression | Diverse (1279){806 EA; 58 AA; 22 EAS; 266 HIS; 127 OTH} | (56) |
| cisplatin | myelosuppression | East Asian (333)[876] | (53) † |
| carbo + paclitaxel | myelosuppression | Japanese (1154) | (63) |
| 5-FU + FOLFOX | myelosuppression | European (221)[791] | (55) |
| thiopurine | myelosuppression | European (175) | (54) |
| thiopurine | myelosuppression | Korean (331)[767] | (52) |
| anthracycline (epirubicin) | myelosuppression | Japanese (318) | (70) |
| paclitaxel | neuropathy | European American (144) | (60) |
| paclitaxel | neuropathy | European (1303) | (59) |
| paclitaxel | neuropathy | European American (855)[154 EA; 117 AA] | (57) |
| paclitaxel/docetaxel | neuropathy | Diverse (1570){1357 EA; 213 AA}[789 EA; 90 AA; 56 OTH] | (39) |
| docetaxel | neuropathy | European American (623) | (58) |
| alkyloid (vincristine) | neuropathy | Diverse (341){209 EA; 43 AFR; 2 EAS; 44 HIS; 23 OTH} | (74) |
| platinating (combination) | neuropathy | Korean (366) | (73) |
| bortezomib | neuropathy | European (469)[114] | (76) |
| antibody (lapatinib) | hepatotoxicity | Diverse American (366){222 EA; 144 OTH}[144 EA; 31 OTH] | (71) |
| antibody (lapatinib) | hepatotoxicity | European American (844)[45] | (69) |
| cisplatin/carboplatin | hepatotoxicity | East Asian (329)[375] | (61) † |
| cisplatin | ototoxicity | European American (511) | (75) |
| cisplatin | ototoxicity | European American (238)[68] | (62) |
| mercaptopurine | dose tolerance | European American (657)[371] | (67) |
| bevacizumab | hypertension | European (824)[149] | (29) |
| anthracycline | cardiotoxicity | European American (280)[diverse: 176] | (64) |
| mercaptopurine | pancreatitis | European (2207)[2122] | (66) |
| asparaginase | pancreatitis | Diverse American (5185){3069 EA; 350 AA; 99 EAS; 1667 OTH}[213 diverse] | (77) |
| glucocorticoid | osteonecrosis | Diverse American (2285){EA:1275; AA:139; EAS:48; other:823}[670] | (65) |
| antimetabolite (methotrexate) | GI toxicity and clearance | European (434)[206] | (68) |
| alkyloid (melphalan) | oral mucositis | European American (972) | (72) |
Replicate is in reference to any study that performs a second association study in another cohort within a given study. Abbreviations: 5-FU (fluorouracil); FOLFOX (folinic acid, fluorouracil, oxaliplatin combination); carb (carboplatin); GI (gastro intestinal); EA (European American); AA (African American); EAS (East Asian); HIS (Hispanic); OTH (other);
(Same cohort, separate evaluations).
Two studies used diverse replication cohorts after beginning with a homogenous discovery set (57,64). Additionally, only two studies maintained a similar degree of diversity in the discovery and replication panels (65,77). We compared inter-ethnic differences in significant GWAS findings and observed that several studies found associations to variants with differing MAF among African, East Asian and European populations. We observed seventeen polymorphisms that reached genome-wide significance and had a fixed allele in at least one of the three populations (Table 2).
Table 2.
Minor Allele Frequency of GWAS significant variants associated with chemotherapeutic toxicity.
| Associated non-uniform variants by ancestry | MAF by Population (HapMap) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug (Class) | SNP | Toxicity | Study Population | Gene | Location (GRCh38.p7) | HCB | YRI | CEU | Reference |
| thiopurine (antimetabolite) | rs79206939 | myelosuppression | East Asian (Korea) | FTO | 16:53826140 | 2.8* | 0 | 0 | (52) |
| 5-FU/FOLFOX | rs16857540 | myelosuppression | European (Spain) | NLGN1 | 3:174182785 | 0 | 37.2 | 15.3 | (55) |
| cis/carboplatin | rs2838566 | hepatotoxicity | East Asian (China) | intergenic | 21:44468699 | 7.1 | 33 | 0 | (61) |
| paclitaxel + epi. (taxane) | rs9501929 | neuropathy | European (USA) | TUBB2A | 6:3157620 | 0 | 28.6 | 4.7 | (59) |
| epi/doxo (anthracyclin) | rs229774 | cardiotoxicity | diverse (Canada) | RARG | 14:83435125 | 0 | 11.1 | 6.6 | (64) |
| melphalan (alkylating) | rs1469167 | oral mucositis | European (USA) | ALDH1A1 | 9:72942091 | 0 | 31.4 | 2.7 | (72) |
| epirubicin (anthracyclin) | rs4149639 | myelosuppression | East Asian (Japan) | TNFRSF1A | 12:6332835 | 11.4 | 22 | 0 | (63) |
| docetaxel (taxane) | rs3747851 | myelosuppression | East Asian (Japan) | DAB2IP | 9:121758981 | 0 | 15.1 | 0.4 | (63) |
| docetaxel (taxane) | rs875858 | neuropathy | European (USA) | VAC14 | 16:70741552 | 0 | 0 | 7.5 | (58) |
| paclitaxel (taxane) | rs17348202 | neuropathy | European (USA) | EPHA4 | 2:221207458 | 0 | 17.7 | 5.8 | (60) |
| glucocorticoid | rs2229288 | osteonecrosis | diverse (USA) | ZFHX3 | 16:72794405 | 0 | 0 | 0.5 | (65) |
| mercaptopurine | rs116855232 | dose tolerance | diverse (USA) | NUDT15 | 13:48045719 | 9.5 | 0 | 0.2 | (67) |
| cisplatin | rs62283056 | ototoxicity | European (USA) | WFS1 | 4: 6274903 | 0.3 | 17.6 | 22.9 | (75) |
| melphalan (alkylating) | rs1426765 | oral mucositis | European (USA) | intergenic | 3:25976116 | 0 | 0.8 | 14.4 | (72) |
| melphalan (alkylating) | rs6804277 | oral mucositis | European (USA) | intergenic | 3:25977271 | 0 | 10 | 12 | (72) |
| melphalan (alkylating) | rs1940228 | oral mucositis | European (USA) | intergenic | 11:103004647 | 0 | 6.1 | 1.7 | (72) |
| melphalan (alkylating) | rs948695 | oral mucositis | European (USA) | intergenic | 11:102990584 | 0 | 32.2 | 1.7 | (72) |
| oxaliplatin (platinum) | rs10486003 | neuropathy | East Asian (Korea) | intergenic | 7:97600466 | 23.3 | 0 | 10.2 | (73) |
Abbreviations: SNP (single nucleotide polymorphism); MAF (minor allele frequency); HCB (Han Chinese in Beijing, China); YRI (Yorubans in Ibadan, Nigeria); CEU (CEPH collection Europeans in Utah, United States of America); PMID# (PubMed Identification number); 5-FU (fluorouracil); FOLFOX (folinic acid, fluorouracil, oxaliplatin combination); epi (epirubicin), doxo (doxorubicin).
Minor Allele Frequency as reported by ExAc (http://exac.broadinstitute.org/); note that HapMap MAF is absent, and a MAF of 5.1% was observed in the Korean cohort used in the cited study.
Of note was a particular SNP from a Korean study (in bold) associated with thiopurine-induced myelosuppression (leukopenia) (52). The coding SNP (rs79206939 p.A134T) in the Fat mass and obesity-associated protein alpha-ketoglutarate dependent dioxygenase gene (FTO) was revealed in a study designed to explore genetic risk factors outside of the thiopurine methyltransferase (TPMT) variants. The variant was found in 9% of patients exhibiting thiopurine-induce leukopenia, and only 1.5% of unaffected patients treated with thiopurine (p = 1.3×10−3). SNPs in TPMT have been implicated in thiopurine toxicities in European populations but failed to replicate in East Asian populations (47). The failed replication is likely a product of low MAF in East Asian populations (20). Similarly, the variant in FTO associated with thiopurine-induced myelosuppression has an MAF of 3% in East Asian populations (5.1% in the Korean discovery cohort), but is fixed as the non-risk allele in European and African populations (Table 2).
However, the majority of common variants are shared across populations (79), suggesting that many GWAS findings may be applicable across populations. For instance, a finding by the Cancer and Leukemia Group B (CALGB) Alliance trials (56) in which a SNP in FYVE, RhoGEF and PH domain containing 4 (FGD4) met significance criteria for association with peripheral neuropathy in a Caucasian discovery sample (p = 3 × 10−6) and was replicated in two independent cohorts: a European cohort (p = 0.01) and an African cohort (p = 0.007). This study highlights the possibility of consistent SNP effects across ancestries. However, despite broadly shared common variation, inter-population divergences in allele frequencies do exist, as do differences in LD driven by population specific demographic histories (79,80). As such, GWAS results cannot be automatically assumed to be broadly applicable across all populations (15).
A few mechanisms exist to explain ancestral differences in SNP-phenotype association. Most simply, a site could be polymorphic with a toxicity-associated allele in one population and be monomorphic (or have very low MAF) in another. Another explanation follows from the presumption that many SNPs do not exert effects themselves but rather tag proximal causal variants in LD with the detected variant. Different ancestral backgrounds have different LD structures, and therefore a common SNP that falls within a haplotype block containing causal variants in one population might not tag for those same variants in another. While it is safe to assume that high-penetrance causal alleles (i.e. loss-of-function alleles from nonsense mutations) will exert their effects regardless of genetic background, more nuanced causal explanations behind the associations between common variants and complex, polygenic phenotypes might reveal genetic background-dependent SNP effects. These effects might therefore be manifest only in individuals with a certain genetic background. Whether ancestral differences in phenotypic associations are predominantly due to differing MAFs, differing LD structures around causal loci, or more complex differences in the causal pathway of the association is unclear, although these explanations are not mutually exclusive (15, 79–81).
Disparities in GWAS Population Representation
Four of the ten toxicities investigated by GWAS were represented by a single study (29,64,65,72), and only one of those four studies incorporated participants of non-European descent (Figure 2A) (65). Of the 28 total studies, 16 utilized cohorts of entirely European descent, compared to 6 that were entirely East Asian, and 6 studies with diverse cohorts (Figure 2B). There were no studies composed entirely of individuals of African descent. Additionally, within the 6 diverse/multi-ancestral studies, half of all participants were of European descent (5817/11861 in discovery sets) (Table 1).
Figure 2. GWAS studies of Chemotherapeutic Toxicity.
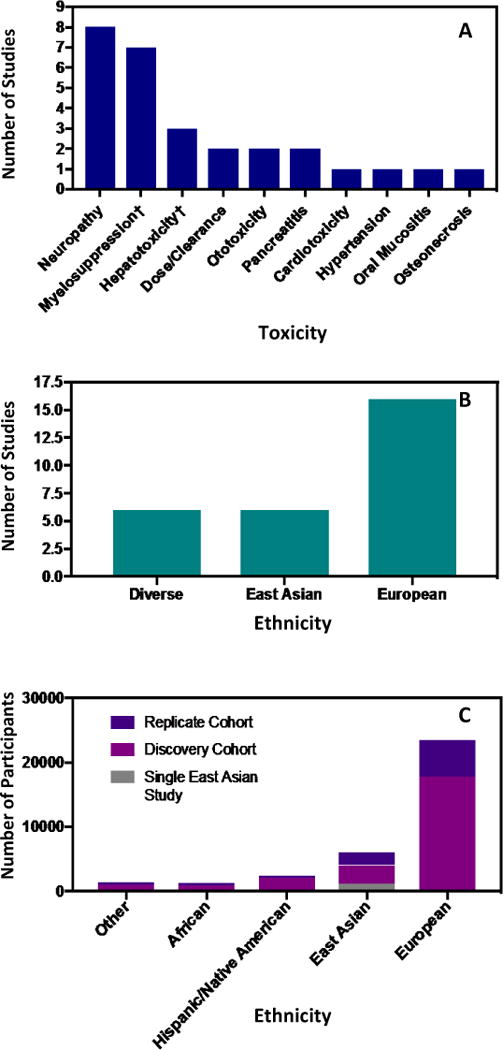
(A) Breakdown of toxicities from GWAS of anticancer agent toxicity studies. 15 of the 28 (54%) studies investigated myelosuppression and neuropathy. Associations to cardiotoxicity, hypertension, pancreatitis, and oral mucositis are all based on single European studies lending to potential population-based bias among these toxicities. Of the four toxicities represented by a single GWAS, only two of the studies investigating osteonecrosis and pancreatitis used broadly diverse panels of participants. († indicates a single cohort that was used to evaluate two separate toxicities.) (B) Breakdown of 28 studies by ancestry. Half of participants within the five multi-ancestral studies were of European descent (>49%), and more than 85% of all participants were either entirely of European descent, or entirely of East Asian descent. (C) Population based breakdown of participants in GWAS of pharmacogenomic anticancer agent toxicity with further breakdown into replication and discovery cohorts. More than half of the participants from the East Asian discovery GWAS come from a single Japanese study.
We evaluated population representation by tallying the ancestry of participants in each study. We found that 70.8% of GWAS participants were of European descent compared to 14.9% from East Asian descent, 6.6% from Hispanic/Native American ancestry, 3.7% from African descent, and 3.9% from all other ancestries. A single Japanese study was noted to represent more than half of the participants from all East Asian discovery cohorts (Figure 2C) (63). Four diverse studies included participants of Hispanic ancestry with one including 1,238 Hispanic individuals in the discovery and replication sets [>10% Native American ancestry as assessed by STRUCTURE] (77). Unfortunately, it was not always apparent which studies may have categorized participants of Hispanic ancestry as European.
Paucity of African Sampling
Sub-Saharan African participants are woefully under-represented, with 920 participants in discovery cohorts and only 1240 (discovery and replication) out of 33,112 total participants, or 3.7% (39,56,57,65,74,77). This may be an artifact of the trend to use currently available phase III clinical trial data-sets, which have tended to use non-diverse cohorts of European descent [<15% non-white participants in Cancer Trials Support Unit (CTSU) and CALGB combined] (82). The paucity of participants of African descent in anticancer agent toxicity GWAS is particularly unfortunate as Sub-Saharan African populations are among those with the highest genetic diversity, and least LD of extant human populations world-wide (83). Such diversity could be highly valuable in the application of fine-mapping in diverse trans-ethnic cohorts, and analyzing a greater numbers of variants could lead to associations with novel pathways (84). Additionally, the majority of participants of African ancestry in these studies are admixed individuals from North America. Admixed African Americans are individuals who share European, African, and in some cases, Native American ancestry. African Americans are not equivalent to Sub-Saharan African populations, nor do they share the same degree of genetic diversity (85).
The pharmacogenetics community has consciously and appropriately avoided using multi-ethnicity or admixed populations in GWAS to avoid false-positive associations (86,87). However, by limiting participants of African descent to admixed and migrant populations from relatively few North American and Western European locales, studies may be restricting associations to limited haplotypes that may be specific to the historical demographics of migrants to these regions. Appropriate statistical methods can be used to address spurious associations when including admixed populations by accounting for ancestry in imputation and regression (87–90). Including diverse participants in cohorts can increase power in GWAS (91), and the proper incorporation of these improved statistical models mitigates confounding due to admixture (92). Although admixed participants lead to greater genetic diversity overall, they cannot be considered a comprehensive solution to the lack of African participants in GWAS. Additional hurdles must be overcome to expand ancestral diversity within GWAS and leverage the genetic diversity that is unique to humans in the African continent, home to more than 15% of the world’s population and a greater proportion of the total human genetic diversity (83,93). It is therefore imperative to undertake bigger efforts to include African populations in future pharmacogenomic anticancer toxicity GWAS as well as in other GWAS.
Pharmacogenomic Challenges
Although associations in pharmacogenomic GWAS tend to have greater effect sizes than traditional disease-associated GWAS (94), several factors have added to challenges that are not as common in traditional GWAS. Most obvious is limited sample sizes of anticancer agent toxicity GWAS; cases and controls in pharmacogenomic studies correspond to patients treated with a specific agent and therefore represent a smaller pool of potential participants than that of typical common diseases. This makes large phase III clinical trials of anticancer agents good resources given the large participation and adequate data collection of dosages, phenotypes, and demographics. However and as stated, the utilization of these readily available datasets may come at the cost of ancestral diversity. “Nonwhite” participants are less likely to consent to pharmacogenomic studies [OR = 0.50, 95% CI = 0.43 to 0.57, P < .001], and participation in pharmacogenomics suffers overall (both “white” and “nonwhite” participants) as racial diversity increases at institutions (82).
Of the 28 studies evaluated, we found that the mean sample size of the GWAS dataset was 879 individuals (including both cases and controls), with a median of 490. The largest study included 5,185 (77), while the smallest study utilized only 144 participants (60). Studies with such small sample sizes are greatly underpowered, leading to a limited ability to detect variants with moderate to low effect sizes (95–97). However, studies with small sample sizes can still provide meaningful insight and do not require ancestrally homogeneous cohorts if proper methods are implemented. For instance, researchers from St. Jude Children’s Research Hospital investigating the association of glucocorticoid treatment and osteonecrosis in a diverse panel of childhood acute lymphoblastic leukemia patients identified many potential gene candidates despite analyzing only 400 cases (65). This highlights the importance of mining biological data from available resources to maximize GWAS utility.
Another challenge lies in evaluating toxicity phenotypes. Most are not quantitative. Myelosuppression and ototoxicity represent exceptions; however the degree of myelosuppression could be missed based on the frequency of measurement. Ototoxicity requires audiometry by a hearing specialist. Toxicities can occur at various times during or after treatment and are sometimes subject to a physician’s best judgment rather than objectively quantifiable means. Unless great care is taken when characterizing participants, such challenges could lead to case-control assignment errors. Furthermore, treatments vary in regimens and doses, and toxicities often lead to dose reduction or treatment termination. The standard of care varies by malignancy type and subtype, which can differentially contribute to manifestations of toxicities. Secondary interventional therapeutics are common among cancer patients. Variability can be observed across geographical regions, institutions, and patient characteristics. Such clinical heterogeneity confounds analyses unless rigorous care is taken during data collection and analysis. A number of studies evaluated in this review displayed heterogeneity of agents within a class, multiple primary agents, or multiple toxic drugs (55,63,68,72,73,76,77). Problems with accurately assessing phenotype and clinical heterogeneity are particularly troublesome when choosing and properly applying replication sets. Replicating the findings of a GWAS requires great care in matching both phenotype criteria and demographics of the subjects being used (98,99). This can also exacerbate problems with the inclusion of diversity in GWAS, as replication sets are typically chosen to reflect the demographics of the discovery set as closely as possible in the hopes of maximizing the probability of replicating observed effects.
These challenges have likely contributed to the scarcity of anticancer agent toxicity GWAS and the limited sample sizes. Disparities in the availability of resources required to overcome these challenges unfortunately exacerbate the insufficient diversity among these studies. Furthermore, the history and wealth of phase III clinical data, and early failures to address potential false positives when utilizing diverse or admixed panels in GWAS have also contributed to the lack of diversity in the relatively few studies that have been performed to date.
Conclusion
We have shown that pharmacogenomic anticancer agent toxicity GWAS suffer from a lack of diversity in the populations studied. A number of strategies to garner greater diversity in GWAS have been suggested in recent years. Institutional changes such as prioritizing funding of non-European and ancestrally diverse studies, and incorporating the importance of utilizing under-represented populations in training programs have been proposed (16). Other recommendations include initiating dialog in under-represented communities, developing educational programs to increase awareness, and employing more strategic means of recruitment (100).
Several barriers exist with regards to ancestral disparities in the realm of biological research and medical care. These barriers include, but are not limited to socio-economic disadvantages, access to care, and geographical proximity to institutions of academic medicine. Fortunately, the incorporation of GWAS has now spread well beyond the initial confines of large academic centers in North America and Europe and has led to several recent studies in China, Japan, and Korea, expanding upon much needed data from East Asian populations. Despite representing the second most studied population after Europeans, East Asians are still proportionally underrepresented. While it is promising that researchers are attempting to include diversity in cohorts, there is a great need to further increase diversity while making a concerted effort to initiate studies including diverse panels on the African continent itself.
Representatives of other populations are almost non-existent in current pharmacogenomic studies of anticancer agent toxicities. It is essential to leverage genetic diversity by including all ancestries in future studies. Existing disparities pose challenges in the implementation of genetic studies that could logically lead to widening health outcome gaps, creating a vicious cycle of inequality. We support making significant efforts to include diverse panels of participants to maximize the potential of discovering associated variants. Further efforts need to be implemented to develop better statistical and computational models to estimate risk in diverse populations, potentially utilizing local chromosomal ancestry (101), fine-scale mapping using multi-ethnic cohorts (84), and incorporating functional data into traditional GWAS (102). We also echo the call to increase the number of non-European studies and biobanks, multi-institute consortia and multisite studies that serve to increase genetic diversity (16,100,103). Finally, it is essential that efforts be made to perform large-scale GWAS in diverse populations across the globe, not merely cities in North America and Western Europe. The long-term persistence of a lack of diversity in GWAS could perpetuate disparities in outcomes.
Acknowledgments
Funding: Research reported in this publication was supported by NIH/NCI R01CA157823 (MED), RO1CA036401 (MED) University of Chicago Women’s Board and the University of Chicago Comprehensive Cancer Center P30 CA14599 (MED).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Howlader N, Noone A, Krapcho M, Garshell J, Miller D, Altekruse S, et al. SEER Cancer Statistics Review, 1975–2012. National Cancer Institute; Bethesda, MD: 2015. pp. 1–101. http://seer.cancer.gov/csr/1975_2012/, based on November 2014 SEER data submission, posted to the SEER web site. [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer. 2013;132:1133–45. doi: 10.1002/ijc.27711. [DOI] [PubMed] [Google Scholar]
- 4.American Cancer Society. Cancer Facts & Figures 2016. Cancer Facts Fig 2016. 2016:1–9. [Google Scholar]
- 5.Bradshaw PT, Stevens J, Khankari N, Teitelbaum SL, Neugut AI, Gammon MD. Cardiovascular Disease Mortality Among Breast Cancer Survivors. Epidemiology. 2016;27:6–13. doi: 10.1097/EDE.0000000000000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostoufi-Moab S, Seidel K, Leisenring WM, Armstrong GT, Oeffinger KC, Stovall M, et al. Endocrine Abnormalities in Aging Survivors of Childhood Cancer: A Report From the Childhood Cancer Survivor Study. J Clin Oncol. 2016;34:3240–7. doi: 10.1200/JCO.2016.66.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hudson MM, Ness KK, Gurney JG, Mulrooney Da, Chemaitilly W, Krull KR, et al. Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA. 2013;309:2371–81. doi: 10.1001/jama.2013.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips SM, Padgett LS, Leisenring WM, Stratton KK, Bishop K, Krull KR, et al. Survivors of childhood cancer in the United States: prevalence and burden of morbidity. Cancer Epidemiol Biomarkers Prev. 2015;24:653–63. doi: 10.1158/1055-9965.EPI-14-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ness KK, Hudson MM, Jones KE, Leisenring W, Yasui Y, Chen Y, et al. Effect of Temporal Changes in Therapeutic Exposure on Self-reported Health Status in Childhood Cancer Survivors. Ann Intern Med. 2016;166:89–98. doi: 10.7326/M16-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brock PR, Knight KR, Freyer DR, Campbell KCM, Steyger PS, Blakley BW, et al. Platinum-induced ototoxicity in children: A consensus review on mechanisms, predisposition, and protection, including a new International Society of Pediatric Oncology Boston ototoxicity scale. J Clin Oncol. 2012;30:2408–17. doi: 10.1200/JCO.2011.39.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Easton DF, Bishop DT, Ford D, Crockford GP, Haites N, Milner B, et al. Genetic linkage analysis in familial breast and ovarian cancer: Results from 214 families. Am J Hum Genet. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- 12.Peshkin BN, Alabek ML, Isaacs C. BRCA1/2 mutations and triple negative breast cancers. Breast Dis. 2010;32:25–33. doi: 10.3233/BD-2010-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schilsky RL. Personalized medicine in oncology: the future is now. Nat Rev Drug Discov. 2010;9:363–6. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 14.Rotimi CN, Jorde LB. N Engl J Med. Vol. 363. Massachusetts Medical Society; 2010. Ancestry and Disease in the Age of Genomic Medicine; pp. 1551–8. [DOI] [PubMed] [Google Scholar]
- 15.Bustamante CD, De La Vega FM, Burchard EG. Genomics for the world. Nature. 2011;475:163–5. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature England. 2016;538:161–4. doi: 10.1038/538161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daly AK. Genome-wide association studies in pharmacogenomics. Nat Rev Genet. 2010;11:241–6. doi: 10.1038/nrg2751. [DOI] [PubMed] [Google Scholar]
- 18.Colombel JF, Ferrari N, Debuysere H, Marteau P, Gendre JP, Bonaz B, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–30. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 19.Collie-Duguid ES, Pritchard SC, Powrie RH, Sludden J, Collier DA, Li T, et al. The frequency and distribution of thiopurine methyltransferase alleles in Caucasian and Asian populations. Pharmacogenetics. 1999;9:37–42. doi: 10.1097/00008571-199902000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Jung YS, Cheon JH, Park JJ, Moon CM, Kim ES, Lee JH, et al. Correlation of genotypes for thiopurine methyltransferase and inosine triphosphate pyrophosphatase with long-term clinical outcomes in Korean patients with inflammatory bowel diseases during treatment with thiopurine drugs. J Hum Genet. 2010;55:121–3. doi: 10.1038/jhg.2009.125. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Lopez E, Gutierrez-Camino A, Astigarraga I, Navajas A, Echebarria-Barona A, Garcia-Miguel P, et al. Vincristine pharmacokinetics pathway and neurotoxicity during early phases of treatment in pediatric acute lymphoblastic leukemia. Pharmacogenomics. 2016;17:731–41. doi: 10.2217/pgs-2016-0001. [DOI] [PubMed] [Google Scholar]
- 22.Ramsey LB, Bruun GH, Yang W, Treviño LR, Vattathil S, Scheet P, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22:1–8. doi: 10.1101/gr.129668.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider BP, Lai D, Shen F, Jiang G, Radovich M, Li L, et al. Charcot-Marie-Tooth gene, SBF2, associated with taxane-induced peripheral neuropathy in African-Americans. Oncotarget. 2016;7:82244–82253. doi: 10.18632/oncotarget.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daly AK. Genome-wide association studies in pharmacogenomics. Nat Rev Genet. 2010;11:241–6. doi: 10.1038/nrg2751. [DOI] [PubMed] [Google Scholar]
- 25.Tian C, Gregersen PK, Seldin MF. Accounting for ancestry: Population substructure and genome-wide association studies. Hum Mol Genet. 2008;17:R143–50. doi: 10.1093/hmg/ddn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Donnell PH, Dolan ME. Cancer pharmacoethnicity: Ethnic differences in susceptibility to the effects of chemotherapy. Clin Cancer Res. 2009;15:4806–14. doi: 10.1158/1078-0432.CCR-09-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Zhong B, Lun X, Lai Y, Bella aE, Yang W, et al. Specific Safety Profile of Bevacizumab in Asian Patients With Advanced NSCLC: A Meta-Analysis. Med. 2015;94:e975. doi: 10.1097/MD.0000000000000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polite BN, Sing A, Sargent DJ, Grothey A, Berlin J, Kozloff M, et al. Exploring racial differences in outcome and treatment for metastatic colorectal cancer: results from a large prospective observational cohort study (BRiTE) Cancer. 2012;118:1083–90. doi: 10.1002/cncr.26394. [DOI] [PubMed] [Google Scholar]
- 29.Schneider BP, Li L, Shen F, Miller KD, Radovich M, O’Neill A, et al. Br J Cancer. Vol. 111. Nature Publishing Group; 2014. Genetic variant predicts bevacizumab-induced hypertension in ECOG-5103 and ECOG-2100; pp. 1241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prandoni P, Lensing AWA, Piccioli A, Bernardi E, Simioni P, Girolami B, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood. 2002;100:3484–8. doi: 10.1182/blood-2002-01-0108. [DOI] [PubMed] [Google Scholar]
- 31.Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122:1712–23. doi: 10.1182/blood-2013-04-460121. [DOI] [PubMed] [Google Scholar]
- 32.Caine GJ, Stonelake PS, Lip GYH, Kehoe ST. The hypercoagulable state of malignancy: pathogenesis and current debate. Neoplasia. 2002;4:465–73. doi: 10.1038/sj.neo.7900263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rickles FR, Falanga A, Montesinos P, Sanz MA, Brenner B, Barbui T. Bleeding and thrombosis in acute leukemia: What does the future of therapy look like? Thromb Res. 2007;120:S99–106. doi: 10.1016/S0049-3848(07)70137-8. [DOI] [PubMed] [Google Scholar]
- 34.Chojnowski K, Wawrzyniak E, Treliński J, Niewiarowska J, Cierniewski C. Assessment of coagulation disorders in patients with acute leukemia before and after cytostatic treatment. Leuk Lymphoma. 1999;36:77–84. doi: 10.3109/10428199909145951. [DOI] [PubMed] [Google Scholar]
- 35.Zangari M, Fink LM, Elice F, Zhan F, Adcock DM, Tricot GJ. Thrombotic events in patients with cancer receiving antiangiogenesis agents. J Clin Oncol. 2009;27:4865–73. doi: 10.1200/JCO.2009.22.3875. [DOI] [PubMed] [Google Scholar]
- 36.Vu K, Luong NV, Hubbard J, Zalpour A, Faderl S, Thomas DA, et al. A retrospective study of venous thromboembolism in acute leukemia patients treated at the University of Texas MD Anderson Cancer Center. Cancer Med. 2014;4:27–35. doi: 10.1002/cam4.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer. 2007;110:2339–46. doi: 10.1002/cncr.23062. [DOI] [PubMed] [Google Scholar]
- 38.Brewer JR, Morrison G, Dolan ME, Fleming GF. Chemotherapy-induced peripheral neuropathy: Current status and progress. Gynecol Oncol. 2016;140:176–83. doi: 10.1016/j.ygyno.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider BP, Li L, Radovich M, Shen F, Miller KD, Flockhart DA, et al. Genome-wide association studies for taxane-induced peripheral neuropathy (TIPN) in ECOG-5103 and ECOG-1199. Clin Cancer Res. 2015;21:5082–91. doi: 10.1158/1078-0432.CCR-15-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egbelakin A, Ferguson MJ, MacGill EA, Lehmann AS, Topletz AR, Quinney SK, et al. Increased Risk of Vincristine Neurotoxicity Associated with Low CYP3A5 Expression Genotype in Children with Acute Lymphoblastic Leukemia. Pediatr Blood Cancer. 2011;56:361–7. doi: 10.1002/pbc.22845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Renbarger JL, McCammack KC, Rouse CE, Hall SD. Effect of race on vincristine-associated neurotoxicity in pediatric acute lymphoblastic leukemia patients. Pediatr Blood Cancer. 2008;50:769–71. doi: 10.1002/pbc.21435. [DOI] [PubMed] [Google Scholar]
- 42.Tamura T, Sasaki Y, Eguchi K, Shinkai T, Ohe Y, Nishio M, et al. Phase I and pharmacokinetic study of paclitaxel by 24-hour intravenous infusion. Japanese J Cancer Res. 1994;85:1057–62. doi: 10.1111/j.1349-7006.1994.tb02906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe A, Taniguchi M, Sasaki S. Induction chemotherapy with docetaxel, cisplatin, fluorouracil and l-leucovorin for locally advanced head and neck cancers: a modified regimen for Japanese patients. Anticancer Drugs. 2003;14:801–7. doi: 10.1097/00001813-200311000-00005. [DOI] [PubMed] [Google Scholar]
- 44.Takei Y, Suzuki M, Ohwada M, Saga Y, Kohno T, Machida S, et al. A feasibility study of paclitaxel and carboplatin therapy in Japanese patients with epithelial ovarian cancer. Oncol Rep. 2003;10:951–5. [PubMed] [Google Scholar]
- 45.Okano S, Tahara M, Zenda S, Fuse N, Yoshino T, Doi T, et al. Induction chemotherapy with docetaxel, cisplatin and s-1 followed by proton beam therapy concurrent with cisplatin in patients with t4b nasal and sinonasal malignancies. Jpn J Clin Oncol. 2012;42:691–6. doi: 10.1093/jjco/hys096. [DOI] [PubMed] [Google Scholar]
- 46.Millward MJ, Boyer MJ, Lehnert M, Clarke S, Rischin D, Goh B-C, et al. Docetaxel and carboplatin is an active regimen in advanced non-small-cell lung cancer: a phase II study in Caucasian and Asian patients. Ann Oncol Off J Eur Soc Med Oncol England. 2003;14:449–54. doi: 10.1093/annonc/mdg118. [DOI] [PubMed] [Google Scholar]
- 47.Han HS, Reis IM, Zhao W, Kuroi K, Toi M, Suzuki E, et al. Racial differences in acute toxicities of neoadjuvant or adjuvant chemotherapy in patients with early-stage breast cancer. Eur J Cancer. 2011;47:2537–45. doi: 10.1016/j.ejca.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 48.Frisina RD, Wheeler HE, Fossa SD, Kerns SL, Fung C, Sesso HD, et al. Comprehensive audiometric analysis of hearing impairment and tinnitus after cisplatin-based chemotherapy in survivors of adult-onset cancer. J Clin Oncol. 2016;34:2712–20. doi: 10.1200/JCO.2016.66.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eiamprapai P, Yamamoto N, Hiraumi H, Ogino-Nishimura E, Kitamura M, Hirano S, et al. Effect of cisplatin on distortion product otoacoustic emissions in Japanese patients. Laryngoscope. 2012;122:1392–6. doi: 10.1002/lary.23336. [DOI] [PubMed] [Google Scholar]
- 50.Whitehorn H, Sibanda M, Lacerda M, Spracklen T, Ramma L, Dalvie S, et al. High prevalence of cisplatin-induced ototoxicity in Cape Town, South Africa. S Afr Med J. 2014;104:288–91. doi: 10.7196/samj.7389. [DOI] [PubMed] [Google Scholar]
- 51.Hertz DL, McLeod HL, McLeod H, Hertz D, McLeod H, Collins F, et al. Using pharmacogene polymorphism panels to detect germline pharmacodynamic markers in oncology. Clin Cancer Res. 2014;20:2530–40. doi: 10.1158/1078-0432.CCR-13-2780. [DOI] [PubMed] [Google Scholar]
- 52.Kim HS, Cheon JH, Jung ES, Park J, Aum S, Park SJ, et al. A coding variant in FTO confers susceptibility to thiopurine-induced leukopenia in East Asian patients with IBD. Gut. 2016 doi: 10.1136/gutjnl-2016-311921. gutjnl-2016-311921. [DOI] [PubMed] [Google Scholar]
- 53.Cao S, Wang S, Ma H, Tang S, Sun C, Dai J, et al. Genome-wide association study of myelosuppression in non-small-cell lung cancer patients with platinum-based chemotherapy. Pharmacogenomics J. 2016;16:41–6. doi: 10.1038/tpj.2015.22. [DOI] [PubMed] [Google Scholar]
- 54.Zabala W, Cruz R, Barreiro-de Acosta M, Chaparro M, Panes J, Echarri A, Esteve M, et al. New genetic associations in thiopurine-related bone marrow toxicity among inflammatory bowel disease patients. Pharmacogenomics. 2013;14:631–40. doi: 10.2217/pgs.13.38. [DOI] [PubMed] [Google Scholar]
- 55.Fernandez-Rozadilla C, Cazier JB, Moreno V, Crous-Bou M, Guinó E, Durán G, et al. Pharmacogenomics in colorectal cancer: a genome-wide association study to predict toxicity after 5-fluorouracil or FOLFOX administration. Pharmacogenomics J. 2012:209–17. doi: 10.1038/tpj.2012.2. [DOI] [PubMed] [Google Scholar]
- 56.Ramsey LB, Panetta JC, Smith C, Yang W, Fan Y, Winick NJ, et al. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood. 2013;121:898–904. doi: 10.1182/blood-2012-08-452839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baldwin RM, Owzar K, Zembutsu H, Chhibber A, Kubo M, Jiang C, et al. A genome-wide association study identifies novel loci for paclitaxel-induced sensory peripheral neuropathy in CALGB 40101. Clin Cancer Res. 2012;18:5099–109. doi: 10.1158/1078-0432.CCR-12-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hertz DL, Owzar K, Lessans S, Wing C, Jiang C, Kelly WK, et al. Pharmacogenetic discovery in CALGB (alliance) 90401 and mechanistic validation of a VAC14 polymorphism that increases risk of docetaxel-induced neuropathy. Clin Cancer Res. 2016;22:4890–4900. doi: 10.1158/1078-0432.CCR-15-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abraham JE, Guo Q, Dorling L, Tyrer J, Ingle S, Hardy R, et al. Replication of genetic polymorphisms reported to be associated with taxane-related sensory neuropathy in patients with early breast cancer treated with paclitaxel. Clin Cancer Res. 2014;20:2466–75. doi: 10.1158/1078-0432.CCR-13-3232. [DOI] [PubMed] [Google Scholar]
- 60.Leandro-García LJ, Inglada-Pérez L, Pita G, Hjerpe E, Leskelä S, Jara C, et al. Genome-wide association study identifies ephrin type A receptors implicated in paclitaxel induced peripheral sensory neuropathy. J Med Genet. 2013;50:599–605. doi: 10.1136/jmedgenet-2012-101466. [DOI] [PubMed] [Google Scholar]
- 61.Cao S, Wang C, Ma H, Yin R, Zhu M, Shen W, et al. Genome-wide Association Study on Platinum-induced Hepatotoxicity in Non-Small Cell Lung Cancer Patients. Sci Rep. 2015;5:11556. doi: 10.1038/srep11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu H, Robinson GW, Huang J, Lim JY-S, Zhang H, Bass JK, et al. Common variants in ACYP2 influence susceptibility to cisplatin-induced hearing loss. Nat Genet. 2015;47:263–6. doi: 10.1038/ng.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Low SK, Chung S, Takahashi A, Zembutsu H, Mushiroda T, Kubo M, et al. Genome-wide association study of chemotherapeutic agent-induced severe neutropenia/leucopenia for patients in Biobank Japan. Cancer Sci. 2013;104:1074–82. doi: 10.1111/cas.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aminkeng F, Bhavsar AP, Visscher H, Rassekh SR, Li Y, Lee JW, et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet. 2015;47:1079–84. doi: 10.1038/ng.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karol SE, Yang W, Van Driest SL, Chang TY, Kaste S, Bowton E, et al. Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2015;126:1770–6. doi: 10.1182/blood-2015-05-643601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heap Ga, Weedon MN, Bewshea CM, Singh A, Chen M, Satchwell JB, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46:1131–4. doi: 10.1038/ng.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang JJ, Landier W, Yang W, Liu C, Hageman L, Cheng C, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33:1235–42. doi: 10.1200/JCO.2014.59.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Treviño LR, Shimasaki N, Yang W, Panetta JC, Cheng C, Pei D, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27:5972–8. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parham LR, Briley LP, Li L, Shen J, Newcombe PJ, King KS, et al. Comprehensive genome-wide evaluation of lapatinib-induced liver injury yields a single genetic signal centered on known risk allele HLA-DRB1*07:01. Pharmacogenomics J. 2016;16:180–5. doi: 10.1038/tpj.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Srinivasan Y, Sasa M, Honda J, Takahashi A, Uno S, Kamatani N, et al. Genome-wide association study of epirubicin-induced leukopenia in Japanese patients. Pharmacogenet Genomics. 2011;21:552–8. doi: 10.1097/FPC.0b013e328348e48f. [DOI] [PubMed] [Google Scholar]
- 71.Spraggs CF, Budde LR, Briley LP, Bing N, Cox CJ, King KS, et al. HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol. 2011;29:667–73. doi: 10.1200/JCO.2010.31.3197. [DOI] [PubMed] [Google Scholar]
- 72.Coleman EA, Lee JY, Erickson SW, Goodwin JA, Sanathkumar N, Raj VR, et al. GWAS of 972 autologous stem cell recipients with multiple myeloma identifies 11 genetic variants associated with chemotherapy-induced oral mucositis. Support Care Cancer. 2014;23:841–9. doi: 10.1007/s00520-014-2406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Won HH, Lee J, Park JO, Park YS, Lim HY, Kang WK, et al. Polymorphic markers associated with severe oxaliplatin-induced, chronic peripheral neuropathy in colon cancer patients. Cancer. 2012;118:2828–36. doi: 10.1002/cncr.26614. [DOI] [PubMed] [Google Scholar]
- 74.Diouf B, Crews KR, Lew G, Pei D, Cheng C, Bao J, et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. Jama. 2015;313:815–23. doi: 10.1001/jama.2015.0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wheeler HE, Gamazon ER, Frisina R, Perez-Cervantes C, El Charif O, Mapes B, et al. Variants in WFS1 and other Mendelian deafness genes are associated with cisplatin-associated ototoxicity. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-2809. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Magrangeas F, Kuiper R, Avet-Loiseau H, Gourraud W, Guerin-Charbonnel C, Ferrer L, et al. A Genome-Wide Association Study Identifies a Novel Locus for Bortezomib-Induced Peripheral Neuropathy in European Multiple Myeloma Patients. Clin Cancer Res. 2016;22:4350–5. doi: 10.1158/1078-0432.CCR-15-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu C, Yang W, Devidas M, Cheng C, Pei D, Smith C, et al. Clinical and Genetic Risk Factors for Acute Pancreatitis in Patients With Acute Lymphoblastic Leukemia. J Clin Oncol. 2016;34:2133–40. doi: 10.1200/JCO.2015.64.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Panofsky A, Bliss C. Am Sociol Rev. Vol. 82. SAGE Publications; 2017. Ambiguity and Scientific Authority; pp. 59–87. [Google Scholar]
- 79.Henn BM, Gravel S, Moreno-Estrada A, Acevedo-Acevedo S, Bustamante CD. Fine-scale population structure and the era of next-generation sequencing. Hum Mol Genet. 2010;19:R221–6. doi: 10.1093/hmg/ddq403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Henn BM, Botigué LR, Bustamante CD, Clark AG, Gravel S. Estimating Mutation Load in Human Genomes. Nat Rev Genet. 2015;16:333–43. doi: 10.1038/nrg3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adeyemo A, Rotimi C. Genetic variants associated with complex human diseases show wide variation across multiple populations. Public Health Genomics. 2010;13:72–9. doi: 10.1159/000218711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dressler LG, Deal AM, Owzar K, Watson D, Donahue K, Friedman PN, et al. Participation in Cancer Pharmacogenomic Studies: A Study of 8456 Patients Registered to Clinical Trials in the Cancer and Leukemia Group B (Alliance) J Natl Cancer Inst. 2015;107:dvj188. doi: 10.1093/jnci/djv188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Campbell MC, Tishkoff SA. AFRICAN GENETIC DIVERSITY: Implications for Human Demographic History, Modern Human Origins, and Complex Disease Mapping. Annu Rev Genomics Hum Genet. 2008;9:403–33. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Asimit JL, Hatzikotoulas K, Mccarthy M, Morris AP, Zeggini E. Trans-ethnic study design approaches for fine-mapping. Eur J Hum Genet. 2016;24:1330–6. doi: 10.1038/ejhg.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. The genetic ancestry of african americans, latinos, and european Americans across the United States. Am J Hum Genet. 2015;96:37–53. doi: 10.1016/j.ajhg.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ziv E, Burchard EG. Human population structure and genetic association studies. Pharmacogenomics. 2003;4:431–41. doi: 10.1517/phgs.4.4.431.22758. [DOI] [PubMed] [Google Scholar]
- 87.Tiwari HK, Barnholtz-Sloan J, Wineinger N, Padilla MA, Vaughan LK, Allison DB. Review and evaluation of methods correcting for population stratification with a focus on underlying statistical principles. Hum Hered. 2008;66:67–86. doi: 10.1159/000119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu EY, Li M, Wang W, Li Y. MaCH-Admix: Genotype Imputation for Admixed Populations. Genet Epidemiol. 2013;37:25–37. doi: 10.1002/gepi.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pulit SL, Voight B, de Bakker PIW. Multiethnic genetic association studies improve power for locus discovery. PLoS One. 2010;5:1–9. doi: 10.1371/journal.pone.0012600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Medina-Gomez C, Felix JF, Estrada K, Peters MJ, Herrera L, Kruithof CJ, et al. Challenges in conducting genome-wide association studies in highly admixed multi-ethnic populations: the Generation R Study. Eur J Epidemiol. 2015;30:317–30. doi: 10.1007/s10654-015-9998-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.POPULATION REFERENCE BUREAU. World Population Data Sheet. World Popul Data Sheet. 2015 2015. [Google Scholar]
- 94.Maranville JC, Cox NJ. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J. 2016;16:388–92. doi: 10.1038/tpj.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spencer CCA, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: Sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477. doi: 10.1371/journal.pgen.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hong EPP, Park JWW. Sample size and statistical power calculation in genetic association studies. Genomics Inform. 2012;10:117–22. doi: 10.5808/GI.2012.10.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15:335–46. doi: 10.1038/nrg3706. [DOI] [PubMed] [Google Scholar]
- 98.Group NW. Replicating genotype–phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 99.Bush WS, Moore JH. Chapter 11: Genome-Wide Association Studies. PLoS Comput Biol. 2012;8:e1002822. doi: 10.1371/journal.pcbi.1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haga SB. Impact of limited population diversity of genome-wide association studies. Genet Med. 2010;12:81–4. doi: 10.1097/GIM.0b013e3181ca2bbf. [DOI] [PubMed] [Google Scholar]
- 101.Wheeler HE, Gorsic LK, Welsh M, Stark AL, Gamazon ER, Cox NJ, et al. Genome-wide local ancestry approach identifies genes and variants associated with chemotherapeutic susceptibility in African Americans. PLoS One. 2011;6:e21920. doi: 10.1371/journal.pone.0021920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet. 2014;94:559–73. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011;475:163–5. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]