

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a common kidney disease caused by mutations in PKD1 or PKD2. Metformin reduces cyst growth in mouse models of PKD1. However, metformin has not been studied in animal models of PKD2, and the cellular mechanism underlying its effectiveness is not entirely clear. This study investigated the effects of metformin on cyst formation in a zebrafish model of polycystin-2 deficiency resulting from morpholino knockdown of pkd2. We added metformin (2.5 to 20 mM) to the embryo media between 4 and 48 hours post fertilisation and observed pronephric cyst formation by using the wt1b promoter-driven GFP signal in Tg(wt1b:GFP) pkd2 morphants. Metformin inhibited pronephric cyst formation by 42–61% compared with the untreated controls. Metformin also reduced the number of proliferating cells in the pronephric ducts, the degree of dorsal body curvature, and the infiltration of leukocytes surrounding the pronephros. Moreover, metformin treatment increased the phosphorylation of adenosine monophosphate-activated protein kinase (AMPK) and enhanced autophagy in the pronephros. Our data suggest that metformin reduces cyst formation through activation of the AMPK pathway and modulation of defective cellular events such as proliferation and autophagy. These results also imply that metformin could have therapeutic potential for ADPKD treatment.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic kidney disease resulting from mutations of PKD1 or PKD2 1. Affected patients may gradually lose their renal function because of progressive cyst formation in the kidneys; ADPKD accounts for 5% to 10% of cases of end-stage kidney disease (ESKD)2. Extrarenal presentations, such as liver cysts, cerebral aneurysms, and colon diverticulosis, can also cause serious complications. Patients with PKD2 mutations have a milder disease course than do those with PKD1 mutations. The ages at which patients with PKD1 and PKD2 mutations develop ESKD differ by almost 20 years3.
Polycystin-1 (encoded by PKD1) is a 4302-amino-acid membrane glycoprotein that is responsible for cell–cell or cell–matrix interactions critical for maintaining the renal tubular morphology4. Polycystin-2 (encoded by PKD2) is a 968-amino-acid cation ion channel involved in the regulation of intracellular calcium homeostasis5, 6. Polycystin-1 and polycystin-2 can act synergistically or independently at the cell membrane, endoplasmic reticulum, and cilia, regulating cell proliferation, fluid secretion, extracellular matrix mechanics, and ciliary functions7.
The mechanism of cyst formation in ADPKD remains incompletely understood. Previous studies have shown that cyclic adenosine monophosphate (cAMP) and mammalian target of rapamycin (mTOR) are the major pathways involved in cell proliferation and fluid secretion in ADPKD8. Recent studies have suggested that enhanced aerobic glycolysis and defective autophagy in ADPKD cells may contribute to its hyperproliferative phenotype9. Furthermore, experimental evidence suggests that inflammation and macrophage infiltration have an additional role in promoting cystogenesis10. These novel discoveries of disease mechanisms have led to the investigation of many targeted therapies11–13. However, no treatment for ADPKD approved by the US Federal Drug Administration is currently available. A possible strategy to accelerate the discovery of new treatment is identifying new uses of existing drugs and repurposing them accordingly.
Metformin, an approved biguanide derivative, has been used for treating type 2 diabetes mellitus for decades14, 15. Previous studies have shown that metformin inhibits gluconeogenesis through the activation of liver kinase B1 and adenosine monophosphate-activated protein kinase (AMPK)16, 17. In recent years, the therapeutic potential of metformin in cancer and polycystic ovary disease has drawn increasing attention18, 19. For example, data from retrospective studies indicate that patients treated with metformin have a lower risk of prostate cancer and other urologic malignancies20, suggesting that metformin could have clinical applications in addition to glycemic control in type 2 diabetes mellitus21.
MacCarty et al. hypothesised that metformin can reduce cyst formation on the basis of its abilities to activate AMPK and suppress cystic fibrosis transmembrane conductance regulator (CFTR) and mTOR22. Accordingly, a previous study demonstrated that metformin inhibited cyst growth in Madin–Darby canine kidney (MDCK) cells and Pkd1 conditional knockout mice23. However, the exact in vivo mechanisms underlying the effect of metformin on cystogenesis are not entirely understood. Furthermore, recent studies have suggested that polycystin-1-deficent and polycystin-2-deficent cells might be different in the degree of AMPK inhibition24. Hence, whether metformin can inhibit cyst growth in the setting of polycystin-2 deficiency and the possible mechanisms of its action remain to be determined.
In this study, we investigated the effects of metformin on the initiation of pronephric cysts using a zebrafish pkd2 model. We observed that metformin reduces pronephric cyst formation through the activation of AMPK and restores autophagy activity. These findings indicate that metformin could play a role in the treatment of early-stage ADPKD.
Results
Metformin prevents cyst initiation in pkd2 morphants
In this study, we used a zebrafish model of PKD2 for evaluating the effects of metformin on cyst initiation25–29. A translation-blocking morpholino oligos (MO) against pkd2 was injected in a transgenic line, in which green fluorescent protein (GFP) expression was driven by the pronephros-specific wt1b promoter. We treated the Tg(wt1b:GFP) pkd2 morphants with different concentrations of metformin in the E3 buffer between 4 and 48 hours post fertilisation (hpf). As illustrated in Fig. 1A, metformin treatment resulted in a significant reduction (42% to 61%) of cyst formation in the Tg(wt1b:GFP) pkd2 morphants compared with untreated controls at 48 hpf (P < 0.01). The frequency of glomerular cyst formation decreased dose-dependently from 71.4% ± 4.6% in the untreated pkd2 morphants to 41.5% ± 6.8% with 2.5 mM metformin and 33.4% ± 5.1%, 28.2% ± 4.6%, and 31.8% ± 4.5% with 5 mM, 10 mM, and 20 mM metformin, respectively (Fig. 1B). In transverse histological sections, we confirmed the rescue effect of metformin treatment on the pronephric cysts in the pkd2 morphants (Fig. 1C). These results indicate that metformin could inhibit the early stage of cyst formation in the zebrafish model of PKD2.
Figure 1.
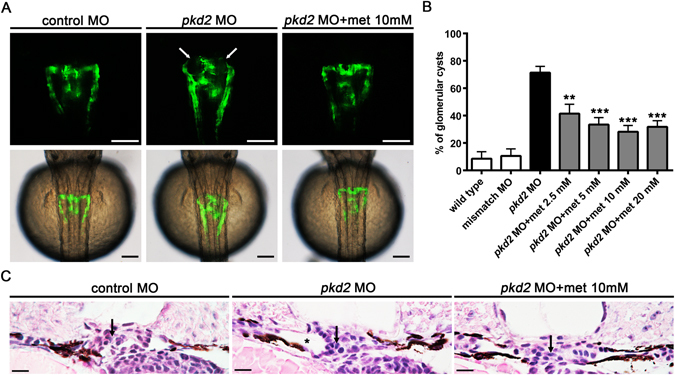
Metformin inhibits pronephric cyst formation in pkd2 morphants. Tg(wt1b:GFP) pkd2 morphants were immersed in embryo media (E3) supplemented with 2.5, 5, 10, or 20 mM metformin until 48 hpf. (A) Representative images of the pronephros in the control embryos, pkd2 morphants, and metformin-treated pkd2 morphants are shown. Note the pronephric cysts (arrows) in the Tg(wt1b:GFP) pkd2 morphant. Photographs from in vivo observation through fluorescence microscopy (dorsal view, anterior to the top) were recorded at 48 hpf. The lower panels show overlays of transmission (grey) and fluorescence (green) images of the upper panels. Scale bar, 100 µm. (B) Comparative frequencies of the cystic phenotype in the control embryos and pkd2 morphants treated with different concentrations of metformin as indicated (n = 45–99 per group). Data represent five independent experiments. **P < 0.01, ***P < 0.001 compared with pkd2 knockdown. (C) Transverse histological sections of the embryos at 48 hpf, illustrating the pronephric cyst (indicated by the mark *) in a pkd2 morphant and the rescue effect of metformin treatment. Arrows indicate the position of glomeruli. H&E stain, scale bar, 10 µm.
Metformin reduces dorsal body curvature and cloaca malformation in pkd2 morphants
We examined the effect of metformin on the curvature phenotype of pkd2 morphants (Fig. 2A–C). Dorsal axis curvature results from an overproduction of type II collagen in the notochord sheath and has been used as a surrogate readout for cystogenesis in pkd2 morphants26. As illustrated in Fig. 2G, metformin partially reduced the frequencies of the overall curvature and severe dorsal curvature (>90°) phenotypes by 14% (114/116 vs. 81/96, P < 0.001) and 15% (67/116 vs. 41/96, P < 0.05), respectively, compared with untreated controls. These data indicate that metformin also ameliorated the defects in extracellular matrix formation involved in the pathogenesis of ADPKD. For comparison, we treated pkd1a/b morphants with 10 mM metformin and the curvature phenotype was also partially suppressed, similar to that observed in pkd2 morphants (Supplementary Fig. S1). However, the small percentages of pronephric cysts in the pkd1a/b morphants preclude us from further studies using this model30.
Figure 2.
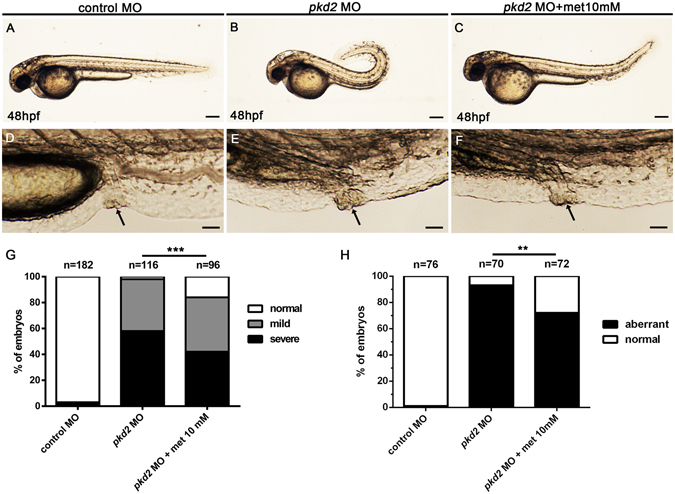
Metformin ameliorates dorsal axis curvature and cloaca malformation in pkd2 morphants. Zebrafish embryos were treated with metformin (10 mM) in supplemented E3 media. (A–C) Representative images showing the common body axis of a control embryo, the severe dorsal curvature of a pkd2 morphant, and the mild dorsal curvature of a metformin-treated pkd2 morphant. Scale bar, 200 µm. (D–F) Representative images showing the cloaca of a control embryo, the cloaca malformation of a pkd2 morphant, and the normal cloaca of a metformin-treated pkd2 morphant. Scale bar, 50 µm. (G) Comparative frequencies of the dorsal curvature phenotype in each group (n = 182, 116, and 96 per group). Data represent four independent experiments. ***P < 0.001 compared with pkd2 morphants. (H) Comparative frequencies of the aberrant cloaca phenotype in each group (n = 76, 70, and 72 per group). Data represent three independent experiments. **P < 0.01 compared with pkd2 morphants.
The cloaca malformation has been shown to correlate the formation of pronephric cysts in several zebrafish models of ciliopathies6, 31–33. In agreement with these studies, we found that the frequency of aberrant cloaca increased in pkd2 morphants and metformin significantly improved the phenotype by 21% (65/70 vs. 52/72, P < 0.01) (Fig. 2D–F,H). These data indicate that metformin consistently suppressed the different phenotypes of pkd2 morphants. However, whether the improvement in cloacal morphology can lead to increased urine flow in metformin-treated pkd2 morphants requires further study of kidney function using rhodamine-dextran filtration assays33–36.
Metformin reduces tubular cell proliferation
We next sought to determine whether metformin affects cell proliferation in the pronephric kidney. We performed double immunostaining using antibodies against phosphohistone H3 (PH3) and the Na/K-ATPase α-1 subunit (α6 F) to mark proliferating cells in the pronephric ducts (Fig. 3A). Significantly fewer proliferating cells were observed in the pronephric ducts of the pkd2 morphants treated with metformin than in those of the untreated pkd2 morphants (Fig. 3B). These data suggest that metformin reduced pronephric cyst formation in the pkd2 morphants through inhibition of epithelial cell proliferation.
Figure 3.

Metformin suppresses pronephric epithelial proliferation in pkd2 morphants. (A) Representative confocal immunofluorescence images showing proliferative cells (arrows) in pkd2 morphants with and without metformin (10 mM) treatment. Embryos were stained using anti-PH3 (red) to mark proliferating cells and anti-α6 F (green) to label the pronephric ducts at 48 hpf. (B) Counts of anti-PH3- and anti-α6F-positive cells in the anterior and posterior pronephric ducts (n = 14–17 per group). *P < 0.05, ***P < 0.001. Data represent two independent experiments. Scale bar, 20 µm.
Metformin reduces leukocyte accumulation
Previous studies have revealed that inflammation contributes to cell proliferation and cyst formation in ADPKD10. To explore the mechanisms by which metformin inhibits cyst growth, we analysed the effect of metformin on leukocyte infiltration in the pkd2 morphants. Whole-mount in situ hybridisation revealed significantly increased l-plastin and mpx expression in the trunk area surrounding the pronephric ducts in the pkd2 morphants compared with mismatched MO controls (Fig. 4A). The pan-leukocyte marker l-plastin is an actin-binding protein preferentially expressed in monocytes/macrophages and mpx is a neutrophil marker37. Metformin treatment caused a significant reduction of leukocyte infiltration in the pronephric area in the pkd2 morphants (Fig. 4B and C). These data indicate that metformin could have an additional anti-inflammatory role that contributes to its therapeutic effects on polycystic kidney disease.
Figure 4.
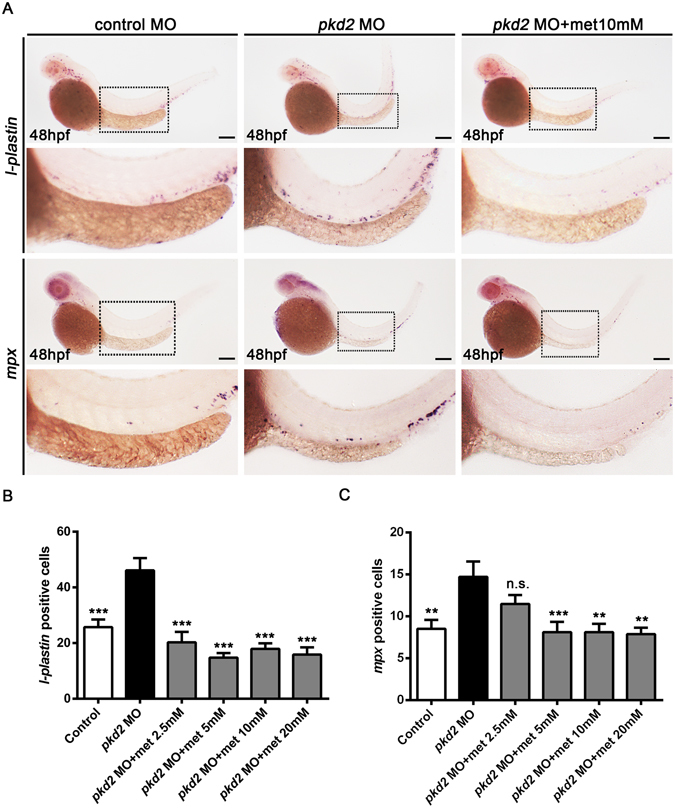
Metformin reduces leukocyte infiltration in pkd2 morphants. (A) Representative images of l-plastin-positive and mpx-positive cells surrounding the pronephric ducts in a control MO-injected embryo, a pkd2 morphant, and a pkd2 morphant treated with metformin (10 mM). Leukocytes were labelled using in situ hybridisation for l-plastin and mpx at 48 hpf. The lower panel shows an enlarged view of the dashed box indicated in the upper image. Scale bar, 200 µm. (B) Counts of the l-plastin-positive cells (within the region of yolk extension as indicated by the dashed box) in the controls and metformin-treated pkd2 morphants (2.5 mM to 20 mM). ***P < 0.001 compared with pkd2 morphants. Data represent two independent experiments, n = 7–11 per group. (C) Counts of the mpx-positive cells. **P < 0.01, ***P < 0.001 compared with pkd2 morphants. n.s. = not significant. Data represent two independent experiments, n = 16–20 per group.
Activation of AMPK by metformin
A previous study revealed that Pkd1 −/− mouse embryonic fibroblasts exhibit a lower level of AMPK phosphorylation than do wild-type cells9. Metformin has been shown to stimulate AMPK in cell and mouse models of ADPKD23, 24. Therefore, we evaluated the effect of metformin on the activation of the AMPK signalling pathway. Western blot analysis using an antibody against p-AMPKα at Thr172 indicated that the p-AMPK/AMPK ratio was significantly higher in the metformin-treated pkd2 morphants than in the untreated controls (Fig. 5A and B, Supplementary Fig. S2). These data are consistent with the hypothesis that AMPK activation inhibits polycystic kidney disease. However, we did not observe a lower baseline expression level of p-AMPK in the pkd2 morphants than in the wild-type controls; this effect could be partly due to the presence of residual maternal pAMPK in the zebrafish embryos38.
Figure 5.

Metformin increases AMPK levels in pkd2 morphants. (A) Representative Western blots for phospho-Thr172 AMPK, total AMPK, and β-actin in total embryo lysates from mismatch MO-injected controls and pkd2 morphants with and without metformin (10 mM) treatment at 48 hpf. (B) Quantification of p-AMPK relative to total AMPK through densitometric analysis of Western blots; n = 9 from five independent experiments. *P < 0.05, n.s. = not significant. Cropped blot images are shown, and all the blots were run under the same experimental conditions. Uncropped blots are shown in Supplementary Fig. S2.
Metformin cannot prevent cyst formation in tsc1a morphants
A study indicated that the AMPK-dependent activation of tuberous sclerosis complex (TSC) 1 and 2 proteins plays a major role in the inhibition of the mTOR signalling pathway during energetic demands39. Therefore, we tested whether metformin prevented cyst initiation in the absence of TSC proteins. Morpholino knockdown of tsc1a induced severe glomerular cysts in zebrafish embryos at 48 hpf, as reported previously (Fig. 6A)40. Metformin apparently did not affect cyst formation in the tsc1a morphants (Fig. 6B). This observation is consistent with an AMPK/TSC-dependent mechanism underlying the prevention of cyst formation by metformin.
Figure 6.

Metformin cannot rescue the cystic phenotype in tsc1a morphants. Tg(wt1b:GFP) tsc1a morphants were immersed in embryo media (E3) containing 2.5 to 20 mM metformin until 48 hpf. (A) Representative images indicating prominent cystic pronephros (arrows) in Tg(wt1b:GFP) tsc1a morphants. Metformin does not affect the formation and severity of pronephric cysts. The upper panels show fluorescence images (dorsal view, anterior to the top). The lower panels show overlays of fluorescence (green) and transmission (grey) images of the upper panels. Scale bar, 100 µm. (B) Comparison of the cystic phenotype in different treatment groups. Data represent two independent experiments (n = 28–46 per group). ***P < 0.001, n.s. = not significant.
Metformin enhances autophagy in the pronephros
Defective autophagy, an AMPK downstream cellular process that degrades cytoplasmic components for energy production, has been associated with the pathogenesis of ADPKD in recent studies41. Therefore, we examined whether metformin increased the autophagy activities in pkd2 morphants. Autophagy activity was assessed by detecting the intracellular levels of microtubule-associated protein light chain 3 (LC3) through immunofluorescence and Western blot analysis42. As illustrated in Fig. 7A, the pkd2 morphants exhibited lower LC3 staining in the pronephric ducts than did the control embryos, and the deficiency was rectified by metformin treatment. Consistent with these findings, the Western blot analysis indicated that the conversion of cytoplasmic LC3-I into LC3-II was significantly lower in the pkd2 morphants than in the control embryos, and metformin treatment restored the autophagy activities (Fig. 7B and Supplementary Fig. S2). Furthermore, we observed that simultaneous knockdown of the autophagy gene atg5 significantly increased the cystic phenotype in the pkd2 morphants (Fig. 7C and D). Although the magnitudes of these differences were moderate and further studies are needed to confirm our findings, these results suggest that the activation of AMPK and restoration of autophagy constitute a plausible mechanism for the inhibition of cyst growth by metformin43.
Figure 7.
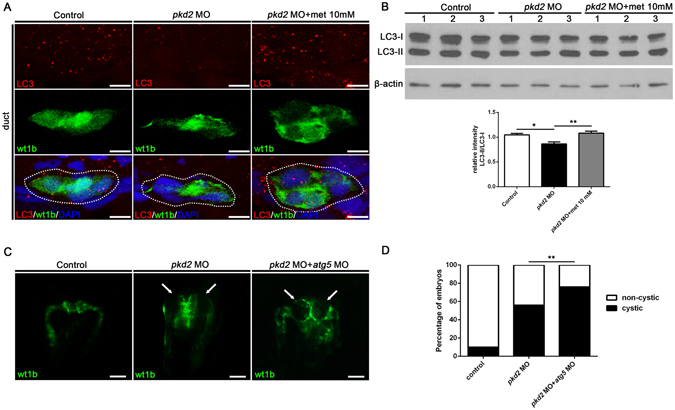
Metformin enhances autophagy in Tg(wt1b:GFP) pkd2 morphants. (A) Representative confocal images of pronephric ducts (green, wt1b:GFP) in control MO-injected embryos, pkd2 morphants, and pkd2 morphants treated with metformin (10 mM) at 48 hpf (n = 4 per group). Transverse cryosections were stained for LC3 (red), and the nuclei were counterstained with DAPI (blue). Note the reduced expression of LC3 (orange dots, red and green merged) in the pkd2 morphant and the rescue effect of metformin treatment. The dotted circle indicates the pronephric duct. Scale bar, 10 µm. (B) Quantification of the expression of LC3-II relative to LC3-I through Western blot analysis of total embryo lysates. ß-actin was used as a loading control; n = 8 from four independent experiments. *P < 0.05 compared with control; **P < 0.01 compared with untreated pkd2 morphants. The uncropped blots are shown in Supplementary Fig. S2. (C) A combined knockdown of atg5 and pkd2 increased the frequency of cystic phenotype in the Tg(wt1b:GFP) pkd2 morphants. Representative images of the pronephros in the control embryos, pkd2 morphants, and atg5/pkd2 double morphants are shown. Arrows indicate the pronephric cysts. (D) Comparison of the cystic phenotype in different groups. Data represent four independent experiments (n = 81, 86, and 87 per group). **P < 0.01.
Discussion
The zebrafish has become an increasingly recognised model for identifying drug candidates for ADPKD treatment44–47. Chemical compound screening in zebrafish pkd2 models indicated that histone deacetylase inhibitors inhibited cyst growth48. Candidate drugs that are effective in other PKD animal models, including rapamycin, roscovitine, and pasireotide, have been successfully validated using the zebrafish system34, 49. In the current study, we demonstrated that metformin activated the phosphorylation of AMPK and prevented cyst formation in a zebrafish pkd2 model. In particular, we demonstrated that metformin inhibited epithelial cell proliferation and restored normal autophagy activity in the pronephric kidney. Furthermore, we demonstrated that metformin did not prevent cyst formation in tsc1a morphants resulting from knocking down the AMPK downstream target TSC1.
Metformin was shown to suppress cyst growth and fluid secretion through the inhibition of mTOR and CFTR in a previous study using MDCK cell cysts, mouse embryonic kidney explant cultures, and Pkd1 mouse models23. In accordance with this study, we found that metformin inhibited early cystogenesis in zebrafish pkd2 morphant embryos, indicating that metformin can inhibit cyst growth in both PKD1 and PKD2 animal models. These results support the hypothesis that metformin reduces cyst formation in the early stages of ADPKD. Interestingly, a previous study suggests that polycystin-2 deficient cell lines are less amenable to metformin and rapamycin than polycystin-1 deficient cell lines due to the less activation of mTOR pathway24. However, we observed a similar suppressive effect of metformin on the curvature phenotype in both pkd1a/b and pkd2 morphants. The discrepancy could be explained by the differences between in vivo and in vitro cystic models, or metformin could have pleiotropic effects on multiple signalling pathways related to the PKD2 deficiency. Further studies will be required to clarify our findings.
One possible mechanism by which metformin prevents cyst formation is through the activation of AMPK and inhibition of cell proliferation. The AMPK signalling pathway plays a crucial role in maintaining normal kidney structure during nephron morphogenesis by affecting cell proliferation and migration50, 51. Activated AMPK restores the cell energy balance by shutting down the ATP-consuming synthesis pathways, and thus causes cells to switch from an anabolic to a catabolic state18. Metformin reduced cell proliferation in an AMPK-dependent manner in MDCK cysts grown in 3D collagen gels23. Similarly, intraperitoneal injection of metformin resulted in a reduction in the number of Ki67-positive epithelial cells in the cystic kidneys of Pkd1 flox/− Ksp-Cre mice at postnatal day 723. Furthermore, forced activation of AMPK by 2-deoxyglucose (2DG) restored normal extracellular signal-regulated kinase (ERK) activity, inhibited glycolysis, and reduced the cystic index and proliferation rate in Pkd1 conditional knockout mice9. Our findings are consistent with these results and suggest that metformin may inhibit cystogenesis through an AMPK-dependent pathway.
Growing evidence substantiates the role of macrophages in promoting cyst growth in ADPKD. Macrophages were demonstrated to stimulate cell proliferation and cyst expansion in coculture experiments within a collagen matrix52. The recruitment and retention of renal macrophages contributes to the increased proliferation during cyst growth in ADPKD10. A previous study showed that metformin treatment reduced inflammatory cytokine production in peripheral blood mononuclear cells obtained from healthy volunteers53. Metformin also suppressed inflammation through activation of AMPK and phosphatase and tensin homolog (PTEN) in vascular smooth muscle cells54. In accordance with these findings, we observed that metformin reduced the recruitment of macrophages in pkd2 morphants, which could be an alternative mechanism through which metformin reduces cell proliferation and cyst formation.
Another significant finding in this study is that metformin corrects the defective autophagy observed in pkd2 morphants. Autophagy is a highly regulated cellular process that degrades and recycles intracellular proteins and organelles in lysosomes during metabolic stress or nutritional deprivation55. Our finding that pkd2 morphants had defective autophagy is consistent with a previous study showing that Pkd1 −/− mouse embryonic fibroblasts had insufficient autophagy activity upon glucose deprivation9. Furthermore, our results suggested that metformin might reduce cyst formation through the enhancement of autophagy in ADPKD56. Autophagy suppression could reduce the senescence of cyst-lining cells and lead to increased proliferation, apoptosis, and cyst growth57. Autophagy has also been shown to regulate the formation and function of primary cilia58. The detailed mechanisms that link autophagy and cyst growth require further study59.
The similarities in the cystic phenotype of pkd2 and tsc1a morphants prompted us to investigate whether metformin could also inhibit cyst formation in this previously described model of tuberous sclerosis40. However, our results demonstrated that metformin could not rescue the cystic phenotype of the tsc1a morphants. This observation is consistent with an AMPK-dependent effect of metformin because TSC1 is a known downstream target of AMPK. Long-term treatment with metformin also failed to suppress renal tumours in a Tsc1 +/− mouse model60. We were unable to further determine the relative contribution of AMPK activation in the inhibition of cystogenesis because of a lack of a proper morpholino or specific antagonist to reduce AMPK activity without interfering with the development of zebrafish embryos38. Therefore, we cannot exclude the possibility that metformin inhibits cystogenesis through an AMPK-independent effect18, 61.
Our data contribute to a growing body of evidence that metabolic abnormalities are crucial in the pathogenesis of ADPKD50, 62. Inhibition of aerobic glycolysis by using 2DG suppressed cell proliferation, leukocyte infiltration, and cyst formation in mouse models of PKD19. Mild to moderate food restriction was demonstrated to activate AMPK and reduce cyst area, renal fibrosis, and inflammation63. These studies support a new therapeutic strategy for ADPKD, which involves reprogramming of cellular metabolism64.
Although our results support the hypothesis that metformin is a potential treatment for ADPKD, metformin could cause lactic acidosis in patients with advanced renal failure18. Therefore, the inhibitory effect of metformin on cyst growth in animal models requires confirmation in future clinical studies. A randomised, double-blind clinical trial evaluating metformin in the treatment of patients with ADPKD was started in 2016 and is expected to be completed in the next few years (ClinicalTrials.gov Identifier: NCT02656017).
In conclusion, metformin reduces cyst formation in pkd2-deficient zebrafish embryos. The data suggest that metformin may prevent cyst formation through activation of the AMPK pathway and modulation of defective cellular events such as proliferation and autophagy. The findings also indicate the therapeutic potential of metformin in treatment of ADPKD at the early stages. Further studies are required to assess whether metformin can improve clinical outcomes in patients with ADPKD.
Methods
Zebrafish maintenance
Animal experiments were approved by the Chang Gung University Institutional Animal Care and Use Committee. The investigation conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Zebrafish and embryos were maintained according to standard procedures65. The embryos were staged according to hours post fertilisation. We used the Tg(wt1b:GFP) line (kindly provided by Prof. Christoph Englert, Fritz Lipmann Institute, Jena, Germany) for in vivo observation of pronephric cysts25–27, 66.
Morpholino injection
Zebrafish embryos at the one- or two-cell stage were microinjected with 0.125 mM antisense MO. MOs were obtained from Gene Tools (Philomath, OR) and had the following sequences: pkd2 ATG-MO (5′-AGGACGAACGCGACTGGAGCTCATC-3′)29, pkd2 5-mismatch MO (5′-AGCACCAACCCGACTGCACCTCATC-3′), tsc1a ATG-MO (5′-CCATAGTTGTGCAGGACAGTGGGCA-3′)40, tsc1a 5-mismatch MO (5′-CCATACTTCTGCAGCACACTGGCCA-3′), pkd1a exon 8 splice-MO MO: (5′-GATCTGAGGACTCACTGTGTGATTT-3′)30, pkd1b exon 45 splice-MO MO: (5′-ACATGATATTTGTACCTCTTTGGTT-3′)30, and atg5 ATG-MO (5′-CACATCCTTGTCATCTGCCATTATC-3′)67.
Drug treatment for zebrafish
Pkd2 morphant embryos (n = 20–30) were incubated in 6-cm Petri dishes with 8 mL of the E3 medium (5 mM NaCl, 0.17 mM KCl, 0.4 mM CaCl2, and 0.16 mM MgSO4) in an incubator at 28.5 °C. Metformin (Sigma) was added to the E3 medium at final concentrations of 5 to 20 mM at 4 hpf, a time point before the development of body curvature and pronephric cysts according to previous studies30, 48. At 48 hpf, the living embryos were anaesthetised with tricaine (0.2 mg/mL) and oriented in 3% methylcellulose (Sigma). The presence of pronephric cysts was determined using fluorescence microscopy25–27. The dorsal body curvature phenotype of pkd2 morphants was categorised as normal, mild (tail angle less than 90°), or severe (tail curvature angle greater than 90°)48.
Immunofluorescence
Embryos were fixed overnight in 4% paraformaldehyde at 4 °C. Immunostaining was performed in whole-mount embryos as described previously68. A rabbit anti-PH3 antibody (1:200, Millipore) was used to label proliferating cells, and a mouse anti-α6 F antibody (1:200, Developmental Studies Hybridoma Bank) was used to label pronephric epithelial cells. LC3, an autophagosome marker, was labelled using a rabbit anti-LC3B antibody (1:200, Novus Biologicals). Secondary antibodies used were the Alexa Fluor 594 goat antirabbit IgG and Alexa Fluor 488 goat antimouse IgG (1:500, Molecular Probes). Proliferating cells in the pronephric ducts were counted in anti-PH3 and anti-α6 F stained whole-mount embryos using fluorescence microscopy. For confocal imaging, embryos were flat-mounted and the alpha 6 F and PH3 signals were recorded in z-series stacks using a Zeiss LSM 510 confocal microscope. For analysis of LC3 staining, the immunostained embryos were embedded in the OTC medium and cryosectioned through the pronephros. The sections were mounted in Vectashield (Vector Laboratories) with DAPI and imaged using confocal microscopy.
Histology
The embryos were fixed in 4% paraformaldehyde overnight at 4 °C and embedded in glycolmethacrylate (JB-4; Polyscience). Serial sections (4 μm) were cut and stained with Hematoxylin and Eosin (H & E).
Whole-mount in situ hybridisation
The embryos were fixed overnight in 4% paraformaldehyde at 4 °C. Whole-mount in situ hybridisation was performed according to published protocols65. Antisense digoxigenin-labelled RNA probes were synthesised from linearised plasmid templates containing l-plastin and mpx cDNAs.
Western blot analysis
Proteins were extracted from whole embryos for Western blot analysis using standard protocols38, 69. Primary antibodies, namely anti-phospho-AMPKα (Thr172) (1:1000, Cell Signalling), anti-AMPKα (1: 1000, Cell Signalling), anti-LC3B (1:10000, Novus Biologicals), and anti-β-actin (AC-15) (1: 10000, Abcam) antibodies, were used overnight at 4 °C. Horseradish peroxidase-conjugated secondary antibodies were used for 1 h at room temperature. The signals were detected through enhanced chemiluminescence.
Statistical analysis
Values are expressed as the mean ± SEM. Comparisons between groups were performed using the Student’s t test or ANOVA followed by Dunnett’s multiple comparison test. Values that were not normally distributed were analysed using the Kruskal–Wallis test followed by Dunnett’s multiple comparison test. Categorical variables were analysed using Fisher’s exact test or the chi-square test. P values less than 0.05 were considered statistically significant. All analyses were performed using GraphPad Prism 5.0 (GraphPad, La Jolla, CA, USA).
Electronic supplementary material
Acknowledgements
The authors thank Yi-Hui Huang for technical assistance. This work was supported by grants from Chang Gung Memorial Hospital (CMRPG3D0651, CMRPG3E2001) and the Ministry of Science and Technology of Taiwan (102-2314-B-182-045, 104-2314-B-182A-114).
Author Contributions
M.-Y.C. and Y.-C. Cheng conceived and designed the experiments; T.-L.M. performed the experiments; C.-C.H., Y.-C.T., Y.-C. Chen, and C.-W.Y. analysed the data; and M.-Y.C. and Y.-C. Cheng wrote the paper. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-07300-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Heyer CM, et al. Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27:2872–2884. doi: 10.1681/ASN.2015050583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ong AC, Devuyst O, Knebelmann B, Walz G. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015;385:1993–2002. doi: 10.1016/S0140-6736(15)60907-2. [DOI] [PubMed] [Google Scholar]
- 3.Cornec-Le Gall E, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–1013. doi: 10.1681/ASN.2012070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talbot JJ, et al. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J Am Soc Nephrol. 2014;25:1737–1748. doi: 10.1681/ASN.2013091026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geng L, et al. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci. 2006;119:1383–1395. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 6.Obara T, et al. Polycystin-2 immunolocalization and function in zebrafish. J Am Soc Nephrol. 2006;17:2706–2718. doi: 10.1681/ASN.2006040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ong AC, Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 2015;88:699–710. doi: 10.1038/ki.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124:2315–2324. doi: 10.1172/JCI72272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rowe I, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19:488–493. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest. 2015;125:2399–2412. doi: 10.1172/JCI80467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres VE, Gansevoort RT, Czerwiec FS. Tolvaptan in autosomal dominant polycystic kidney disease. N Engl J Med. 2013;368:1259. doi: 10.1056/NEJMc1300762. [DOI] [PubMed] [Google Scholar]
- 12.Walz G, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 13.Serra AL, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 14.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viollet B, et al. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front Biosci (Landmark Ed) 2009;14:3380–3400. doi: 10.2741/3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viollet B, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HG, et al. Metformin inhibits P-glycoprotein expression via the NF-kappaB pathway and CRE transcriptional activity through AMPK activation. Br J Pharmacol. 2011;162:1096–1108. doi: 10.1111/j.1476-5381.2010.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayyid RK, Fleshner NE. Potential role for metformin in urologic oncology. Investig Clin Urol. 2016;57:157–164. doi: 10.4111/icu.2016.57.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollak M. Overcoming Drug Development Bottlenecks With Repurposing: Repurposing biguanides to target energy metabolism for cancer treatment. Nat Med. 2014;20:591–593. doi: 10.1038/nm.3596. [DOI] [PubMed] [Google Scholar]
- 22.McCarty MF, Barroso-Aranda J, Contreras F. Activation of AMP-activated kinase as a strategy for managing autosomal dominant polycystic kidney disease. Med Hypotheses. 2009;73:1008–1010. doi: 10.1016/j.mehy.2009.05.043. [DOI] [PubMed] [Google Scholar]
- 23.Takiar V, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci USA. 2011;108:2462–2467. doi: 10.1073/pnas.1011498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mekahli D, et al. Polycystin-1 but not polycystin-2 deficiency causes upregulation of the mTOR pathway and can be synergistically targeted with rapamycin and metformin. Pflugers Arch. 2014;466:1591–1604. doi: 10.1007/s00424-013-1394-x. [DOI] [PubMed] [Google Scholar]
- 25.Chang MY, et al. Inhibition of the P2X7 receptor reduces cystogenesis in PKD. J Am Soc Nephrol. 2011;22:1696–1706. doi: 10.1681/ASN.2010070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Corre S, Eyre D, Drummond IA. Modulation of the secretory pathway rescues zebrafish polycystic kidney disease pathology. J Am Soc Nephrol. 2014;25:1749–1759. doi: 10.1681/ASN.2013101060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arif Pavel M, et al. Function and regulation of TRPP2 ion channel revealed by a gain-of-function mutant. Proc Natl Acad Sci USA. 2016;113:E2363–2372. doi: 10.1073/pnas.1517066113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schottenfeld J, Sullivan-Brown J, Burdine RD. Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development. 2007;134:1605–1615. doi: 10.1242/dev.02827. [DOI] [PubMed] [Google Scholar]
- 29.Sun Z, et al. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 30.Mangos S, et al. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech. 2010;3:354–365. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bubenshchikova E, et al. Wtip and Vangl2 are required for mitotic spindle orientation and cloaca morphogenesis. Biol Open. 2012;1:588–596. doi: 10.1242/bio.20121016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slanchev K, Putz M, Schmitt A, Kramer-Zucker A, Walz G. Nephrocystin-4 is required for pronephric duct-dependent cloaca formation in zebrafish. Hum Mol Genet. 2011;20:3119–3128. doi: 10.1093/hmg/ddr214. [DOI] [PubMed] [Google Scholar]
- 33.Rothschild SC, Francescatto L, Drummond IA, Tombes RM. CaMK-II is a PKD2 target that promotes pronephric kidney development and stabilizes cilia. Development. 2011;138:3387–3397. doi: 10.1242/dev.066340. [DOI] [PubMed] [Google Scholar]
- 34.Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol. 2008;23:2095–2099. doi: 10.1007/s00467-008-0898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu W, et al. The Joubert Syndrome Protein Inpp5e Controls Ciliogenesis by Regulating Phosphoinositides at the Apical Membrane. J Am Soc Nephrol. 2017;28:118–129. doi: 10.1681/ASN.2015080906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kramer-Zucker AG, et al. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development. 2005;132:1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- 37.de Jong JL, Zon LI. Use of the zebrafish system to study primitive and definitive hematopoiesis. Annu Rev Genet. 2005;39:481–501. doi: 10.1146/annurev.genet.39.073003.095931. [DOI] [PubMed] [Google Scholar]
- 38.Mendelsohn BA, Kassebaum BL, Gitlin JD. The zebrafish embryo as a dynamic model of anoxia tolerance. Dev Dyn. 2008;237:1780–1788. doi: 10.1002/dvdy.21581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choo AY, et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38:487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiBella LM, Park A, Sun Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum Mol Genet. 2009;18:595–606. doi: 10.1093/hmg/ddn384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Rechter S, et al. Autophagy in renal diseases. Pediatr Nephrol. 2016;31:737–752. doi: 10.1007/s00467-015-3134-2. [DOI] [PubMed] [Google Scholar]
- 42.Lenoir O, Tharaux PL, Huber TB. Autophagy in kidney disease and aging: lessons from rodent models. Kidney Int. 2016;90:950–964. doi: 10.1016/j.kint.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 43.Zhu P, Sieben CJ, Xu X, Harris PC, Lin X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum Mol Genet. 2017;26:158–172. doi: 10.1093/hmg/ddx045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16:299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- 45.Sussman CR, et al. Phosphodiesterase 1A modulates cystogenesis in zebrafish. J Am Soc Nephrol. 2014;25:2222–2230. doi: 10.1681/ASN.2013040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou X, et al. SIRT2 regulates ciliogenesis and contributes to abnormal centrosome amplification caused by loss of polycystin-1. Hum Mol Genet. 2014;23:1644–1655. doi: 10.1093/hmg/ddt556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu M, et al. Resveratrol delays polycystic kidney disease progression through attenuation of nuclear factor kappaB-induced inflammation. Nephrol Dial Transplant. 2016;31:1826–1834. doi: 10.1093/ndt/gfw058. [DOI] [PubMed] [Google Scholar]
- 48.Cao Y, et al. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci USA. 2009;106:21819–21824. doi: 10.1073/pnas.0911987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tietz Bogert PS, et al. The zebrafish as a model to study polycystic liver disease. Zebrafish. 2013;10:211–217. doi: 10.1089/zeb.2012.0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menezes LF, et al. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4alpha as a disease modifier. PLoS Genet. 2012;8:e1003053. doi: 10.1371/journal.pgen.1003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vasilyev A, Liu Y, Hellman N, Pathak N, Drummond IA. Mechanical stretch and PI3K signaling link cell migration and proliferation to coordinate epithelial tubule morphogenesis in the zebrafish pronephros. PLoS One. 2012;7:e39992. doi: 10.1371/journal.pone.0039992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swenson-Fields KI, et al. Macrophages promote polycystic kidney disease progression. Kidney Int. 2013;83:855–864. doi: 10.1038/ki.2012.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buldak L, et al. Metformin affects macrophages’ phenotype and improves the activity of glutathione peroxidase, superoxide dismutase, catalase and decreases malondialdehyde concentration in a partially AMPK-independent manner in LPS-stimulated human monocytes/macrophages. Pharmacol Rep. 2014;66:418–429. doi: 10.1016/j.pharep.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 54.Kim SA, Choi HC. Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem Biophys Res Commun. 2012;425:866–872. doi: 10.1016/j.bbrc.2012.07.165. [DOI] [PubMed] [Google Scholar]
- 55.Klionsky DJ, Emr SD. Cell biology - Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hardie DG. AMPK and autophagy get connected. EMBO J. 2011;30:634–635. doi: 10.1038/emboj.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ravichandran K, Edelstein CL. Polycystic kidney disease: a case of suppressed autophagy? Semin Nephrol. 2014;34:27–33. doi: 10.1016/j.semnephrol.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 58.Pampliega O, Cuervo AM. Autophagy and primary cilia: dual interplay. Curr Opin Cell Biol. 2016;39:1–7. doi: 10.1016/j.ceb.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen N, Eritja N, Lock R, Debnath J. Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3-kinase in three-dimensional culture. Oncogene. 2013;32:2543–2554. doi: 10.1038/onc.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J, Kalogerou M, Gallacher J, Sampson JR, Shen MH. Renal tumours in a Tsc1+/− mouse model show epigenetic suppression of organic cation transporters Slc22a1, Slc22a2 and Slc22a3, and do not respond to metformin. Eur J Cancer. 2013;49:1479–1490. doi: 10.1016/j.ejca.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 61.Cameron AR, et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ Res. 2016;119:652–665. doi: 10.1161/CIRCRESAHA.116.308445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Menezes LF, Lin CC, Zhou F, Germino GG. Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine. 2016;5:183–192. doi: 10.1016/j.ebiom.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warner G, et al. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27:1437–1447. doi: 10.1681/ASN.2015020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rowe I, Boletta A. Defective metabolism in polycystic kidney disease: potential for therapy and open questions. Nephrol Dial Transplant. 2014;29:1480–1486. doi: 10.1093/ndt/gft521. [DOI] [PubMed] [Google Scholar]
- 65.Westerfield, M. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio), 4th ed. University of Oregon Press, Eugene, OR (2000).
- 66.Perner B, Englert C, Bollig F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev Biol. 2007;309:87–96. doi: 10.1016/j.ydbio.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 67.Lee E, et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014;10:572–587. doi: 10.4161/auto.27649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krens SF, et al. Distinct functions for ERK1 and ERK2 in cell migration processes during zebrafish gastrulation. Dev Biol. 2008;319:370–383. doi: 10.1016/j.ydbio.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 69.Santoriello C, et al. Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis Model Mech. 2009;2:56–67. doi: 10.1242/dmm.001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.