

Abstract
Defects in genes encoding the isoforms of the laminin alpha subunit have been linked to various phenotypic manifestations, including brain malformations, muscular dystrophy, ocular defects, cardiomyopathy, and skin abnormalities. We report here a severe defect of neuromuscular transmission in a consanguineous patient with a homozygous variant in the laminin alpha-5 subunit gene (LAMA5). The variant c.8046C>T (p.Arg2659Trp) is rare and has a predicted deleterious effect. The affected individual, who also carries a rare homozygous sequence variant in LAMA1, had muscle weakness, myopia, and facial tics. Magnetic resonance imaging of brain showed mild volume loss and periventricular T2 prolongation. Repetitive nerve stimulation revealed 50% decrement of compound muscle action potential amplitudes and 250% facilitation immediately after exercise, Endplate studies identified a profound reduction of the endplate potential quantal content and endplates with normal postsynaptic folding that were denuded or partially occupied by small nerve terminals. Expression studies revealed that p.Arg2659Trp caused decreased binding of laminin alpha-5 to SV2A and impaired laminin-521 cell-adhesion and cell projection support in primary neuronal cultures. In summary, this report describing severe neuromuscular transmission failure in a patient with a LAMA5 mutation expands the list of phenotypes associated with defects in genes encoding alpha-laminins.
Keywords: congenital myasthenic syndrome (CMS), LAMA5, laminin α5, presynaptic
1 INTRODUCTION
Mutations in more than 20 genes have been found to account for multiple forms of congenital myasthenic syndromes (CMS), which are diseases characterized by muscle weakness and fatigability (Engel, Shen, Selcen, & Sine, 2015).
Laminins are extracellular matrix (ECM) heterotrimeric glycoproteins composed of subunits α, β, and γ (Aumailley et al., 2005).
Laminins provide both mechanical support and signaling, and participate in important cellular processes such as development, maintenance and regeneration of the nerve terminal and muscle fiber. The assembly of the laminins isoforms, −α2, −α4, α5, −β2, and −γ1 in laminins α2β2γ1, α4β2γ1, and α5β2γ1 is essential for the neuromuscular junction (NMJ) since these are the only laminin variants selectively expressing at the small portion of the basal lamina that covers the adult motor endplate (Patton, Miner, Chiu, & Sanes, 1997).
Laminin β2 plays a fundamental role in the organization of the NMJ. Mice lacking laminin β2 appear normal at birth, but later develop progressive weakness and proteinuria and die within the first weeks of life (Noakes, Gautam, Mudd, Sanes, & Merlie, 1995).
Laminin α4 appears to play a role in the precise alignment of pre- and postsynaptic specialized structures since α4-deficient mice show a peculiar loss of precise apposition of presynaptic active zones to secondary synaptic clefts at the NMJ (Patton et al., 2001).
The role that laminin α5 plays at the NMJ has been difficult to establish because mice lacking laminin α5 die at late embryonic stages due to multiple developmental abnormalities and placental insufficiency (Miner, Cunningham, & Sanes, 1998).
However, a conditional mutant where laminin α5 was selectively removed showed delay of postsynaptic maturation and complete arrest of postsynaptic development was observed in double mutants lacking laminins α4 and α5 (Nishimune et al., 2008). Importantly, in spite that the conditional laminin α5 mouse does not display any overt phenotype, presynaptic differentiation appears to be primarily affected since in this animal model motor nerves only partially cover the postsynaptic membrane during the initial period of postnatal development.
We describe here an individual with a severe CMS attributed to a homozygous mutation in the gene encoding the laminin α5 subunit (LAMA5) (MIM 601033), which showed selective presynaptic ultrastructural changes of the NMJ reminiscent of those found at early stages of differentiation in the conditional Lama5 mutant mouse.
2 CLINICAL REPORT
The proband is a 24-year-old woman who was born full-term to a consanguineous couple of second cousins. She was born hypotonic with weak sucking and crying, but no arthrogryposis was noted at birth. After birth she required tracheostomy and mechanical ventilation for respiratory failure and at the age of 13, she underwent corrective surgery for severe scoliosis. She had weakness that worsened with respiratory infections but she had no episodes of dyspnea or apnea. Currently, she requires mechanical ventilation for most parts of the day and gastric tube for feeding. Her parents and three siblings are healthy, but an older brother died of muscle weakness and respiratory failure. Serum creatinine was normal and antibodies against the acetylcholine receptor (AChR) and the muscle specific kinase (MuSK) were negative. A recent physical examination showed minor dysmorphic features including elongated face, elevated hard palate, and mild facial and body hirsutism (Supporting Information Figure S1). Her mentation was normal, but the cranial nerve examination revealed bilateral ptosis, and weakness of the bilateral inferior oblique, facial, tongue, and soft palate muscles. She also had facial tics (Supporting Information Video 1). She had only moderate weakness of neck and proximal limb muscles. Deep tendon reflexes were depressed and the remainder of the neurologic examination was normal. The ophthalmologic examination showed normal pupillary response to light and moderate myopia without retinal abnormalities. Tests of parasympathetic and sudomotor sympathetic function were normal (Supporting Information Table S1). A brain MRI revealed mild volume loss for age and periventricular T2 prolongation (Figure 1a,b). Repetitive stimulation of the right spinal accessory nerve at 2 Hz revealed 55% decrement of the compound muscle action potential (CMAP) amplitudes. There was severe reduction of CMAP amplitudes with more than 250% increment of CMAP amplitudes immediately after 30 s of maximal muscle activation (Figure 1c,d).
FIGURE 1.
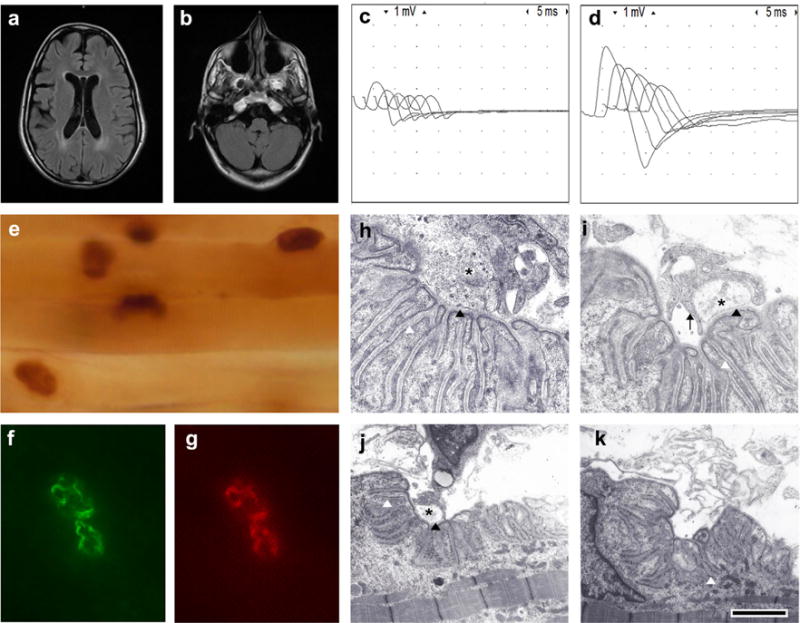
Clinical and pathologic findings: (a) Axial FLAIR demonstrating mild gray and white matter volume loss for age and periventricular T2 prolongation. (b) Axial FLAIR showing mild loss of volume in the cerebellum and brainstem without cerebellar cysts or cerebellar dysplasia. (c) Repetitive stimulation before exercise. (d) Immediately after exercise for 30 s there is 250% increase in the amplitude of the first CMAP of the train. (e) Acetylcholinesterase reaction in teased longitudinal muscle fiber bundles showing normal configuration of endplates. (f) An example of an endplate exposed to a goat anti-laminin α5 antibody and a donkey anti-goat FITC secondary antibody showing normal expression of laminin α5. (g) The same endplate as in b counterstained with rhodamine-αBGT demonstrating normal expression of AChRs. (h) An example of EM of a normal NMJ from a control muscle biopsy showing normal size of the nerve terminal (black asterisk), normal postsynaptic folds, and normal width of the primary and secondary synaptic clefts (black and white arrowheads respectively). (i) A NMJ from the patient showing a small nerve terminal (asterisk) partially encased by Schwann cell projections (black arrow), which do not invade the primary synaptic cleft (black arrowhead). Notice also that the postsynaptic folds and width of the primary synaptic clefts are normal. (j) NMJ from the patient showing similar findings as in i. (k) An example of a NMJ from the patient denuded of its nerve terminal. The postsynaptic folds and the secondary synaptic cleft (white arrowhead) are normal. Artifacts of AChE staining have been electronically subtracted. The calibration mark represents 100 μm in e, 10 μm in f and g, 1 μm in H and I, and 2 μm in j and k.
To study the mechanism of failure of neuromuscular transmission we performed an anconeus muscle biopsy at age 7, which included microelectrode recordings and electron microscopy (EM) of the NMJ as previously described (Maselli, Mass, Distad, & Richman, 1991). Light microscopy revealed mild angular atrophy of type II fibers and type I predominance.
Microelectrode recordings showed severe reduction of the average quantal content of end plate potentials (EPPs) evoked by nerve stimulation at 1 Hz, which was largely due to the fact that at most endplates, nerve stimulation failed to elicit EPPs (Supporting Information Table S2). The averaged EPP quantal content of 2.14 ± 0.74 calculated at three endplates after the perfusion of the muscle with 250 μM 3,4 diaminopyridine (3,4 DAP) was about four times higher than the average EPP quantal content of 0.54 ± 0.21 calculated at seven endplates before the addition of 3,4 DAP. In contrast with the severe abnormalities of the EPPs the amplitudes, frequencies and decay time constants of miniature endplate potentials (MEPPs) were normal.
The most distinctive findings of the electron microscopy (EM) of the NMJ were: (1) endplates with normal postsynaptic folding that were partially occupied by small nerve terminals or were denuded of their nerve endings; (2) encasement of nerve terminals by projections of the Schwann cell that did not invade the synaptic space; and (3) moderate reduction of the density of synaptic vesicles (SVs) in the nerve terminals (Figure 1h–k). The morphometric analysis of the NMJ confirmed that in the patient in comparison with controls, the postsynaptic folding expressed as the endplate index (ratio of postsynaptic membrane length/presynaptic membrane length) was not decreased. Furthermore, the number of secondary folds per mm of primary cleft was increased suggesting an increased complexity of the postsynaptic folding pattern. By contrast, the average nerve terminal area and density of SVs were diminished and many endplates were denuded of their nerve endings. In spite of these morphometric changes the width of the primary synaptic cleft was not reduced in the patient in comparison with controls (Supporting Information Table S3). Additional endplate studies showed endplates of normal size and normal expression of acetylcholinesterase (AChE), laminin α5, and AChRs. (Figure 1e–g).
3 MATERIALS AND METHODS
To investigate the genetic bases of the proband’s condition; the coding regions of several candidate genes including CHRNA1, CHRNB1, CHRND, CHRNE, RAPSN, MUSK, DOK7, and CHAT were sequenced. No pathogenic variants were found. Next, whole exome sequencing (WES) was performed as described in the Supporting Information Method section. The study was approved by the institutional review board of the University of California Davis and all participating individuals signed a consent form.
4 RESULTS
No plausible candidate variants were identified by WES under a compound heterozygous model. Analysis under a homozygous recessive model identified 15 candidate genes (Supporting Information Table S4), but only the variants in LAMA1 (rs532162048), p.Asp210Asn; and LAMA5 (rs201012962), p.Arg2659Trp, were considered potentially relevant as the other genes did not appear to play a direct role in synaptic transmission (Supporting Information Table S5). Both variants were present in the parents in the heterozygous state and absent in the non-affected sister (DNA from two brothers was not available). The homozygous variant in LAMA1 (MIM 615960) was considered unlikely to be causal of the CMS because the protein product of LAMA1, laminin α1, is not expressed at the adult NMJ, and because biallelic mutations in LAMA1 are associated with the so-called Poretti-Boltshauser syndrome (PBS), which does not comprise neuromuscular involvement (Aldinger et al., 2014). Moreover, other frequent clinical features of PBS, such as cerebellar dysplasia with cysts, and retinal dystrophy were not present in the proband. Nevertheless, we cannot rule out that some of the clinical features of our patient, including, myopia, facial tics, and brain MRI abnormalities, which have previously been described in PBS (Vilboux et al., 2016), may result from the LAMA1 variation.
The p.Arg2659Trp substitution in LAMA5, located in the domain II of the protein involved in the coil-coiled structure, was considered a priority candidate variant because it is rare (maximum alternate allele frequency in any subpopulation in ExAC = 0.00044), is relatively conserved in mammals (Supporting Information Figure S2), and is predicted to be damaging by PolyPhen-2 HumVar (score = 0.978), SIFT (score = 0.001), and CADD v1.3 (score = 24.9). Furthermore, laminin α5 expresses at the embryonic and adult NMJ, and a conditional mouse mutant where laminin α5 is removed shows developmental abnormalities of the NMJ (Nishimune et al., 2008).
To elucidate the effect of p.Arg2659Trp, we introduced it in a human laminin α5 clone and tested its impact in three important biological process involving laminins: signaling, cell adhesion, and neurite outgrowth support.
Laminins containing the α5 form a high affinity complex with the SV protein SV2 (Son et al., 2000), which in turn binds to synaptotagmin, a Ca2+ sensor protein that is implicated in SV exocytosis (Schivell, Batchelor, & Bajjalieh, 1996). It is also thought that the SV2/laminin α5 complex may help define sites of vesicle retrieval for endocytosis (Carlson, Valdez, & Sanes, 2010). Thus, giving the important role that the SV2/laminin α5 interaction may play in SV trafficking we tested the binding of LAMA5 p.Arg2659Trp to SV2A using dot blotting as previously described (Arredondo et al., 2014). Figure 2a,b, which summarizes the results of three different transfection experiments, shows that although the p.Arg2659Trp substitution did not prevent the association of LAMA5 with SV2A, in comparison with wild type (WT), the binding of the mutant LAMA5 to SV2A was reduced by 18% (p < 0.05).
FIGURE 2.
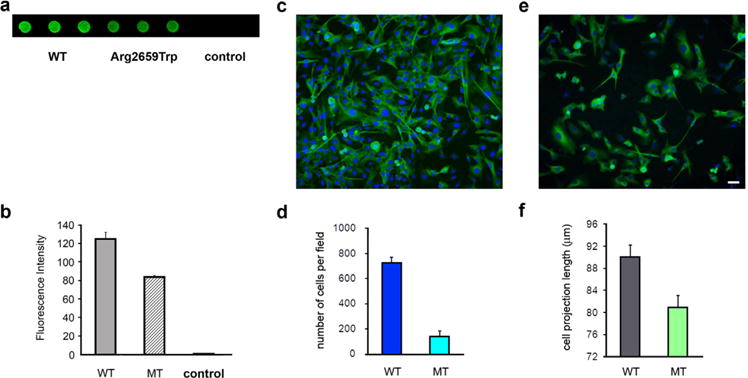
Analysis of the interaction of LAMA5 mutant with SV2A and the ability of the LAMA5 mutant to support cell adhesion and βIII tubulin-positive cell outgrow. (a) Dot blot analysis showing decreased immunoreactivity against SV2A in immobilized lysates from HEK cells transfected with mutant LAMA5 cDNA (wells 4–6) in comparison with lysates from cells transfected with WT LAMA5 cDNA (wells 1–3). (b) Bar graph summarizing measurements of fluorescence intensity in arbitrary units. Control group represents lysates of untransfected cells. (c) Example of normal cell density and neurite outgrowth in a slide coated with WT laminin. (d) Summary bar graph of four transfection experiments showing that cell adhesion to slides coated with laminin-521 containing the LAMA5-Trp2659 was only about 25% of slides coated with the WT laminin-521. (e) Example of low cell density and shorter neurite outgrow in a slide coated with mutant laminin (f) Bar graph showing that βIII tubulin-positive cell projections were overall shorter in slides coated with mutant laminin in comparison with those coated with WT laminin. The calibration mark represents 25 μm in c and e.
Next we tested the effect of p.Arg2659Trp on cell adhesion and neurite outgrowth as described in the Supporting Information Method section. Figure 2c,d shows that the density of cells that adhered to slides coated with lysates from cells transfected with the mutant laminin-521 was only about 25% of that of cells that attached to slides coated with lysates from cells transfected with WT laminin-521. There was also difference in the average length of βIII tubulin-positive cell projections, as those in slides coated with mutant laminin were shorter than those in slides coated with WT laminin [80.94 ± 1.67 μm (n = 774) vs. 90.02 ± 2.13 μm (n = 478), p < 0.001 (Student’s t-test)] (Figure 2e,f).
5 DISCUSSION
The deficient support of cell adhesion and neurite outgrowth by the mutant laminin α5 provides a plausible explanation for the ultrastructural findings of underdeveloped nerve terminals observed at the NMJs of the patient. However, since laminin α5 is widely expressed in adult tissues, it is unclear why the proband’s phenotype does not include abnormalities of other organs such as the lung, heart, and kidneys that have the highest levels of expression of laminin α5.
The selective involvement of the NMJ in our case may be accounted by two separate, but not mutually exclusive explanations. The first explanation is that a defective laminin alpha-5 may be sufficient to provide effective ECM support to tissues like lung, heart, and kidneys, but may fail to maintain normal function at more active metabolic sites such as at the NMJ. This interpretation is supported by the finding that in the conditional laminin alpha-5 mouse model, none of the defects observed in knockout embryos (e.g., exencephaly, and abnormalities in craniofacial anatomy, lung, kidney, and hair) are observed (Nishimune et al., 2008). Nevertheless, in this model there is marked developmental delay of the NMJ, which suggests that the NMJ is particularly vulnerable to defects of laminin α5. The second explanation is that a defective laminin α5 may affect a fundamental signaling pathways operating at the NMJ but not in other tissues.
While the interaction between laminin α5 with its primary receptor integrin α3β1 takes place at many organs, including lung, kidney, and skin (Kikkawa, Sanzen, & Sekiguchi, 1998; Pouliot, Saunders, & Kaur, 2002) the interaction between laminin α5 with SV2A and synaptotagmin via SVA2A is restricted to the NMJ.
Laminins containing the α5 subunit bind to the luminal domain of SV2, presumably at highly conserved N-linked glycosylation sites. After fusion of the SV to the presynaptic membrane the luminal side of the vesicle becomes exposed to the ECM enabling the interaction of SV2 with laminin α5. This interaction may provide an activity-dependent mechanism for anchoring the presynaptic membrane to the ECM, particularly during early stages of nerve development and regeneration (Son et al., 2000). Thus, an impaired interaction of the mutant laminin α5 with SV2A may explain the underdeveloped nerve terminals seen in this patient, but not the modest signs of central nervous system involvement found in this patient. Furthermore, given the multiple interactions of laminin α5 with other proteins a complete expression study would require the analysis of the interaction with many other proteins of the NMJ.
The other presynaptic deficit observed in our patient involved a reduction of the probability of release similar to that observed in the Lambert Eaton Myasthenic Syndrome (LEMS) (García, Mynlieff, Sanders, Beam, & Walrond, 1996). The underlying LEMS-like defect in our patient, similar to that seen in recently described CMS (Supporting Information Table S6), was supported by three different lines of evidence. First, there was an increase of more than 250% of the CMAP amplitude immediately after exercise similar to that seen in LEMS. Second, there was a 400% increased of EPP quantal content after exposure of the anconeus muscle in vitro to 3,4 DAP. Third, there was an excellent clinical response to 3,4 DAP.
A disruption of the interaction of laminin α5 with SV2A may also explain the LEMS-like defect as several studies have proposed that the SV2 isoforms regulate presynaptic Ca2+ dynamics, (Chang & Südhof, 2009; Janz et al., 1999) which has direct impact on the probability of SV release. Furthermore, the N-terminal region of the three SV2 isoforms binds to synaptotagmin-1, which is a Ca2+ sensor protein primarily involved in SV exocytosis (Perin et al., 1991).
In summary, we identified homozygous variants in LAMA5 and LAMA1 in a patient with a complex phenotype reminiscent of PBS, except for the presence of severe failure of neuromuscular transmission due to the LAMA5 mutation that reduces binding of laminin α5 to SV2A and laminin 521 support of cell adhesion and cell outgrowth. This results in a severe CMS characterized by underdeveloped nerve terminals and a severe presynaptic failure of neuromuscular transmission. The condition responds well to a combined treatment with pyridostigmine sulfate and 3,4 DAP.
Supplementary Material
Acknowledgments
The authors thank the proband of the study for participating in this research and funding from NIH (Grant R01NS049117-01) to Dr. Ricardo A. Maselli, and the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 2UM1HG006493 to Drs. Debbie Nickerson, Michael Bamshad, and Suzanne Leal.
Funding information
National Institutes of Health, Grant number: R01NS049117-01; National Human Genome Research Institute, Grant number: 2UM1HG006493
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
CONFLICT OF INTEREST
None.
References
- Aldinger KA, Mosca SJ, Tétreault M, Dempsey JC, Ishak GE, Hartley T, Phelps IG, Lamont RE, O’Day DR, Basel D, Gripp KW, Baker L, Stephan MJ, Bernier FP, Boycott KM, Majewski J, University of Washington Center for Mendelian Genomics; Care4-Rare Canada. Parboosingh JS, Innes AM, Doherty D. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. American Journal of Human Genetics. 2014;95:227–234. doi: 10.1016/j.ajhg.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arredondo J, Lara M, Ng F, Gochez DA, Lee DC, Logia SP, Nguyen J, Maselli RA. COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylcholinesterase impair the interaction of ColQ with proteins of the basal lamina. Human Genetics. 2014;133:599–616. doi: 10.1007/s00439-013-1391-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biology. 2005;5:326–332. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Carlson SS, Valdez G, Sanes JR. Presynaptic calcium channels and α3-integrins are complexed with synaptic cleft laminins, cytoskeletal elements and active zone components. Journal of Neurochemistry. 2010;115:654–666. doi: 10.1111/j.1471-4159.2010.06965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Südhof TC. SV2 renders primed synaptic vesicles competent for Ca2+ -induced exocytosis. Journal of Neuroscience. 2009;29:883–897. doi: 10.1523/JNEUROSCI.4521-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurology. 2015;4:420–434. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García KD, Mynlieff M, Sanders DB, Beam KG, Walrond JP. Lambert–Eaton sera reduce low-voltage and high-voltage activated Ca2+ currents in murine dorsal root ganglion neurons. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9264–9269. doi: 10.1073/pnas.93.17.9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz R, Südhof TC, Hammer RE, Unni V, Siegelbaum SA, Bolshakov VY. Essential roles in synaptic plasticity for synaptogyrin I and synaptophysin I. Neuron. 1999;24:687–700. doi: 10.1016/s0896-6273(00)81122-8. [DOI] [PubMed] [Google Scholar]
- Kikkawa Y, Sanzen N, Sekiguchi K. Isolation and characterization of laminin-10/11 secreted by human lung carcinoma cells. laminin-10/11 mediates cell adhesion through integrin alpha3 beta1. The Journal of Biological Chemistry. 1998;273:15854–15859. doi: 10.1074/jbc.273.25.15854. [DOI] [PubMed] [Google Scholar]
- Maselli RA, Mass DP, Distad BJ, Richman DP. Anconeus muscle: A human muscle preparation suitable for in-vitro microelectrode studies. Muscle and Nerve. 1991;14:1189–1192. doi: 10.1002/mus.880141208. [DOI] [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis exencephaly, syndactyly, and placentopathy in mice lacking the laminin 5 chain. The Journal of Cell Biology. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune H, Valdez G, Jarad G, Moulson CL, Müller U, Miner JH, Sanes JR. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. The Journal of Cell Biology. 2008;182:1201–1215. doi: 10.1083/jcb.200805095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin β2. Nature. 1995;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. The Journal of Cell Biology. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BL, Cunningham JM, Thyboll J, Kortesmaa J, Westerblad H, Edström L, Sanes JR. Properly formed but improperly localized synaptic specializations in the absence of laminin alpha4. Nature Neuroscience. 2001;6:597–604. doi: 10.1038/88414. [DOI] [PubMed] [Google Scholar]
- Perin MS, Johnston PA, Ozcelik T, Jahn R, Francke U, Südhof TC. Structural and functional conservation of synaptotagmin (p65) in Drosophila and humans. The Journal of Biological Chemistry. 1991;266:615–622. [PubMed] [Google Scholar]
- Pouliot N, Saunders NA, Kaur P. Laminin 10/11: An alternative adhesive ligand for epidermal keratinocytes with a functional role in promoting proliferation and migration. Experimental Dermatology. 2002;11:387–397. doi: 10.1034/j.1600-0625.2002.110501.x. [DOI] [PubMed] [Google Scholar]
- Schivell AE, Batchelor RH, Bajjalieh SM. Isoform-specific, calcium-regulated interaction of the synaptic vesicle proteins SV2 and synaptotagmin. The Journal of Biological Chemistry. 1996;271:27770–27775. doi: 10.1074/jbc.271.44.27770. [DOI] [PubMed] [Google Scholar]
- Son YJ, Scranton TW, Sunderland WJ, Baek SJ, Miner JH, Sanes JR, Carlson SS. The synaptic vesicle protein SV2 is complexed with an alpha5-containing laminin on the nerve terminal surface. The Journal of Biological Chemistry. 2000;275:451–460. doi: 10.1074/jbc.275.1.451. [DOI] [PubMed] [Google Scholar]
- Vilboux T, Malicdan MC, Chang YM, Guo J, Zerfas PM, Stephen J, Cullinane AR, Bryant J, Fischer R, Brooks BP, Zein WM, Wiggs EA, Zalewski CK, Poretti A, Bryan MM, Vemulapalli M, Mullikin JC, Kirby M, Anderson SM, NISC Comparative Sequencing Program. Huizing M, Toro C, Gahl WA, Gunay-Aygun M. Cystic cerebellar dysplasia and biallelic LAMA1 mutations: A lamininopathy associated with tics, obsessive compulsive traits and myopia due to cell adhesion and migration defects. Journal of Medical Genetics. 2016;53:318–329. doi: 10.1136/jmedgenet-2015-103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.