

Klebsiella pneumoniae is a serious human pathogen associated with resistance to multiple antibiotics and high mortality. K. variicola and K. quasipneumoniae are closely related organisms that are generally considered to be less-virulent opportunistic pathogens. We used a large, comprehensive, population-based strain collection and whole-genome sequencing to investigate infections caused by these organisms in our hospital system. We discovered that K. variicola and K. quasipneumoniae isolates are often misidentified as K. pneumoniae by routine clinical microbiology diagnostics and frequently cause severe life-threatening infections similar to K. pneumoniae. The presence of KPC in K. variicola and K. quasipneumoniae strains as well as NDM-1 metallo-beta-lactamase in one K. variicola strain is particularly concerning because these genes confer resistance to many different beta-lactam antibiotics. The sharing of plasmids, as well as evidence of homologous recombination, between these three species of Klebsiella is cause for additional concern.
KEYWORDS: clinical microbiology, KPC, Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola, MALDI, MLST, NDM-1, pathogenesis, whole-genome sequencing, bioinformatics, clinical methods
ABSTRACT
Klebsiella pneumoniae is a major threat to public health, causing significant morbidity and mortality worldwide. The emergence of highly drug-resistant strains is particularly concerning. There has been a recognition and division of Klebsiella pneumoniae into three distinct phylogenetic groups: Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. K. variicola and K. quasipneumoniae have often been described as opportunistic pathogens that have less virulence in humans than K. pneumoniae does. We recently sequenced the genomes of 1,777 extended-spectrum-beta-lactamase (ESBL)-producing K. pneumoniae isolates recovered from human infections and discovered that 28 strains were phylogenetically related to K. variicola and K. quasipneumoniae. Whole-genome sequencing of 95 additional non-ESBL-producing K. pneumoniae isolates recovered from patients found 12 K. quasipneumoniae strains. Matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis initially identified all patient isolates as K. pneumoniae, suggesting a potential pitfall in conventional clinical microbiology laboratory identification methods. Whole-genome sequence analysis revealed extensive sharing of core gene content and plasmid replicons among the Klebsiella species. For the first time, strains of both K. variicola and K. quasipneumoniae were found to carry the Klebsiella pneumoniae carbapenemase (KPC) gene, while another K. variicola strain was found to carry the New Delhi metallo-beta-lactamase 1 (NDM-1) gene. K. variicola and K. quasipneumoniae infections were not less virulent than K. pneumoniae infections, as assessed by in-hospital mortality and infection type. We also discovered evidence of homologous recombination in one K. variicola strain, as well as one strain from a novel Klebsiella species, which challenge the current understanding of interrelationships between clades of Klebsiella.
IMPORTANCE Klebsiella pneumoniae is a serious human pathogen associated with resistance to multiple antibiotics and high mortality. K. variicola and K. quasipneumoniae are closely related organisms that are generally considered to be less-virulent opportunistic pathogens. We used a large, comprehensive, population-based strain collection and whole-genome sequencing to investigate infections caused by these organisms in our hospital system. We discovered that K. variicola and K. quasipneumoniae isolates are often misidentified as K. pneumoniae by routine clinical microbiology diagnostics and frequently cause severe life-threatening infections similar to K. pneumoniae. The presence of KPC in K. variicola and K. quasipneumoniae strains as well as NDM-1 metallo-beta-lactamase in one K. variicola strain is particularly concerning because these genes confer resistance to many different beta-lactam antibiotics. The sharing of plasmids, as well as evidence of homologous recombination, between these three species of Klebsiella is cause for additional concern.
INTRODUCTION
Edwin Klebs first described Klebsiella pneumoniae organisms in 1875 while examining the airways of patients who died from pneumonia, and Carl Friedlander formally described the species in 1882 (1). Ever since, Klebsiella pneumoniae has been increasingly recognized as a cause of significant human morbidity and mortality (2, 3). Of great concern to public health, many community-acquired and health care-associated outbreaks of invasive K. pneumoniae disease have been reported (4, 5). Neonates, the elderly, and immunocompromised individuals are at greatest risk for poor outcomes (5). The ability of this organism, and other closely related species, to undergo chromosomal recombination and exchange plasmids enables them to readily alter the repertoire of virulence factors and antimicrobial resistance genes (6).
Over the past 2 decades, many related Klebsiella species have been identified and classified (7–10). Previously referred to as phylogroups KpII and KpIII, Klebsiella quasipneumoniae (10) and Klebsiella variicola (9) likely diverged from a common ancestor of K. pneumoniae six million years ago (11, 12). Whereas K. quasipneumoniae is typically recovered from the gastrointestinal tracts of healthy humans, K. variicola organisms are frequently isolated from agricultural sources such as plants, surface water, sewage, soil, and mucosal surfaces of plant-eating livestock (7, 13, 14). Recovery of K. quasipneumoniae and K. variicola strains from patients is generally thought to be representative of colonization (8, 10); however, opportunistic infections have been reported (15–17). The pathogenicity of these organisms in human infection has not been thoroughly studied.
The objective of this study was to investigate the population genetic structure of K. variicola and K. quasipneumoniae strains recovered from human infections in our health care system (Houston Methodist Hospital System). Whole-genome sequencing demonstrated that K. pneumoniae, K. quasipneumoniae, and K. variicola strains share chromosomal and mobile genes encoding virulence factors and antimicrobial resistance mechanisms. Importantly, we identified for the first time K. variicola strains carrying the Klebsiella pneumoniae carbapenemase (KPC) and New Delhi metallo-beta-lactamase 1 (NDM-1) antimicrobial resistance genes, as well as a K. quasipneumoniae strain carrying the KPC antimicrobial resistance gene. Furthermore, we describe the misidentification of K. variicola and K. quasipneumoniae as K. pneumoniae by matrix-assisted laser desorption ionization—time of flight mass spectrometry (MALDI-TOF MS) and low-resolution sequence typing. We provide detail on the diversity and severity of human infections caused by these pathogens, including one novel strain related to K. variicola. These data provide new insight into the pathogenesis of the underrecognized human pathogens K. quasipneumoniae and K. variicola.
RESULTS
Whole-genome sequencing reveals that K. variicola and K. quasipneumoniae are misidentified by MALDI-TOF MS as K. pneumoniae.
To study the population genetic structure of Klebsiella pneumoniae isolates recovered from patients in our health care system (Houston Methodist Hospital System) between 2011 and 2015, we recently sequenced the genomes of 1,777 extended-spectrum-beta-lactamase (ESBL)-producing strains (18). All isolates in this comprehensive population-based collection were identified as K. pneumoniae using MALDI-TOF MS in our diagnostic microbiology laboratory (19, 20). We unexpectedly discovered that 28 strains were phylogenetically allied with K. variicola (13 strains) and K. quasipneumoniae (15 strains) (Table 1 and Fig. 1). Thus, in our health care system, ESBL-producing K. variicola and K. quasipneumoniae cause approximately 2% of human infections attributed to K. pneumoniae. To determine the possible additional presence of K. variicola and K. quasipneumoniae among non-ESBL-producing K. pneumoniae, we sequenced the genomes of 95 strains recovered in 2017. We discovered that 12 non-ESBL-producing strains were also phylogenetically allied with K. quasipneumoniae (12.6% of non-ESBL-producing isolates) (Table 1 and Fig. 1). The K. variicola and K. quasipneumoniae strains were recovered from multiple anatomic sites, including blood, drains, respiratory specimens, tissue, and urine (Table 1).
TABLE 1 .
Summary of the clinical characteristics of the K. variicola and K. quasipneumoniae strains
| Strain | MALDI-TOF MS identification |
Genomic identification |
ESBL producing |
Collection date (mo/yr) |
Sample source |
Associated patient mortality |
|---|---|---|---|---|---|---|
| KPN325 | K. pneumoniae | K. variicola | Yes | 6/2012 | Urine | No |
| KPN349 | K. pneumoniae | K. variicola | Yes | 7/2012 | Wound | No |
| KPN458 | K. pneumoniae | K. variicola | Yes | 10/2012 | Blood | No |
| KPN700 | K. pneumoniae | K. variicola | Yes | 3/2013 | Blood | No |
| KPN771 | K. pneumoniae | K. variicola | Yes | 5/2013 | Drain | No |
| KPN807 | K. pneumoniae | K. variicola | Yes | 6/2013 | Urine | No |
| KPN1264 | K. pneumoniae | K. variicola | Yes | 2/2014 | Respiratory | No |
| KPN1401 | K. pneumoniae | K. variicola | Yes | 4/2014 | Respiratory | Yes |
| KPN1415 | K. pneumoniae | K. variicola | Yes | 4/2014 | Respiratory | Yes |
| KPN1481 | K. pneumoniae | K. variicola | Yes | 6/2014 | Urine | No |
| KPN1556 | K. pneumoniae | K. variicola | Yes | 7/2014 | Wound | No |
| KPN1705 | K. pneumoniae | K. variicola | Yes | 8/2014 | Wound | No |
| KPN1751 | K. pneumoniae | K. variicola | Yes | 10/2014 | Urine | Yes |
| KPN560 | K. pneumoniae | K. quasipneumoniae | Yes | 12/2012 | Urine | No |
| KPN712 | K. pneumoniae | K. quasipneumoniae | Yes | 4/2013 | Respiratory | No |
| KPN1132 | K. pneumoniae | K. quasipneumoniae | Yes | 12/2013 | Urine | No |
| KPN1398 | K. pneumoniae | K. quasipneumoniae | Yes | 4/2014 | Respiratory | No |
| KPN1470 | K. pneumoniae | K. quasipneumoniae | Yes | 5/2014 | Drain | No |
| KPN1533 | K. pneumoniae | K. quasipneumoniae | Yes | 8/2014 | Urine | No |
| KPN1648 | K. pneumoniae | K. quasipneumoniae | Yes | 9/2014 | Wound | No |
| KPN1673 | K. pneumoniae | K. quasipneumoniae | Yes | 9/2014 | Wound | No |
| KPN1688 | K. pneumoniae | K. quasipneumoniae | Yes | 8/2014 | Blood | Yes |
| KPN1711 | K. pneumoniae | K. quasipneumoniae | Yes | 9/2014 | Wound | No |
| KPN1715 | K. pneumoniae | K. quasipneumoniae | Yes | 10/2014 | Respiratory | No |
| KPN1962 | K. pneumoniae | K. quasipneumoniae | Yes | 2/2015 | Urine | No |
| KPN2096 | K. pneumoniae | K. quasipneumoniae | Yes | 3/2015 | Urine | No |
| KPN2105 | K. pneumoniae | K. quasipneumoniae | Yes | 4/2015 | Drain | No |
| KPN2119 | K. pneumoniae | K. quasipneumoniae | Yes | 4/2015 | Urine | Yes |
| NEK11 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK12 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK19 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK36 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK42 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK47 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK56 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Blood | No |
| NEK59 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Respiratory | No |
| NEK66 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK67 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Urine | No |
| NEK118 | K. pneumoniae | K. quasipneumoniae | No | 2/2017 | Blood | No |
| NEK122 | K. pneumoniae | K. quasipneumoniae | No | 3/2017 | Urine | No |
FIG 1 .
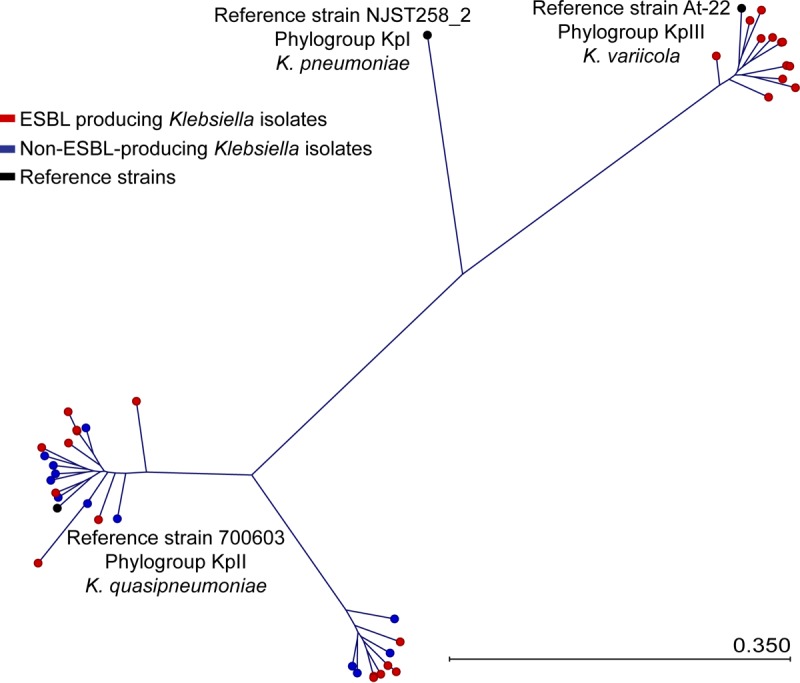
Phylogenetic tree of K. variicola and K. quasipneumoniae from human infections. Polymorphisms were called against the K. pneumoniae reference genome NJST258_2 (KpI). ESBL-producing Klebsiella isolates are represented with red circles, while non-ESBL-producing Klebsiella are represented with blue circles. The K. variicola clade (KpIII) is at the top right, with the At-22 reference strain indicated by a black circle. The two clades of K. quasipneumoniae (KpII) are present at the bottom left, with the reference genome 700603 indicated by the black circle. The outlier Klebsiella KPN1705 strain is not shown. The scale bar indicates the mean number of nucleotide substitutions per site.
Multilocus sequence typing and capsule genotyping misclassify K. variicola and K. quasipneumoniae isolates as K. pneumoniae.
Over the years, K. variicola and K. quasipneumoniae strains have been misclassified as K. pneumoniae, resulting in the deposition of misidentified reference strains in public repositories, including the public K. pneumoniae multilocus sequence typing (MLST) schema (21–24). We used the whole-genome sequence data to generate a K. pneumoniae MLST assignment for each strain. In our collection of 13 ESBL-producing K. variicola isolates, 6 (46%) had a defined K. pneumoniae MLST type and 3 (23%) had a novel combination of known K. pneumoniae MLST alleles (Table 2). Among the 15 ESBL-producing K. quasipneumoniae isolates, 12 (80%) had a defined K. pneumoniae MLST type, and 1 (7%) had a novel combination of known alleles (Table 2). Similarly, among the 12 non-ESBL-producing K. quasipneumoniae strains, 3 (25%) had a defined K. pneumoniae MLST type, and 6 (50%) had a novel combination of known alleles (Table 2). Consistent with the extensive genetic diversity observed within each Klebsiella clade (Fig. 2 and 3), multiple different MLST designations were found for K. variicola and K. quasipneumoniae strains in our collection (Table 2).
TABLE 2 .
Genomic characteristics of the K. variicola and K. quasipneumoniae strainsa
| Strain | Species | MLST | Capsule | nif | KPC | CTX-M | NDM-1 | SHV-OKP- LEN core |
SHV-OKP- LEN plasmid |
TEM |
|---|---|---|---|---|---|---|---|---|---|---|
| KPN325 | K. variicola | 1174 | KL33* | + | − | − | − | LEN-24 | SHV-12 | − |
| KPN349 | K. variicola | 468 | KL60 | + | − | − | − | LEN-24 | SHV-5 | − |
| KPN458 | K. variicola | NF | KL19 | + | − | − | − | LEN-24 | SHV-12 | − |
| KPN700 | K. variicola | 681 | KL143* | + | − | − | − | LEN-2 | − | − |
| KPN771 | K. variicola | 681 | KL143* | + | − | − | − | LEN-2 | − | − |
| KPN807 | K. variicola | NF | KL105 | + | − | CTX-M-15 | − | OKP-B-16 | − | TEM-166 |
| KPN1264 | K. variicola | 454 | KL109* | − | − | − | − | LEN-24 | SHV-30 | − |
| KPN1401 | K. variicola | NF | KL19 | + | KPC-2 | − | − | LEN-20 | − | − |
| KPN1415 | K. variicola | NF | KL19 | + | KPC-2 | − | − | LEN-4 | − | TEM-79 |
| KPN1481 | K. variicola | 906 | KL10 | + | − | CTX-M-15 | NDM-1 | LEN-16 | − | − |
| KPN1556 | K. variicola | NF | KL53* | + | − | − | − | LEN-24 | SHV-12 | − |
| KPN1705 | K. variicola | NF | KL153 | + | − | − | − | LEN-24 | SHV-30 | − |
| KPN1751 | K. variicola | 1456 | KL60 | + | − | − | − | LEN-24 | SHV-12, SHV-66 | − |
| KPN560 | K. quasipneumoniae | 1602 | KL53 | − | − | CTX-M-15 | − | OKP-B-19 | TEM-198 | |
| KPN712 | K. quasipneumoniae | NF | KL101 | − | − | − | − | OKP-B-3 | SHV-12 | − |
| KPN1132 | K. quasipneumoniae | 476 | KL57 | − | − | − | − | OKP-B-15 | SHV-12 | − |
| KPN1398 | K. quasipneumoniae | 2133 | KL106* | + | − | CTX-M-15 | − | OKP-B-15 | − | TEM-166 |
| KPN1470 | K. quasipneumoniae | 138 | KL1 | + | − | CTX-M-15 | − | OKP-B-2 | − | TEM-33 |
| KPN1533 | K. quasipneumoniae | 2351 | KL114 | + | − | − | − | OKP-A-5 | SHV-12 | − |
| KPN1648 | K. quasipneumoniae | 2351 | KL114 | + | − | CTX-M-3 | − | OKP-A-5 | − | TEM-30 |
| KPN1673 | K. quasipneumoniae | 1887 | KL121 | + | − | − | − | OKP-B-8 | SHV-12 | − |
| KPN1688 | K. quasipneumoniae | 1887 | KL121 | + | − | − | − | OKP-B-8 | SHV-12 | − |
| KPN1711 | K. quasipneumoniae | NF | KL1 | + | KPC-2 | − | − | OKP-A-7 | − | − |
| KPN1715 | K. quasipneumoniae | 1887 | KL121* | + | − | − | − | OKP-B-8 | SHV-12 | − |
| KPN1962 | K. quasipneumoniae | 196 | KL46 | − | − | − | − | OKP-A-5 | − | TEM-198 |
| KPN2096 | K. quasipneumoniae | 414 | KL123 | − | − | − | − | OKP-B-8 | − | − |
| KPN2105 | K. quasipneumoniae | 978 | KL125 | + | − | CTX-M-3 | − | OKP-A-3 | − | − |
| KPN2119 | K. quasipneumoniae | 978 | KL125 | + | − | CTX-M-3 | − | OKP-A-3 | − | − |
| NEK11 | K. quasipneumoniae | 138 | KL1 | + | − | − | − | OKP-B-2 | − | − |
| NEK12 | K. quasipneumoniae | NF | KL16 | + | − | − | − | OKP-B-5 | − | − |
| NEK19 | K. quasipneumoniae | NF | KL16 | + | − | − | − | OKP-B-3 | − | TEM-198 |
| NEK36 | K. quasipneumoniae | 283 | KL10 | + | − | − | − | OKP-B-15 | − | − |
| NEK42 | K. quasipneumoniae | NF | KL33 | + | − | − | − | OKP-A-11 | − | − |
| NEK47 | K. quasipneumoniae | NF | KL158 | + | − | − | − | OKP-A-7 | − | − |
| NEK56 | K. quasipneumoniae | 894 | KL58 | − | − | − | − | OKP-B-15 | − | − |
| NEK59 | K. quasipneumoniae | NF | KL128 | + | − | − | − | OKP-A-10 | − | − |
| NEK66 | K. quasipneumoniae | NF | KL139 | − | − | − | − | OKP-B-1 | − | − |
| NEK67 | K. quasipneumoniae | 477 | KL15 | + | − | − | − | OKP-B-3 | − | − |
| NEK118 | K. quasipneumoniae | NF | KL13 | + | − | − | − | OKP-A-11 | − | − |
| NEK122 | K. quasipneumoniae | NF | KL56 | + | − | − | − | OKP-B-5 | − | − |
The presence or absence of select antimicrobial resistance genes, MLST, capsule genotype, presence or absence of nif genes, and presence of SHV-LEN-OKP beta-lactamase in core genome and plasmids. Capsule loci that appear to be variants are indicated with an asterisk.
FIG 2 .
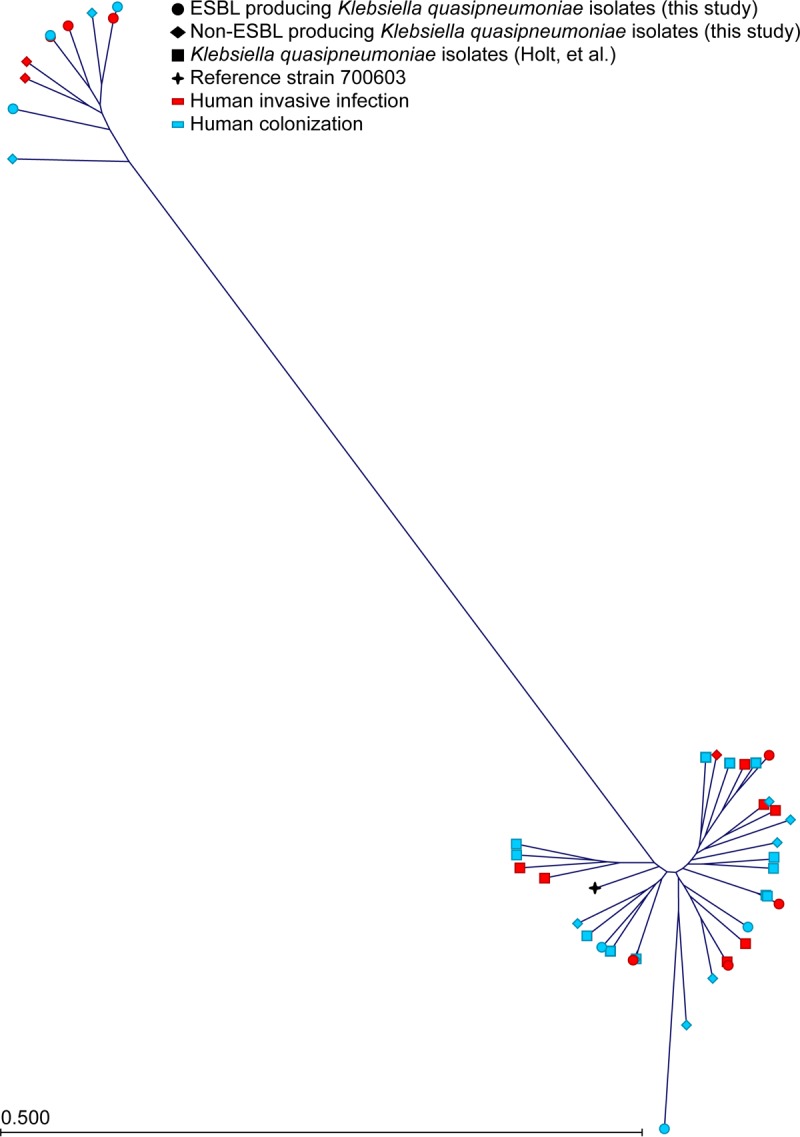
Phylogenetic tree of K. quasipneumoniae from humans. Polymorphisms in the K. quasipneumoniae strains were called against the 700603 reference genome (black cross). The clade KpIIA is at the top left, while the KpIIB clade is at the bottom right. ESBL-producing strains sequenced for this study are represented by circles. Non-ESBL-producing strains of both clades identified in our collection are represented by diamonds. Strains previously sequenced by Holt et al. (8) are represented by squares. Strains associated with human invasive infection sites are indicated in red, and human colonization is indicated in blue.
FIG 3 .
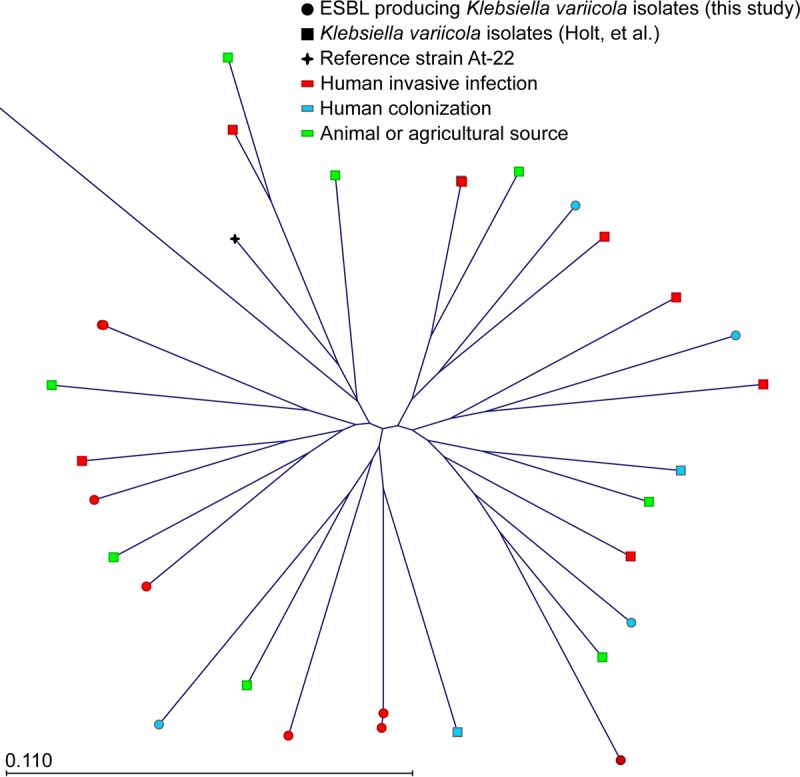
Phylogenetic tree of K. variicola from humans and animals. Polymorphisms in the K. variicola strains were called against the At-22 reference genome (black cross). All K. variicola isolates identified were ESBL producers. There is one outlier Klebsiella, KPN1705, which projects beyond the border of the figure to the top left. Strains sequenced for this study are represented by circles, while those previously sequenced by Holt et al. (8) are represented by squares. Strains associated with human invasive infection sites are indicated in red, human colonization is indicated in blue, and animal-associated isolates are indicated in green.
Capsule serotype has been used for close to a century to classify Klebsiella species in general (25). Capsule genotype, as assessed by sequencing the genes in the capsule locus, has been used more recently as a molecular tool to subclassify K. pneumoniae strains (26–28). Among our collection of K. variicola and K. quasipneumoniae isolates, every strain had a K. pneumoniae capsule locus detected (Table 2). Again, consistent with the extensive genetic diversity observed within each clade, multiple different capsule genotypes were found, including some strains with novel combinations of capsule genes (Table 2). Taken together, these data demonstrate that the majority of K. variicola and K. quasipneumoniae strains recovered from human patients would be misidentified by typing methods such as K. pneumoniae MLST and capsule genotyping.
Whole-genome sequencing reveals extensive genetic diversity within and between Klebsiella clades.
To begin assessing phylogenetic relationships between K. pneumoniae, K. variicola, and K. quasipneumoniae strains, single nucleotide polymorphisms (SNPs) were determined relative to the ST258 K. pneumoniae reference genome NJST258_2 (GenBank accession number CP006918.1) (6). Consistent with a previous report using strains collected from a variety of human, animal, and environmental sources (8), each Klebsiella species formed a distinct clade, with K. quasipneumoniae divided into two subclades (Fig. 1). Phylogenetic analysis demonstrated that the ESBL-producing and non-ESBL-producing K. quasipneumoniae strains are intermingled on the cladogram, suggesting that they are derived from a common genetic pool (Fig. 2). However, this does not imply that our ESBL-producing and non-ESBL-producing K. quasipneumoniae strains are identical, because both groups include strains with a diverse array of different MLST, capsule, and plasmid genotypes (Tables 2 and 3).
TABLE 3 .
Plasmid replicons carried by the K. variicola and K. quasipneumoniae strainsa
| Strain | FIBK_Kpn3 | FIIK | FII_1 |
|---|---|---|---|
| KPN325 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| KPN349 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| KPN458 | |||
| KPN700 | FIBK_1_Kpn3_JN233704_92 | ||
| KPN771 | |||
| KPN807 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| KPN1264 | FIBK_1_Kpn3_JN233704_92 | FIIK_2_CP000966_pKp91_67 | FII_1_pKP91_CP000966_17 |
| KPN1401 | _FIIK_1_CP000648 | FII_1_pSFO_AF401292_16 | |
| KPN1415 | _FIIK_1_CP000648 | FII_3_AF401292_pSFo157_18 | |
| KPN1481 | FIIK_2_CP000966_pKp91_67 | FII_1_pKP91_CP000966_17 | |
| KPN1556 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| KPN1705 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| KPN1751 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| KPN560 | |||
| KPN712 | |||
| KPN1132 | FIBK_1_Kpn3_JN233704_92 | ||
| KPN1398 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| KPN1470 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| KPN1533 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| KPN1648 | FIBK_1_Kpn3_JN233704_92 | ||
| KPN1673 | FIBK_1_Kpn3_JN233704_92 | ||
| KPN1688 | FIBK_1_Kpn3_JN233704_92 | ||
| KPN1711 | FIBK_1_Kpn3_JN233704_92 | FIIK_2_CP000966_pKp91_67* | FII_1_pKP91_CP000966_17 |
| KPN1715 | FIBK_1_Kpn3_JN233704_92 | ||
| KPN1962 | FIBK_1_Kpn3_JN233704_92 | FII29_1_pUTI89_CP003035_15 | |
| KPN2096 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| KPN2105 | _FIIK_1_CP000648 | ||
| KPN2119 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| NEK11 | FIBK_1_Kpn3_JN233704_92 | ||
| NEK12 | FIBK_1_Kpn3_JN233704_92 | ||
| NEK19 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| NEK36 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| NEK42 | _FIIK_1_CP000648 | ||
| NEK47 | FIBK_1_Kpn3_JN233704_92 | ||
| NEK56 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | |
| NEK59 | FIBK_1_Kpn3_JN233704_92 | ||
| NEK66 | FIBK_1_Kpn3_JN233704_92 | ||
| NEK67 | FIBK_1_Kpn3_JN233704_92 | _FIIK_1_CP000648 | FII_1_pKP91_CP000966_17 |
| NEK118 | FIBK_1_Kpn3_JN233704_92 | ||
| NEK122 |
Presence or absence of the three most common plasmid replicons found in the Klebsiella isolates in this study.
Phylogenetic comparison of our K. quasipneumoniae isolates recovered from infected patients with publicly available sequence data from strains collected from humans (8) showed much genetic intermixing (Fig. 2). Similarly, analysis of K. variicola strains recovered from our patients with invasive infection (Table 1) or colonized humans and animals (8) also revealed no clear phylogenetic segregation (Fig. 3). Taken together, these data suggest that K. quasipneumoniae and K. variicola strains recovered from infected patients and colonized humans or animals are derived from a common genetic pool.
One outlier strain, KPN1705, was present in the K. variicola population (Fig. 3). The KPN1705 strain was allied phylogenetically with K. variicola, yet it was 251,390 SNPs distant from the K. variicola At-22 reference genome. This was striking compared to the average distance to the At-22 reference of 38,056 SNPs (range, 31,777 to 45,299) in our other K. variicola strains. Since hybrid strains of Klebsiella with large chromosomal recombination events have been described previously (8), we compared the reads from strain KPN1705 against At-22 (K. variicola), NJST258_2 (K. pneumoniae), and 700603 (K. quasipneumoniae) reference genomes. The SNP distribution was uniform across the three reference genomes, which made the possibility of KPN1705 being the result of a single large recombination event less likely (data not shown). To further evaluate its phylogeny, we determined that KPN1705 was a single locus variant of the K. pneumoniae ST1155. The only other ST1155 strain reported in the literature is designated 10982 and is proposed to represent a novel species (22). Strain 10982 was isolated from a perianal swab from an intensive care unit (ICU) patient in Maryland in 2005. Thus, our strain KPN1705 appears most closely related to a novel Klebsiella species that colonizes the gastrointestinal (GI) tract of humans and may be capable of causing human disease.
SHV-LEN-OKP core chromosomal beta-lactamases are not restricted by Klebsiella species.
The SHV-LEN-OKP beta-lactamases are core chromosomal genes of Klebsiella that have been suggested to be differentiators of Klebsiella species: K. pneumoniae (SHV restricted), K. quasipneumoniae (OKP restricted), and K. variicola (LEN restricted) (10, 29, 30). However, one potential confounder is that the SHV beta-lactamase genes can also be carried on plasmids (31). We next used the whole-genome sequence data to assess SHV-LEN-OKP gene content in our 40 K. variicola and K. quasipneumoniae strains. Among our collection, 7/13 (54%) K. variicola strains carried SHV on plasmids in addition to the chromosomal LEN gene (Table 2). Similarly, 7/15 (47%) K. quasipneumoniae strains carried SHV on a plasmid in addition to the chromosomal OKP gene. Only 1/12 (8%) non-ESBL-producing K. quasipneumoniae isolate carried SHV on a plasmid in addition to the chromosomal OKP genes. The presence of SHV on plasmids, sometimes in multiple copies, may complicate the identification and analysis of Klebsiella strains.
Unexpectedly, we discovered one K. variicola strain (designated KPN807; Table 2) carrying a chromosomal copy of OKP rather than the expected LEN gene that is typical of K. variicola strains. This OKP gene was carried within a 7-kb segment of chromosomal DNA that had a higher sequence identity to K. quasipneumoniae than K. variicola, suggesting recombination of this region (Fig. 4).
FIG 4 .

Comparison of the 7-kb region of recombination in strain KPN807. KPN807 assembled contigs are compared to the At-22 K. variicola reference genome (top) and the 700603 K. quasipneumoniae reference genome (bottom). The consensus is indicated by a red bar, with SNPs marked as colored vertical lines. KPN807 has a marked increase in SNP density in the center of the region relative to At-22 and the flanking regions relative to 700603, suggesting that this 7-kb region of the chromosome has recombined into the K. variicola KPN807 from a K. quasipneumoniae isolate.
K. variicola and K. quasipneumoniae have similar antimicrobial resistance and plasmid replicon content to K. pneumoniae, including KPC and NDM-1.
The emergence of multidrug resistance among K. pneumoniae strains is a cause of public concern. A table of the MIC values for the 40 K. variicola and K. quasipneumoniae strains is provided in Table S1 in the supplemental material. Next, we used the whole-genome sequence data to compare plasmid replicons carried by the different Klebsiella species. There were many plasmid replicons and antimicrobial resistance genes among the ESBL-producing K. variicola and K. quasipneumoniae isolates that were also identified in K. pneumoniae (Table 3) (18). In general, plasmid replicons tended to be associated with genomically allied strains (Fig. 5). In particular, the FIBk, FIIk, and FII replicons were detected in the majority of our K. variicola and K. quasipneumoniae isolates. Several strains also possessed the KPC-2 carbapenemase and CTX-M beta-lactamase genes associated with multiple drug resistance. Of note, KPC-positive K. variicola and K. quasipneumoniae strains have not been previously reported in the United States.
FIG 5 .

Beta-lactamase gene content of K. pneumoniae, K. variicola, and K. quasipneumoniae. The phylogenetic relationship between strains based upon the presence of beta-lactamase genes is shown on a rectangular cladogram. Polymorphisms were called against the K. pneumoniae reference genome NJST258_2. K. quasipneumoniae are represented by circles, while K. variicola are represented by squares. ESBL-producing Klebsiella are shown in red, while non-ESBL producing Klebsiella are shown in blue. To the right of the cladogram, the common beta-lactamase genes are shown. The first column lists the core genome SHV-OKP-LEN beta-lactamase: LEN in red, OKP-A in green, and OKP-B in blue. No SHV was found in the core chromosome. The most common allele(s) for OKP and LEN is represented with a darker shade of the primary color. The second column represents the plasmid SHV-OKP-LEN beta-lactamase content, with the most common allele SHV-12 in purple and all others in gray. The third column indicates the CTX-M-15 alleles in teal, with CTX-M-3 in gray. The fourth column shows the TEM alleles, with the most common allele TEM-198 in orange and all others in gray. The fifth and sixth columns represent the presence of KPC-2 and NDM-1 alleles in black, respectively.
MIC testing for the Klebsiella strains in this study. Download TABLE S1, XLSX file, 0.1 MB (55.3KB, xlsx) .
Copyright © 2017 Long et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Unexpectedly, one K. variicola strain (designated KPN1481; Table 3) carried a plasmid, pKPN1481-1, with the New Delhi metallo-beta-lactamase 1 (NDM-1) gene (GenBank accession numbers CP020847 to CP020852). This represents the first NDM-1-producing K. variicola strain to be reported. To better characterize the plasmid and strain containing the NDM-1 gene, it was sequenced to closure using single molecule real-time (SMRT) sequencing. Results confirmed the presence of the NDM-1 gene on pKPN1481-1, a 342-kb plasmid (18). Alignment of pKPN1481-1 to NDM-1 gene-containing plasmids from K. pneumoniae strains recovered from our health care system and elsewhere revealed extensive similarity (18, 32, 33). Importantly, the 10,273-bp region containing the NDM-1 gene was identical in sequence to four NDM-1 plasmids carried by K. pneumoniae isolates in our collection.
Gene content comparison between K. pneumoniae, K. variicola, and K. quasipneumoniae.
We compared the gene content between our ESBL-producing K. variicola strains, K. quasipneumoniae strains, and 12 representative K. pneumoniae strains from our previously published collection (18). We identified a total of 30,075 unique genes present in the pangenome. A Klebsiella core genome was identified consisting of 2,800 unique genes that were present in >95% of the isolates. Most of the accessory genes were present in less than 15% of the isolates (23,334 genes; 77.5% of the total). A binary tree of accessory gene content demonstrates that the accessory gene content is largely differentiated by the three major clades, suggesting a clade-specific core genome for each ESBL Klebsiella species as well as the presence of unique mobile genetic content (Fig. 6). This finding is further reinforced when considering the core genome of the K. variicola and ESBL-producing K. quasipneumoniae in our study. The ESBL-producing K. quasipneumoniae strains have a core genome of 3,338 unique genes, while the K. variicola strains have a core genome of 3,960 genes. This difference may be in part a reflection of the division of K. quasipneumoniae into two distinct clades. The presence of nif operon genes, which facilitate nitrogen fixation, has been associated with agricultural isolates of Klebsiella. We found nif genes present in 12/13 (92.3%) ESBL-producing K. variicola human isolates, 11/15 (73.3%) ESBL-producing K. quasipneumoniae isolates, and 10/12 (83.3%) non-ESBL-producing K. quasipneumoniae isolates, suggesting that nif genes may persist in isolates found in human infections. The presence or absence of select genes is included in Tables S2 and S3.
FIG 6 .
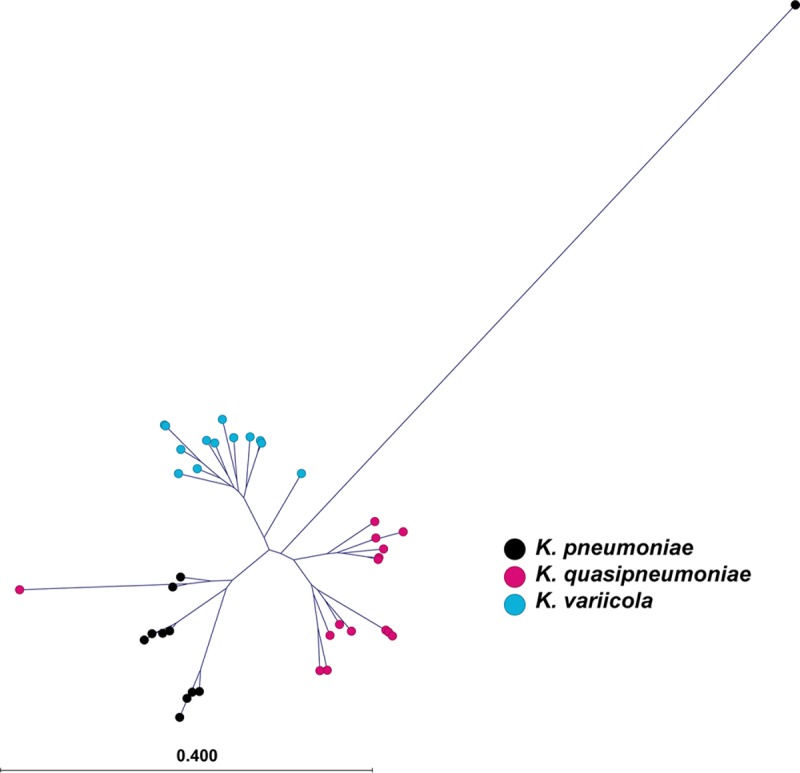
Binary accessory genome tree of K. pneumoniae, K. variicola, and K. quasipneumoniae. The phylogenetic relationship between strains based upon the presence or absence of genes in the pangenome is shown. The three clades are visible, with the K. pneumoniae strains (black circles), K. variicola strains (blue circles), and K. quasipneumoniae strains (magenta circles) indicated. The K. pneumoniae strains separate into two clades based upon their MLST type, while the K. quasipneumoniae strains separate into the two distinct clades representing K. quasipneumoniae subsp. quasipneumoniae and K. quasipneumoniae subsp. similipneumoniae. One strain of K. quasipneumoniae (KPN1470) is allied with K. pneumoniae, due to similarities in accessory genome content.
Presence or absence of select genes in ESBL-producing Klebsiella strains in this study. Download TABLE S2, XLSX file, 4.3 MB (4.4MB, xlsx) .
Copyright © 2017 Long et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Presence or absence of select genes in non-ESBL-producing K. quasipneumoniae strains. Download TABLE S3, XLSX file, 7 MB (7.1MB, xlsx) .
Copyright © 2017 Long et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Human infections caused by K. variicola and K. quasipneumoniae are similar in site and mortality to those caused by K. pneumoniae.
Although a few cases of opportunistic infection have been published, most reports suggest that K. variicola and K. quasipneumoniae most commonly occupy agricultural niches or benignly colonize the gastrointestinal tract (8, 10, 34). However, among patients in Houston, TX, with infections caused by ESBL-producing organisms, K. pneumoniae, K. variicola, and K. quasipneumoniae were associated with similar infection types and outcomes (Table 1). That is, ESBL-producing K. pneumoniae, K. variicola, and K. quasipneumoniae strains caused a similar ratio of invasive infections relative to recovery from the urinary tract (42.5%, 60.0%, and 54.5%, respectively; not significantly different by chi-square test). Also, no significant difference in mortality was observed between the three clades (16.0%, 13.3%, and 18.0%, respectively; not significantly different by the chi-square test).
DISCUSSION
K. pneumoniae is a well-known cause of life-threatening infections. However, there is conflicting evidence of the pathogenicity of the closely related organisms K. variicola and K. quasipneumoniae. They are most often associated with agricultural niches (7, 9) or thought to represent colonization rather than infection (8). At present, K. variicola and K. quasipneumoniae are viewed most commonly as benign endosymbionts of plants, colonizers of the gastrointestinal tracts of animals and humans that consume these foods, or occasional opportunistic pathogens. However, a recent report from Stockholm, Sweden, suggests that these organisms may be an underrecognized cause of bacteremia, including fatal infections (23). Among patients at our health care system, K. variicola and K. quasipneumoniae strains caused a minority of Klebsiella infections. However, when ESBL-producing K. variicola and K. quasipneumoniae infections did occur, they were as virulent as ESBL-producing K. pneumoniae strains, causing invasive infections and mortality at rates statistically similar to those of K. pneumoniae strains.
Our whole-genome sequence data provide clues to the similar virulence of K. variicola and K. quasipneumoniae strains relative to K. pneumoniae strains. The core genome contents of strains in the three clades are similar, despite the extent of single nucleotide polymorphism and divergence between the three clades over millions of years of evolution. Also, consistent with previous reports (8), we observed extensive sharing of plasmid replicons between the clades. The plasmids associated with these replicons often carry multiple genes encoding virulence factors or antimicrobial resistance mechanisms, and they readily exchange genetic material between one another (35). The first time detection of K. variicola and K. quasipneumoniae strains carrying the KPC gene in the United States or K. variicola carrying the NDM-1 gene anywhere is particularly concerning for the potential propagation of multidrug resistance and increased virulence capacity among various Klebsiella species. Notably, previous studies have suggested that homologous recombination between strains in the different clades does not occur (10). However, strain KPN807, a K. variicola isolate, clearly carries a blaOKP gene (OKP-B-15) rather than the expected blaLEN gene. The chromosomally encoded blaOKP gene in strain KPN807 is flanked on both sides by two additional genes with high identity to the homologous region of a K. quasipneumoniae reference genome (strain 700603; GenBank accession number CP014696.2). BLAST search of this 7-kb region did not reveal its presence in the genome of any other species or plasmids that could serve as possible third party donors. This 7-kb region of recombination is bordered on one side by the lacY and lacZ genes, which have been implicated previously in bacterial recombination events (36). Thus, the K. variicola isolate KPN807 may have acquired the blaOKP gene from K. quasipneumoniae by homologous recombination. Similarly, Holt et al. reported one K. quasipneumoniae strain that likely underwent a 735-kb homologous recombination with a K. pneumoniae strain (8). Taken together, these data suggest that the possibility of homologous recombination between K. pneumoniae, K. variicola, and K. quasipneumoniae is underestimated. The extent to which homologous recombination shapes the virulence of these organisms warrants further investigation.
The inability of conventional clinical microbiology laboratory techniques to distinguish K. variicola and K. quasipneumoniae from K. pneumoniae may contribute to our underestimation of their potential for causing serious human infections (37). Their biochemical phenotypes are closely overlapping. Adonitol fermentation has been suggested to be a useful classifier (K. pneumoniae is reportedly adonitol positive, and K. quasipneumoniae and K. variicola are reportedly adonitol negative), but the phenotype is unstable and not consistent between all strains within the three species (38). Other biochemical tests, such as indole positivity, are similarly flawed (38). The presence of the nitrogen-fixing nif operon has been classically associated with agricultural Klebsiella isolates; however many of our strains isolated from human infections still carried nif genes (8, 39). Misidentification has also propagated into the Klebsiella pneumoniae MLST and other genotyping schema. Many of our K. variicola and K. quasipneumoniae strains had perfect MLST allele and capsule locus genotype matches within the K. pneumoniae schemes (Table 1). Similarly, K. pneumoniae reference strains Kp5-1 and Kp342 are clearly genomically allied with K. variicola strains yet were identified as K. pneumoniae before K. variicola was recognized as a distinct species (10). The K. quasipneumoniae reference strain 700603 was originally identified as an ATCC-type strain of K. pneumoniae and is still present in the ATCC catalog as a K. pneumoniae subsp. pneumoniae (https://www.atcc.org/Products/All/700603.aspx). In addition to type strain misidentification, omission of these pathogens from reference databases may contribute to their misidentification by MALDI-TOF MS methods (15, 24, 40). The 40 strains recovered by our clinical microbiology laboratory, which uses MALDI-TOF MS as the primary identification method for Gram-negative rods, were reported as K. pneumoniae. Although K. variicola is included in the current Bruker microorganism reference library (v6.0.0.0), the earlier library versions used when these isolates were recovered in 2011 to 2015 contained at least one K. variicola strain misassigned as a K. pneumoniae strain. Consistent with this idea, all K. variicola strains, except for the outlier KPN1705, were correctly identified when reanalyzed using the current database in 2017. The Klebsiella 10982-like strain KPN1705 is still identified as a K. pneumoniae by the current Bruker database (v6.0.0.0), despite its genomic similarity to K. variicola. Similarly, K. quasipneumoniae is not included in the current Bruker microorganism reference library (v6.0.0.0) or in previous versions. As a direct result, the Bruker MALDI-TOF MS will not identify K. quasipneumoniae, and all of these strains are misidentified as K. pneumoniae. As 12 of 95 (12.6%) of our non-ESBL-producing K. pneumoniae strains tested from 2017 were actually K. quasipneumoniae, the full extent of K. quasipneumoniae-caused human disease may be vastly underestimated. Commercially available molecular methods also misidentify these pathogens as K. pneumoniae or fail to identify them altogether (41, 42). Finally, lab-developed multiplex PCR assays that detect specific core chromosomal beta-lactamases have been cited as potential methods of differentiation (30). Although the blaSHV, blaOKP, and blaLEN genes are closely associated with the chromosomes of K. pneumoniae, K. quasipneumoniae, and K. variicola, respectively, our data clearly reveal the presence of blaSHV on plasmids in strains belonging to all three clades and one strain that acquired a different chromosomally encoded beta-lactamase gene by homologous recombination.
These data provide new insight into the natural history and pathogenesis of K. variicola and K. quasipneumoniae, as well as the novel 10982-like Klebsiella species. Larger studies using comprehensive population-based strain collections are needed to confirm the extent of potential recombination between these Klebsiella and its potential impact on the virulence of these important human pathogens.
MATERIALS AND METHODS
Collection of K. pneumoniae strains.
We previously sequenced the genomes of 1,777 ESBL-producing Klebsiella strains recovered from patients with infections in our health care system (Houston Methodist Hospital) from 2011 to 2015 (18). To determine the possible presence or absence of K. variicola and K. quasipneumoniae strains among non-ESBL-producing isolates, we sequenced the genomes of an additional 95 Klebsiella strains collected in 2017. All strains were identified as Klebsiella pneumoniae by MALDI-TOF MS. Clinical significance was evaluated by review of the electronic medical record. Strains were cryopreserved at the time of recovery by transferring colonial material to Todd-Hewitt broth containing 20% glycerol and storing at −80°C. This study was approved by the institutional review board at Houston Methodist Hospital and Research Institute (protocol IRB1010-0199).
Whole-genome sequencing of Klebsiella.
To prepare whole-genome sequencing libraries, the cryopreserved stocks were grown on tryptic soy agar containing 5% sheep blood. Genomic DNA was extracted using standard methods (Qiagen, Valencia, CA), and NexteraXT libraries were prepared using the manufacturer’s protocols (Illumina, San Diego, CA) and sequenced on an Illumina MiSeq or NextSeq instrument.
Bioinformatic analysis of strains.
The single nucleotide polymorphism calling pipeline and additional bioinformatic pipelines were described previously (18). Strains from Holt et al. were downloaded from European Nucleic Acid Archive (accession number ERP000165). BLAST was performed using the NCBI BLAST toolkit as well as CLC Genomics Workbench v.10.1. Visualization of SNP distribution was performed using CLC Genomics Workbench v.10.1. FASTQ files were assembled into contigs using Spades v3.9.0, and contigs were annotated using Prokka v1.12 (43, 44). Gene content analysis was performed using Roary v3.6.1 (45). The 12 strains of ESBL-producing K. pneumoniae included for pangenome comparison are KPN1, KPN2, KPN9, KPN11, KPN12, KPN17, KPN18, KPN133, KPN1998, KPN2000, KPN2108, and KPN2129 (18). Assembly of SNPs into phylogenetic trees was accomplished with the scripts prephix v3.3.0, phrecon v4.6.0, and FastTreeMP v2.1 (46). Prephix and phrecon are available from GitHub (https://github.com/codinghedgehog).
MALDI-TOF MS identification.
Identification of isolates was performed in the Houston Methodist clinical microbiology laboratory using a Bruker Biotyper MALDI-TOF MS as part of the standard clinical microbiology practice as described previously (19). Briefly, colonial material from agar plates or pelleted cells from liquid blood cultures were transferred to the target plate, dried at room temperature for approximately 2 min, and covered with alpha-cyano-4-hydroxycinnamic acid (HCCA) matrix. Spectra were collected using the default instrument settings and interpreted using the research use only microorganism reference library on the Biotyper (Bruker, Billerica, MA). At the time of isolation in 2011 to 2015, Bruker microorganism reference library versions starting with 4.0.0.0 were used. The strains were reanalyzed in 2017 using version 6.0.0.0, which was installed in January 2017.
Accession number(s).
The genomes of the strains sequenced for this study have been deposited in the NCBI database under BioProject PRJNA376414 and PRJNA386693.
ACKNOWLEDGMENTS
This work was supported by funds from the Fondren Foundation and the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Service (contract HHSN272201400027C).
We acknowledge Kathryn Holt for graciously providing sequence data and Kathryn Stockbauer for help in preparing the manuscript.
Footnotes
This paper was submitted via the mSphereDirect™ pathway.
Contributor Information
Sarah E. F. D'Orazio, University of Kentucky.
Christopher Doern, Virginia Commonwealth University.
Carey-Ann D. Burnham, Washington University School of Medicine in St. Louis.
REFERENCES
- 1.Friedlaender C. 1882. Über die Schizomyceten bei der acuten fibrösen Pneumonie. Archiv Pathol Anat 87:319–324. doi: 10.1007/BF01880516. [DOI] [Google Scholar]
- 2.Wyres KL, Holt KE. 2016. Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol 24:944–956. doi: 10.1016/j.tim.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Li B, Zhao Y, Liu C, Chen Z, Zhou D. 2014. Molecular pathogenesis of Klebsiella pneumoniae. Future Microbiol 9:1071–1081. doi: 10.2217/fmb.14.48. [DOI] [PubMed] [Google Scholar]
- 4.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program Group, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satlin MJ, Chen L, Patel G, Gomez-Simmonds A, Weston G, Kim AC, Seo SK, Rosenthal ME, Sperber SJ, Jenkins SG, Hamula CL, Uhlemann AC, Levi MH, Fries BC, Tang YW, Juretschko S, Rojtman AD, Hong T, Mathema B, Jacobs MR, Walsh TJ, Bonomo RA, Kreiswirth BN. 2017. Multicenter clinical and molecular epidemiological analysis of bacteremia due to carbapenem-resistant Enterobacteriaceae (CRE) in the CRE epicenter of the United States. Antimicrob Agents Chemother 61:e02349-16. doi: 10.1128/AAC.02349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeLeo FR, Chen L, Porcella SF, Martens CA, Kobayashi SD, Porter AR, Chavda KD, Jacobs MR, Mathema B, Olsen RJ, Bonomo RA, Musser JM, Kreiswirth BN. 2014. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proc Natl Acad Sci U S A 111:4988–4993. doi: 10.1073/pnas.1321364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brisse S, Verhoef J. 2001. Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. Int J Syst Evol Microbiol 51:915–924. doi: 10.1099/00207713-51-3-915. [DOI] [PubMed] [Google Scholar]
- 8.Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D, Jenney A, Connor TR, Hsu LY, Severin J, Brisse S, Cao H, Wilksch J, Gorrie C, Schultz MB, Edwards DJ, Nguyen KV, Nguyen TV, Dao TT, Mensink M, Minh VL, Nhu NT, Schultsz C, Kuntaman K, Newton PN, Moore CE, Strugnell RA, Thomson NR. 2015. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci U S A 112:E3574–E3581. doi: 10.1073/pnas.1501049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenblueth M, Martínez L, Silva J, Martínez-Romero E. 2004. Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst Appl Microbiol 27:27–35. doi: 10.1078/0723-2020-00261. [DOI] [PubMed] [Google Scholar]
- 10.Brisse S, Passet V, Grimont PA. 2014. Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int J Syst Evol Microbiol 64:3146–3152. doi: 10.1099/ijs.0.062737-0. [DOI] [PubMed] [Google Scholar]
- 11.Haeggman S, Löfdahl S, Paauw A, Verhoef J, Brisse S. 2004. Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother 48:2400–2408. doi: 10.1128/AAC.48.7.2400-2408.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fevre C, Passet V, Weill F-X, Grimont PAD, Brisse S. 2005. Variants of the Klebsiella pneumoniae OKP chromosomal beta-lactamase are divided into two main groups, OKP-A and OKP-B. Antimicrob Agents Chemother 49:5149–5152. doi: 10.1128/AAC.49.12.5149-5152.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zadoks RN, Griffiths HM, Munoz MA, Ahlstrom C, Bennett GJ, Thomas E, Schukken YH. 2011. Sources of Klebsiella and Raoultella species on dairy farms: be careful where you walk. J Dairy Sci 94:1045–1051. doi: 10.3168/jds.2010-3603. [DOI] [PubMed] [Google Scholar]
- 14.Martínez-Romero E, Silva-Sanchez J, Barrios H, Rodríguez-Medina N, Martínez-Barnetche J, Téllez-Sosa J, Gómez-Barreto RE, Garza-Ramos U. 2015. Draft genome sequences of Klebsiella variicola plant isolates. Genome Announc 3:e0105-15. doi: 10.1128/genomeA.01015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berry GJ, Loeffelholz MJ, Williams-Bouyer N. 2015. An investigation into laboratory misidentification of a bloodstream Klebsiella variicola infection. J Clin Microbiol 53:2793–2794. doi: 10.1128/JCM.00841-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seki M, Gotoh K, Nakamura S, Akeda Y, Yoshii T, Miyaguchi S, Inohara H, Horii T, Oishi K, Iida T, Tomono K. 2013. Fatal sepsis caused by an unusual Klebsiella species that was misidentified by an automated identification system. J Med Microbiol 62:801–803. doi: 10.1099/jmm.0.051334-0. [DOI] [PubMed] [Google Scholar]
- 17.Breurec S, Melot B, Hoen B, Passet V, Schepers K, Bastian S, Brisse S. 2016. Liver abscess caused by infection with community-acquired Klebsiella quasipneumoniae subsp. quasipneumoniae. Emerg Infect Dis 22:529–531. doi: 10.3201/eid2203.151466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long SW, Olsen RJ, Eagar TN, Beres SB, Zhao P, Davis JJ, Brettin T, Xia F, Musser JM. 2017. Population genomic analysis of 1,777 extended-spectrum beta-lactamase-producing Klebsiella pneumoniae isolates, Houston, Texas: unexpected abundance of clonal group 307. mBio 8:e00489-17. doi: 10.1128/mBio.00489-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wimmer JL, Long SW, Cernoch P, Land GA, Davis JR, Musser JM, Olsen RJ. 2012. Strategy for rapid identification and antibiotic susceptibility testing of Gram-negative bacteria directly recovered from positive blood cultures using the Bruker MALDI Biotyper and the BD Phoenix system. J Clin Microbiol 50:2452–2454. doi: 10.1128/JCM.00409-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez KK, Olsen RJ, Musick WL, Cernoch PL, Davis JR, Land GA, Peterson LE, Musser JM. 2013. Integrating rapid pathogen identification and antimicrobial stewardship significantly decreases hospital costs. Arch Pathol Lab Med 137:1247–1254. doi: 10.5858/arpa.2012-0651-OA. [DOI] [PubMed] [Google Scholar]
- 21.Garza-Ramos U, Silva-Sánchez J, Martínez-Romero E, Tinoco P, Pina-Gonzales M, Barrios H, Martínez-Barnetche J, Gómez-Barreto RE, Tellez-Sosa J. 2015. Development of a Multiplex-PCR probe system for the proper identification of Klebsiella variicola. BMC Microbiol 15:64. doi: 10.1186/s12866-015-0396-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hazen TH, Zhao L, Sahl JW, Robinson G, Harris AD, Rasko DA, Johnson JK. 2014. Characterization of Klebsiella sp. strain 10982, a colonizer of humans that contains novel antibiotic resistance alleles and exhibits genetic similarities to plant and clinical Klebsiella isolates. Antimicrob Agents Chemother 58:1879–1888. doi: 10.1128/AAC.01605-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maatallah M, Vading M, Kabir MH, Bakhrouf A, Kalin M, Nauclér P, Brisse S, Giske CG. 2014. Klebsiella variicola is a frequent cause of bloodstream infection in the Stockholm area, and associated with higher mortality compared to K. pneumoniae. PLoS One 9:e113539. doi: 10.1371/journal.pone.0113539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodrigues C, Novais Â, Sousa C, Ramos H, Coque TM, Cantón R, Lopes JA, Peixe L. 2017. Elucidating constraints for differentiation of major human Klebsiella pneumoniae clones using MALDI-TOF MS. Eur J Clin Microbiol Infect Dis 36:379–386. doi: 10.1007/s10096-016-2812-8. [DOI] [PubMed] [Google Scholar]
- 25.Julianelle LA. 1926. A biological classification of Encapsulatus pneumoniae (Friedlander’s bacillus). J Exp Med 44:113–128. doi: 10.1084/jem.44.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan YJ, Lin TL, Chen CT, Chen YY, Hsieh PF, Hsu CR, Wu MC, Wang JT. 2015. Genetic analysis of capsular polysaccharide synthesis gene clusters in 79 capsular types of Klebsiella spp. Sci Rep 5:15573. doi: 10.1038/srep15573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brisse S, Passet V, Haugaard AB, Babosan A, Kassis-Chikhani N, Struve C, Decré D. 2013. wzi gene sequencing, a rapid method for determination of capsular type for Klebsiella strains. J Clin Microbiol 51:4073–4078. doi: 10.1128/JCM.01924-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyres KL, Wick RR, Gorrie C, Jenney A, Follador R, Thomson NR, Holt KE. 2016. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb Genom 2:e000102. doi: 10.1099/mgen.0.000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tärnberg M, Nilsson LE, Monstein HJ. 2009. Molecular identification of (bla)SHV, (bla)LEN and (bla)OKP beta-lactamase genes in Klebsiella pneumoniae by bi-directional sequencing of universal SP6- and T7-sequence-tagged (bla)SHV-PCR amplicons. Mol Cell Probes 23:195–200. doi: 10.1016/j.mcp.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Fonseca EL, Ramos ND, Andrade BG, Morais LL, Marin MF, Vicente AC. 2017. A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn Microbiol Infect Dis 87:315–317. doi: 10.1016/j.diagmicrobio.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Matthew M, Hedges RW, Smith JT. 1979. Types of beta-lactamase determined by plasmids in Gram-negative bacteria. J Bacteriol 138:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doi Y, Hazen TH, Boitano M, Tsai YC, Clark TA, Korlach J, Rasko DA. 2014. Whole-genome assembly of Klebsiella pneumoniae coproducing NDM-1 and OXA-232 carbapenemases using single-molecule, real-time sequencing. Antimicrob Agents Chemother 58:5947–5953. doi: 10.1128/AAC.03180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villa L, Poirel L, Nordmann P, Carta C, Carattoli A. 2012. Complete sequencing of an IncH plasmid carrying the blaNDM-1, blaCTX-M-15 and qnrB1 genes. J Antimicrob Chemother 67:1645–1650. doi: 10.1093/jac/dks114. [DOI] [PubMed] [Google Scholar]
- 34.de Melo MES, Cabral AB, Maciel MAV, da Silveira VM, de Souza Lopes AC. 2011. Phylogenetic groups among Klebsiella pneumoniae isolates from Brazil: relationship with antimicrobial resistance and origin. Curr Microbiol 62:1596–1601. doi: 10.1007/s00284-011-9903-7. [DOI] [PubMed] [Google Scholar]
- 35.Zurfluh K, Poirel L, Nordmann P, Klumpp J, Stephan R. 2015. First detection of Klebsiella variicola producing OXA-181 carbapenemase in fresh vegetable imported from Asia to Switzerland. Antimicrob Resist Infect Control 4:38. doi: 10.1186/s13756-015-0080-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shpakovskii GV, Berlin IUA. 1983. Structure of a recombination site in the transducing bacteriophage lambda plac5 DNA. Bioorg Khim 9:711–713. (In Russian.) [PubMed] [Google Scholar]
- 37.Bowers JR, Lemmer D, Sahl JW, Pearson T, Driebe EM, Wojack B, Saubolle MA, Engelthaler DM, Keim P. 2016. KlebSeq, a diagnostic tool for surveillance, detection, and monitoring of Klebsiella pneumoniae. J Clin Microbiol 54:2582–2596. doi: 10.1128/JCM.00927-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alves MS, Dias RC, de Castro AC, Riley LW, Moreira BM. 2006. Identification of clinical isolates of indole-positive and indole-negative Klebsiella spp. J Clin Microbiol 44:3640–3646. doi: 10.1128/JCM.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fouts DE, Tyler HL, DeBoy RT, Daugherty S, Ren Q, Badger JH, Durkin AS, Huot H, Shrivastava S, Kothari S, Dodson RJ, Mohamoud Y, Khouri H, Roesch LF, Krogfelt KA, Struve C, Triplett EW, Methé BA. 2008. Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Genet 4:e1000141. doi: 10.1371/journal.pgen.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Veen SQ, Claas EC, Kuijper EJ. 2010. High-throughput identification of bacteria and yeast by matrix-assisted laser desorption ionization-time of flight mass spectrometry in conventional medical microbiology laboratories. J Clin Microbiol 48:900–907. doi: 10.1128/JCM.02071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ledeboer NA, Lopansri BK, Dhiman N, Cavagnolo R, Carroll KC, Granato P, Thomson R Jr, Butler-Wu SM, Berger H, Samuel L, Pancholi P, Swyers L, Hansen GT, Tran NK, Polage CR, Thomson KS, Hanson ND, Winegar R, Buchan BW. 2015. Identification of Gram-negative bacteria and genetic resistance determinants from positive blood culture broths by use of the Verigene Gram-negative blood culture multiplex microarray-based molecular assay. J Clin Microbiol 53:2460–2472. doi: 10.1128/JCM.00581-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salimnia H, Fairfax MR, Lephart PR, Schreckenberger P, DesJarlais SM, Johnson JK, Robinson G, Carroll KC, Greer A, Morgan M, Chan R, Loeffelholz M, Valencia-Shelton F, Jenkins S, Schuetz AN, Daly JA, Barney T, Hemmert A, Kanack KJ. 2016. Evaluation of the FilmArray blood culture identification panel: results of a multicenter controlled trial. J Clin Microbiol 54:687–698. doi: 10.1128/JCM.01679-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nurk S, Bankevich A, Antipov D, Gurevich A, Korobeynikov A, Lapidus A, Prjibelsky A, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, McLean J, Lasken R, Clingenpeel SR, Woyke T, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling genomes and mini-metagenomes from highly chimeric reads, p 158–170. In Deng M, Jiang R, Sun F, Zhang X (ed), Research in computational molecular biology. RECOMB 2013. Lecture Notes in Computer Science, vol 7821. Springer, Berlin, Germany. doi: 10.1007/978-3-642-37195-0_13. [DOI] [Google Scholar]
- 44.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 45.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MIC testing for the Klebsiella strains in this study. Download TABLE S1, XLSX file, 0.1 MB (55.3KB, xlsx) .
Copyright © 2017 Long et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Presence or absence of select genes in ESBL-producing Klebsiella strains in this study. Download TABLE S2, XLSX file, 4.3 MB (4.4MB, xlsx) .
Copyright © 2017 Long et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Presence or absence of select genes in non-ESBL-producing K. quasipneumoniae strains. Download TABLE S3, XLSX file, 7 MB (7.1MB, xlsx) .
Copyright © 2017 Long et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.