

Abstract
Selective recognition of the E-box sequences on muscle gene promoters by heterodimers of myogenic basic helix–loop–helix (bHLH) transcription factors, such as MyoD, with the ubiquitous bHLH proteins E12 and E47 is a key event in skeletal myogenesis. However, homodimers of MyoD or E47 are unable of binding to and activating muscle chromatin targets, suggesting that formation of functional MyoD/E47 heterodimers is pivotal in controlling muscle transcription. Here we show that p38 MAPK, whose activity is essential for myogenesis, regulates MyoD/E47 heterodimerization. Phosphorylation of E47 at Ser140 by p38 induces MyoD/E47 association and activation of muscle-specific transcription, while the nonphosphorylatable E47 mutant Ser140Ala fails to heterodimerize with MyoD and displays impaired myogenic potential. Moreover, inhibition of p38 activity in myocytes precludes E47 phosphorylation at Ser140, which results in reduced MyoD/E47 heterodimerization and inefficient muscle differentiation, as a consequence of the impaired binding of the transcription factors to the E regulatory regions of muscle genes. These findings identify a novel pro-myogenic role of p38 in regulating the formation of functional MyoD/E47 heterodimers that are essential for myogenesis.
Keywords: E protein, MRF, muscle differentiation, p38 MAPK, transcription
Introduction
MyoD is a master regulator of skeletal muscle development belonging to the family of basic helix–loop–helix (bHLH) myogenic transcription factors, collectively referred to as muscle regulatory factors (MRFs), which include MyoD (Davis et al, 1987), Myf5 (Braun et al, 1989), myogenin (Edmonson and Olson, 1989) and MRF4 (Rhodes and Konieczny, 1989). MyoD (as the other MRFs) induces muscle-specific transcription upon binding to E-box consensus sequence(s) (CANNTG) within muscle-specific promoters via its basic domain (Lassar et al, 1989; Blackwell and Weintraub, 1990; Davis et al, 1990). Homodimers of MyoD are transcriptionally inactive, presenting a very poor capacity for muscle E-box binding; however, MyoD acquires high affinity for the muscle E box upon heterodimerization through the HLH domain with ubiquitous bHLH proteins, referred to as E proteins (Murre et al, 1989a; Blackwell and Weintraub, 1990; Davis et al, 1990; Lassar et al, 1991). Among the E proteins that have been shown to serve as partners for the MRFs are E12 and E47, encoded by the E2A gene, with E47 being expressed at significantly higher levels than E12 in C2C12 myoblasts (Watada et al, 1995). The formation of the active MyoD/E heterodimer complex is negatively regulated by Id family members during myoblast proliferation. The Id proteins have an HLH domain but lack the amino-terminal associated basic region necessary for DNA binding (Christy et al, 1991). Id inhibits MyoD activity in vivo by forming either transcriptionally inactive complexes of MyoD-Id or by forming heterodimers with E-proteins and effectively blocking the formation of active MyoD/E protein complexes (Jen et al, 1992).
Homodimers of E47, like those of MyoD, cannot bind to and activate muscle chromatin targets. More specifically, E47 homodimers are incapable of activating transcription other than from B-cell-specific promoters (Shen and Kadesch, 1995). Selective and productive recognition of E boxes on muscle promoters requires heterodimerization of MyoD with E12 or E47, rendering the formation of this functional dimer the key event in skeletal myogenesis (Murre et al, 1989a; Blackwell and Weintraub, 1990; Lassar et al, 1991; Kadesch, 1992). Thus, deciphering the molecular mechanisms regulating the formation and functional activity of the MyoD/E protein transcription factor is central for the understanding of muscle differentiation.
p38 MAP kinase (MAPK) plays a fundamental role in the transition of myoblasts to differentiated myocytes (Cuenda and Cohen, 1999; Zetser et al, 1999; Wu et al, 2000). p38 was previously shown to induce the activity of the muscle coactivator MEF2 and to stimulate MyoD activity through an unknown indirect mechanism, independently of MEF2 (Zetser et al, 1999; Wu et al, 2000). Recently, p38 has been shown to target the SWI/SNF complex to muscle promoters (Simone et al, 2004). As both MEF2 and SWI/SNF are ubiquitously expressed proteins, the question of how p38 exerts its specific pro-myogenic effects still remains. In this study, we unambiguously demonstrate that phosphorylation of E47 by p38 is required for the formation of the functional MyoD/E47 heterodimer in vitro and in vivo. In particular, we show that Ser140 of E47 is a key regulatory site phosphorylated by p38 in MyoD-directed muscle differentiation. These findings identify a novel pro-myogenic role of p38 MAPK in regulating the formation of functional MyoD/E47 heterodimers that are essential for myogenesis.
Results
p38 activation promotes MyoD/E47–DNA binding activity and E-box-mediated muscle transcription
The regulation of the functional MyoD/E protein heterodimer formation is pivotal in controlling muscle gene transcription. On the other hand, p38 MAPK is necessary for myogenesis since blockade of its activity in C2C12 myoblasts by SB203580 (assessed by in vitro kinase assay) abrogates both muscle-specific gene expression (myogenin and myosin heavy chain, MHC) and myoblast fusion in differentiation-promoting conditions (DM) (Figure 1A, compare GM to DM). We analyzed p38 activity and MyoD/E47 heterodimer binding to muscle E boxes during myogenesis, by Western blotting using an anti-phospho-p38-specific antibody and by electrophoretic mobility shift assay (EMSA) using specific antibodies for MyoD and E47, respectively, observing that both events are timely and specifically induced in C2C12 myoblasts undergoing differentiation (Figure 1B). These results suggested a correlation between activation of p38 and MyoD/E47–E-box complex formation in differentiating myoblasts. Next, we investigated the effect of p38 inhibition on MyoD/E47 heterodimer-mediated muscle-specific transcription upon transfection of the muscle-specific MCK-luc construct. Abrogation of p38 activity selectively inhibited muscle creatine kinase (MCK) promoter activity during myoblast differentiation (Figure 1C). Coexpression of MyoD and E47 in NIH3T3 fibroblasts resulted in a higher MCK-dependent luciferase activity than expression of MyoD alone, while no MCK activity was obtained by cotransfecting E47 alone (Figure 1D, left; Supplementary Figure 1A). MyoD activity (when transfected alone) can be ascribed to its association with endogenous E47 (Supplementary Figure 1A). Importantly, overexpression of a constitutively active form of MKK6, MKK6(b)E (from now on referred to as MKK6), a p38 activating kinase, increased MyoD-dependent MCK-luc reporter activity in an SB203580-dependent manner (Supplementary Figure 1). An interaction between p38 and MEF2C, a myogenic coactivator, was suggested by previous studies (Wu et al, 2000), and the MCK promoter contains two MEF2 binding sites (Amacher et al, 1993), suggesting that the activation of the MCK promoter by differentiation-induced p38 could be mediated by MEF2. To exclude this possibility, we used a reporter containing four E boxes from the MCK promoter, which can exclusively be activated by heterodimers of the MRFs. SB203580 treatment reduced the MKK6-induced MyoD/E47-mediated activity from the muscle E-box promoter in fibroblasts (Figure 1D, right; Supplementary Figure 1), and the activity from the E box in differentiating myocytes (Figure 1E). MKK6-activated p38 also induced the formation of the MyoD/E47–E-box complex, as shown by EMSA, while no protein–DNA complex was detected in cells transfected with MyoD or E47 alone (Figure 1F), in agreement with the reported low affinity of the MyoD or E47 homodimers for the muscle E box (Lassar et al, 1991). These results indicated that p38 induces the formation of the functional MyoD/E47–DNA binding activity necessary for muscle-specific transcription.
Figure 1.
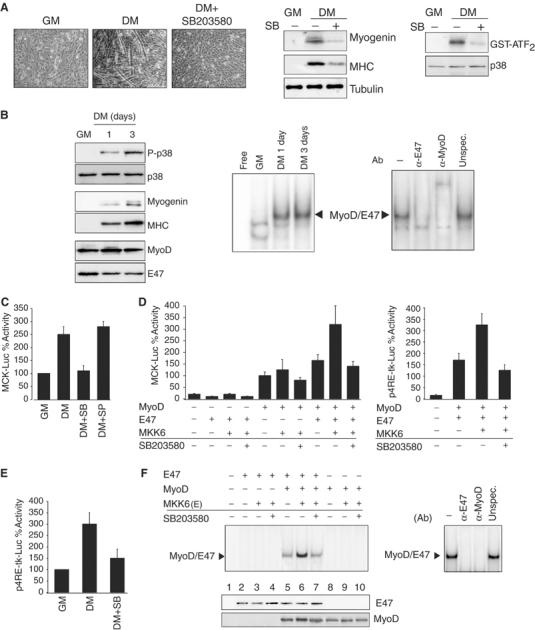
p38 activation promotes MyoD/E47–DNA binding activity and E-box-mediated myogenic transcription. (A) Inhibition of p38 MAPK activity abrogates myoblast differentiation and fusion. C2C12 myoblasts were cultured in growth medium (GM) and then shifted to differentiation medium (DM) for 3 days in the presence or absence of SB203580 (10 μM). Left: Effect of SB203580 on myoblast fusion in DM. Low-power views of representative fields are shown. Magnifications are × 100. Middle: p38 activity from cell extracts corresponding to the above treatments was analyzed by in vitro kinase assay using GST-ATF2 as a substrate. Right: The effect of inhibition of p38 by SB203580 treatment on myogenic differentiation was analyzed by Western blotting using antibodies against myogenin and MHC. (B) p38 activity and MyoD/E47–E-box complex formation are concomitantly induced in DM, compared to GM. Left: p38 phosphorylation is induced during myoblast differentiation. C2C12 myoblasts were cultured in GM and then shifted to DM for 1 and 3 days. Activation of p38 was determined by Western blotting of whole-cell extracts (WCE) using a specific anti-phospho-p38 antibody (P-p38). C2C12 myogenic differentiation was assessed by Western blotting using antibodies against myogenin and MHC. MyoD and E47 levels were also analyzed in GM and DM. Right: MyoD/E47–E-box binding activity is induced during myoblast differentiation. Nuclear extracts (NE) of C2C12 myoblasts cultured in GM and DM for 1 and 3 days were prepared and analyzed by EMSA using the E-box double-stranded oligonucleotide (corresponding to the right E box of the MCK promoter) as a probe. The arrowhead indicates specific MyoD/E47–E-box complex formation during C2C12 differentiation. Right: The specificity of the MyoD/E47 heterodimer (corresponding to C2C12 cells after 24 h in DM) was analyzed by EMSA, after incubating NE with specific antibodies against MyoD or E47, or with an irrelevant antibody. (C) MCK transcriptional activity during myoblast differentiation depends on p38 MAPK activity. C2C12 cells were transfected with pMCK-Luc and after 24 h cells were either maintained in GM or shifted to DM for an additional 48 h. Cells in DM were treated or not with the p38 MAPK inhibitor SB203580 (SB) or with the JNK MAPK inhibitor SP600125 (SP) (20 μM), and luciferase activity was measured subsequently. (D) p38 MAPK activity induces MyoD/E47-dependent transcription from the MCK or 4RE-tk promoters. NIH3T3 fibroblasts were transfected with MCK-Luc (left) or 4RE-tk-luc (right) promoter-reporter plasmids with MyoD and/or E47 expression vectors. When indicated, MKK6 expression plasmid (+) or empty vector (−) was included in the transfected DNA. Cells were maintained for 48 h in GM, in the presence or absence of SB203580, and luciferase activity was measured subsequently. All transfection experiments were normalized to β-galactosidase activity. (E) SB203580 treatment inhibits the reporter activity of p4RE-tk-luc in C2C12 differentiating cells. C2C12 cells were transfected with p4RE-tk-luc and analyzed as in panel C. (F) MyoD/E47–DNA binding activity is induced by ectopic activation of p38 MAPK. Left: NIH3T3 cells were transiently transfected with E47 (lanes 2–7) and/or MyoD (lanes 5–10), or empty vector (lane 1), and MKK6(E) (lanes 3, 4, 6, 7, 9 and 10) expression plasmids, in the absence or presence of SB203580. Protein–DNA binding was analyzed by EMSA after incubation of NE with the labeled E box from the MCK promoter (as in (B)). Right: The composition of the MyoD/E47–E-box binding activity (corresponding to lane 6) was analyzed by EMSA, after incubating NE with specific antibodies against MyoD or E47, or with an irrelevant antibody. Western blotting against ectopic E47 and MyoD proteins was performed to show equal expression of transfected plasmids.
p38 increases the formation of the MyoD/E47 heterodimer rather than its binding to DNA
We have shown that MKK6-activated p38 increases the formation of the MyoD/E47–DNA complex. p38 may be increasing either the affinity between E47 and MyoD or the binding of the heterodimer to the E-box-containing DNA. To test these possibilities, we used a construct (MyoD∼E47) that expresses the MyoD protein tethered to the E47 protein (Neuhold and Wold, 1993). Since cells transfected with this construct will contain ‘preformed' MyoD/E47 heterodimers, we reasoned that if p38 enhances MyoD/E47 dimer formation, transfection of MKK6 into NIH3T3 cells should have no effect on the DNA binding activity of a MyoD∼E47 fusion protein. MKK6 induced neither the DNA binding of a MyoD∼E47 tethered protein (Neuhold and Wold, 1993) (originating ‘preformed' MyoD/E47 heterodimers, as evidenced by using specific MyoD and E47 antibodies in EMSA) (Figure 2A and B) nor the MyoD∼E47-dependent luciferase activity (Figure 2C), contrary to the induction observed when MyoD and E47 were transfected separately (Figures 2A and 1D, right), indicating that p38 induced specifically the formation of the MyoD/E47 heterodimer rather than its binding to DNA. Early studies had shown that MRFs could dimerize with E proteins in vitro in the absence of DNA (Lassar et al, 1991). Immunoprecipitation of MyoD followed by immunoblotting with E47 demonstrated the association between MyoD and E47 in differentiating, but not in proliferating, myoblasts (Figure 2D); this association was prevented by SB203580 treatment, but was resistant to DNaseI activity (Figure 2E), indicating that MyoD/E47 heterodimerization during myogenic differentiation in vivo depends on p38 activity but not on DNA.
Figure 2.
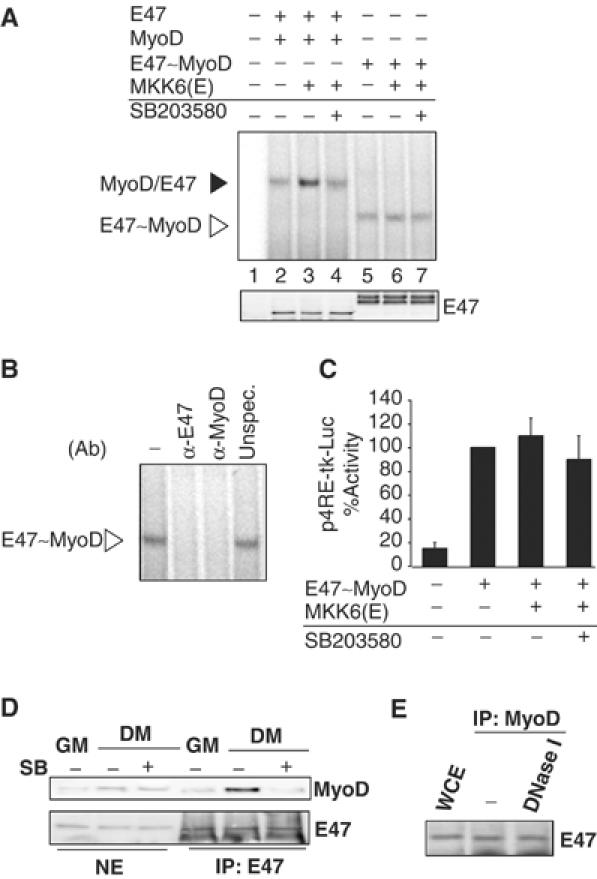
p38 induces MyoD/E47 heterodimerization rather than its binding to DNA. (A) Effect of MKK6-activated p38 on MyoD and E47 heterodimerization. NIH3T3 cells were transiently transfected with expression plasmids for E47 and MyoD (lanes 2–4), or MyoD∼E47 (lanes 5–7), or empty vector (lane 1), and MKK6(E) (lanes 3, 4, 6 and 7), in the absence or presence of SB203580. Protein–DNA binding activity was analyzed by EMSA, as in Figure 1F. (B) Specificity of the E-box–MyoD∼E47 binding activity. The composition of the MyoD∼E47 tethered dimer binding to the E box (from lane 6) was analyzed by EMSA, after incubating NE with specific antibodies against MyoD or E47, or with an irrelevant antibody. Western blotting against ectopic E47 and MyoD protein was performed to show equal expression of transfected plasmids. (C) The transcriptional activity of the MyoD∼E47 tethered dimer is not potentiated by activated p38. MyoD∼E47 expression plasmid was cotransfected with the 4RE-tk-luc reporter plasmid in NIH3T3 cells. When indicated, MKK6 expression plasmid (+) or empty vector (−) was included in the transfection experiment in the presence or absence of SB203580. Cells were assayed for luciferase expression 48 h following transfection under GM conditions. All transfection experiments were normalized to β-galactosidase activity. In the case of mixtures of MyoD+E47 monomers compared with tethered dimers, molar normalization was based on the monomer protein-coding sequences (i.e. 1 U of MyoD plus 1 U of E47 compared with 1 U of MyoD∼E47). (D) p38 activity is necessary for MyoD and E47 dimerization during C2C12 cell differentiation. C2C12 cells were cultured in GM or differentiated for 1 day in DM, in the absence or presence of SB203580, and NE were immunoprecipitated with an anti-E47 antibody. Immunoblotting was performed with an anti-MyoD antibody. NE were subjected to immunoblotting with antibodies against MyoD and E47, as control of endogenous protein levels. (E) MyoD heterodimerizes with E47 in the absence of DNA. C2C12 cells were differentiated for 1 day in DM, and MyoD was immunoprecipitated from WCE, and the immunoprecipitate was divided in two fractions: one was left untreated and the other was subjected to 30 min of DNaseI treatment. Both fractions were analyzed for the presence of E47 by Western blotting.
Phosphorylation of E47, but not of MyoD, by p38 is necessary for MyoD/E47 association in vitro
The mechanism by which p38 increases MyoD/E47 heterodimer formation could be direct (on either or both proteins) and/or indirect (by activating additional signaling molecules). We examined whether MyoD and E47 proteins may serve as direct substrates for p38 by in vitro phosphorylation analysis using GST fusion proteins and purified MKK6-activated p38 in the presence of [γ-32P]ATP. While the level of GST-MyoD labeling was very low, GST-E47 exhibited intense labeling, which augmented with a higher amount of the fusion protein (Figure 3A). GST-ATF2, but not GST alone, was phosphorylated (Figure 3A), as expected. These results demonstrated the ability of p38 to efficiently phosphorylate E47, but not MyoD, in vitro; that is, E47 is a better in vitro phosphorylation substrate than MyoD for p38.
Figure 3.
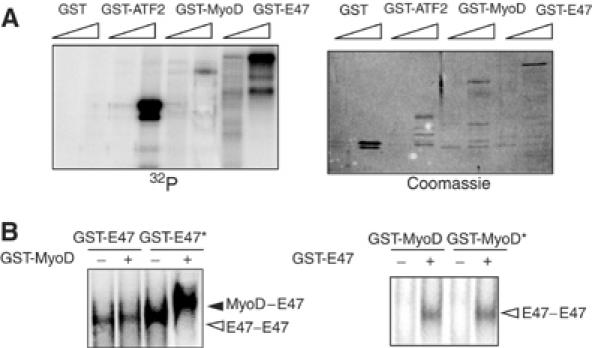
E47, but not MyoD, phosphorylation by p38 is required for the formation of the MyoD/E47 heterodimer. (A) E47 is a better substrate than MyoD for p38 phosphorylation in vitro. Two different amounts of purified GST-E47, GST-MyoD, GST-ATF2 and GST alone (200 and 1000 ng) were incubated with 200 ng of MKK6-activated p38α. All incubations were performed in the presence of [γ-32P]ATP, and labeled proteins separated by SDS–PAGE and detected by autoradiography. GST-ATF2 and GST alone were used as positive and negative p38 MAPK phosphorylation control substrates, respectively. Coomassie staining confirmed equal loading of the purified recombinant proteins. (B) Phosphorylation of E47 by p38 is necessary for the formation of the E47/MyoD heterodimer–E-box complex. Left: GST-E47 (200 ng) was in vitro phosphorylated by activated p38 in the presence or absence of ATP. Phosphorylated (E47*) and unphosphorylated (E47) forms of E47 were incubated with 200 ng of GST-MyoD (or 200 ng of GST, as control) and proteins were allowed to form homo- or heterodimers prior to the addition of the 32P-labeled E-box probe, and analyzed by EMSA. Right: A similar assay was performed by incubating p38-phosphorylated (MyoD*) and unphosphorylated (MyoD) forms of MyoD with GST-E47, followed by EMSA. Arrowheads indicate the formation of the MyoD/E47 heterodimer (black) and E47/E47 homodimers (white) binding to the E box. No MyoD/MyoD homodimers were found bound to the E box, irrespective of the phosphorylation status of MyoD.
The dimerization capacities of MyoD and/or E47 were compared before and after in vitro phosphorylation by p38. First, GST-E47 was incubated with MKK6-activated p38 in the presence or absence of ATP, and the phosphorylated (GST-E47*) or unphosphorylated (GST-E47) E47 proteins were further incubated with GST alone or GST-MyoD, and subjected to EMSA. Unphosphorylated GST-E47 bound the E box only as an homodimer, even in the presence of MyoD (Figure 3B, left); phosphorylated GST-E47* showed increased homodimer formation when compared to unphosphorylated E47 (alone or mixed with MyoD) (Figure 3B, left). However, unphosphorylated GST-MyoD dimerized with phosphorylated GST-E47*, as evidenced by the appearance of a slower migrating band corresponding to the MyoD/E47 heterodimer (Figure 3B, right). The reverse experiment with phosphorylated (GST-MyoD*) or unphosphorylated (GST-MyoD) MyoD proteins and GST-E47 confirmed that the direct physical association of MyoD and E47 in vitro requires phosphorylation of E47, but not MyoD, by p38 (Figure 3B, right).
p38 phosphorylates E47 at Ser140 in vitro and in vivo
Analysis of the primary amino-acid sequence of the E47 protein revealed 13 MAPK phosphorylation consensus sites (SP/TP) (Figure 4A). Sequential E47 deletion mutants containing a gradient of SP/TP sites (from 0 to 13) were generated and found to be phosphorylated by p38 to a similar extent. The fact that the shorter E47 deletion (GST-2*), containing two potential N-terminal phosphorylation sites (serines 135 and 140), showed the same labeling as the full-length E47 (Figure 4A) suggested that these two serines might be the ones phosphorylated by p38. To confirm this hypothesis, Ser135 and Ser140 were individually mutated to alanine in the GST-E47 full-length fusion protein and the mutant proteins subjected to p38 phosphorylation analysis in vitro. E47 mutated at Ser135 (S135A) retained the phosphorylation level of wild-type (WT) E47 by p38 (Figure 4B), while mutation of Ser140 (S140A) generated a protein that was refractory to p38-directed γ-ATP labeling, although it was capable of binding to p38, as shown by pull-down assays (Figure 4C). This result demonstrated that Ser140 is required for phosphorylation of E47 by p38, but not for the interaction between both molecules.
Figure 4.
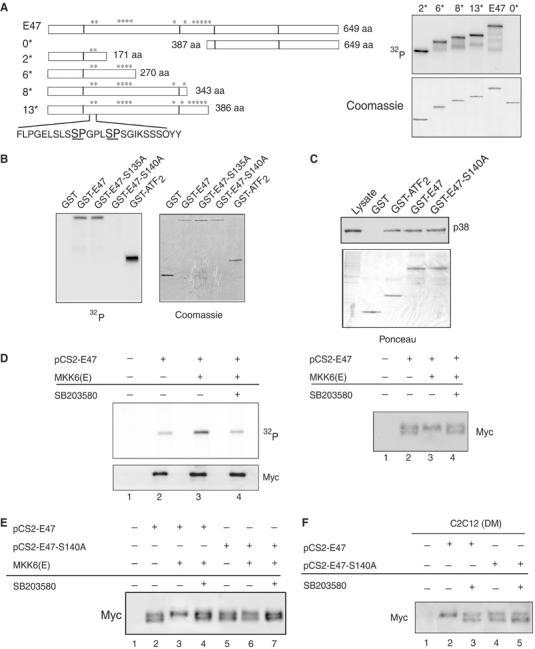
p38 phosphorylates E47 at Ser140 in vitro and during myoblast differentiation in vivo. (A) Phosphorylation of E47 deletion mutants by p38 in vitro. Equal amounts of purified full-length E47 (E47) or deletion mutants (GST-0*, GST-2*, GST-6*, GST-8*, GST-13*) (see left panel) were incubated with 200 ng of activated p38. All incubations were performed in the presence of [γ-32P]ATP, and labeled proteins separated by SDS–PAGE and detected by autoradiography (right). GST numbering indicates the number of potential p38 phosphorylation sites in the E47 deletion fragments. The C-terminal fragment of E47, GST-0* (containing no p38 consensus sites), was used as a negative control for the phosphorylation assays. Amino-acid sequence including Ser135 and Ser140 is shown. Coomassie staining showed that equal amounts of substrate proteins were used in the in vitro phosphorylation assays. (B) E47 is phosphorylated at Ser140 in vitro. Purified full-length E47 or E47 mutated at serines 135 or 140 were incubated with 200 ng of activated p38, and analyzed for its p38 phosphorylation in vitro, as in (A). Coomassie staining confirmed equal loading of the different purified proteins. (C) p38 MAPK interacts with full-length and mutated E47 in vitro. 293T cell lysates were incubated with the indicated GST fusion proteins bound to glutathione–Sepharose beads. Total cellular proteins (lysate) and bound cellular proteins were analyzed using an anti-p38 antibody. The lysate lane contains 1/10 of the input used in incubations. Ponceau staining showed that comparable amounts of GST proteins were used in the pull-down assays. (D) E47 is phosphorylated by p38 in vivo. Left: 293T cells were transfected with a myc-tagged E47 expression vector, pCS2-E47, together with either an empty vector (−) or MKK6 expression plasmid, as indicated, and metabolically labeled with [32P]orthophosphate. Myc-tagged proteins were immunoprecipitated from cell extracts, separated using SDS–PAGE and blotted. Phosphorylated proteins were visualized by autoradiography. The lower panel shows immunoblotting of the membrane with an anti-Myc (9E10) antibody to confirm equal expression of E47-transfected plasmids in all lanes. Right: NE from 293T cells (transfected as above) were prepared and the differential mobility of the hypo- and hyperphosphorylated E47 proteins was resolved by SDS–PAGE under special separation conditions, as explained in Materials and methods. Myc-E47 protein was visualized by immunoblotting of the membrane with anti-Myc (9E10) antibody. (E) Mutation of Ser140 to alanine impairs phosphorylation of E47 by p38 in vivo. 293T cells were transfected with a myc-tagged expression vector for E47 WT, pCS2-E47, or Ser140-mutated E47, pCS2-E47-S140A, together with either an empty vector (−) or the MKK6 expression plasmid, as indicated. E47 proteins were resolved as in panel D. (F) Phosphorylation of E47 protein by differentiation-induced p38 in C2C12 cells in DM. C2C12 cells were transfected with the myc-tagged expression vectors pCS2-E47 and pCS2-E47-S140A. At 12 h after transfection, cells were shifted to DM for 1 day, in the absence or presence of SB203580, and NE obtained and separated, as in panel D.
Our next aim was to investigate whether E47 is phosphorylated by p38 in vivo, and whether Ser140 is the specific E47 phosphorylation site. In vivo phosphorylation of E47 (WT and S140A) was assayed either by in vivo radioactive labeling with [32P]orthophosphate or by differential electrophoretic mobility of phosphorylated proteins in Myc-E47- and Myc-E47-S140A-293T-transfected cells, respectively (Figure 4D–F). We found that the WT Myc-E47 protein presented basal levels of phosphorylation (Figure 4D, left, lane 2), being detected as a doublet, corresponding to slow- and fast-migrating forms of the protein (Figure 4D, right, lane 2). Ectopic expression of MKK6 increased the in vivo labeling of Myc-E47 (Figure 4D, left, lane 3) and resulted in the exclusive detection of the slow-migrating form of the protein (Figure 4D, right, lane 3). Incubation of cells with SB203580 reversed the MKK6-induced effects (Figure 4D, right and left, lane 4). Similar to the WT protein, the Myc-E47-S140A mutant was detected as a doublet (Figure 4E, left, lanes 2 and 5); however, E47-S140A remained refractory to MKK6-activated p38 in vivo (as shown by the invariable presence of the doublet) (Figure 4E, left, lanes 3 and 6). These results demonstrated that E47 is phosphorylated by p38 in vivo, and that Ser140 is the specific E47 phosphorylation site.
E47 is phosphorylated at Ser140 by muscle differentiation-induced p38
Our next aim was to investigate if E47 is phosphorylated by differentiation-induced p38 during myogenesis. C2C12 myoblasts were transfected with expression vectors for Myc-E47 or Myc-E47-S140A, and shifted to differentiation-promoting conditions (DM) for 48 h, with or without SB203580 treatment, and the electrophoretic mobility of E47 analyzed as above. In the absence of SB203580, WT Myc-E47 is detected exclusively as a slow-migrating, hyperphosphorylated species in C2C12 myocytes after 2 days in DM, while treatment of cells with SB203580 resulted in the appearance of a fast-migrating, hypophosphorylated form of Myc-E47, that is, a doublet (Figure 4F, lanes 2 and 3). In contrast, E47 mutated at Ser140 was detected as a doublet in differentiating C2C12 cells, regardless of the SB203580 treatment (Figure 4F, lanes 4 and 5). Myc-E47 appeared as a single, slow-migrating species in proliferating C2C12 myoblasts, with or without SB205880 treatment (Supplementary Figure 2A). Moreover, in differentiated C2C12 myocytes, endogenous E47 is detected as unique, slow-migrating form, which shifted to a fast-migrating form upon SB203580 cell treatment, indicating that endogenous E47 is phosphorylated by muscle differentiation-induced p38 (Supplementary Figure 2B). Altogether, these results confirmed that the E47 protein is phosphorylated at Ser140 in vivo by p38 during myogenesis.
Ser140 of E47 is a regulatory site for MyoD/E47 heterodimerization and muscle-specific gene transcription
We evaluated whether Ser140 of E47 is a regulatory site for the formation of the functional MyoD/E47 dimer. p38-phosphorylated GST-E47* was able to heterodimerize with GST-MyoD in vitro, whereas no heterodimer was formed between GST-MyoD and p38-unphosphorylatable E47-S140A* (Figure 5A). These data demonstrate that the direct physical association of MyoD and E47 in vitro requires Ser140 phosphorylation of E47 by p38. Next, we aimed to analyze whether this mechanism was also required for MyoD/E47 heterodimer formation in vivo. NIH3T3 cells were cotransfected with expression vectors for MyoD and for E47 or E47-S140A, with or without MKK6, in the absence or presence of SB203580, and formation of the heterodimer and binding to the E box were analyzed by EMSA. Overexpression of MKK6 significantly increased the level of dimerization between MyoD and WT E47 in vivo (Figure 5B), while it had no effect on MyoD/E47-S140A dimer formation in the same cells (Figure 5B). Furthermore, MKK6 induced MyoD/E47-mediated, but not MyoD/E47-S140A-mediated, MCK- and 4RE-dependent transcriptional activities, in an SB203580-depending fashion (Figure 5C). These results demonstrated that Ser140 of E47 is a regulatory site for MyoD/E47-mediated muscle-specific transcription.
Figure 5.
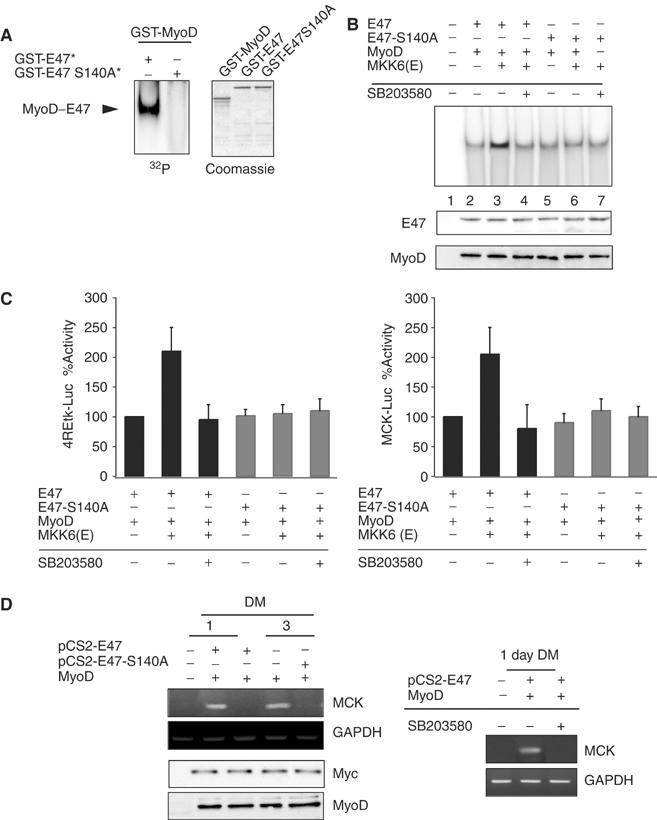
Ser140 of E47 is a regulatory p38 site in muscle differentiation in vitro and in vivo. (A) Mutation of Ser140 of E47 abrogates MyoD/E47 heterodimer formation in vitro. In all, 200 ng of GST-E47 or GST-E47-S140A was in vitro phosphorylated by activated p38 (GST-E47* and GST-E47-S140A*, respectively) and incubated with 200 ng of GST-MyoD, followed by addition of the 32P-labeled E-box probe, and analyzed by EMSA, as in Figure 3B. The arrowhead indicates the formation of the MyoD/E47 heterodimer. Coomassie staining confirmed equal amounts of the purified recombinant proteins. (B) Top: Inability of activated p38 to induce MyoD/E47-S140A heterodimer binding activity in vivo. NIH3T3 cells were transiently transfected with expression plasmids for MyoD and for WT or serine 140-mutated E47, with or without MKK6(E), or with vector alone (lane 1), in the absence or presence of SB203580, as indicated. Protein–DNA binding was analyzed by EMSA as in Figure 1F. Bottom: Western blotting was performed to show equal amounts of MyoD- and E47-transfected plasmids. (C) Influence of E47 Ser140 phosphorylation by p38 on 4RE-tk and MCK promoter activities. NIH3T3 cells were cotransfected with 4RE-tk-luc (left) or MCK-Luc (right) promoter-reporter plasmids, and WT or mutated E47 and MyoD expression plasmids. When indicated, MKK6 expression plasmid (+) or empty vector (−) was included in the transfection experiment with the presence or absence of SB203580. Cells were assayed for luciferase expression 48 h following transfection under GM conditions. All transfection experiments were normalized to β-galactosidase activity. (D) Inability of E47-S140 to induce muscle-specific expression from MyoD-expressing NIH3T3 fibroblasts in differentiation-promoting conditions. Left: NIH3T3 fibroblasts were transfected with expression vectors for MyoD and for Myc-E47 or Myc-E47-S140A, or with an empty vector (−), and switched to DM for 1 and 3 days. MCK and GAPDH mRNA expression was analyzed by RT–PCR using specific primers. Ectopic expression of MyoD and E47 (WT and mutated) proteins was analyzed by Western blotting using anti-MyoD and anti-Myc antibodies, respectively. Right: Inhibition of p38 activity abrogates MCK mRNA expression from MyoD/E47-transfected fibroblasts. NIH3T3 fibroblasts were transfected with expression vectors for MyoD and for Myc-E47, and cultured in DM for 1 day, in the absence or presence of SB203580. MCK and GAPDH mRNA expression was analyzed by RT–PCR (as in (D), left).
Overexpression of E47-S140A abrogates MyoD-mediated conversion of fibroblasts into myocytes
Ectopic expression of MyoD family members in non-muscle cells results in the expression of muscle-specific genes under differentiation-promoting conditions (DM) (Tapscott et al, 1988; Murre et al, 1989b; Davis et al, 1990; Lassar et al, 1991). Based on the above results, it is conceivable that overexpression of the nonphosphorylatable mutant form of E47 should be sufficient for reducing or even preventing MyoD-mediated muscle-specific gene expression in non-muscle cells in DM. For this purpose, NIH3T3 fibroblasts were transfected with MyoD and E47 (WT or S140A) to comparable levels of expression, transferred to DM for 1 and 3 days, and the expression of the endogenous MCK gene analyzed as an index of muscle conversion. As shown in Figure 5D, MCK mRNA levels were induced in MyoD- and E47-expressing fibroblasts after 1 and 3 days in DM, but not in cells expressing MyoD and E47-S140A (Figure 5D, left); moreover, the expression of MCK in MyoD- and E47-transfected NIH3T3 cells was inhibited by SB203580 (Figure 5D, right). These results demonstrated that the conversion of non-muscle cells to the myogenic lineage by MyoD is abrogated by the nonphosphorylatable E47 mutant; furthermore, they proved that MyoD/E47-mediated myogenic conversion requires p38 activity.
Inhibition of p38 activity downregulates MyoD/E47 heterodimer formation and binding to muscle chromatin targets during myogenesis
Altogether, our results are consistent with a model in which phosphorylation of E47 by p38 is required for MyoD/E47 association and subsequent binding to muscle E boxes during myogenesis (see Figure 1B). Therefore, blockade of p38 activity should inhibit formation of cellular MyoD/E47 heterodimers on muscle promoters in vivo. SB203580 treatment (2 h) of C2C12 differentiated cells did not alter significantly the endogenous MyoD and E47 protein levels (Figure 6A, left); however, the formation of the MyoD/E47 heterodimer was dramatically reduced in SB203580-treated cells (Figure 6A, right). Furthermore, chromatin immunoprecipitation (ChIP) assays showed that MyoD and E47 are specifically associated with the promoter regions of the MCK and MHC genes (Figure 6B, left), confirming that the MyoD/E47 heterodimer binds to E boxes from both promoter regions in differentiating cells in vivo. In contrast, SB203580 treatment prevented full MyoD and E47 binding to the E boxes on both muscle promoters (Figure 6B, right). Therefore, p38 activity is necessary for efficient MyoD and E47 binding to the E boxes on both muscle promoters in vivo.
Figure 6.
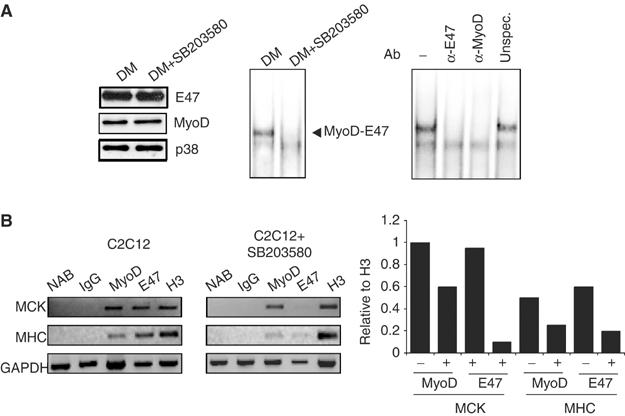
p38 activity is required for MyoD/E47 heterodimer binding to muscle promoters in myocytes in vivo. (A) Inhibition of p38 activity downregulates MyoD/E47 heterodimer formation and binding to DNA. C2C12 cells differentiated for 24 h in DM were further treated (or not) with 10 μM SB203580 for 2 h. NE and WCE were prepared for EMSA and Western blotting analyses, respectively. Left: The influence of SB203580 treatment on MyoD, E47 and p38 expression levels in C2C12 differentiating cells was analyzed by immunoblotting using specific antibodies. Right: MyoD/E47–DNA binding activity in C2C12 is inhibited by SB203580 treatment. NE from C2C12 cells subjected to the indicated cell treatments were analyzed by EMSA using the MCK-E box as a probe. The arrowhead indicates specific MyoD/E47–E-box complexes. The specificity of the MyoD/E47 heterodimer (corresponding to C2C12 cells after 24 h in DM) was analyzed by EMSA, after incubating NE with specific antibodies against MyoD or E47, or with an irrelevant antibody. (B) Influence of p38 inhibition on ChIP of two muscle regulatory regions with antibodies against MyoD and E47. Chromatin derived from C2C12 cells treated as in panel A was immunoprecipitated with control IgG, MyoD, E47 or histone 3 (H3) antibodies, and subjected to PCR with primers corresponding to the MCK and MHC promoter regions containing MyoD binding sites (E boxes) (primer sequences are reported in Materials and methods). NAB, no antibody. This is a representative experiment of three, with similar results. Bottom: Graph represents experimental values of the shown experiment relative to H3 immunoprecipitation.
Discussion
The binding of the MyoD transcription factor to the E-box sequences present in the promoters of muscle genes exclusively as a heterodimer with E47 constitutes the crucial event required for the initiation of muscle-specific transcription. Homodimers of MyoD or of E47 can neither bind to nor activate muscle-specific genes (Murre et al, 1989a; Lassar et al, 1991). Thus, elucidating the mechanism(s) regulating MyoD heterodimerization is central to understanding how myogenesis is initiated. Here we demonstrate that p38 MAPK, whose activity is essential for myogenesis (Cuenda and Cohen, 1999; Zetser et al, 1999; Wu et al, 2000), regulates MyoD/E47 heterodimerization by phosphorylating E47. In particular, we show that phosphorylation of E47 at Ser140 by p38 induces MyoD/E47 dimerization, subsequent binding to E box and activation of muscle-specific gene transcription, while nonphosphorylatable E47 (E47-Ser140Ala) fails to associate with MyoD and displays reduced myogenic potential. Moreover, inhibition of p38 activity precludes E47 phosphorylation at Ser140 in differentiating myocytes, correlating with a reduction of MyoD/E47 heterodimer formation, impaired binding to muscle regulatory regions and inefficient muscle differentiation. These findings unambiguously demonstrate a novel mechanism of p38 in regulating formation of functional MyoD/E47 heterodimers that are essential for myogenesis.
Despite the fact that MyoD and E47 proteins are expressed in muscle cells and could potentially form homodimers capable of binding to the muscle E box, we have exclusively detected the MyoD/E47 heterodimer form in differentiating myocytes, in agreement with previous studies (Murre et al, 1989a; Blackwell and Weintraub, 1990; Lassar et al, 1991). Moreover, we have demonstrated that E47 phosphorylated by p38 exhibits a higher ability to associate with MyoD in vitro and in vivo. Experiments using a tethered MyoD∼E47 protein further showed that E47 phosphorylation by p38 increased its affinity for MyoD rather than the binding of the heterodimer to DNA. In particular, our results have revealed that E47 is phosphorylated exclusively at Ser140 by p38 in vitro and in vivo, resulting in increased MyoD/E47 association and MyoD-mediated muscle-specific transcription. These observations led us to propose that p38, by phosphorylating Ser140 of E47, effectively increases the number of functional MRF-E protein complexes in cells. Indeed, phosphorylation of E47 by p38 at Ser140 enhanced MyoD/E47 heterodimer formation, while the nonphosphorylatable E47 mutant Ser140Ala was refractory to the pro-dimerizing effect of p38. Moreover, conversion of non-muscle cells to the myogenic lineage by artificial expression of MyoD was prevented by nonphosphorylatable E47-S140A.
The bHLH domain of MRFs and E proteins is considered the minimum structure required for myogenic activity, with the basic region necessary for DNA binding and the HLH motif essential for dimerization, while other domains of these transcription factors control other functions such as transcriptional activation (Murre et al, 1989a; Davis et al, 1990; Voronova and Baltimore, 1990; Ellenberger et al, 1994), fitting with an established model of transcription factors being composed of independently functioning modules. Studies by Lu and Sloan (2002) demonstrated that this assumption is not completely accurate. By using forced heterodimers of MyoD/E47, these authors showed that dimers containing complete bHLH domains but lacking other regions of E47 (in particular, E47 proteins lacking N-terminus) have only 20% of the DNA binding ability and transcriptional transactivating activity of WT dimers, suggesting that regions of E47 outside of the bHLH domain may also be targets for regulation. Interestingly, our study has identified Ser140, located at the N-terminus of E47, as a functional p38 phosphoacceptor site required for full MyoD/E47–DNA complex formation, reinforcing the idea that the N-terminal region may affect dimerization and DNA binding of the bHLH proteins. At present, only the crystal structure of bHLH is available (Ellenberger et al, 1994; Ma et al, 1994), while N-terminal region of E47 is not well characterized. It is conceivable that changes in the protein structure of the N-terminal region will affect the configuration of the C-terminal domain. Two possibilities might be considered: (1) the N-terminal region of E47 sequesters the C-terminal dimerization/DNA binding domain and phosphorylation relieves this effect, or (2) there is a second dimerization interface located in the N-terminal region of E47 that is sensitive to phosphorylation. These predictions will require determining the complete structure of E47. It cannot be excluded, however, that this post-translational modification of E47 may additionally affect the efficiency with which the MyoD/E47 transcription factor interacts with other components of the transcriptional machinery to activate target muscle genes.
Phosphorylation was previously associated to the homodimer–heterodimer equilibrium of bHLH proteins. Hypophosphorylation of two serines N-terminal to the bHLH domain in E47 allowed DNA binding by E47 homodimers in B cells (Sloan et al, 1996). However, no information about the ligand, its receptor or any of the intracellular signal transducers participating in the hypophosphorylation status of E47 during B-cell development was provided. Even though casein kinase II and mos kinase were also proposed to influence MyoD dimerization in vitro, their role in regulating in vivo myogenesis has never been elucidated (Johnson et al, 1996; Lenormand et al, 1997). p38 was recently shown to inactivate E47 transcriptional activity (Page et al, 2004), although no clear mechanism or functional implication in myogenesis was demonstrated. Here we provide a novel mechanism demonstrating that p38 MAPK (whose activity is necessary for myogenesis) regulates MyoD/E47 association in vitro and in vivo. In particular, we show that phosphorylation of E47 at Ser140 by p38 induces MyoD/E47 dimerization, subsequent binding to E box and activation of muscle-specific gene transcription.
p38 was previously shown to induce the activity of the muscle coactivator MEF2 and, recently, to target the SWI/SNF complex to muscle promoters through the functional MyoD transcription factor (Wu et al, 2000; Simone et al, 2004). Based on these studies and on our present findings, we propose that p38 may complex two timely and closely linked muscle transcription mechanisms. By binding to and phosphorylating E47, p38 promotes formation of the functional MyoD/E47 heterodimer (this study), which will allow subsequent recruitment of the SWI/SNF complex on myogenic promoters.
In summary, our findings provide compelling evidence that p38 is a key determinant in regulating the formation of functional MyoD/E47 heterodimers that are essential for skeletal myogenesis, and identify a novel role of p38 in regulating the initiation of muscle differentiation.
Materials and methods
Cell culture
C2C12, NIH3T3 and 293T cell lines were cultured in growth medium (GM): DMEM containing 10% of FBS. To induce differentiation, GM was replaced by DM: DMEM supplemented with 2% HS.
Reagents
The p38 kinase inhibitor SB203580 and JNK inhibitor SP600125 (JNK inhibitor II) (Calbiochem) were used at a final concentration of 10 and 20 μM, respectively. Antibodies used were as follows: anti-p38 MAP kinase (sc-535, Santa Cruz Biotechnology), anti-phospho-p38 MAP kinase Thr180/Tyr182 (Cell Signaling), anti-Myogenin (Developmental Studies Hybridoma Bank), anti-MyoD (sc-760, Santa Cruz Biotechnology), anti-E47 (sc-763, Santa Cruz Biotechnology), anti-Myc clone 9E10 (Developmental Studies Hybridoma Bank), anti-Myc (sc-789, Santa Cruz Biotechnology), anti-MHC clone MF20 (Developmental Studies Hybridoma Bank) and anti α-tubulin clone DM1A (Sigma).
Plasmid constructs
The following plasmids were used (details are provided as Supplementary data): pEMSV-MyoD, pcDNA3-MKK6(b)E, pcDNA3-HA-p38, pcDNA3-E47 and pECE-E47∼MyoD, p4RE-tk-Luc and pMCK-Luc. pGEX-E47-S140A, pGEX-E47-S135A, pcDNA3-E47-S140A and pCS2-E47-S140A (containing the indicated substitutions of serines by alanines) were generated by oligonucleotide-directed mutagenesis. Plasmids for GST-E47 fusion proteins (full length and deletion mutants) and for Myc-tagged E47 proteins (full length and mutants) were generated by standard PCR techniques.
Transfections
C2C12, NIH3T3 and 293T cells were transiently transfected with the different plasmids by the standard calcium phosphate precipitation method. Details of the procedure are described in Supplementary data.
Western blot analysis
Cells were lysed to obtain WCE (using immunoprecipitation buffer) or NE (see EMSA, below). WCE were resolved by SDS–PAGE, transferred to PVDF membranes and incubated overnight at 4°C with indicated primary antibodies. Details of buffer and procedure are described in Supplementary data.
Purification of GST fusion proteins and pull-down assays
pGEX, pGEX-E47 (full length and corresponding deletions), pGEX-E47-S135A, pGEX-E47-S140A and pGEX-ATF2 fusion proteins were expressed in Escherichia coli BL21 (DE3) and purified using glutathione–Sepharose beads (Amersham Pharmacia Biotech). Details of the procedure are described in Supplementary data.
p38 kinase assay
p38 was immunoprecipitated from WCE with an anti-p38 antibody and immunocomplexes were recovered with protein A–Sepharose and washed, as described (Suelves et al, 2004).
In vitro kinase assays
Activated p38 was incubated with purified GST fusion proteins as described (Suelves et al, 2004).
In vivo phosphorylation assays
293T cells were transfected with different myc-tagged expression plasmids. Cells were labeled 36 h after transfection during 3 h with 0.5 mCi [32P]orthophosphate per milliliter in the absence or presence of 10 μM SB203580. Details of the procedure are described in Supplementary data.
Immunoprecipitation assays
Cells were washed in ice-cold PBS and subsequently lysed in ice-cold immunoprecipitation buffer (see above). MyoD protein was immunoprecipitated using anti-MyoD (Sc-760). For DNaseI analysis, see Supplementary data.
Electrophoretic mobility shift assays with nuclear extracts
NE were obtained from C2C12 cells and NIH3T3 cells. Details of EMSA are described in Supplementary data.
EMSA with purified proteins
Purified GST proteins were phosphorylated by activated p38 in the presence or absence of ATP, and subjected to EMSA. Details of the procedure are described in Supplementary data.
RNA isolation and RT–PCR analysis
Total RNA (5 μg) was analyzed by reverse transcription–polymerase chain reaction (RT–PCR). DNA primers and details of the procedure are described in Supplementary data.
Chromatin immunoprecipitation assays
Antibodies for ChIP assays are MyoD and E47 purchased from Santa Cruz (sc-760 and sc-763) and histone H3 from Abcam. Details of ChIP analysis (including DNA primers and PCR conditions) are described in Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Methods
Acknowledgments
We sincerely thank Drs V Sartorelli and A Nebreda for helpful comments and critical reading of the manuscript. We are also thankful to Drs F Miralles, C Caelles, C Murre, V Ruiz, S Tapscott, PL Puri, K Walsh, B Wold, E Olson, J Han, T Ellenberger, S Sloan, A Cano, G Caretti and M Di Padova for generously providing us with reagents and/or for helpful discussions. We appreciate the help received from all PMC lab members throughout this work. This project was supported by SAF2001-0482, MDA, SAF2004-06983, Marató-TV3, FIS-PI021873, AFM and AI-HI2002-134.
References
- Amacher SL, Buskin JN, Hauschka SD (1993) Multiple regulatory elements contribute differentially to muscle creatine kinase enhancer activity in skeletal and cardiac muscle. Mol Cell Biol 13: 2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TK, Weintraub H (1990) Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science 250: 1104–1110 [DOI] [PubMed] [Google Scholar]
- Braun T, Buschhausen-Deker G, Bober E, Tannich E, Arnold HH (1989) A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J 8: 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy BA, Sanders LK, Lau LF, Copeland NG, Jenkins NA, Nathans D (1991) An Id-related helix–loop–helix protein encoded by a growth factor-inducible gene. Proc Natl Acad Sci USA 88: 1815–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Cohen P (1999) Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol Chem 274: 4341–4346 [DOI] [PubMed] [Google Scholar]
- Davis RL, Cheng PF, Lassar AB, Weintraub H (1990) The MyoD DNA binding domain contains a recognition code for muscle-specific gene activation. Cell 60: 733–746 [DOI] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987–1000 [DOI] [PubMed] [Google Scholar]
- Edmonson DG, Olson EN (1989) A gene with homology to the myc similarity region of MyoD1 is expressed during myogenesis and is sufficient to activate the muscle differentiation program. Genes Dev 3: 628–640 [DOI] [PubMed] [Google Scholar]
- Ellenberger T, Fass D, Arnaud M, Harrison SC (1994) Crystal structure of transcription factor E47: E-box recognition by a basic region helix–loop–helix dimer. Genes Dev 8: 970–980 [DOI] [PubMed] [Google Scholar]
- Jen Y, Weintraub H, Benezra R (1992) Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev 6: 1466–1479 [DOI] [PubMed] [Google Scholar]
- Johnson SE, Wang X, Hardy S, Taparowsky EJ, Konieczny SF (1996) Casein kinase II increases the transcriptional activities of MRF4 and MyoD independently of their direct phosphorylation. Mol Cell Biol 16: 1604–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch T (1992) Helix–loop–helix proteins in the regulation of immunoglobulin gene transcription. Immunol Today 13: 31–36 [DOI] [PubMed] [Google Scholar]
- Lassar AB, Buskin JN, Lockshon D, Hauschka SD, Weintraub H, Davis RL, Apone S (1989) MyoD is a sequence-specific DNA binding protein requiring a region of myc homology to bind to the muscle creatine kinase enhancer. Cell 58: 823–831 [DOI] [PubMed] [Google Scholar]
- Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H (1991) Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 66: 305–315 [DOI] [PubMed] [Google Scholar]
- Lenormand JL, Benayoun B, Guillier M, Vandromme M, Leibovitch MP, Leibovitch SA (1997) Mos activates myogenic differentiation by promoting heterodimerization of MyoD and E12 proteins. Mol Cell Biol 17: 584–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sloan SR (2002) The basic helix–loop–helix domain of the E47 transcription factor requires other protein regions for full DNA binding activity. Biochem Biophys Res Commun 290: 1521–1528 [DOI] [PubMed] [Google Scholar]
- Ma PC, Rould MA, Weintraub H, Pabo CO (1994) Crystal structure of MyoD bHLH domain–DNA complex: perspectives on DNA recognition and implications for transcriptional activation. Cell 77: 451–459 [DOI] [PubMed] [Google Scholar]
- Murre C, McCaw PS, Baltimore D (1989a) A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, ad myc proteins. Cell 56: 777–783 [DOI] [PubMed] [Google Scholar]
- Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, Weintraub H, Baltimore D (1989b) Interactions between heterologous helix–loop–helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 58: 537–544 [DOI] [PubMed] [Google Scholar]
- Neuhold LA, Wold B (1993) HLH forced dimers: tethering MyoD to E47 generates a dominant positive myogenic factor insulated from negative regulation by Id. Cell 74: 1033–1042 [DOI] [PubMed] [Google Scholar]
- Page JL, Wang X, Sordillo LM, Johnson SE (2004) MEKK1 signaling through p38 leads to transcriptional inactivation of E47 and repression of skeletal myogenesis. J Biol Chem 279: 30966–30972 [DOI] [PubMed] [Google Scholar]
- Rhodes SJ, Konieczny SF (1989) Identification of MRF4: a new member of the muscle regulatory factor gene family. Genes Dev 3: 2050–2061 [DOI] [PubMed] [Google Scholar]
- Shen CP, Kadesch T (1995) B-cell-specific DNA binding by an E47 homodimer. Mol Cell Biol 15: 4518–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL (2004) p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 36: 738–743 [DOI] [PubMed] [Google Scholar]
- Sloan SR, Shen CP, McCarrick-Walmsley R, Kadesch T (1996) Phosphorylation of E47 as a potential determinant of B-cell-specific activity. Mol Cell Biol 16: 6900–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suelves M, Lluís F, Ruiz V, Nebreda AR, Muñoz-Cánoves P (2004) Phosphorylation of MRF4 transactivation domain by p38 mediates repression of specific myogenic genes. EMBO J 23: 365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB (1988) MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 242: 405–411 [DOI] [PubMed] [Google Scholar]
- Voronova A, Baltimore D (1990) Mutations that disrupt DNA binding and dimer formation in the E47 helix–loop–helix protein map to distinct domains. Proc Natl Acad Sci USA 87: 4722–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watada H, Kajimoto Y, Umayahara Y, Matsuoka T, Morishima T, Yamasaki Y, Kawamori R, Kamada T (1995) Ubiquitous, but variable, expression of two alternatively spliced mRNAs encoding mouse homologues of transcription factors E47 and E12. Gene 153: 255–259 [DOI] [PubMed] [Google Scholar]
- Wu Z, Woodring P, Bhakta K, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL (2000) p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 20: 3951–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetser A, Gredinger E, Bengal E (1999) p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol Chem 274: 5193–5200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Methods