

Abstract
BET3 is a component of TRAPP, a complex involved in the tethering of transport vesicles to the cis-Golgi membrane. The crystal structure of human BET3 has been determined to 1.55-Å resolution. BET3 adopts an α/β-plait fold and forms dimers in the crystal and in solution, which predetermines the architecture of TRAPP where subunits are present in equimolar stoichiometry. A hydrophobic pocket within BET3 buries a palmitate bound through a thioester linkage to cysteine 68. BET3 and yeast Bet3p are palmitoylated in recombinant yeast cells, the mutant proteins BET3 C68S and Bet3p C80S remain unmodified. Both BET3 and BET3 C68S are found in membrane and cytosolic fractions of these cells; in membrane extractions, they behave like tightly membrane-associated proteins. In a deletion strain, both Bet3p and Bet3p C80S rescue cell viability. Thus, palmitoylation is neither required for viability nor sufficient for membrane association of BET3, which may depend on protein–protein contacts within TRAPP or additional, yet unidentified modifications of BET3. A conformational change may facilitate palmitoyl extrusion from BET3 and allow the fatty acid chain to engage in intermolecular hydrophobic interactions.
Keywords: α/β-plait, BET3 protein, protein palmitoylation, TRAPP complex, vesicle tethering
Introduction
Intracellular targeting and transport of proteins in eukaryotic cells depend on a variety of proteins and protein complexes involved in the coating, budding, release, uncoating, tethering and fusion of vesicles. Vesicle budding commonly relies on formation of a protein coat around the bud and is initiated by the binding of a GTPase on the exit membrane. Subsequently, the GTPase interacts with a GTPase-activating protein, leading to GTP hydrolysis and the recruitment of coat-forming proteins to the vesicle bud. These coat-forming proteins interact with proteins on the membrane, initially leading to polymerization and stabilization of the bud and subsequently to dissection of the vesicle from the membrane (Barlowe, 1995; Springer et al, 1999). Vesicle fusion to the target compartments is triggered by membrane proteins termed soluble N-ethyl-sensitive factor (NSF) attachment protein (SNAP) receptors or SNAREs that have to be located on both the vesicle (as v-SNARE) and the target membranes (as t-SNARE) and which interact with a number of soluble factors. In addition to the interaction with SNAPs, binding of an AAA ATPase, NSF and membrane-specific multi-protein complexes to the vesicle is essential for tethering and fusion (reviewed by Hay and Scheller, 1997; Nichols and Pelham, 1998; Jahn et al, 2003; Bonifacino and Glick, 2004).
The attachment of vesicles to their target membrane is mediated by a Rab GTPase and tethering factors. These tethers are multi-component complexes which are transport process-specific (Lowe, 2000; Whyte and Munro, 2002). For example, a complex called exocyst is involved in the tethering of exocytotic vesicles to the plasma membrane (TerBush and Novick, 1995). Along with the conserved oligomeric Golgi (COG) and Golgi-associated retrograde protein (GARP) complexes, the exocyst has been classified as a quadrefoil tethering complex (Whyte and Munro, 2002). The TRAPP complexes, involved in vesicle transport to the Golgi, are unrelated to the quadrefoil complexes. TRAPP has been reported to act as a guanine nucleotide exchange factor for the Rab GTPase Ypt1p (Jones et al, 2000; Wang et al, 2000).
The TRAPP I complex, necessary for tethering ER-derived vesicles to the Golgi, comprises approximately equimolar amounts of seven proteins (Bet3p, Bet5p, Trs20p, Trs23p, Trs31p, Trs33p and Trs85p; yeast nomenclature) whose sequences are highly conserved from yeast to plants and mammals (Sacher et al, 2000, 2001; Sacher and Ferro-Novick, 2001). The TRAPP II complex that guides vesicles inside the Golgi comprises TRAPP I and three additional proteins: Trs65p, Trs120p and Trs130p. The individual deletion of many TRAPP complex components is lethal (Sacher et al, 2000), highlighting their importance for vesicle transport and, possibly, other cellular processes. A tagged human BET3 has been used to purify a human TRAPP complex and to identify its subunits (Gavin et al, 2002).
BET3 was identified by a synthetic lethal screen as a yeast gene with a role in targeting and fusion of ER-derived vesicles to the Golgi membrane (Rossi et al, 1995). The bet3-1 mutant showed a defect in secreting invertase, α-factor and carboxypeptidase Y, leading to accumulation of these proteins at the ER-to-Golgi stage of transport prior to SNARE complex formation. Sequence analysis identified two novel motifs, [44]LX2#GX2#GX2LXE[57] and [133]G#2XGXL[139] (where X represents any amino acid and # corresponds to a hydrophobic residue; human BET3 numbering), conserved between Bet3p and two other TRAPP subunits: Trs31p and Trs33p (Sacher et al, 2000). The bet3-1 mutant, in which the TRAPP complex fails to form, involves alteration of the second conserved glycine residue (G52) in the first motif, underscoring the importance of this motif in Bet3p function (Figure 1).
Figure 1.
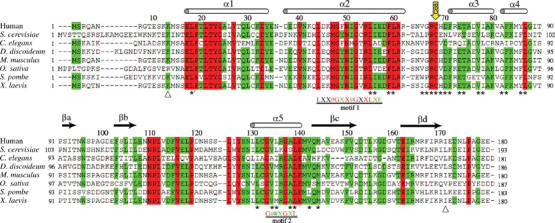
Structure-based multiple sequence alignment of BET3 homologs (Gönczy et al, 2000; Glöckner et al, 2002; Klein et al, 2002; Mammalian Gene Collection (MGC) Program Team, 2002; Wood et al, 2002). The secondary structure assignment for human BET3 is represented by cylinders (α-helix) and arrows (β-strand) above the aligned sequences. The triangles below the sequence alignment denote the extent of the assigned human BET3 structure described in this paper (residues 13–171 with respect to the wild-type protein sequence). Fully conserved residues are highlighted in red and additional, highly conserved residues (identical in at least six out of eight aligned sequences) are shaded green. Residues marked by an asterisk below the aligned sequences denote the 27 residues lining the hydrophobic pocket with any atom within 6 Å of the palmitoyl group (including L18 from the adjacent molecule in the BET3 dimer). A yellow arrow points towards the residue C68. The multiple sequence alignment was prepared using CLUSTAL (Higgins et al, 1992).
BET3 is a central subunit of TRAPP that has been used to precipitate the intact tethering complex both from yeast and from human cells (Sacher et al, 1998; Gavin et al, 2002). As the first step towards a structural characterization of TRAPP, here we present the crystal structure of human BET3, a 20-kDa protein comprising 180 amino acids with 54% identity to its yeast ortholog (Sacher et al, 1998). We show BET3 to be dimeric in the crystal and in solution and identify a covalent modification with a palmitate deeply buried within a hydrophobic channel of the protein. These findings have important implications for the architecture of the TRAPP I complex and its mode of Golgi membrane attachment.
Results and discussion
Structure analysis and quality of the model
For affinity purification of BET3, the protein was labeled with a His6 and a StrepII tag, fused to its N- and C-termini, respectively (corresponding to a construct comprising 200 amino acids). The fusion protein had a calculated molecular mass (Mr) of 22 582 kDa, but eluted from the size-exclusion chromatography column at an Mr of approximately 42 kDa (data not shown).
The structure of BET3 was determined by the single-wavelength anomalous diffraction (SAD) technique and refined using data to 1.55-Å resolution. In general, the quality of the electron density map is good for 151 of the expected 200 residues in the fusion protein, the exception being the loop which connects residues 114 and 121 for which the electron density is weak and segmental flexibility must therefore be assumed. Additionally, the N-terminal 21 and the C-terminal 20 residues, including the affinity tags, are not visible in the electron density map and the structure described here is 12 and nine residues, respectively, shorter than the wild-type protein. The side chains of eight residues (K13, E35, E57, K84, F115, L118, N121 and R170) have weak density associated with them and a further seven side chains (K32, K46, R62, R67, N109, E117 and D120) are disordered and have been truncated to the last visible side chain atom in the final model. In the Ramachandran diagram, all residues of the model are located in the most favorable or additionally allowed regions (89.5 and 10.5%, respectively).
BET3 structure
The BET3 monomer has overall dimensions of approximately 36 × 42 × 46 Å and is constructed from four β-strands and five α-helices which harbor 16 and 39%, respectively, of the residues of the total wild-type polypeptide chain (Figure 2A). In addition, there are two segments of 310-helix (residues 120–122 and 154–156). The relationship between the elements of secondary structure and their positions in the sequence is presented in the alignment of representative BET3 homologs in Figure 1.
Figure 2.
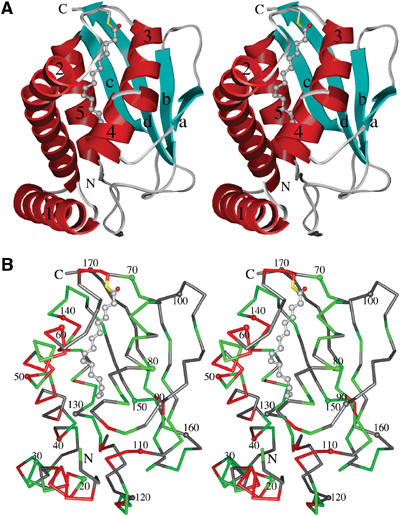
Stereo diagrams of human BET3. (A) Schematic representation of the BET3 monomer with the helices and strands colored red and cyan, respectively, and labeled. The palmitate molecule covalently linked to residue C68 through a thioester bond is shown in atom colors (carbon, grey; oxygen, red; sulfur, yellow). (B) Cα trace with every tenth residue dotted and numbered. The pattern of sequence conservation across members of the BET3 family is also illustrated using the coloring scheme outlined in Figure 1. Unless otherwise stated, molecular drawings were prepared using CHIMERA (Huang et al, 1996).
The secondary structural elements of BET3 are arranged in an α/β-plait topology constructed by a twisted, antiparallel, four-stranded β-sheet on one side, with the helices forming the other side of the structural motif. The pattern of sequence conservation between BET3 homologs indicates that both identical and highly conserved residues are primarily associated with the α-helical face of the molecule (Figure 2B), implying that the β-sheet simply acts as a structural scaffold. Additionally, residues 110–119 in the flexible loop connecting βb to α5 are highly conserved, which suggests that they most likely represent a binding motif for one of the other components of the TRAPP complex. Database searches using VAST (Madej et al, 1995; Gibrat et al, 1996) and DALI (Holm and Sander, 1993) reveal only limited topological similarities between BET3 and other proteins in the Protein Data Bank (PDB). Specifically, these relate to overlaps of elements of the α/β-plait fold with members of the (α+β) class of proteins. Overall, these similarities do not suggest that the PDB contains any protein with a fold significantly similar to BET3. The recently released structures of mouse BET3 (PDB entries 1VPG, 1WC8, 1WC9) show this protein to share a closely similar fold with human BET3.
BET3 dimer
BET3 forms a dimer around the crystallographic two-fold axis, primarily involving interactions between α1 and, to a lesser extent, α2, α4 and the N-terminal segment of α5 (Figure 3A). Analysis of the molecular surface buried between these subunits shows that the overall accessible surface area (Lee and Richards, 1971) of the BET3 monomer is approximately 9000 Å2 and that 18% (∼1500 Å2) of the subunit surface area is buried in the crystallographic dimer. Furthermore, the residues located at the dimer interface are highly conserved among the sequences of BET3 homologs (see Figures 1 and 2B). A dimeric arrangement is consistent with the biophysical data reported in this paper and with previous studies, which suggested that there are at least two copies of Bet3p and the other protein subunits in the fully assembled TRAPP complex (Sacher et al, 2000).
Figure 3.
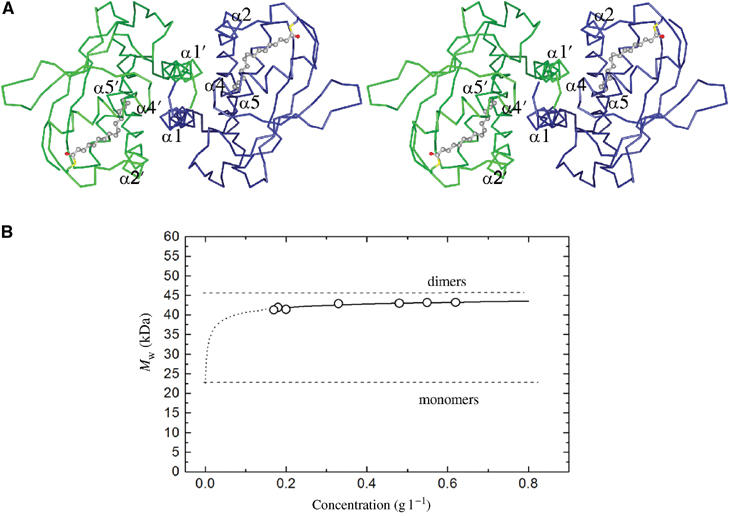
BET3 dimer in the crystal and in solution. (A) Cα trace of the BET3 dimer viewed down the crystallographic two-fold axis. Secondary structural elements interacting at the dimer interface are labeled. The two subunits are shown in different colors. (B) Concentration dependence of the relative molecular mass (Mr) of BET3 in aqueous buffer from analytical ultracentrifugation, confirming a dimeric quaternary structure. The dotted line represents a monomer–dimer equilibrium fit with a Kd of 448 nM.
The dimeric structure of BET3 adopted in the crystal has been additionally confirmed by analytical ultracentrifugation (Figure 3B). The molecular mass (Mr) obtained from sedimentation equilibrium experiments ranged between 43.2 kDa at a protein concentration of 0.62 g l−1 and 41.2 kDa at 0.17 g l−1. These values are somewhat lower than the expected value for the dimeric protein, and the positive concentration dependence of Mr is typical for an equilibrium between monomers and dimers. Based on the partial concentrations of both species at all Mr values, an equilibrium dissociation constant (Kd) of 448±34 nM was calculated. Given this affinity, we cannot be certain that in cells BET3 is always present as a dimer, but a dimeric structure seems very likely in compartments enriched in BET3.
BET3 palmitoylation
A striking feature of the BET3 structure is the presence of a hydrophobic pocket within the core of the α-helical face that is approximately 20 Å in length and is formed by the C-terminal end of α2, the loop connecting α2 to α3 and helices α3 to α5 (see Figure 2A). Additionally, within the dimer, the N-terminal end of α1 blocks the base of the pocket adjacent to the subunit interface in the symmetry-related molecule (see Figure 3A). An elongated electron density feature spans the pocket (Figure 4A) in the structures of both the wild-type and the selenomethionine-incorporated protein (data not shown), and is therefore unlikely to be an artifact of crystallization. The shape of the electron density is consistent with it being a palmitate molecule covalently attached to the protein through a thioester linkage to the fully conserved residue C68 (C-S separation 1.9 Å), corresponding to C80 in Bet3p. There are 27 highly conserved and predominantly hydrophobic residues that line the pocket with any atom within 6 Å of the S-palmitoyl group (Figure 4B). In all, 20 are identical in at least six of the eight aligned BET3 sequences, whereas conservative substitutions are seen at the remaining seven positions: V65 (L/I/W), G66 (P/S), H69 (E/N/S/A), I78 (L/V), A82 (G), Y86 (F), and L135 (I/M) (see Figure 1, human BET3 sequence numbering). The recently published structure of mouse BET3 (Kim et al, 2005) also shows a fatty acid moiety occupying this site. Palmitate is the most commonly found S-linked fatty acid (Dunphy and Linder, 1998), implying that palmitoylation of BET3 is a natural modification likely to play a functional role. The occlusion of the fatty acid chain inside a hydrophobic crevice of the protein explains why BET3 could be purified as a soluble protein from recombinant yeast cells.
Figure 4.
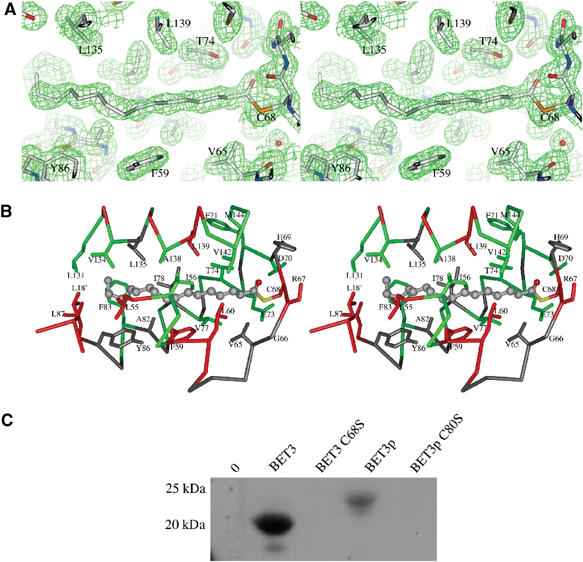
Covalent modification of BET3 with palmitate. (A) A portion of the (2m∣Fobs∣−D∣Fcalc∣)αcalc electron density map centered on the palmitoyl chain and contoured at 1.2σ (prepared using PyMOL; DeLano, 2003). The separation between the C1-atom position of the palmitoyl group and the side chain Sγ of C68 is approximately 1.9 Å, clearly indicating that a thioester bond has formed between these two entities. (B) The side chains of the 27 residues lining the hydrophobic pocket with at least one atom residing within 6 Å of the palmitate molecule are shown using the same coloring scheme as outlined in Figure 1. In all, 20 of these residues are identical in at least six of the eight aligned BET3 sequences, whereas conservative substitutions are seen at the remaining seven positions: V65 (L/I/W), G66 (P/S), H69 (E/N/S/A), I78 (L/V), A82 (G), Y86 (F), L135 (I/M). (C) Metabolic labeling with [3H]palmitate. AH22ura3 yeast strain with and without BET3 plasmids was grown in the presence of [3H]palmitic acid. BET3 and Bet3p were purified and samples were analyzed by SDS–PAGE and fluorography.
The palmitoylation of BET3 in vivo was shown with metabolic labeling of yeast cells expressing BET3, Bet3p and the corresponding cysteine-to-serine mutants (BET3 C68S, Bet3p C80S). Wild-type BET3 and Bet3p purified from these cultures were palmitoylated as shown by fluorography, whereas no palmitic acid was attached to the mutant proteins (Figure 4C). Differences in intensity are due to the higher expression level of BET3 compared to Bet3p.
The crystal structure of one of the other constituents of the TRAPP complex, the monomeric murine SEDL, an ortholog of yeast Trs20p, has previously been reported (Jang et al, 2002). SEDL shares a folding topology identical to the N-terminal regulatory domain of two SNAREs, yeast Ykt6p and mouse Sec22b, despite undetectable sequence homology between these proteins. This has led to the suggestion that SEDL serves regulatory and/or adaptor functions through multiple protein–protein interactions. All three proteins are involved in ER-to-Golgi vesicle transport, and the structural similarities suggest that they may interact with different partners that belong to the same family of proteins. Interestingly, the N-terminal domain of Ykt6p has been implicated in the palmitoylation of the fusion factor Vac8 (Dietrich et al, 2004). Ykt6p presents palmitoyl-CoA via its N-terminal domain to Vac8, while transfer to Vac8's SH4 domain occurs spontaneously. Furthermore, Trs20p, the SEDL ortholog in yeast, has been reported to interact with Bet3p on the basis of a genome-wide yeast two-hybrid analysis (Ito et al, 2000). This leads to the intriguing possibility that Trs20p (SEDL) exhibits the acyltransferase activity for Bet3p (BET3) palmitoylation.
Possible function of BET3 palmitoylation
Palmitoylation most commonly contributes to the membrane localization of proteins that would otherwise be cytoplasmic, but has also been shown to modulate protein–protein interactions (Dunphy and Linder, 1998). In theory, a single palmitoyl group should be sufficient for membrane association (Peitzsch and McLaughlin, 1993). Since previous studies have shown that TRAPP resides within a Triton X-100-resistant fraction of the Golgi (Sacher et al, 2000), we addressed the question whether BET3 palmitoylation is involved in its membrane anchoring. Preparations of cytosolic and membrane fractions from yeast cells expressing BET3 and BET3 C68S, respectively, were analyzed by immunoblotting. Wild-type and mutant BET3 were detectable both in the cytosol and the membrane (Figure 5A). By loading crude cell lysates on a sucrose cushion, it was demonstrated that BET3 as well as BET3 C68S actually are membrane associated and do not form aggregates (not shown). The membrane-bound forms of the proteins cannot be released by extraction with high salt, urea, carbonate and Triton X-100, a feature associated with tightly membrane-associated proteins (Figure 5B). These data show that BET3 is tightly bound to membranes, but that this interaction is only in part mediated through palmitoylation. We take this to suggest that the tight binding to an anchor protein or another modification is additionally required and sufficient to mediate the membrane association of BET3. Since BET3 from the soluble cytosolic pool was purified for crystallization, we cannot exclude the possibility that an additional modification is present in membrane-bound BET3.
Figure 5.
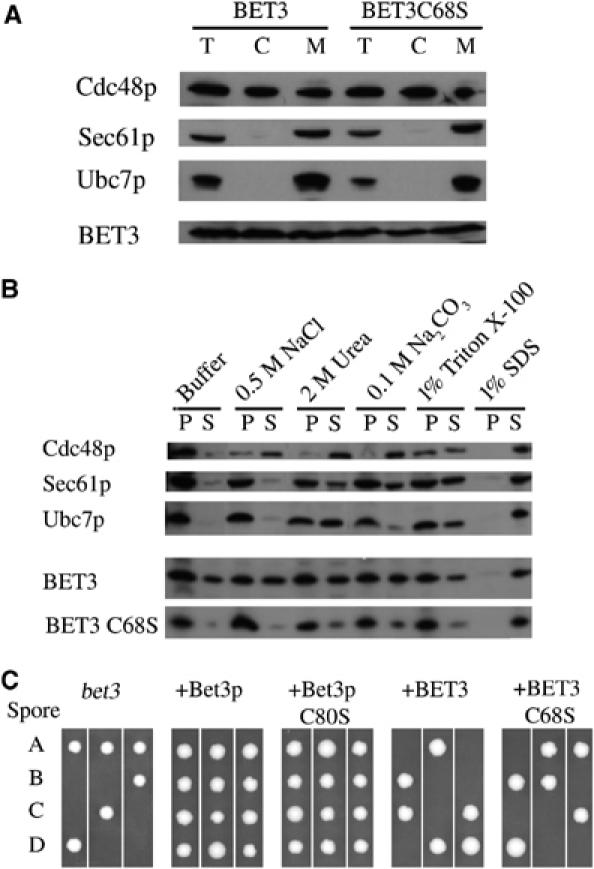
(A) Membrane preparations from yeast expressing BET3 and BET3 C68S. Cleared lysates were subjected to ultracentrifugation. Total cell lysate (T), cytosolic (C), and membrane (M, 10-fold concentrated) fractions were analyzed by SDS–PAGE and immunoblotting. (B) Membrane extractions of BET3 and BET3 C68S. Membranes were treated with buffer, 0.5 M NaCl, 2 M urea, 0.1 M Na2CO3, 1% Triton X-100, or 1% SDS for 30 min on ice. Samples were separated into pellet (P) and supernatant (S) fractions by centrifugation and analyzed by SDS–PAGE and immunoblotting. The integral membrane protein Sec61p and the peripheral membrane-associated proteins Cdc48p and Ubc7p served as reference. (C) Tetrad analysis of yeast Y25984 bet3 deletion strain (EUROSCARF; MAT a/α; his3Δ1/his3Δ1; leu2Δ0/leu2Δ0; lys2Δ0/LYS2; MET15/met15Δ0; ura3Δ0/ura3Δ0; YKR068c∷kanMX4/YKR068c). Sporulation was induced in cells with and without BET3 plasmids, and spores were dissected on YPD plates of individual tetrads.
To address the question if palmitoylation of BET3 is essential for protein function in vivo, BET3 constructs were transformed in a yeast bet3 deletion strain, and sporulation of these transformants was induced. Loss of Bet3p is lethal and results in only two viable spores if no complementation can be achieved through the transformed constructs. In contrast to human SEDL which complements Trs20p (Gecz et al, 2003), BET3 fails to rescue haploid cells with a bet3 deletion despite the high degree of sequence conservation. Since not only Bet3p but also Bet3p C80S complement a bet3 knockout (Figure 5C), palmitoylation of Bet3p does not appear to be essential for cell viability. However, the expression of BET3 under the control of a copper promoter, even when not induced, might lead to a high intracellular level of BET3. A fine-regulatory effect of the palmitoylation on protein stabilization or protein–protein interactions may remain undetected in these cells, when they survive in the absence of a fully functional secretory system.
There is some structural similarity between the palmitate-binding α-helical face of BET3 and the nonspecific lipid-transfer proteins (LTPs) from plants and mammals (Shin et al, 1995) although their complete domain folds do not match. The arrangement of three out of the four helices in barley LTP are similar in BET3 (αA, αD and αC in barley LTP are equivalent to α2, α4 and α5 in BET3; Figure 6A) and the bent helix αA in barley LTP is in a position similar to the kinked helix α2 in BET3. In barley LTP, the binding pocket can contract or expand to accommodate lipids or fatty acids of various sizes (Lerche et al, 1997; Lerche and Poulsen, 1998). These conformational changes are accompanied by a bending motion in αA and structural adjustments at the C-terminus and helix αC.
Figure 6.
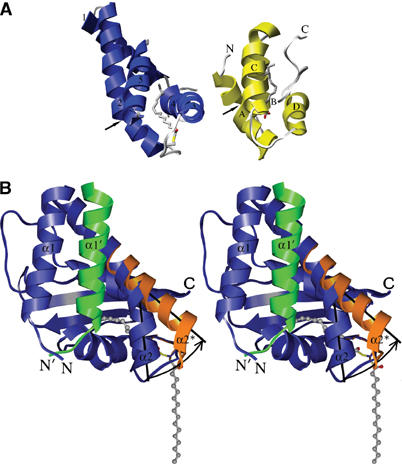
Proposed mechanism for the extrusion of the palmitoyl group from the hydrophobic pocket. (A) Structural similarity between the α-helical face of BET3 (blue) and the nonspecific LTP from barley (yellow), here shown with bound palmitate (Lerche and Poulsen, 1998). Helices A, D and C in barley LTP adopt an arrangement similar to helices 2, 4 and 5 in BET3. The arrows point to the kink in helix α2 of BET3 and to a corresponding position within helix αA of the LTP, where a conformational change is observed when the pocket is adjusted for the binding of differently sized lipid-like ligands. (B) The BET3 monomer is shown in blue and α1′ from the other subunit of the dimer, that blocks the base of the hydrophobic pocket, is highlighted in green. The palmitoyl group is likely to play a role in membrane association but is, however, completely buried within the α-helical face of the molecule. A rotation of approximately 30° in α2 about a hinge region close to G52, coupled with a maximum displacement of approximately 9 Å, would open the pocket and allow the palmitoyl group to slide out of the pocket. An arrow represents the proposed trajectory of α2, and its new position (α2*) is highlighted in orange. Subsequently, it is envisaged that the pocket will close to exclude the bulk solvent.
In the crystal structure of BET3, the palmitoyl group is completely buried within the α-helical face of the molecule. Based on the structural similarity between BET3 and LTP, we propose a model for the extrusion of the buried palmitoyl group from the hydrophobic pocket. The mechanism, not unlike the structural adjustments described for the LTPs (Lerche et al, 1997), involves a rotation of approximately 30° in α2 about a hinge region close to residue G52, coupled with a maximum displacement of approximately 9 Å that would open the pocket and allow the palmitoyl group to slide out of the pocket (Figure 6B). We envisage that this conformational change in α2 will alleviate the extended hydrogen-bonding pattern seen within the putative hinge region of this highly kinked helix and is likely to be triggered by interactions with one or more of the other constituents of the TRAPP complex. Interestingly, the two novel motifs, [44]LX2#GX2#GX2LXE[57] and [133]G#2XGXL[139] (Figure 1; human BET3 numbering), conserved between Bet3p and two other TRAPP components, Trs31p and Trs33p (Sacher et al, 2000), are sandwiched together at the interface between α2 and α5 at the exact position of the proposed hinge region. The bet3-1 mutant that fails to form the TRAPP complex involves alteration of G52 in the first motif, which will perturb the structure of the protein within the proposed hinge region. This suggests an important functional role of the helix kink. The palmitate liberated from the protein in this way would be free to bind to another hydrophobic site such as a membrane bilayer or a binding pocket of another protein. The empty hydrophobic pocket of BET3 could now be available for intermolecular interactions with apolar moieties such as membrane anchors.
Conclusion
The three-dimensional structure of recombinant human BET3, presented here, provides important insights into the molecular basis of TRAPP complex formation and its localization to the cis-Golgi membrane. BET3 adopts an α/β-plait topology and a dimeric quaternary organization in solution and is covalently modified with a palmitate moiety attached to residue C68 which is strictly conserved among all BET3 orthologs, suggesting a functional role. We present a model that delineates a mechanism for extraction of the palmitoyl chain from a crevice on the surface of soluble BET3. This will allow subsequent modulation of interaction with the target membrane or binding other molecules. The characterization of Cys-to-Ser mutants of BET3 showed that palmitoylation does not seem to be essential for cell viability and is not required for membrane recruitment of BET3. Since BET3 behaves as a tightly membrane-bound protein, this attachment may be mediated by an additional modification not present in the crystal structure or through interactions with one of the other components of the TRAPP complex. The hydrophobic cavity of BET3 might also represent a binding pocket for acylated interacting proteins. Finally, a dimeric BET3 defines the stoichiometry of TRAPP, since, for symmetry reasons, it necessitates the recruitment of further dimeric subunits or of pairs of monomeric TRAPP components such as SEDL (Jang et al, 2002).
Cloning and expression
The coding sequence of the human BET3 gene AF041432 (GenBank) was expressed in the yeast Saccharomyces cerevisiae under the control of the inducible CUP1 promoter of the yeast metallothionein gene. The BET3 gene was PCR-amplified from the IMAGE clone p998M1228 with gene-specific primers, using the Expand High Fidelity PCR System (Roche, Penzberg, Germany). BamHI and NotI restriction sites were introduced into the 5′-end of the forward and reverse primer, respectively. The PCR product was cut at the BamHI/NotI site and directionally cloned into the BamHI and NotI sites of the yeast expression vector pYEXTHS-BN (Holz et al, 2002). The yeast BET3 gene YKR068c was PCR amplified from AH22ura3 genomic DNA and cloned accordingly.
Site-directed mutagenesis was performed according to the QuikChange™ mutagenesis protocol essentially as described by Wang and Malcolm (1999). The mutations C68S in BET3 and C80S in Bet3p were introduced using the primers 5′-gctcggtcaaatgttgggaggAgccatgactt tcgggaaactgcg-3′ and 5′-gctagaacggcattgccacgcAgtgagaattt agtgaagacaagc-3′, respectively, and reverse complementary oligonucletides. The sequence of mutant clones was verified by DNA sequencing. Standard media and protocol were used for transformation and tetrad analysis (Ausubel et al, 1994).
The expression cassette of the resulting plasmid pYEXTHS-BN/BET3 comprised the regulated CUP1 promoter, the His6 tag, the coding sequence of BET3 and the StrepII tag sequence. The plasmid was used to transform S. cerevisiae strain AH22ura3 (MATa, ura3Δ, leu2-23,112, his4-519, can1). The resulting transformants were selected on SD-ura medium (2% dextrose, 0.67% yeast nitrogen base (Difco), 40 mg l−1 leucine and 40 mg l−1 histidine). A yeast transformant harboring the BET3 expression plasmid (clone 0500000092, internal ID) was grown at 28°C in WMVIII minimal medium supplemented with histidine in a BioFlow3000 3-l fermenter to an optical density (OD600) of 8. Standard fermentation conditions were as described (Prinz et al, 2003).
For the production of selenomethionine (SeMet)-labeled protein, the yeast cells were grown in pre-cultures containing 3 mg l−1 SeMet. The cultures were grown in BioFlow110 1-l fermenters at 28°C and pH 6.0. 0.5 l WMVIII media containing 50 ml of a 100 × amino-acid solution (Studts and Fox, 1999) and 3 mg l−1 SeMet were inoculated as described above and fed-batch fermentation was performed for 19 h. Thereafter, 50 mg l−1 SeMet and 1 mM CuSO4 were added to induce gene expression and protein labeling.
Protein purification
The cells were suspended in 40% (w/v) lysis buffer containing 140 mM phosphate buffer, pH 9.0, 300 mM NaCl, 2 mM β-mercaptoethanol and 1 mM PMSF and disrupted by six passages through a high-pressure homogenizer Emulsiflex-05 (Avenstin, Canada) at 6°C and 2000 bar. The cleared lysate (adjusted to pH 7.4) was then subjected to metal-affinity chromatography and StrepTactin-affinity chromatography using an Äkta Explorer (Pharmacia Amersham Biosciences, Germany) at room temperature. BD Talon Super flow matrix (Clonetech, Germany) was used for the metal-affinity chromatography. After application of the protein solution, the column was washed with several volumes of 50 mM phosphate buffer, pH 8.0, 150 mM NaCl, and the protein was eluted with 50 mM phosphate buffer, pH 7.0, 150 mM NaCl, 200 mM imidazol. Absorption at 280 nm, conductivity and pressure were monitored. BET3 was further purified by chromatography using a StrepTactin MacroPrep (IBA, Germany) column after equilibration with 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA. After application, the column was washed with four volumes of the same buffer, and subsequently the protein was eluted using 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 2 mM DTT and 2.5 mM D-desthiobiotin (Sigma, Germany). Samples were taken throughout the procedure and analyzed by SDS–PAGE and Western blot.
Metabolic labeling with [3H]palmitate
Strains with BET3 encoding plasmids were grown in WMVIII supplemented with histidine. The AH22ura3 strain was grown in YPD. Cultures of 25 ml were inoculated at OD600 0.5 and grown to a density of 2.5. To the cells, 500 μCi [3H]palmitate (40–60 Ci mmol−1, Amersham Bioscience, Germany), 1 μg ml−1 cerulenin (Sigma) and 1 mM CuSO4 were added and cultures were incubated overnight. Cells were harvested and lysed with glass beads in 50 mM Tris, pH 7.5, 5 mM EDTA, 0.5% SDS and 1 mM PMSF. Lysates were cleared by centrifugation (10 min, 13 000 g) and diluted 5-fold with 20 mM Tris, pH 8, 150 mM NaCl, 1 mM PMSF and a pulldown was performed using StrepTactin MacroPrep resin (IBA, Germany) for 1 h at 4°C. Samples were washed with the same buffer and boiled in SDS loading buffer containing 2.5 mM D-desthioboitin. Proteins were separated by SDS–PAGE, the gel was fixated with 10% acetic acid and soaked with Amplify (Pharmacia), dried and subjected to fluorography.
Membrane preparation and extraction
Yeast microsomes were prepared from a crude protein extract obtained by glass bead lysis essentially as described (Panzner et al, 1995). For extractions, membranes were treated with 50 mM Tris, pH 7.5, 5 mM EDTA, 1 mM PMSF, or 0.5 M NaCl, 2 M urea, 0.1 M Na2CO3, 1% Triton X-100, or 1% SDS and incubated on ice for 30 min. Samples were separated into pellet and supernatant (30 min, 14 000 g) and supernatants were subjected to acetone precipitation. Fractions were analyzed by SDS–PAGE and immunoblotting.
BET3 proteins were detected using Penta-His-HRP conjugate (Qiagen). Bands on immunoblots were visualized with enhanced chemiluminescence (ECL, Amersham Bioscience).
Analytical ultracentrifugation and quaternary structure analysis
The molecular mass of BET3 in aqueous buffer was determined using an analytical ultracentrifuge XL-A (Beckman, Palo Alto, CA) equipped with absorbance optics. Samples of approximately 70 μl protein at different concentrations (0.17–0.62 g l−1) were centrifuged in externally loaded six-channel cells against buffer (15 mM Tris–HCl, pH 8.5, 50 mM NaCl, 0.1 mM EDTA, 0.02% NaN3, 2 mM DTT) for 2 h at 26 000 r.p.m. (overspeed), followed by 26 h at an equilibrium speed of 22 000 r.p.m. at 10°C. The radial concentration distributions of each sample at sedimentation equilibrium were recorded at three different wavelengths between 275 and 290 nm and fitted globally to equation (1)
![]()
with
![]()
using POLYMOLE (Behlke et al, 1997). In these equations, ρ is the solvent density, ν is the partial specific volume, ω is the angular velocity, R is the gas constant and T the absolute temperature. Ar is the radial absorbance and Arm represents the corresponding value at the meniscus position. When proteins adopt a monomer–dimer equilibrium, the molecular mass M can be treated as a weight average parameter (Mw). In the case of BET3, this value is a composite of the molecular mass values Mm (here 22 584) or Md and the partial concentrations of monomers, cm, and dimers, cd, according to equation (3).
![]()
from which the equilibrium constant Kd=cm2/cd can be determined.
Crystallization and structure determination
Native BET3 crystals were obtained by the sitting-drop method of vapor diffusion using a 96-well Greiner plate (Crystal Quick™ low profile) at 20°C and drops containing 400 nl of protein (16 mg ml−1) plus 400 nl of reservoir solution equilibrated against 75 μl of reservoir solution. All pipetting steps were carried out using semi-automated dispensing systems (Mueller et al, 2001). These crystals grew optimally using 22% PEG 400 as the precipitant in 100 mM Bis–Tris buffer, pH 5.5. Data were collected from a single, flash-frozen native crystal (100 K) to 1.55-Å resolution using a MAR345 imaging plate at the Protein Structure Factory beamline BL-14.2 of Free University of Berlin at BESSY (Berlin, Germany). All data were reduced with DENZO and SCALEPACK (Otwinowski and Minor, 1997). The crystal used for data collection had unit cell parameters a=72.9 Å, c=56.8 Å and belonged to space group P3121 with a monomer in the asymmetric unit. Subsequently, crystals for SeMet-incorporated BET3 (21.9 mg ml−1) were obtained using 25% Jeffamine M600 as the precipitant in 100 mM Na–Hepes buffer, pH 7.0, and peak-wavelength data (0.9797 Å) were collected to 2.5-Å resolution from a single, flash-frozen crystal (100 K). The SeMet crystal was nonisomorphous with respect to the native protein, with unit cell dimensions of a=77.0 Å and c=57.7 Å in P3121. The data collection statistics for the native and SeMet-incorporated BET3 crystals are presented in Table I.
Table 1.
X-ray data collection statistics
| SeMet-BET3 | BET3 | |
|---|---|---|
| Wavelength (Å) | 0.9797 | 0.9786 |
| Resolution (Å) | 30–2.50 (2.59–2.50) | 30–1.55 (1.58–1.55) |
| Total observations | 152 879 (44 037) | 86 840 (3241) |
| Unique observations | 13 261 (1330) | 24 935 (1126) |
| Completeness (%) | 100 (99.8) | 97.0 (89.2) |
| 〈I/σ(I)〉 | 30.5 (6.4) | 20.3 (2.0) |
| Rsyma | 0.071 (0.508) | 0.055 (0.480) |
| aRsym=∑hkl ∑i∣I−〈Ii〉∣/∑hkl ∑iIi, where Ii is the intensity of a given measurement and the sums are over all measurements and reflections. Values in parentheses refer to the highest resolution shell. | ||
The structure of human BET3 was determined by the SAD approach using the selenium peak wavelength data. Selenium sites were located and initial phases were calculated using SOLVE (Terwilliger and Berendzen, 1999). The phases were subsequently improved by maximum-likelihood density modification implemented in RESOLVE (Terwilliger, 2000). This led to an initial model in which 36 residues were placed and an additional 36 built without side chains (accounting for 72/200 residues). The initial model was extended manually using the program O (Jones et al, 1991) and the Se positions as a guide, and led to a model in which 96 residues were placed and an additional 30 built without side chains. After preliminary refinement of the partial SeMet-BET3 structure, the model was placed into the nonisomorphous unit cell of the high-resolution native protein, subsequently refined using REFMAC (Murshudov et al, 1997) and then used as a starting model for autotracing of additional amino acids using the program ARP/wARP (Perrakis et al, 1999). Several rounds of iterative model building and refinement followed and difference density in the map was assigned as a palmitate molecule conjugated to the protein through a thioester bond to C68. The final model (comprising 159 amino acids, 123 water molecules and a palmitate molecule), refined using data between 30- and 1.55-Å resolution, has an average B-factor of 21 Å2 and an R- and free R-factor of 20.8 and 24.3%, respectively, with good geometry. The refinement statistics are summarized in Table II. The coordinates and diffraction amplitudes have been deposited in the Protein Data Bank with accession code 1SZ7.
Table 2.
Refinement statistics
| Resolution (Å) | 30.0–1.55 |
| Rworka | 0.208 |
| Rfreeb | 0.243 |
| r.m.s.d. bond distances (Å) | 0.012 |
| r.m.s.d bond angles (°) | 1.328 |
| Total number of non-H atoms | 1230 |
| Av. protein B value (Å2) | 21 |
| Number of solvent molecules | 123 |
| Av. solvent B value (Å2) | 33 |
| Number of ligand atoms | 17 |
| Av. ligand B value (Å2) | 25 |
| aRwork=∑∣∣F(obs)∣−∣F(calc)∣∣/∑∣F(obs)∣ for the 95% of the reflection data used in refinement. | |
| bRfree=∑∣∣F(obs)∣−∣F(calc)∣∣/∑∣F(obs)∣ for the remaining 5%. | |
Acknowledgments
We thank Patrick Umbach, Anja Koch, Nabila Ibrahim and Ralf Rydschevsky (all from the Protein Structure Factory, Berlin) for their kind help. This work was supported by funding from the German Federal Ministry for Education and Research (BMBF) through the ‘Leitprojektverbund Proteinstrukturfabrik' and the Fonds der Chemischen Industrie.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1994) Current Protocols in Molecular Biology. New York, USA: Greene Publishing Associates and Wiley-Interscience [Google Scholar]
- Barlowe C (1995) COPII: a membrane coat that forms endoplasmic reticulum-derived vesicles. FEBS Lett 369: 93–96 [DOI] [PubMed] [Google Scholar]
- Behlke J, Ristau O, Schönfeld HJ (1997) Nucleotide-dependent complex formation between the Escherichia coli chaperonins GroEL and GroES studied under equilibrium conditions. Biochemistry 36: 5149–5156 [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116: 153–166 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2003) The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific LLC [Google Scholar]
- Dietrich LE, Gurezka R, Veit M, Ungermann C (2004) The SNARE Ykt6 mediates protein palmitoylation during an early stage of homotypic vacuole fusion. EMBO J 23: 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy JT, Linder ME (1998) Signalling functions of protein palmitoylation. Biochim Biophys Acta 1436: 245–261 [DOI] [PubMed] [Google Scholar]
- Gavin A-C, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Küster B, Neubauer G, Superti-Furga G (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147 [DOI] [PubMed] [Google Scholar]
- Gecz J, Shaw MA, Bellon JR, de Barros Lopes M (2003) Human wild-type SEDL protein functionally complements yeast Trs20p but some naturally occurring SEDL mutants do not. Gene 320: 137–344 [DOI] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH (1996) Surprising similarities in structure comparison. Curr Opin Struct Biol 6: 377–385 [DOI] [PubMed] [Google Scholar]
- Glöckner G, Eichinger L, Szafranski K, Pachebat JA, Bankier AT, Dear PH, Lehmann R, Baumgart C, Parra G, Abril JF, Guigo R, Kumpf K, Tunggal B, Cox E, Quail MA, Platzer M, Rosenthal A, Noegel AA (2002) Sequence and analysis of chromosome 2 of Dictyostelium discoideum. Nature 418: 79–85 [DOI] [PubMed] [Google Scholar]
- Gönczy P, Echeverri C, Oegema K, Coulson A, Jones SJ, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlu N, Bork P, Hyman AA (2000) Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature 408: 331–336 [DOI] [PubMed] [Google Scholar]
- Hay JC, Scheller RH (1997) SNAREs and NSF in targeted membrane fusion. Curr Opin Cell Biol 9: 505–512 [DOI] [PubMed] [Google Scholar]
- Higgins DG, Bleasby AJ, Fuchs R (1992) CLUSTAL V: improved software for multiple sequence alignment. Comput Appl Biosci 8: 189–191 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Holz C, Hesse O, Bolotina N, Stahl U, Lang C (2002) A micro-scale process for high-throughput expression of cDNAs in the yeast Saccharomyces cerevisiae. Protein Expr Purif 25: 372–378 [DOI] [PubMed] [Google Scholar]
- Huang CC, Couch GS, Pettersen EF, Ferrin TE (1996) Chimera: an extensible molecular modeling application constructed using standard components. Pacific Symp Biocomput 1: 724 [Google Scholar]
- Ito T, Tashiro K, Muta S, Ozawa R, Chiba T, Nishizawa M, Yamamoto K, Kuhara S, Sakaki Y (2000) Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci USA 97: 1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Lang T, Südhof TC (2003) Membrane fusion. Cell 112: 519–533 [DOI] [PubMed] [Google Scholar]
- Jang SB, Kim Y-G, Cho Y-S, Suh P-G, Kim K-H, Oh B-H (2002) Crystal structure of SEDL and its implications for a genetic disease spondyloepiphyseal dysplasia tarda. J Biol Chem 277: 49863–49869 [DOI] [PubMed] [Google Scholar]
- Jones S, Newman C, Liu F, Segev N (2000) The TRAPP complex is a nucleotide exchanger for Ypt1 and Ypt31/32. Mol Biol Cell 11: 4403–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kim Y-G, Sohn EJ, Seo J, Lee K-J, Lee H-S, Hwang I, Whiteway M, Sacher M, Oh B-H (2005) Crystal structure of bet3 reveals a novel mechanism for Golgi localization of tethering factor TRAPP. Nature Struct Mol Biol 12: 38–45 [DOI] [PubMed] [Google Scholar]
- Klein SL, Strausberg RL, Wagner L, Pontius J, Clifton SW, Richardson P (2002) Genetic and genomic tools for Xenopus research: the NIH Xenopus initiative. Dev Dyn 225: 384–391 [DOI] [PubMed] [Google Scholar]
- Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55: 379–400 [DOI] [PubMed] [Google Scholar]
- Lerche MH, Kragelund BB, Bech LM, Poulsen FM (1997) Barley lipid transfer protein complexed with palmitoyl-CoA: the structure reveals a hydrophobic binding site that can expand to fit both large and small lipid-like ligands. Structure 5: 291–306 [DOI] [PubMed] [Google Scholar]
- Lerche MH, Poulsen FM (1998) Solution structure of barley lipid transfer protein complexed with palmitate. Two different binding modes of palmitate in the homologous maize and barley nonspecific lipid transfer proteins. Protein Sci 7: 2490–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M (2000) Tethers and TRAPPs. Curr Biol 10: R407–R409 [DOI] [PubMed] [Google Scholar]
- Madej T, Gibrat JF, Bryant SH (1995) Threading a database of protein cores. Proteins Struct Funct Genet 23: 356–369 [DOI] [PubMed] [Google Scholar]
- Mammalian Gene Collection (MGC) Program Team (2002) Generation and initial analysis of more than 15 000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA 99: 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller U, Nyarsik L, Horn M, Rauth H, Przewieslik T, Saenger W, Lehrach H, Eickhoff H (2001) Development of a technology for automation and miniaturisation of protein crystallisation. J Biotechnol 285: 7–14 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Pelham HR (1998) SNAREs and membrane fusion in the Golgi apparatus. Biochim Biophys Acta 1404: 9–31 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Panzner S, Dreier L, Hartmann E, Kostka S, Rapoport TA (1995) Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell 81: 561–570 [DOI] [PubMed] [Google Scholar]
- Peitzsch RM, McLaughlin S (1993) Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry 32: 10436–10443 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Prinz B, Schultchen J, Rydzewski R, Holz C, Boettner M, Stahl U, Lang C (2003) Establishing a versatile fermentation and purification procedure for human proteins expressed in the yeasts Saccharomyces cerevisiae and Pichia pastoris for structural genomics. J Struct Funct Genom 5: 29–44 [DOI] [PubMed] [Google Scholar]
- Rossi G, Kolstad K, Stone S, Palluault F, Ferro-Novick S (1995) BET3 encodes a novel hydrophilic protein that acts in conjunction with yeast SNAREs. Mol Biol Cell 6: 1769–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher M, Barrowman J, Schieltz D, Yates JR III, Ferro-Novick S (2000) Identification and characterization of five new subunits of TRAPP. Eur J Cell Biol 79: 71–80 [DOI] [PubMed] [Google Scholar]
- Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, Ferro-Novick S (2001) TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Mol Cell 7: 433–442 [DOI] [PubMed] [Google Scholar]
- Sacher M, Ferro-Novick S (2001) Purification of TRAPP from Saccharomyces cerevisiae and identification of its mammalian counterpart. Methods Enzymol 329: 234–241 [DOI] [PubMed] [Google Scholar]
- Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, Schieltz D, Yates JR III, Abeliovich H, Ferro-Novick S (1998) TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J 17: 2494–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DH, Lee JY, Hwang KY, Kim KK, Suh SW (1995) High-resolution crystal structure of the non-specific lipid-transfer protein from maize seedlings. Structure 3: 189–199 [DOI] [PubMed] [Google Scholar]
- Springer S, Spang A, Schekman R (1999) A primer on vesicle budding. Cell 97: 145–148 [DOI] [PubMed] [Google Scholar]
- Studts JM, Fox BG (1999) Application of fed-batch fermentation to the preparation of isotopically labeled or selenomethionyl-labeled proteins. Protein Expr Purif 16: 109–119 [DOI] [PubMed] [Google Scholar]
- TerBush DR, Novick P (1995) Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae. J Cell Biol 130: 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D 56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Malcolm BA (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange™ site-directed mutagenesis. BioTechniques 26: 680–682 [DOI] [PubMed] [Google Scholar]
- Wang W, Sacher M, Ferro-Novick S (2000) TRAPP stimulates guanine nucleotide exchange on Ypt1p. J Cell Biol 151: 289–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JR, Munro S (2002) Vesicle membrane complexes in membrane traffic. J Cell Sci 115: 2627–2637 [DOI] [PubMed] [Google Scholar]
- Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, Basham D, Bowman S, Brooks K, Brown D, Brown S, Chillingworth T, Churcher C, Collins M, Connor R, Cronin A, Davis P, Feltwell T, Fraser A, Gentles S, Goble A, Hamlin N, Harris D, Hidalgo J, Hodgson G, Holroyd S, Hornsby T, Howarth S, Huckle EJ, Hunt S, Jagels K, James K, Jones L, Jones M, Leather S, McDonald S, McLean J, Mooney P, Moule S, Mungall K, Murphy L, Niblett D, Odell C, Oliver K, O'Neil S, Pearson D, Quail MA, Rabbinowitsch E, Rutherford K, Rutter S, Saunders D, Seeger K, Sharp S, Skelton J, Simmonds M, Squares R, Squares S, Stevens K, Taylor K, Taylor RG, Tivey A, Walsh S, Warren T, Whitehead S, Woodward J, Volckaert G, Aert R, Robben J, Grymonprez B, Weltjens I, Vanstreels E, Rieger M, Schäfer M, Müller-Auer S, Gabel C, Fuchs M, Düsterhöft A, Fritz C, Holzer E, Moestl D, Hilbert H, Borzym K, Langer I, Beck A, Lehrach H, Reinhardt R, Pohl TM, Eger P, Zimmermann W, Wedler H, Wambutt R, Purnelle B, Goffeau A, Cadieu E, Dreano S, Gloux S, Lelaure V, Mottier S, Galibert F, Aves SJ, Xiang Z, Hunt C, Moore K, Hurst SM, Lucas M, Rochet M, Gaillardin C, Tallada VA, Garzon A, Thode G, Daga RR, Cruzado L, Jimenez J, Sanchez M, del Rey F, Benito J, Dominguez A, Revuelta JL, Moreno S, Armstrong J, Forsburg SL, Cerutti L, Lowe T, McCombie WR, Paulsen I, Potashkin J, Shpakovski GV, Ussery D, Barrell BG, Nurse P, Cerrutti L (2002) The genome sequence of Schizosaccharomyces pombe. Nature 415: 871–880 [DOI] [PubMed] [Google Scholar]