

ABSTRACT
The rapid detection of pathogenic strains in food products is essential for the prevention of disease outbreaks. It has already been demonstrated that whole-metagenome shotgun sequencing can be used to detect pathogens in food but, until recently, strain-level detection of pathogens has relied on whole-metagenome assembly, which is a computationally demanding process. Here we demonstrated that three short-read-alignment-based methods, i.e., MetaMLST, PanPhlAn, and StrainPhlAn, could accurately and rapidly identify pathogenic strains in spinach metagenomes that had been intentionally spiked with Shiga toxin-producing Escherichia coli in a previous study. Subsequently, we employed the methods, in combination with other metagenomics approaches, to assess the safety of nunu, a traditional Ghanaian fermented milk product that is produced by the spontaneous fermentation of raw cow milk. We showed that nunu samples were frequently contaminated with bacteria associated with the bovine gut and, worryingly, we detected putatively pathogenic E. coli and Klebsiella pneumoniae strains in a subset of nunu samples. Ultimately, our work establishes that short-read-alignment-based bioinformatics approaches are suitable food safety tools, and we describe a real-life example of their utilization.
IMPORTANCE Foodborne pathogens are responsible for millions of illnesses each year. Here we demonstrate that short-read-alignment-based bioinformatics tools can accurately and rapidly detect pathogenic strains in food products by using shotgun metagenomics data. The methods used here are considerably faster than both traditional culturing methods and alternative bioinformatics approaches that rely on metagenome assembly; therefore, they can potentially be used for more high-throughput food safety testing. Overall, our results suggest that whole-metagenome sequencing can be used as a practical food safety tool to prevent diseases or to link outbreaks to specific food products.
KEYWORDS: fermentation, food-borne pathogens, metagenomics
INTRODUCTION
In recent years, high-throughput sequencing (HTS) has become an important tool in food microbiology (1). HTS enables in-depth characterization of food-related microbial isolates, via whole-genome sequencing (WGS), and it facilitates culture-independent analysis of mixed microbial communities in foods, via metagenomic sequencing.
WGS has provided invaluable insights into the genetics of starter cultures (2, 3), and it is routinely used in epidemiology to identify outbreak-associated foodborne pathogens isolated from clinical samples, through comparison of the single-nucleotide polymorphism (SNP) profiles of outbreak strain genomes versus nonoutbreak strain genomes (4–6). Metagenomic sequencing enables elucidation of the roles of microorganisms during food production (7–9), and it can be used to track microorganisms of interest through the food production chain, as illustrated by Yang et al. (10), who used whole-metagenome shotgun sequencing to track pathogenic species in the beef production chain. Indeed, metagenomic sequencing can be used to detect pathogens in foods to monitor outbreaks of foodborne illnesses (11), but few studies have done so, because of the limited taxonomic resolution achievable using these methods. Typically, 16S rRNA gene sequencing provides genus-level taxonomic resolution (12), and although sub-genus-level classification is achievable using species classifiers (13) or oligotyping (14, 15), these methods cannot accurately discriminate between strains. Similarly, tools for metagenome sequence classification usually provide species-level resolution (16). However, strain-level resolution is necessary for the accurate identification of pathogens in food products (17). Leonard et al. successfully achieved strain-level resolution of Shiga toxin-producing Escherichia coli (STEC) strains in spinach samples using metagenome shotgun sequencing (18). However, the bioinformatics methods used in that study were based on metagenome assembly, which is a computationally demanding process (19, 20), and alternative strain-level identification methods are needed.
Since 2016, several short-read-alignment-based software applications that can achieve strain-level characterization of microorganisms from metagenome shotgun sequencing data, including MetaMLST (20), StrainPhlAn (21), and PanPhlAn (19), have been released. All three applications are considerably faster than metagenome-assembly-based methods. To date, these programs have not been employed to detect pathogens in food products, but there is strong evidence to suggest that they have considerable potential for this purpose. MetaMLST accurately predicted that the strain responsible for the 2011 German E. coli outbreak belonged to E. coli sequence type 678 (ST678) (20); similarly, PanPhlAn accurately predicted that the strain was a Shiga toxin producer (19), based on an analysis of the gut metagenomes of infected patients (22). StrainPhlAn has so far not been used for epidemiological purposes, but a recent study demonstrated that it can be used to predict the phylogenetic relatedness of bacterial strains from different samples (21).
MetaMLST aligns sequencing reads against a housekeeping gene database to identify sequence types present in metagenomic samples, based on multilocus sequence typing (MLST). The MetaMLST database contains all currently known sequence types, but it can be updated as required to include newly identified sequence types. MetaMLST does not require any prior knowledge of the microbial composition of samples, and it can simultaneously detect the sequence types of different species. PanPhlAn aligns sequencing reads against a species-specific pangenome database, constructed from reference genomes, to functionally characterize strains present in metagenomic samples. PanPhlAn allows users to generate customizable pangenome databases for any species. StrainPhlAn extracts species-specific marker genes from sequencing reads and aligns the markers against reference genomes to identify the strains present in metagenomic samples. StrainPhlAn requires output from MetaPhlAn2, and the two programs use the same database.
In this study, we describe the characterization of nunu, a traditional Ghanaian fermented milk product (FMP), at the genus, species, and strain levels, using a combination of 16S rRNA gene sequencing and whole-metagenome shotgun sequencing. Nunu is produced by the spontaneous fermentation of raw cow milk in calabashes or plastic or metal containers under ambient conditions, and it is usually consumed after 24 to 36 h (23). At present, little is known about nunu's microbiology, relative to other FMPs such as kefir and yoghurt (24). Previously, a number of potentially pathogenic bacteria, including Enterobacter, Escherichia, and Klebsiella, were detected in nunu by culture-based methods (25). Here, we carry out the first culture-independent analysis of a number of nunu samples. In addition to detecting the presence of a variety of lactic acid bacteria typical of fermented dairy products, MetaMLST, PanPhlAn, and StrainPhlAn all indicated the presence of pathogenic E. coli and Klebsiella pneumoniae in a subset of the samples. We also demonstrated that these tools could accurately predict the presence of pathogenic strains in foods by testing them on food metagenomes that had been spiked with Shiga toxin-producing E. coli. Ultimately, our work establishes that short-read-alignment-based methods can be used for the detection of pathogens in foods.
RESULTS
Analysis of nunu samples by 16S rRNA gene sequencing.
Nunu samples were collected from producers with hygiene practice training (n = 5) and producers without hygiene practice training (n = 5). The 16S rRNA gene sequencing analysis revealed that there were no significant differences in the alpha diversity of nunu samples from trained versus untrained producers (see Fig. S1a in the supplemental material), although there was a clear separation in the beta diversity results for the two groups (Fig. S1b).
The 16S rRNA data were also analyzed to determine bacterial compositions (Fig. 1a). At the family level, all of the samples were dominated by Lactobacillales; at the genus level, most samples were dominated by Streptococcus, although sample 1t2am was dominated by Lactococcus. Enterococcus was detected at ≥3% relative abundance in 4/10 samples (1 trained and 3 untrained), and the level was highest in sample 2u6am, where Enterococcus was present at 19% relative abundance. In addition, Staphylococcus was detected in all 10 samples, although its abundance was ≤1% in each case. The detection of staphylococci was consistent with a corresponding culture-dependent analysis of the samples (see the supplemental material). Importantly, Enterobacteriales were also prevalent. Enterobacter was detected at ≥1% relative abundance in 9/10 samples (4 samples from trained producers and 5 from untrained producers), and the level was highest in sample 2u8am, where Enterobacter was present at 23% relative abundance. Escherichia or Shigella was detected at ≥1% relative abundance in 8/10 samples (4 trained and 4 untrained), and the level was highest in sample 1t7am, where the species were present at 17% relative abundance; this finding was again consistent with culture-dependent analysis of the samples (see the supplemental material).
FIG 1.

Analysis of nunu samples based on 16S rRNA gene sequencing. (a) Heat map showing the 25 most abundant bacterial genera across the nunu samples. (b) Bar plot showing genera that were differentially abundant in the two groups.
The Kruskal-Wallis test indicated that there were significant differences in the relative abundances of Macrococcus (P = 0.01), which was more abundant in samples from trained producers, and Streptococcus (P = 0.02), which was more abundant in samples from untrained producers (Fig. 1b). No other genera had significantly different abundances.
Species-level compositional analysis of nunu samples, as revealed by shotgun sequencing.
MetaPhlAn2-based analysis of shotgun metagenomic data provided results that were generally consistent with those derived from amplicon sequencing. Eleven species accounted for >90% of the microbial composition of every sample (Fig. 2). At the species level, most samples were dominated by Streptococcus infantarius, although sample 1t2am was dominated by Lactococcus lactis. Enterococcus faecium was detected at ≥1% relative abundance in 4/10 samples (2 trained and 2 untrained), and the level was highest in sample 1t2am, where the species was present at 22% relative abundance. High levels of Enterobacteriales were again apparent. Enterobacter cloacae were detected in sample 1t8am, where it was present at 1% relative abundance. Escherichia coli was detected at ≥7% relative abundance in 2/10 samples (2 trained), and the level was highest in sample 1t7am, where the species was present at 13% relative abundance. Klebsiella pneumoniae was detected at ≥3% relative abundance in 7/10 samples (4 trained and 3 untrained), and the level was highest in sample 1t8am, where the species was present at 71% relative abundance. In contrast, Klebsiella was not detected by amplicon sequencing; this discrepancy might be due to similarities in the 16S rRNA genes from these genera (26).
FIG 2.

Species-level microbial compositions of nunu samples, as determined by MetaPhlAn2.
The Kruskal-Wallis test indicated that there were significant differences in the relative abundances of Macrococcus caseolyticus (P = 0.01), which was more abundant in samples from trained producers, and Streptococcus infantarius (P = 0.01), which was more abundant in samples from untrained producers (Fig. S2). No other species had significantly different abundances.
Investigation of the functional potential of the nunu microbiota.
SUPER-FOCUS was used to provide an overview of the functional potential of the nunu metagenome. As expected, a significant proportion of the metagenome was assigned to housekeeping functions such as carbohydrate metabolism, nucleic acid metabolism, and protein metabolism (Fig. 3). However, SUPER-FOCUS also detected high levels of functions associated with horizontal gene transfer (HGT) and virulence in nunu. The level 1 subsystem of phages, prophages, and transposable elements was present at ≥1% average relative abundance in both groups, although it was significantly more abundant in nunu samples from trained producers (P = 0.047). Similarly, the level 1 subsystem of virulence was present at ≥3.5% average relative abundance in both groups.
FIG 3.

Average abundances of the SUPER-FOCUS level 1 functions that were detected in nunu samples.
HUMAnN2 was used to provide more comprehensive insights into the functional potential of the nunu metagenome. Unsurprisingly, the 25 most abundant genetic pathways were associated with carbohydrate metabolism, nucleic acid metabolism, and protein metabolism (Fig. 4a). Multidimensional scaling (MDS) analysis of all of the normalized HUMAnN2 pathway abundances suggested that there were differences in the overall functional potentials of the groups (Fig. S3), and we detected significant differences in the relative abundances of some individual pathways (Table S1). Notably, we observed that histidine degradation pathways were more abundant in trained samples (P = 0.047) (Fig. 4b). Furthermore, histidine decarboxylase genes were detected only in trained samples. Several other undesirable genetic pathways were detected in both groups. For example, putrescine biosynthesis pathways and polymyxin resistance genes cooccurred in 7/10 samples (Fig. 4c), and these pathways were all attributed to E. cloacae, E. coli, K. pneumoniae, or a combination of these three species. We detected several other antibiotic resistance genes, including β-lactamase genes and methicillin resistance genes, in both groups (Fig. S4). In addition, we found HGT-associated genes, including plasmid maintenance genes and transposition genes, in both groups.
FIG 4.

HUMAnN2 analysis. (a) Heat map showing the 25 most abundant MetaCyc pathways detected across the 10 nunu metagenomic samples. (b) Bar plot showing differences in the abundances of the MetaCyc pathways HISDEG-PWY and HISTSYN-PWY in nunu samples from trained producers and nunu samples from untrained producers. (c) Bar plots showing the relative contributions of E. cloacae, E. coli, and K. pneumoniae to the MetaCyc pathways PWY-6305 (putrescine biosynthesis) and PWY0-1338 (polymyxin resistance).
Application of strain-level analysis to characterize enteric bacteria in nunu.
Leonard et al. previously used metagenomic sequencing to detect E. coli in spinach that had been intentionally spiked with E. coli O157:H7 strain Sakai (11). We downloaded the metagenomic reads from that study (16 samples), and we subjected them to StrainPhlAn, MetaMLST, and PanPhlAn analysis to confirm that these tools can accurately detect pathogens in food samples; MetaMLST was used for multilocus sequence typing, StrainPhlAn was used for phylogenetic identification, and PanPhlAn was used for functional characterization. MetaMLST accurately detected E. coli ST11 in 7/16 spinach samples (Table 1). StrainPhlAn detected E. coli strains in 5/16 samples, and it showed that the E. coli strain in each of those samples was closely related to E. coli O157:H7 strain Sakai (Fig. 5). PanPhlAn detected Shiga toxin genes in 15/16 samples (Table 1), and it indicated that the E. coli strain in each of those samples was most closely related to E. coli O157:H7 strain Sakai. Thus, overall, PanPhlAn was the most sensitive method in this instance, since it was able to detect STEC in almost all of the samples, whereas the other tools detected STEC in less than one-half of the samples. In a follow-up study, Leonard et al. spiked spinach with 12 different Shiga toxin-producing E. coli strains, and they detected single strains in 17 samples (18). We downloaded the metagenomic reads from the 17 samples and ran PanPhlAn, and we were able to identify Shiga toxin genes in all 17 samples (Table S2).
TABLE 1.
Results of MetaMLST and PanPhlAn analysis of spinach metagenomes spiked with E. coli O157:H7 Sakai
| ENA accession no. | No. of reads | E. coli abundance (%) | No. of copiesa |
Sequence type | Confidence (%) | |
|---|---|---|---|---|---|---|
| stxA2 | stxB2 | |||||
| SRR2177250 | 9,365,812 | 5.28412 | 1 | 1 | Unknown | NA |
| SRR2177251 | 17,562,542 | 4.31712 | 1 | 1 | 11 | 99.97 |
| SRR2177280 | 11,707,292 | 21.16364 | 1 | 1 | 100001 | 99.97 |
| SRR2177281 | 10,580,532 | 2.84187 | 1 | 1 | Unknown | NA |
| SRR2177282 | 6,155,636 | 60.51406 | 1 | 1 | 11 | 100 |
| SRR2177283 | 13,120,244 | 10.11327 | 1 | 1 | 11 | 100 |
| SRR2177284 | 7,500,056 | 2.05064 | NA | NA | Unknown | NA |
| SRR2177285 | 14,482,370 | 66.69813 | 1 | 1 | 11 | 100 |
| SRR2177286 | 14,035,970 | 69.17834 | 1 | 1 | 11 | 100 |
| SRR2177287 | 12,242,348 | 5.62746 | 1 | 1 | Unknown | NA |
| SRR2177288 | 8,303,788 | 10.75005 | 1 | 1 | 11 | 100 |
| SRR2177357 | 14,621,672 | 8.02047 | 1 | 1 | 11 | 100 |
| SRR2177358 | 10,684,052 | 3.18652 | 1 | 1 | Unknown | NA |
| SRR2177359 | 4,964,436 | 1.17146 | 1 | 1 | Unknown | NA |
| SRR2177360 | 12,729,834 | 1.81229 | 1 | 0 | Unknown | NA |
| SRR2177361 | 11,946,092 | 0.70921 | 0 | 1 | Unknown | NA |
NA, not applicable.
FIG 5.
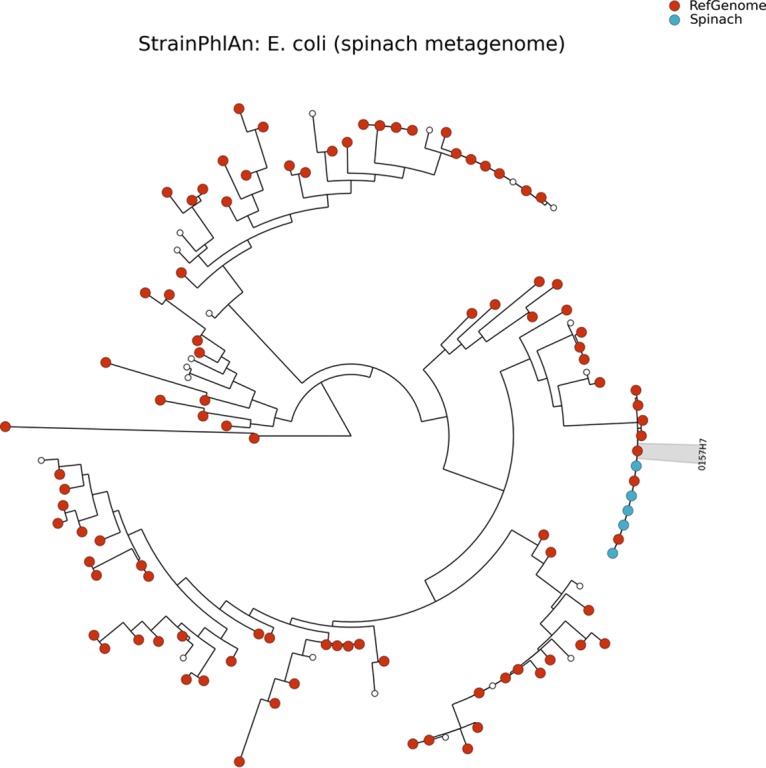
StrainPhlAn analysis of the spinach metagenome.
Having established the relative merits of these tools, we subsequently employed all three strategies to identify the strains of E. coli and K. pneumoniae present in the nunu samples. With regard to E. coli, MetaMLST detected a novel E. coli sequence type in sample 1t7am (Table 2). StrainPhlAn detected 24 E. coli marker genes in the samples, and a phylogenetic tree generated by aligning the markers against 118 E. coli reference genomes (listed in Table S3) revealed that the E. coli strain in one sample, sample 1t7am, was closely related to E. coli O139:H28 E24377A (Fig. 6a). PanPhlAn detected E. coli strains in two samples, namely, samples 1t7am and 1t8am. MDS analysis indicated that the strains from the two samples were functionally distinct from one another. Notably, a ShET2 enterotoxin-encoding gene was identified in the E. coli strain from sample 1t7am; the same gene was found in E. coli O139:H28 E24377A. With regard to K. pneumoniae, MetaMLST detected the known K. pneumoniae sequence type ST39 in sample 2u3am. Apparently novel K. pneumoniae sequence types were identified in six other samples (Table 1). StrainPhlAn detected 38 K. pneumoniae marker genes in the samples, and a phylogenetic tree constructed by aligning the markers against 40 K. pneumoniae reference genomes (listed in Table S4) revealed that the K. pneumoniae strains in two samples, samples 1t8am and 2u3am, were closely related to K. pneumoniae KpQ3 (Fig. 6b). In contrast, the K. pneumoniae strain in sample 1t7am was most closely related to K. pneumoniae UCICRE 7. MDS analysis of the PanPhlAn output showed that five of the detected K. pneumoniae strains were functionally similar to one another (Fig. 6c). However, two of the detected K. pneumoniae strains, from samples 1t6am and 1t7am, appeared to be functionally distinct from the others. In addition, PanPhlAn indicated that sample 1t6am might have contained multiple strains, since an unusually large number of K. pneumoniae gene families (5,746 families) was detected. A TEM β-lactamase gene was found in sample 1t2am using PanPhlAn; furthermore, an OXA-48 carbapenemase gene was detected in sample 2u8am, and the same gene was found in K. pneumoniae KpQ3.
TABLE 2.
Results of MetaMLST analysis of nunu metagenomic samples
| Sample | Species | Sequence type | Confidence (%) |
|---|---|---|---|
| 1t2am | Klebsiella pneumoniae | 100001 | 98.7 |
| 1t6am | Klebsiella pneumoniae | 100002 | 100 |
| 1t7am | Escherichia coli | 100001 | 100 |
| 1t7am | Klebsiella pneumoniae | 100003 | 99.9 |
| 1t8am | Klebsiella pneumoniae | 100004 | 100 |
| 2u3am | Klebsiella pneumoniae | 39 | 100 |
| 2u6am | Klebsiella pneumoniae | 100005 | 99.91 |
| 2u8am | Klebsiella pneumoniae | 100006 | 99.91 |
FIG 6.

Strain-level analysis. (a and b) Phylogenetic trees showing the relationships between E. coli strains (a) and K. pneumoniae strains (b) detected in the nunu metagenomic samples and their respective reference genomes, as predicted by StrainPhlAn. Nodes highlighted in gray indicate reference genomes of particular interest. (c) MDS showing the functional similarities between strains detected in the nunu metagenomic samples, as predicted by PanPhlAn. Reference genomes are shown in dark gray.
Finally, we compared the time taken to process 10 nunu metagenome samples using the short-read alignment tools versus the metagenome assembler IDBA-UD (Fig. S5). In each case, we observed that all of the short-read alignment tools were faster than IDBA-UD. It is important to note that additional bioinformatics analyses (such as contig binning and SNP analysis) are required to achieve strain-level identification from assembled metagenomes, which emphasizes the superior speed of the short-read alignment tools.
DISCUSSION
Foodborne pathogens are responsible for millions of cases of disease each year in the United States alone (27). High-throughput sequencing can potentially be used to detect pathogenic strains in food products to prevent the occurrence of disease outbreaks. A recent proof-of-concept study demonstrated that whole-metagenome shotgun sequencing accurately detected STEC strains in spiked spinach samples (18). However, that study used whole-metagenome-assembly-based approaches to achieve strain-level taxonomic resolution of the STEC in the samples. Whole-metagenome assembly is a computationally intensive, time-consuming process, as illustrated by Nurk et al., who recently reported that metagenome assembly can take between 1.5 h and 6 h, with a memory footprint ranging from 7.3 GB to 234.5 GB, depending on the chosen assembler, for processing of a single human gut metagenomic sample (28). Thus, the application of more rapid, less intensive bioinformatic tools for strain detection is desirable. In this study, we demonstrate that the short-read-alignment-based programs MetaMLST, StrainPhlAn, and PanPhlAn can accurately identify pathogens in food products.
We validated the accuracy of each approach by processing spinach metagenome data from samples that had been spiked with the STEC O157:H7 Sakai in a previous study (11). We observed that PanPhlAn was the most sensitive approach. Indeed, PanPhlAn was able to identify STEC in every sample where it was present at >2% relative abundance, whereas the other approaches worked best when STEC was present at high relative abundances. However, none of the tools detected E. coli O157:H7 Sakai in every sample tested. The observation of false-negative results indicates that the tools are not entirely accurate. It is likely that increased sequencing depth and/or longer sequencing read lengths would reduce the false-negative rate. We recommend that these tools be used to supplement data from metagenome sequence classifiers like MetaPhlAn2, which did detect E. coli in each sample. Therefore, we subsequently used the strain-level analysis tools in combination with other metagenomic approaches to assess the safety of nunu, a traditional Ghanaian fermented milk product.
Nunu is produced through the spontaneous fermentation of raw cow milk in calabashes or other containers at ambient temperature for 24 to 36 h (23). The crude nature of the nunu production process has raised food safety concerns (25). Indeed, several potentially pathogenic microorganisms were previously detected in nunu samples by microbial culturing (25). This resulted in some nunu producers receiving hygiene practice training to improve food safety. However, our work suggests that there is little difference in the prevalence of pathogens in nunu samples from trained versus untrained producers. One reason for this may be that it is difficult for the nunu producers to adhere to the training recommendations, which are not appropriate for the rural production conditions. During training, the producers were advised to pasteurize the milk before cooling it and adding a starter culture. They were advised to stir the mixture after 4 to 6 h of incubation in a covered container and to refrigerate the product. Lack of access to specific heat controls and electricity, as well as the variance from the traditional method, which does not use a starter culture, are reasons why the training is not followed.
The 16S rRNA gene sequencing revealed that the samples were dominated by Lactobacillales. However, we also detected high abundances of Enterobacteriales, including Enterobacter and Escherichia, in both groups. Subsequently, whole-metagenome shotgun sequencing showed that most samples were dominated by Streptococcus infantarius, a species that had been identified previously in other African dairy products (29, 30). Of concern, S. infantarius has been linked to several human diseases, including bacteremia (31), endocarditis (32), and colon cancer (33). Aside from S. infantarius, two other potentially pathogenic species, namely, Escherichia coli and Klebsiella pneumoniae, were identified in a subset of samples.
Overall, our findings indicate that nunu samples from trained producers and untrained producers were contaminated with fecal material. Cattle feces can be a major source of bacterial contaminants in raw cow milk (34) and thus our results are not entirely surprising, but the remarkable abundance of such microorganisms in nunu is worrying. It had been hoped that nunu could be used to supplement traditional cereal-based weaning foods to improve infant nutrition. However, qualitative research among mothers and health workers highlighted safety concerns, which are valid, as we have shown here. In particular, the presence of E. coli and K. pneumoniae in nunu is a concern; therefore, we employed strain-level metagenomics for further characterization of these bacteria.
In terms of E. coli, strain-level analysis indicated that the E. coli strain in one sample was an enterotoxin producer and was closely related to E. coli O139:H28 E24377A, a strain that was linked to an outbreak of waterborne diarrhea in India (35). In terms of K. pneumoniae, strain-level analysis indicated that the K. pneumoniae strains in two samples were antibiotic resistant and were closely related to K. pneumoniae KpQ3, a strain that was linked to nosocomial outbreaks among burn unit patients. Thus, strain-level analysis suggested that there were likely pathogens in some of the samples. Interestingly, PanPhlAn also suggested that there were functionally distinct strains of both species in nunu samples from different producers. Perhaps this indicates multiple incidences or sources of contamination. Undoubtedly, our work highlights an urgent need to further improve hygiene practices during nunu production, and pasteurization of the starting milk and use of starter-based fermentation systems represent obvious solutions.
In conclusion, our work suggests that short-read-alignment-based strain detection tools can be used to detect pathogens in other foods, apart from nunu or spinach, and they might also be useful for tracing the sources of foodborne disease outbreaks back to particular foods. Such tools represent a significant improvement over 16S rRNA gene sequencing, which is often limited to genus-level identification, or metagenome read classification tools, which are limited to species-level identification (16). In addition, they are faster and less computationally intensive than metagenome-assembly-based strain detection methods, making them more relevant to real-life scenarios that require rapid testing of many food samples. With DNA sequencing costs continuing to decrease, the approach outlined here is an affordable option for food safety testing.
MATERIALS AND METHODS
Sampling.
Five nunu samples were collected from producers with hygiene practice training, and another five samples were collected from producers without hygiene practice training. The identity of the samples from trained and untrained individuals was masked until the sequencing analysis was completed. The samples from the trained group were labeled 1t2am, 1t6am, 1t7am, 1t8am, and 2t2am, and the samples from the untrained group were labeled 1u6am, 2u2am, 2u3am, 2u6am, and 2u8am. All samples were collected in the morning and placed on ice for transport to the laboratory. Sample aliquots (4 ml) were then mixed with glycerol to a final concentration of 20% and were stored at −20°C prior to DNA extraction. DNA was extracted from the samples at the Animal Research Institute (Accra, Ghana) and then sent to Scotland to comply with international laws on the import of animal samples (import license form AB117).
Microbiological analysis.
Basic microbiological culture analysis was carried out in Ghana. The plate-count technique was used to estimate the total viable bacterial counts of the nunu samples on milk plate count agar (Lab M, UK). Bacterial counts were compared for plates growing aerobically or anaerobically at 30°C for 36 to 72 h. Anaerobic plates were incubated in airtight canisters containing CO2Gen sachets (Oxoid, UK), which created an anaerobic atmosphere. Following incubation, colonies were counted using an SC6+ electronic colony counter (Stuart Scientific, UK). The presence of specific pathogens in the nunu samples was determined by streaking nunu directly onto selective agar plates to visually assess bacterial growth. The following selective agars were used: blood agar (Merck, Germany) for Staphylococcus, MacConkey agar (Merck, Germany) for enterobacteria, de Man-Rogosa-Sharpe (MRS) agar (Oxoid, UK) for Lactobacillus species, and Salmonella-Shigella agar (Oxoid, UK). Any plates with mixed growth were repurified by streaking onto selected secondary agars. Lactose-fermenting colonies identified on MacConkey agar were subcultured on eosin methylene blue agar (EMBA) (Scharlau Chemie, Spain) to isolate and to identify E. coli. Additionally, Staphylococcus colonies from blood agar were subcultured on mannitol salt agar (MSA) (Oxoid, UK) to isolate and to identify Staphylococcus aureus. The following biochemical tests were used to confirm bacterial identification: the motility indole urea (MIU) test, the catalase test, the triple sugar iron (TSI) test, and the indole methyl red Voges-Proskaeur citrate (IMViC) test. Cellular morphology was determined by Gram staining and microscopic examination.
DNA extraction and next-generation sequencing.
Briefly, 1 ml of each thawed sample was diluted in 9 ml of sterile phosphate-buffered saline (PBS), mixed thoroughly using a vortex mixer, and centrifuged for 10 min at 8,000 to 10,000 × g. The bacterial cell pellets were resuspended in 432 μl of sterile distilled water and 48 μl of 0.5 M EDTA and mixed thoroughly by a combination of vortex mixing and use of a sterile pipette tip, and the suspension was frozen. The frozen samples were thawed on the bench, refrozen, and finally thawed (giving a total of two freeze-thaw cycles) before DNA extraction using the Promega Wizard genomic DNA extraction kit (Promega, Madison, WI, USA), according to the manufacturer's protocol. The freeze-thaw cycles were carried out to maximize bacterial cell lysis. Following extraction, the DNA pellets were air dried for about 60 min, stored sealed under airtight conditions, and transported from the Animal Research Institute (Accra, Ghana) to the Rowett Institute at the University of Aberdeen for further analysis.
DNA extracts were quantified using the Qubit high-sensitivity DNA assay (BioSciences, Dublin, Ireland), and 16S rRNA gene sequencing libraries were prepared from extracted DNA using the 16S metagenomic sequencing library preparation protocol from Illumina, with minor modifications (36). Samples were sequenced on the Illumina MiSeq system in the Teagasc sequencing facility with a V2 kit (2 × 250 cycles), in accordance with standard Illumina sequencing protocols. Whole-metagenome shotgun libraries were prepared in accordance with the Nextera XT DNA library preparation guide from Illumina (36). Samples were sequenced on the Illumina MiSeq system in the Teagasc sequencing facility with a V3 kit (2 × 300 cycles), in accordance with standard Illumina sequencing protocols.
Bioinformatics.
Raw 16S rRNA gene sequencing reads were quality filtered using PRINSEQ (37). Denoising, operational taxonomic unit (OTU) clustering, and chimera removal were performed using USearch 7 (64 bit) (38), as described by Doyle et al. (34). OTUs were aligned using PyNAST (39). Alpha diversity and beta diversity were calculated using Qiime 1.8.0 (40). Taxonomy was assigned using a BLAST search (41) against the SILVA small subunit (SSU) 119 database (42).
Raw whole-metagenome shotgun sequencing reads were filtered on the basis of quality and quantity and trimmed to 200 bp with a combination of Picard Tools (https://github.com/broadinstitute/picard) and SAMtools (43). MetaPhlAn2 was used to characterize the microbial compositions of samples at the species level (44). MetaMLST (20), PanPhlAn (19), and StrainPhlAn (21) were used to characterize the microbial compositions of the samples at the strain level. GraPhlAn (45) was used to construct phylogenetic trees from the StrainPhlAn output. SUPER-FOCUS (46) and HUMAnN2 (47) were used to determine the microbial metabolic potential of samples. IDBA-UD (48) was used for metagenome assembly.
Statistical analysis.
Statistical analysis was performed in R 3.2.2 (49). The Kruskal-Wallis test was performed using the compareGroups package, and the resulting P values were for multiple comparisons. Principal-coordinate analysis (PCoA) analysis of 16S rRNA gene sequencing data was performed using the phyloseq package (50). MDS was performed using the vegan package. Data visualization was performed using the ggplot2 package.
Accession number(s).
Sequence data have been deposited in the European Nucleotide Archive (ENA) under project accession number PRJEB20873.
Supplementary Material
ACKNOWLEDGMENTS
We thank the researchers at the Animal Research Institute (Accra, Ghana) for their help with sample collection, storage, and processing.
This research was funded by Science Foundation Ireland in the form of a center grant (APC Microbiome Institute grant SFI/12/RC/2273). Research in the Cotter laboratory is also funded by Science Foundation Ireland through grant 11/PI/1137. O.O. is funded by Science Foundation Ireland through a Starting Investigator Research Grant (grant 13/SIRG/2160). K.D. is a PhD student funded by the Ghana Educational Trust Fund. The Rowett Institute receives funding from the Scottish Government (RESAS). The Rowett Institute receives funding from the Scottish Government strategic research portfolio (RAFE).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01144-17.
REFERENCES
- 1.Walsh AM, Crispie F, Claesson MJ, Cotter PD. 2017. Translating omics to food microbiology. Annu Rev Food Sci Technol 8:113–134. doi: 10.1146/annurev-food-030216-025729. [DOI] [PubMed] [Google Scholar]
- 2.Zheng J, Zhao X, Lin XB, Gänzle M. 2015. Comparative genomics Lactobacillus reuteri from sourdough reveals adaptation of an intestinal symbiont to food fermentations. Sci Rep 5:18234. doi: 10.1038/srep18234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun Z, Harris HM, McCann A, Guo C, Argimón S, Zhang W, Yang X, Jeffery IB, Cooney JC, Kagawa TF, Liu W, Song Y, Salvetti E, Wrobel A, Rasinkangas P, Parkhill J, Rea MC, O'Sullivan O, Ritari J, Douillard FP, Ross RP, Yang R, Briner AE, Felis GE, de Vos WM, Barrangou R, Klaenhammer TR, Caufield PW, Cui Y, Zhang H, O'Toole PW. 2015. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat Commun 6:8322. doi: 10.1038/ncomms9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H. 2011. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 6:e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dallman TJ, Byrne L, Ashton PM, Cowley LA, Perry NT, Adak G, Petrovska L, Ellis RJ, Elson R, Underwood A, Green J, Hanage WP, Jenkins C, Grant K, Wain J. 2015. Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin Infect Dis 61:305–312. doi: 10.1093/cid/civ318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwong JC, Mercoulia K, Tomita T, Easton M, Li HY, Bulach DM, Stinear TP, Seemann T, Howden BP. 2016. Prospective whole-genome sequencing enhances national surveillance of Listeria monocytogenes. J Clin Microbiol 54:333–342. doi: 10.1128/JCM.02344-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Filippis F, Genovese A, Ferranti P, Gilbert JA, Ercolini D. 2016. Metatranscriptomics reveals temperature-driven functional changes in microbiome impacting cheese maturation rate. Sci Rep 6:21871. doi: 10.1038/srep21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quigley L, O'Sullivan DJ, Daly D, O'Sullivan O, Burdikova Z, Vana R, Beresford TP, Ross RP, Fitzgerald GF, McSweeney PLH, Giblin L, Sheehan JJ, Cotter PD. 2016. Thermus and the pink discoloration defect in cheese. mSystems 1:e00023-16. doi: 10.1128/mSystems.00023-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh AM, Crispie F, Kilcawley K, O'Sullivan O, O'Sullivan MG, Claesson MJ, Cotter PD. 2016. Microbial succession and flavor production in the fermented dairy beverage kefir. mSystems 1:e00052-16. doi: 10.1128/mSystems.00052-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Noyes NR, Doster E, Martin JN, Linke LM, Magnuson RJ, Yang H, Geornaras I, Woerner DR, Jones KL, Ruiz J, Boucher C, Morley PS, Belk KE. 2016. Use of metagenomic shotgun sequencing technology to detect foodborne pathogens within the microbiome of the beef production chain. Appl Environ Microbiol 82:2433–2443. doi: 10.1128/AEM.00078-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonard SR, Mammel MK, Lacher DW, Elkins CA. 2015. Application of metagenomic sequencing to food safety: detection of Shiga toxin-producing Escherichia coli on fresh bagged spinach. Appl Environ Microbiol 81:8183–8191. doi: 10.1128/AEM.02601-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allard G, Ryan FJ, Jeffery IB, Claesson MJ. 2015. SPINGO: a rapid species-classifier for microbial amplicon sequences. BMC Bioinformatics 16:324. doi: 10.1186/s12859-015-0747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, Sogin ML. 2013. Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol 4:1111–1119. doi: 10.1111/2041-210X.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stellato G, Utter DR, Voorhis A, De Angelis M, Eren AM, Ercolini D. 2017. A few Pseudomonas oligotypes dominate in the meat and dairy processing environment. Front Microbiol 8:264. doi: 10.3389/fmicb.2017.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindgreen S, Adair KL, Gardner PP. 2016. An evaluation of the accuracy and speed of metagenome analysis tools. Sci Rep 6:19233. doi: 10.1038/srep19233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stasiewicz MJ, den Bakker HC, Wiedmann M. 2015. Genomics tools in microbial food safety. Curr Opin Food Sci 4:105–110. doi: 10.1016/j.cofs.2015.06.002. [DOI] [Google Scholar]
- 18.Leonard SR, Mammel MK, Lacher DW, Elkins CA. 2016. Strain-level discrimination of Shiga toxin-producing Escherichia coli in spinach using metagenomic sequencing. PLoS One 11:e0167870. doi: 10.1371/journal.pone.0167870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, Segata N. 2016. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods 13:435–438. doi: 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
- 20.Zolfo M, Tett A, Jousson O, Donati C, Segata N. 2017. MetaMLST: multi-locus strain-level bacterial typing from metagenomic samples. Nucleic Acids Res 45:e7. doi: 10.1093/nar/gkw837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asnicar F, Manara S, Zolfo M, Truong DT, Scholz M, Armanini F, Ferretti P, Gorfer V, Pedrotti A, Tett A, Segata N. 2017. Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems 2:e00164-16. doi: 10.1128/mSystems.00164-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ. 2013. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- 23.Akabanda F, Owusu-Kwarteng J, Tano-Debrah K, Glover RL, Nielsen DS, Jespersen L. 2013. Taxonomic and molecular characterization of lactic acid bacteria and yeasts in nunu, a Ghanaian fermented milk product. Food Microbiol 34:277–283. doi: 10.1016/j.fm.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 24.Marsh AJ, Hill C, Ross RP, Cotter PD. 2014. Fermented beverages with health-promoting potential: past and future perspectives. Trends Food Sci Technol 38:113–124. doi: 10.1016/j.tifs.2014.05.002. [DOI] [Google Scholar]
- 25.Akabanda F, Owusu-Kwarteng J, Glover R, Tano-Debrah K. 2010. Microbiological characteristics of Ghanaian traditional fermented milk product, nunu. Nat Sci 8:178–187. [Google Scholar]
- 26.Fukushima M, Kakinuma K, Kawaguchi R. 2002. Phylogenetic analysis of Salmonella, Shigella, and Escherichia coli strains on the basis of the gyrB gene sequence. J Clin Microbiol 40:2779–2785. doi: 10.1128/JCM.40.8.2779-2785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scallan E, Hoekstra R, Mahon B, Jones T, Griffin P. 2015. An assessment of the human health impact of seven leading foodborne pathogens in the United States using disability adjusted life years. Epidemiol Infect 143:2795–2804. doi: 10.1017/S0950268814003185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdelgadir W, Nielsen DS, Hamad S, Jakobsen M. 2008. A traditional Sudanese fermented camel's milk product, Gariss, as a habitat of Streptococcus infantarius subsp. infantarius. Int J Food Microbiol 127:215–219. [DOI] [PubMed] [Google Scholar]
- 30.Jans C, Kaindi DWM, Böck D, Njage PMK, Kouamé-Sina SM, Bonfoh B, Lacroix C, Meile L. 2013. Prevalence and comparison of Streptococcus infantarius subsp. infantarius and Streptococcus gallolyticus subsp. macedonicus in raw and fermented dairy products from East and West Africa. Int J Food Microbiol 167:186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beck M, Frodl R, Funke G. 2008. Comprehensive study of strains previously designated Streptococcus bovis consecutively isolated from human blood cultures and emended description of Streptococcus gallolyticus and Streptococcus infantarius subsp. coli. J Clin Microbiol 46:2966–2972. doi: 10.1128/JCM.00078-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrero IA, Rouse MS, Piper KE, Alyaseen SA, Steckelberg JM, Patel R. 2002. Reevaluation of Streptococcus bovis endocarditis cases from 1975 to 1985 by 16S ribosomal DNA sequence analysis. J Clin Microbiol 40:3848–3850. doi: 10.1128/JCM.40.10.3848-3850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biarc J, Nguyen IS, Pini A, Gossé F, Richert S, Thiersé D, Van Dorsselaer A, Leize-Wagner E, Raul F, Klein J-P, Schöller-Guinard M. 2004. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S. bovis). Carcinogenesis 25:1477–1484. doi: 10.1093/carcin/bgh091. [DOI] [PubMed] [Google Scholar]
- 34.Doyle CJ, Gleeson D, O'Toole PW, Cotter PD. 2017. Impacts of seasonal housing and teat preparation on raw milk microbiota: a high-throughput sequencing study. Appl Environ Microbiol 83:e02694-16. doi: 10.1128/AEM.02694-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamhankar AJ, Nerkar SS, Khadake PP, Akolkar DB, Apurwa SR, Deshpande U, Khedkar SU, Stålsby-Lundborg C. 2015. Draft genome sequence of enterotoxigenic Escherichia coli strain E24377A, obtained from a tribal drinking water source in India. Genome Announc 3:e00225-15. doi: 10.1128/genomeA.00225-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clooney AG, Fouhy F, Sleator RD, O'Driscoll A, Stanton C, Cotter PD, Claesson MJ. 2016. Comparing apples and oranges? Next generation sequencing and its impact on microbiome analysis. PLoS One 11:e0148028. doi: 10.1371/journal.pone.0148028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 39.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 42.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. 2015. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12:902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 45.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silva GGZ, Green KT, Dutilh BE, Edwards RA. 2016. SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 32:354–361. doi: 10.1093/bioinformatics/btv584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methé B, Schloss PD, Gevers D, Mitreva M, Huttenhower C. 2012. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng Y, Leung HC, Yiu S-M, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 49.R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 50.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.