

ABSTRACT
Biotin prototrophy is a rare, incompletely understood, and industrially relevant characteristic of Saccharomyces cerevisiae strains. The genome of the haploid laboratory strain CEN.PK113-7D contains a full complement of biotin biosynthesis genes, but its growth in biotin-free synthetic medium is extremely slow (specific growth rate [μ] ≈ 0.01 h−1). Four independent evolution experiments in repeated batch cultures and accelerostats yielded strains whose growth rates (μ ≤ 0.36 h−1) in biotin-free and biotin-supplemented media were similar. Whole-genome resequencing of these evolved strains revealed up to 40-fold amplification of BIO1, which encodes pimeloyl-coenzyme A (CoA) synthetase. The additional copies of BIO1 were found on different chromosomes, and its amplification coincided with substantial chromosomal rearrangements. A key role of this gene amplification was confirmed by overexpression of BIO1 in strain CEN.PK113-7D, which enabled growth in biotin-free medium (μ = 0.15 h−1). Mutations in the membrane transporter genes TPO1 and/or PDR12 were found in several of the evolved strains. Deletion of TPO1 and PDR12 in a BIO1-overexpressing strain increased its specific growth rate to 0.25 h−1. The effects of null mutations in these genes, which have not been previously associated with biotin metabolism, were nonadditive. This study demonstrates that S. cerevisiae strains that carry the basic genetic information for biotin synthesis can be evolved for full biotin prototrophy and identifies new targets for engineering biotin prototrophy into laboratory and industrial strains of this yeast.
IMPORTANCE Although biotin (vitamin H) plays essential roles in all organisms, not all organisms can synthesize this vitamin. Many strains of baker's yeast, an important microorganism in industrial biotechnology, contain at least some of the genes required for biotin synthesis. However, most of these strains cannot synthesize biotin at all or do so at rates that are insufficient to sustain fast growth and product formation. Consequently, this expensive vitamin is routinely added to baker's yeast cultures. In this study, laboratory evolution in biotin-free growth medium yielded new strains that grew as fast in the absence of biotin as in its presence. By analyzing the DNA sequences of evolved biotin-independent strains, mutations were identified that contributed to this ability. This work demonstrates full biotin independence of an industrially relevant yeast and identifies mutations whose introduction into other yeast strains may reduce or eliminate their biotin requirements.
KEYWORDS: Saccharomyces cerevisiae, adaptive laboratory evolution, biotin, prototrophy, reverse metabolic engineering, vitamin biosynthesis, whole-genome sequencing
INTRODUCTION
Biotin (vitamin H or B7) functions as an essential prosthetic group of enzymes in all three domains of life. The yeast Saccharomyces cerevisiae harbors four groups of biotin-dependent enzymes: (i) pyruvate carboxylases (Pyc1 and Pyc2), which catalyze anaplerotic formation of oxaloacetate (1, 2); (ii) acetyl-coenzyme A (CoA) carboxylases (Acc1 and Hfa1), which generate malonyl-CoA, the key precursor for lipid synthesis (3); (iii) urea amidolyase (Dur1 and Dur2), which releases ammonia from urea (4); and (iv) the aminoacyl-tRNA synthetase cofactor Arc1, which is the only biotin-dependent protein in S. cerevisiae that is not a carboxylase (5). Biotin is covalently linked to these enzymes by the biotin-protein ligase Bpl1 (6).
While biotin prototrophy is widespread among prokaryotes and plants, animals and most fungi cannot synthesize this vitamin. The final four conserved steps in prokaryotic biotin biosynthesis are initiated by conversion of pimeloyl-CoA to 7-keto-8-aminopelargonic acid (KAPA) by KAPA synthase (BioF), after which DAPA aminotransferase (BioA) transaminates KAPA to 7,8-diaminopelargonic acid (DAPA). Subsequently, dethiobiotin synthetase (BioD) converts DAPA to dethiobiotin. Finally, sulfur insertion by biotin synthase (BioB) yields biotin (7) (Fig. 1). Two pathways for prokaryotic pimeloyl-CoA synthesis have been described. Some bacteria, including Escherichia coli, convert three molecules of malonyl-CoA to pimeloyl-CoA (8). Others, including Bacillus subtilis, generate this precursor by oxidative cleavage of fatty-acyl molecules (9; reviewed in reference 10).
FIG 1.
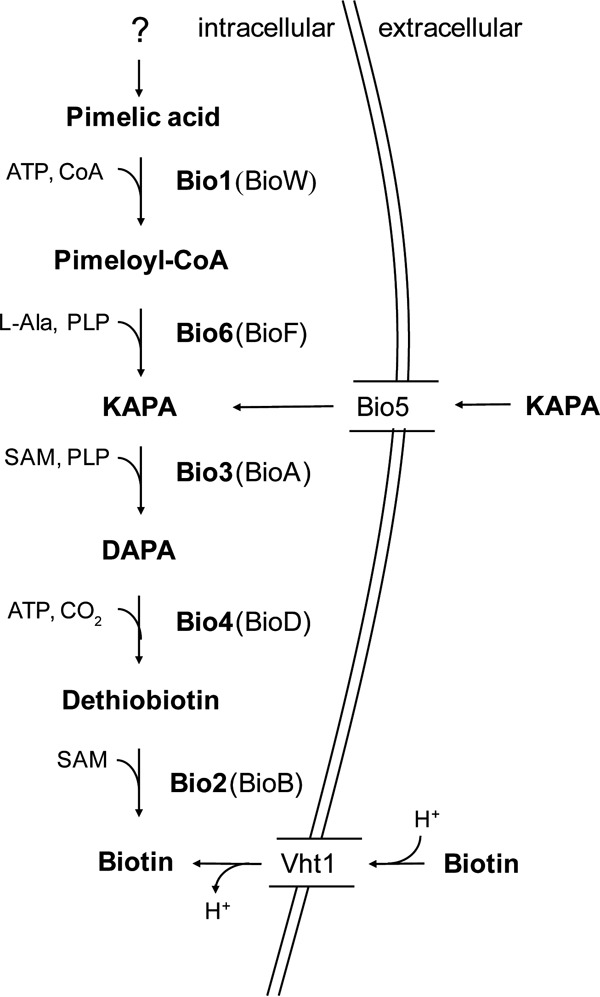
Biotin biosynthesis in S. cerevisiae. BIO genes encode the following enzymes: Bio1, pimeloyl-CoA synthetase; Bio6, KAPA synthetase; Bio3, DAPA aminotransferase; Bio4, dethiobiotin synthase; Bio2, biotin synthase; SAM, S-adenosylmethionine; and PLP, pyridoxal phosphate. The protein names in parentheses indicate the corresponding bacterial enzymes. In S. cerevisiae, biotin and its precursor, KAPA, can be imported via the proton symporter Vht1 and the KAPA permease Bio5, respectively.
Biotin prototrophy is rare among yeasts and has a convoluted evolutionary history, with yeast strains of the same species sometimes exhibiting different biotin requirements. Most yeast strains isolated from nature, as well as laboratory strains, such as S. cerevisiae S288C, are biotin auxotrophs (11). Starting from pimelic acid, biotin biosynthesis in prototrophic yeast strains follows the prokaryotic pathway (Fig. 1), but the yeast pathway for synthesis of this key precursor has not yet been fully elucidated (10, 12, 13). Biotin biosynthesis genes in yeast are assumed to have been at least partially acquired from anaerobic bacteria by horizontal gene transfer, followed by gene duplication and neofunctionalization events (14, 15).
Most biotin-auxotrophic S. cerevisiae strains contain the genes encoding the last three enzymes of the biotin biosynthesis pathway (BIO3, BIO4, and BIO2, which are orthologs of E. coli bioA, bioD, and bioB). Some other yeasts, such as Schizosaccharomyces pombe and Pichia pastoris, contain only the biotin synthase (BIO2) gene, an ortholog of prokaryotic bioB genes. In 2005, Wu et al. discovered BIO6, an ortholog of bacterial bioF genes, in biotin-prototrophic sake strains of S. cerevisiae (16). Hall and Dietrich completed the S. cerevisiae gene set for conversion of pimelic acid to biotin by discovering BIO1, which encodes pimeloyl-CoA synthetase and, in strains that carry either gene, is located adjacent to BIO6. Sequence similarity suggests that BIO6 evolved by duplication and neofunctionalization of BIO3, thereby converting a DAPA synthase into a KAPA synthetase. A similar evolutionary relationship was proposed for BIO1 and YJR154W (14).
Lack or loss of pathways for de novo vitamin biosynthesis in microbes has been proposed to reflect an evolutionary trade-off between fitness in natural environments where biotin is scarce and energy costs involved in biotin synthesis (17). Consistent with this notion, biotin-prototrophic bacteria, archaea, and plants all harbor transporters for biotin uptake (18). S. cerevisiae imports biotin via the high-affinity proton symporter Vht1 (19). Some strains additionally harbor the Bio5 transporter, which imports the precursor KAPA, thus enabling biotin synthesis from exogenous KAPA (15, 20, 21). In S. cerevisiae strains that carry a BIO5 gene, it is tightly linked to BIO3 and BIO4 in a gene cluster on chromosome XIV (ChrXIV) (14).
Most synthetic media for S. cerevisiae are routinely supplied with biotin. Vitamin addition increases production costs and decreases the shelf life of the media. Biotin supplementation adds a delicate step in medium preparation, since the high pH required for dissolving biotin negatively affects its stability, thereby increasing the risk of batch-to-batch variations. Moreover, biotin is an expensive vitamin. It has been estimated that, at a reactor volume of 150 m3, costs for large-scale industrial fermentation processes may be on the order of $1,000 per fermentation (22). Clearly, the availability of fast-growing biotin-prototrophic strains could benefit process economics.
Several studies have focused on engineering microorganisms for biotin prototrophy. Expression of heterologous BIO genes in the biotin-auxotrophic yeast P. pastoris reduced medium costs in fed-batch-based production processes, even though the engineered biotin-prototrophic strain grew more slowly than the reference strain (21). Enhancing biotin synthesis in solventogenic clostridia improved production titers of acetone, butanol, and ethanol, predominantly by increasing cellular viability and performance (23).
The goal of this study was to identify key genetic determinants of biotin prototrophy in S. cerevisiae. To this end, the haploid strain S. cerevisiae CEN.PK113-7D, a popular model for systems biology and metabolic-engineering research (22, 24–26), was subjected to laboratory evolution in biotin-free medium. Evolved biotin-prototrophic cell lines were further characterized by whole-genome resequencing and by reverse engineering of identified mutations in the parental strain.
RESULTS
Laboratory evolution of S. cerevisiae CEN.PK113-7D for full biotin prototrophy.
After inoculation of biotin-free SMD (see Materials and Methods) with a biotin-depleted preculture of strain CEN.PK113-7D, it took ca. 20 days before slow growth, at a specific growth rate of ca. 0.01 h−1, was observed. At a final cell number in the shake flasks of 4 × 1011 cells · liter−1 and a specific growth rate of 0.01 h−1 (i.e., a doubling time of 69 h), this implies an initial concentration of growing cells of at least 2 × 109 cells · liter−1. These results confirm an earlier report that strain CEN.PK113-7D is not completely auxotrophic for biotin but can grow at very low rates in the absence of this vitamin (22).
To explore the evolvability of full biotin prototrophy, i.e., a phenotype with identical specific growth rates in the presence and absence of biotin, strain CEN.PK113-7D was grown in accelerostats. These are continuous cultures in which the dilution rate is increased over time (27). To select for fast-growing biotin-prototrophic mutants in triplicate glucose-limited accelerostat cultures on biotin-free SMD, the dilution rate was feedback controlled based on the CO2 concentration in the off gas (see Fig. S2 in the supplemental material). A fourth laboratory evolution experiment was performed in a sequential batch reactor (SBR) in which automated empty-refill cycles were based on the CO2 concentration in the off gas, leaving ca. 5% of the culture as an inoculum for each subsequent batch cycle (28) (Fig. 2). After 48 to 77 days of accelerostat cultivation, dilution rates of up to 0.27 h−1 were reached (Fig. S2). Single cell lines were isolated at the end of each of the accelerostat experiments and named IMS0478, IMS0480, and IMS0481 (Table 1). The SBR experiment was terminated when, after 47 days and 11 cycles, multicellular aggregates appeared in the culture due to the SBR setup-inherent empty-refill selection procedure, which is prone to unintended selection of cells developing a clumping phenotype, enabling their persistence in the bioreactor without further improvement of their growth rate under selective conditions (29). At this point, the specific growth rate, as estimated from CO2 production profiles, had reached 0.22 h−1 (Fig. 2). A nonaggregating single-colony isolate from the SBR culture was named IMS0496. All four single-cell isolates showed high specific growth rates in biotin-free SMD, ranging from 0.25 h−1 (strain IMS0478) to 0.36 h−1 (strain IMS0481) (Table 2). This value is close to the specific growth rate of CEN.PK113-7D in biotin-supplemented SMD (0.39 to 0.40 h−1) (30–33). Biotin-supplemented cultures of the evolved strains showed an average specific growth rate of 0.35 h−1, indicating that the evolved strain IMS0481 had acquired full biotin prototrophy (Table 2).
FIG 2.

Laboratory evolution of S. cerevisiae CEN.PK113-7D cells in a sequential batch reactor (SBR) for improved growth in biotin-free synthetic medium. Shown are off gas CO2 (percent) profiles during an SBR experiment in which automated empty-refill cycles were based on the CO2 concentration in the off gas, leaving ca. 5% of the culture as an inoculum for each subsequent batch cycle (28). CO2 production in the initial cycle reflects depletion of biotin pools in the inoculum. Specific growth rates (μ) were calculated from the exponential increase of the off gas CO2 concentration in each cycle. Graphs representing the increase of dilution rates in accelerostats over time are depicted in Fig. S2 in the supplemental material.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Relevant genotype | Description/use | Reference |
|---|---|---|---|
| CEN.PK113-7D | MATa | Reference strain | 39 |
| CEN.PK113-5D | MATa ura3-52 | Uracil-auxotrophic reference strain | 39 |
| S288C | MATa | Marker strain for CHEF analysis | YGSCa |
| IMX585 | MATa can1::CAS9-tagA-loxP-natNT2-loxP | CEN.PK113-7D expressing Cas9 | 34 |
| IMS0478 | MATa, evolved | CEN.PK113-7D evolved for biotin prototrophy (accelerostat A) | This study |
| IMS0480 | MATa, evolved | CEN.PK113-7D evolved for biotin prototrophy (accelerostat B) | This study |
| IMS0481 | MATa, evolved | CEN.PK113-7D evolved for biotin prototrophy (accelerostat C) | This study |
| IMS0496 | MATa, evolved | CEN.PK113-7D evolved for biotin prototrophy (sequential batch reactor) | This study |
| IME327 | MATa ura3-52 pUDE446 | CEN.PK113-5D pUDE446 (BIO1-BIO6) | This study |
| IME329 | MATa ura3-52 pUDE448 | CEN.PK113-5D pUDE448 (BIO6) | This study |
| IME331 | MATa ura3-52 pUDE450 | CEN.PK113-5D pUDE450 (BIO1) | This study |
| IME334 | MATa ura3-52 p426GPD | CEN.PK113-5D p426GPD (empty) | This study |
| IMK129 | MATa ura3-52 tpo1::loxP-kanMX-loxP | CEN.PK113-5D; tpo1::loxP-kanMX-loxP | This study |
| IMZ694 | MATa ura3-52 tpo1::loxP-kanMX-loxP pUDE450 | IMK129 (tpo1Δ) pUDE450 (BIO1) | This study |
| IMZ695 | MATa ura3-52 tpo1::loxP-kanMX-loxP p426GPD | IMK129 (tpo1Δ) p426GPD (empty) | This study |
| IMK163 | MATa ura3-52 pdr12::loxP-kanMX-loxP | CEN.PK113-5D; pdr12::loxP-kanMX-loxP | This study |
| IMZ704 | MATa ura3-52 pdr12::loxP-kanMX-loxP pUDE450 | IMK163 (pdr12Δ) pUDE450 (BIO1) | This study |
| IMZ705 | MATa ura3-52 pdr12::loxP-kanMX-loxP p426GPD | IMK163 (pdr12Δ) p426GPD (empty) | This study |
| IMK773 | MATa ura3-52 tpo1::loxP-kanMX-loxP pdr12::hphNT1 | CEN.PK113-5D tpo1::loxP-kanMX-loxP pdr12::hphNT1 | This study |
| IMZ701 | MATa ura3-52 tpo1::loxP-kanMX-loxP pdr12::hphNT1 pUDE450 | IMK773 (tpo1Δ, pdr12Δ) pUDE450 (BIO1) | This study |
| IMZ702 | MATa ura3-52 tpo1::loxP-kanMX-loxP pdr12::hphNT1 p426GPD | IMK773 (tpo1Δ pdr12Δ) p426GPD (empty) | This study |
YGSC, Yeast Genetic Stock Center, Berkeley, CA.
TABLE 2.
Specific growth rates of laboratory-evolved biotin-prototrophic S. cerevisiae strains and of strains carrying defined mutations grown in shake flask cultures containing synthetic media with or without biotin supplementationa
| Strain | Relevant genotype | Growth rate (h−1) in SMD |
|
|---|---|---|---|
| Without biotin | With biotin | ||
| CEN.PK113-7D | Haploid laboratory strain | <0.01 | 0.39 ± 0.01b |
| IMS0478 | CEN.PK113-7D evolved for biotin prototrophy (accelerostat A) | 0.25 ± 0.00 | 0.34 ± 0.02 |
| IMS0480 | CEN.PK113-7D evolved for biotin prototrophy (accelerostat B) | 0.31 ± 0.00 | 0.34 ± 0.00 |
| IMS0481 | CEN.PK113-7D evolved for biotin prototrophy (accelerostat C) | 0.36 ± 0.00 | 0.41 ± 0.00 |
| IMS0496 | CEN.PK113-7D evolved for biotin prototrophy (SBR) | 0.32 ± 0.01 | 0.31 ± 0.02 |
| IME327 | CEN.PK113-5D + pUDE446 (2μ BIO1-BIO6) | 0.16 ± 0.01 | 0.32 ± 0.01 |
| IME329 | CEN.PK113-5D + pUDE448 (2μ BIO6) | No growth | 0.30 ± 0.00 |
| IME331 | CEN.PK113-5D + pUDE450 (2μ BIO1) | 0.15 ± 0.02 | 0.32 ± 0.01 |
| IME334 | CEN.PK113-5D + p426GPD (empty plasmid) | No growth | 0.34 ± 0.01 |
| IMZ694 | tpo1Δ, pUDE450 (2μ BIO1) | 0.23 ± 0.00 | 0.32 ± 0.00 |
| IMZ695 | tpo1Δ p426GPD (empty plasmid) | No growth | 0.33 ± 0.01 |
| IMZ704 | pdr12Δ pUDE450 (2μ BIO1) | 0.25 ± 0.00 | 0.37 ± 0.00 |
| IMZ705 | pdr12Δ p426GPD (empty plasmid) | No growth | 0.40 ± 0.00 |
| IMZ701 | pdr12Δtpo1Δ pUDE450 (2μ BIO1) | 0.25 ± 0.01 | 0.36 ± 0.01 |
| IMZ702 | pdr12Δtpo1Δ p426GPD (empty plasmid) | No growth | 0.37 ± 0.00 |
Cells were grown in aerobic 100-ml shake flask cultures. The data represent averages ± standard deviations of the mean of measurements of independent duplicate cultures.
Reference 32.
Massive amplification of BIO1 and BIO6 and increased expression of BIO genes in evolved biotin-prototrophic strains.
To investigate the molecular basis of the acquired biotin prototrophy, expression levels and copy numbers of BIO genes were measured by quantitative PCR (qPCR) analysis and whole-genome sequencing, respectively. In shake-flask cultures on biotin-supplemented SMD, transcript levels of BIO genes were lower than those of the reference gene, ACT1, in all strains tested (Fig. 3A; see Table S2 in the supplemental material). Transcript levels of BIO genes in the evolved strains resembled those in the parental strain, except for BIO1, whose transcript levels were 4- to 8-fold higher in the evolved strains. Growth in SMD without biotin resulted in transcriptional upregulation of BIO genes (Fig. 3B; see Table S2 in the supplemental material). As observed in biotin-supplemented cultures, BIO1 was expressed at higher levels than the other BIO genes, reaching transcript levels that were 1.5- to 24-fold higher than those of ACT1 (Fig. 3B).
FIG 3.

mRNA levels of BIO genes in strains evolved for biotin prototrophy. (A) Transcript levels in cultures grown on SMD with biotin. Shown are transcript levels of BIO1, BIO2, BIO3, BIO4, and BIO6 in the parent strain, CEN.PK113-7D (hatched bars), and in the evolved strains IMS0478 (white bars), IMS0480 (gray bars), IMS0481 (black bars), and IMS0496 (cross-hatched bars) relative to ACT1 expression levels. (B) Transcript levels in cultures grown on SMD without biotin. Shown are transcript levels of BIO1, BIO2, BIO3, BIO4, and BIO6 in the evolved strains IMS0478 (white bars), IMS0480 (gray bars), IMS0481 (black bars), and IMS0496 (cross-hatched bars) relative to ACT1 expression levels. All qPCR experiments were carried out on duplicate cultures, with analytical triplicates for each culture. Relative expression levels were determined according to the ΔΔCT method (45). The error bars represent the SEM of duplicate analyses.
Analysis of sequencing read depths revealed massive amplification of BIO1 and BIO6 in the evolved strains, with copy numbers ranging from 8 to 43 (Table 3). The similar amplifications of BIO1 and BIO6 observed in this analysis were consistent with their physical linkage (22). The evolved strains harbored one or two copies of BIO2, BIO3, or BIO4, except for the slowest-growing strain, IMS0478, in which BIO3 and BIO4 were amplified 8- and 6-fold, respectively.
TABLE 3.
Estimated copy numbers of BIO1, BIO2, BIO3, BIO4, and BIO6 in the evolved biotin-prototrophic strains and the parent strain, CEN.PK113-7D
| Strain | Gene copy no. per straina |
||||
|---|---|---|---|---|---|
| BIO1 | BIO2 | BIO3 | BIO4 | BIO6 | |
| CEN.PK113-7D | 1 | 1 | 1 | 1 | 1 |
| IMS0478 | 8 | 2 | 8 | 6 | 8 |
| IMS0480 | 42 | 2 | 1 | 1 | 43 |
| IMS0481 | 19 | 2 | 1 | 1 | 19 |
| IMS0496 | 19 | 2 | 2 | 1 | 20 |
Copy numbers were calculated from sequence coverage depths of four strains independently evolved for biotin prototrophy in accelerostats (IMS0478, IMS0480, and IMS0481) and in a sequential batch reactor setup (IMS0496). All five genes indicated occur as single-copy genes in the parental strain CEN.PK113-7D (24).
Amplification of BIO1 and BIO6 involves chromosomal duplications and rearrangements and formation of neochromosomes.
In three of the four evolved biotin-prototrophic strains, read depth analysis revealed a duplication of ChrI (Fig. 4), which, in the parental strain, carries the BIO1 and BIO6 genes (22). Strain IMS0478, which also showed increased copy numbers of other BIO genes, carried additional duplications of ChrIX, the left arm of ChrVIII, and an amplified region close to the telomere of the right arm of ChrXIV, where the BIO3-BIO4-BIO5 cluster is located (Fig. 4A).
FIG 4.
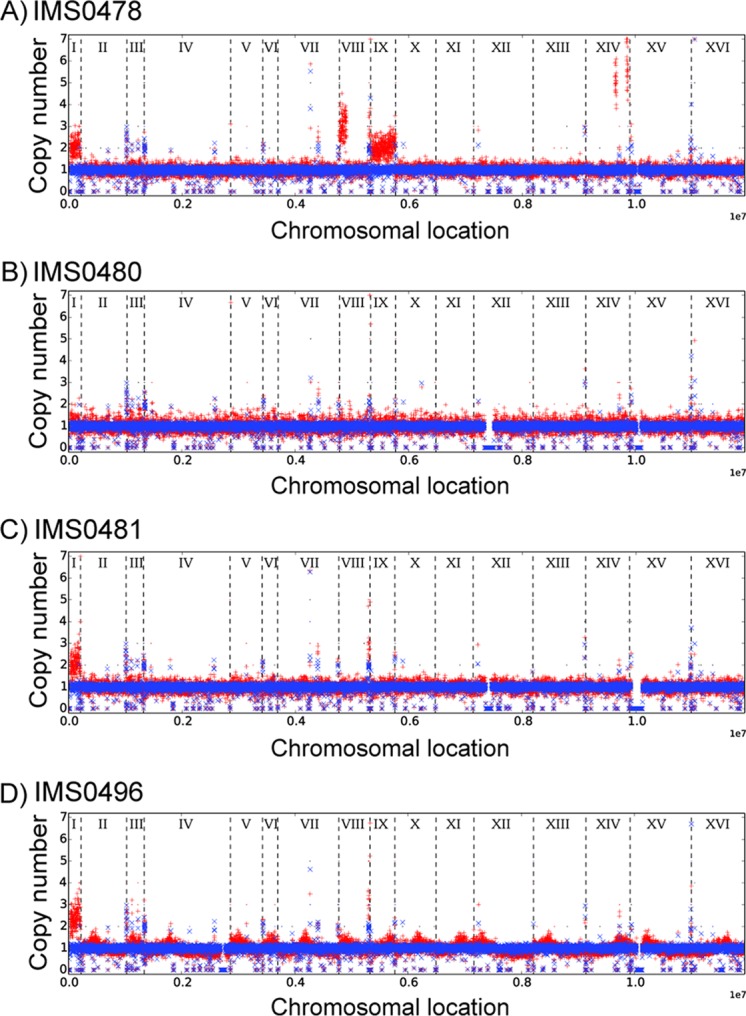
Chromosomal copy number variations in yeast strains evolved for full biotin prototrophy. Strains IMS0478, IMS0480, and IMS0481 were evolved in accelerostats, while strain IMS0496 was evolved in a sequential batch reactor. Copy numbers of chromosomes and chromosomal regions were calculated from sequence data with the Magnolya algorithm (67). The results for the parental strain, S. cerevisiae CEN.PK113-7D, and for the evolved strains are indicated in blue and red, respectively. Individual chromosomes, indicated by Roman numerals, are separated by dashed lines.
Electrophoretic karyotyping revealed strong differences in chromosome sizes between the evolved strains, as well as their common unevolved parental strain, while strains IMS0478, IMS0480, and IMS0481 contained additional, supernumerary chromosomes (neochromosomes) (Fig. 5A). Southern hybridization analysis showed that copies of the BIO1 gene occurred on multiple chromosomes and neochromosomes in the evolved strains (Fig. 5B). This experiment confirmed that S. cerevisiae strain S288C, whose genomic DNA was used as a size marker for chromosome identification, lacks the BIO1 gene (Fig. 5B) (16, 22).
FIG 5.
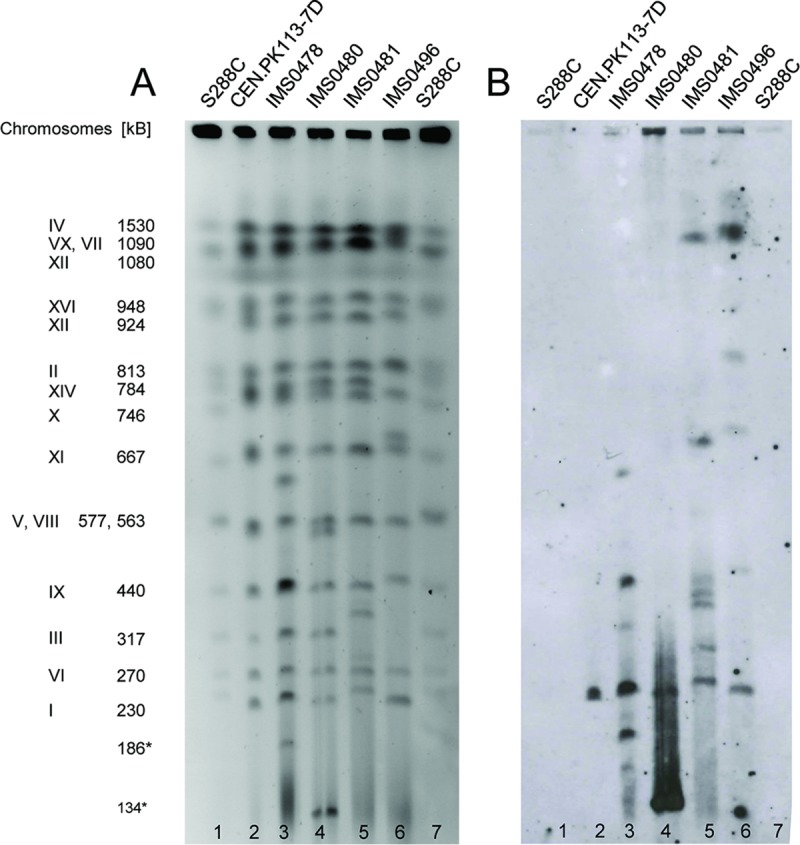
Karyotyping and chromosomal localization of BIO1 in evolved biotin-prototrophic yeast strains. (A) Pulsed-field gel electrophoresis (PFGE) of the chromosomes of evolved biotin-prototrophic strains IMS0478, IMS0480, IMS0481, and IMS0496 and the parent strain, S. cerevisiae CEN.PK113-7D. Chromosome numbers and sizes (kilobases) were obtained using S. cerevisiae S288C as a reference strain. (B) Southern blot of the PFGE gel. Hybridization with BIO1 probe revealed copies of BIO1 on multiple chromosomes in the evolved strains.
Increased copy numbers of BIO1 play a pivotal role in acquired biotin prototrophy.
To investigate if the high copy numbers of BIO1 and BIO6 contributed to the acquired biotin prototrophy of the evolved strains, the genes were expressed from multicopy plasmids under the control of strong, constitutive promoters in a nonevolved, biotin-auxotrophic strain. Overexpression of BIO1, together with BIO6, a situation that mimicked the amplification of both genes in the evolved strains, enabled growth in SMD without biotin at a specific growth rate of 0.15 h−1 (Table 2). However, the same growth rate was obtained when only BIO1 was overexpressed. Further, growth in biotin-free SMD was not observed in a strain that overexpressed only BIO6. These results indicated that increased copy numbers of BIO1, but not of BIO6, contributed to the acquired biotin prototrophy of evolved strains.
Mutations in the membrane transporter genes TPO1 and PDR12 contribute to fast growth in biotin-free medium.
Although overexpression of BIO1 in a nonevolved strain background enabled growth in biotin-free medium, the specific growth rates of the resulting strains were only about half of those observed in the fast-growing evolved strains. To further analyze the genetic basis for fast growth in biotin-free medium, the genome sequences of the evolved biotin-prototrophic strains were analyzed in more detail. The four independently evolved strains harbored 6 to 11 single-nucleotide changes within coding regions relative to the parental strain (see Table S1 in the supplemental material). Three out of the four evolved strains harbored a nonsynonymous or nonsense mutation in TPO1, which encodes a plasma membrane polyamine transporter (34). TPO1 was the only gene that harbored single-nucleotide polymorphisms (SNPs) in more than one strain. However, a nonsense mutation in the ABC transporter gene PDR12 in strain IMS0481 coincided with a nonsense mutation in WAR1 in strain IMS0480, which encodes a transcriptional activator of PDR12 (35) (see Table S1). Inactivation of either PDR12 or WAR1 causes absence of Pdr12 from the plasma membrane (35). Pdr12 has been shown to export a wide range of monocarboxylic acids (C3 to C7) (33, 36). To investigate a possible role of the mutations in PDR12 and/or TPO1 in biotin prototrophy, pdr12Δ, tpo1Δ, and pdr12Δ tpo1Δ mutations were introduced in strains overexpressing BIO1. Deletion of PDR12 or TPO1 did not lead to biotin prototrophy in a strain with a single wild-type BIO1 gene (Table 2). In contrast, deletion of either of these transporter genes greatly increased the specific growth rates of strains that overexpressed BIO1 in biotin-free SMD, from 0.15 h−1 in a strain without transporter deletion to up to 0.25 h−1 in a pdr12Δ strain (Table 2). A combination of both deletions did not lead to a further increase of the specific growth rate (Table 2).
A possible explanation for the observed impact of mutations in TPO1 and PDR12 is that they prevent export of an essential intermediate in the biotin biosynthesis pathway by the encoded transporters. Pimelic acid, a C7-dicarboxylic acid, is the substrate for Bio1, which, based on the amplification of BIO1 observed in evolved strains, may catalyze a rate-controlling reaction in biotin synthesis. For many organic acids, a role of Pdr12 in their export from S. cerevisiae cells has been inferred from a strongly increased sensitivity of pdr12Δ to the acid at pH values below its pKa due to weak organic acid uncoupling (33, 36, 37). Pimelic acid supplementation of cultures grown at pH 4.5, which is below the pKa values of pimelic acid (pKa,1 = 4.71; pKa,2 = 5.58), did not reveal increased sensitivity of PDR12 and/or TPO1 single- or double-deletion mutants (see Fig. S1 in the supplemental material). However, growth of the reference strain, CEN.PK113-7D, at pH 4.5 was not inhibited by pimelic acid concentrations of up to 100 mM (data not shown), indicating that pimelic acid uptake rates were too low to cause weak organic acid uncoupling. The absence of increased sensitivity in the deletion mutants, therefore, neither supports nor excludes a possible role of Tpo1 and/or Pdr12 in pimelic acid export.
DISCUSSION
Although the genome of S. cerevisiae CEN.PK113-7D contains a full set of BIO genes (BIO1, BIO2, BIO3, BIO4, BIO5, and BIO6) (22), the specific growth rate of the strain in biotin-free synthetic medium with glucose was at least 30-fold lower than in cultures supplemented with this vitamin. Although data obtained with this typical laboratory strain (38) cannot be directly extrapolated to natural isolates, such very low growth rates may well be relevant for survival in natural environments where biotin is scarce. Independent laboratory evolution experiments with strain CEN.PK113-7D yielded four evolved strains, three of which grew equally fast in biotin-free medium and in biotin-supplemented cultures. Such complete biotin prototrophy is rare among natural biotin-prototrophic strains, whose growth on biotin-free medium has been reported to vary from weak to vigorous but typically is slower than in biotin-containing medium (10, 14, 16).
The mutations that were shown to contribute to full biotin prototrophy of the laboratory-evolved strains provide new insights into the genetic basis of the phenotype. Up to 40-fold amplification of the clustered BIO1 and BIO6 genes in the evolved strains was shown to reflect a key role of the copy number of BIO1, but not BIO6, in biotin prototrophy. Introduction of a multicopy vector carrying BIO1 in a nonevolved strain enabled it to grow on biotin-free medium at a growth rate ca. 40% of that observed in the fastest-growing evolved strains. These results indicate that the pimeloyl-CoA synthetase Bio1 exerts a high degree of metabolic control over biotin biosynthesis in the nonevolved strain. Comparison of the predicted protein sequence of BIO1 from strain CEN.PK113-7D did not identify amino acids that were not also found in BIO1 genes of other S. cerevisiae strains, indicating that the requirement for BIO1 amplification was unlikely to be due to a CEN.PK-specific, inferior BIO1 allele. Three of the four evolved strains contained a duplication of ChrI, which carries the native copies of BIO1 and BIO6, while additional copies of these genes were found on neochromosomes resulting from translocation events. The plasticity of the yeast genome under selective growth conditions is further illustrated in one of the evolved strains (IMS0478) by the additional presence of two copies of ChrIX, as well as of the left arm of ChrVIII. These results confirm that chromosomal rearrangements and copy number variation are key mechanisms for genetic adaptation in short-term laboratory evolution experiments (39, 40). The biotin-prototrophic sake strain of S. cerevisiae in which BIO6 was first discovered also contains copies of BIO6 on multiple chromosomes (16). This genotypic similarity indicates that the genetic adaptations that enable sake yeasts to grow at the very low biotin concentrations in sake mash (41) at least partially overlap those seen in the present laboratory evolution study.
Surprisingly, loss-of-function mutations in the membrane transporter genes TPO1 and PDR12, as well as in WAR1, which encodes a positive regulator of PDR12 (35, 42), had a strong positive effect on biotin prototrophy. Neither the polyamine transporter Tpo1 (34, 43) nor the monocarboxylate exporter Pdr12 (36) has previously been associated with biotin synthesis. Although in the case of Tpo1 direct mediation of monocarboxylic acid transport has not been demonstrated, both transporters have been implicated in the process, including, in the case of Pdr12, the transport of the C7-monocarboxylate heptatonic acid (37). If Pdr12 and/or Tpo1 export the C7-dicarboxylate pimelic acid, the precursor of biotin biosynthesis, the resulting decrease in the intracellular pimelate concentration could reduce the flux through the rate-controlling pimeloyl-CoA synthetase (Bio1) reaction. Alternatively, Pdr12 and/or Tpo1 might catalyze the export of other key intermediates of biotin synthesis, such as the aminated compounds KAPA and DAPA. A mutation in one of the evolved strains in the KAPA transporter gene BIO5 could also be consistent with this hypothesis.
In the case of Tpo1, an alternative explanation for its impact on biotin prototrophy is related to its role in spermidine homeostasis. Supplementation of this compound in a spermidine-deficient S. cerevisiae strain has been shown to cause an up to 14-fold upregulation of BIO genes (44). At a pH of 5, which was used in the present laboratory evolution, Tpo1 has been reported to primarily catalyze polyamine export (45). Moreover, inactivation of TPO1 has been shown to lead to increased intracellular spermidine concentrations (46, 47). By enhancing the previously demonstrated induction of BIO genes upon biotin depletion (16, 48, 49), accumulation of polyamines could contribute to increased growth rates in biotin-free medium.
Elimination of vitamin requirements of S. cerevisiae could simplify the design and scaling up of fermentation processes and improve process economics. The demonstration that an S. cerevisiae strain that contains a basic complement of BIO genes can be evolved for complete biotin prototrophy opens up perspectives for the development of industrial S. cerevisiae strains that are completely prototrophic for this and, potentially, other vitamins. However, the extensive genomic rearrangements in the evolved strains complicate their direct use as metabolic-engineering platforms. Overexpression of BIO1, combined with deletion of TPO1 or PDR12, was sufficient to reach specific growth rates in biotin-free medium that were only 40% lower than those observed in biotin-supplemented cultures. Null mutations in TPO1 and PDR12 had a similar effect on the specific growth rates of BIO1-overexpressing strains in biotin-free medium, but their effects were not additive (Table 2). While inactivation of PDR12 strongly increases the sensitivity of S. cerevisiae to several apolar carboxylic acids (36), null mutations in TPO1 have been reported to confer an increased tolerance for industrially relevant inhibitors, which in turn has been shown to result in higher productivities of industrial products (46, 50). Inactivation of TPO1, therefore, appears to be the preferred intervention in strategies for eliminating biotin requirements in industrial yeast strains.
A further systematic analysis of the other mutations in evolved biotin-prototrophic strains, combined with overexpression and/or codon optimization of BIO1 and other BIO genes, might allow additional improvements of rationally engineered biotin-prototrophic S. cerevisiae strains. In addition, the fast-growing biotin-prototrophic strains described in this study provide interesting experimental platforms for unraveling the elusive biochemistry of pimelate biosynthesis in S. cerevisiae and other yeasts (10, 12, 13).
MATERIALS AND METHODS
Strains, media, and maintenance.
The S. cerevisiae strains used and constructed in this study (Table 1) belong to the CEN.PK lineage (38), with the exception of S. cerevisiae S288C (38, 51). Yeast strains were grown on synthetic medium (SM) or YP medium (10 g · liter−1 Bacto yeast extract, 20 g · liter−1 Bacto peptone). Synthetic medium with urea as the nitrogen source (SM-urea) contained 38 mM urea and 38 mM K2SO4 instead of (NH4)2SO4. After autoclaving SM at 121°C for 20 min or sterile filtration of SM-urea using 0.2-μm bottle top filters (Thermo Scientific, Waltham, MA), synthetic media were supplemented with 1 ml · liter−1 of a filter-sterilized vitamin solution [0.05 g · liter−1 d-(+)-biotin, 1.0 g · liter−1 d-calcium pantothenate, 1.0 g · liter−1 nicotinic acid, 25 g · liter−1 myoinositol, 1.0 g · liter−1 thiamine hydrochloride, 1.0 g · liter−1 pyridoxol hydrochloride, 0.20 g · liter−1 4-aminobenzoic acid). Biotin-free SM was prepared by omitting biotin from this solution. Unless specifically indicated, “SM” specifies synthetic medium with (NH4)2SO4 as the nitrogen source, while synthetic medium with urea as the nitrogen source is abbreviated as “SM-urea.” After autoclaving concentrated glucose solutions at 110°C for 20 min, glucose was added to SM, SM-urea, and YP media to a final concentration of 20 g · liter−1, yielding SMD, SMD-urea, and YPD, respectively. Shake flasks (500 ml) containing 100 ml medium, as well as 50 ml Cellreactor filter top tubes (Greiner Bio-One B.V., Alphen a/d Rijn, The Netherlands) containing 25 ml medium, were incubated at 30°C and 200 rpm in an Innova incubator (Brunswick Scientific, Edison, NJ). Solid media contained 1.5% Bacto agar and, when indicated, 200 mg · liter−1 G418 or 200 mg · liter−1 hygromycin. Selection and counterselection of the amdSYM cassette were performed as described previously (52). E. coli strains were grown in LB (10 g · liter−1 Bacto tryptone, 5 g · liter−1 Bacto yeast extract, 5 g · liter−1 NaCl) supplemented with 100 mg · liter−1 ampicillin. Yeast and E. coli cultures were stored at −80°C after addition of (30% [vol/vol]) glycerol to stationary-phase shake flask cultures.
Molecular biology techniques.
PCR amplification of DNA fragments with Phusion Hot Start II high-fidelity polymerase (Thermo Scientific) and desalted or PAGE-purified oligonucleotide primers (Sigma-Aldrich, St. Louis, MO) was performed according to the manufacturers' instructions. DreamTaq polymerase (Thermo Scientific) was used for diagnostic PCR. The oligonucleotide primers used in this study are listed in Table 4. Amplified DNA fragments were separated by electrophoresis on 1% (wt/vol) agarose gels (Thermo Scientific) in Tris-acetate-EDTA (TAE) buffer (Thermo Scientific) at 90 V for 35 min and purified with a GenElute PCR Clean-Up kit (Sigma-Aldrich). Plasmids were isolated from yeast cultures with a Zymoprep Yeast Plasmid Miniprep II kit (Zymo Research, Irvine, CA) and from E. coli with a Sigma GenElute Plasmid kit (Sigma-Aldrich). Yeast genomic DNA was isolated using a YeaStar Genomic DNA kit (Zymo Research) or with an SDS-lithium acetate (LiAc) protocol (53). Yeast strains were transformed by the lithium acetate method (54), and eight of the resulting colonies were restreaked three consecutive times on biotin-supplemented selective medium, followed by analytical PCR to verify their genotype. E. coli DH5α was used for chemical transformation (55) or for electroporation. Electroporation was done in a 2-mm cuvette (1652086; Bio-Rad, Hercules, CA) using a Gene Pulser Xcell electroporation system (Bio-Rad). After isolation, plasmids were verified by restriction analysis and analytical PCR.
TABLE 4.
Primers used in this study
| Product | Primer | Sequence (5′ to 3′) |
|---|---|---|
| Construction of plasmids pUD416 and pUD418 | ||
| pPYK1 promoter fragment with TagB | 8907 | CACCTTTCGAGAGGACGATGCCCGTGTCTAAATGATTCGACCAGCCTAAGAATGTTCAACACCTCGGATCGTCGGTTGTG |
| 7428 | TGTGATGATGTTTTATTTGTTTTGATTGG | |
| pPGK1 promoter fragment with TagA | 8908 | ACTATATGTGAAGGCATGGCTATGGCACGGCAGACATTCCGCCAGATCATCAATAGGCACACTGTAATTGCTTTTAGTTGTGTATTTTTAG |
| 5917 | TGTTTTATATTTGTTGTAAAAAGTAGATAATTACTTCC | |
| BIO1 + tBIO1 | 8915 | CCAATCAAAACAAATAAAACATCATCACAATGAACACAAAATCACTCGACTTTTATG |
| 8914 | GTGCCTATTGATGATCTGGCGGAATGTCTGCCGTGCCATAGCCATGCCTTCACATATAGTTTTACTTCTTTCTTTACCTTTTTTTTCTTTTAC | |
| BIO6 + tBIO6 | 8916 | GGAAGTAATTATCTACTTTTTACAACAAATATAAAACAATGTGTGAACATCAATTAACCCAAG |
| 7467 | TTTACAATATAGTGATAATCGTGGACTAGAGCAAGATTTCAAATAAGTAACAGCAGCAAATATTACTCTCCTAAACTGAGCACAAGTTTC | |
| Fusion of TagA-pPGK1 + BIO6-sga1flank | 3847 | ACTATATGTGAAGGCATGGCTATGG |
| 4187 | TTACAATATAGTGATAATCGTGGACTAGAG | |
| Fusion of TagB-pPYK + BIO1-TagA | 4691 | CACCTTTCGAGAGGACGATG |
| 3275 | GTAAGGCGGTCTAGTAGTTATCCGTG | |
| Construction of plasmids pUDE446 to pUD450 | ||
| TagI-BIO6-TagA fragment | 9367 | TATTCACGTAGACGGATAGGTATAGCCAGACATCAGCAGCATACTTCGGGAACCGTAGGCTATTACTCTCCTAAACTGAGCACAAGTTTC |
| 3847 | ACTATATGTGAAGGCATGGCTATGG | |
| TagA-BIO1-TagB fragment | 4691 | CACCTTTCGAGAGGACGATG |
| 3275 | GTGCCTATTGATGATCTGGCGGAATG | |
| TagB-pMBP1-Amp-2u-URA3-TagI fragment | 9369 | GTTGAACATTCTTAGGCTGGTCGAATCATTTAGACACGGGCATCGTCCTCTCGAAAGGTGCGCGCTTGGCGTAATCATGGTC |
| 9366 | GCCTACGGTTCCCGAAGTATGCTGCTGATGTCTGGCTATACCTATCCGTCTACGTGAATACGGTACCCAATTCGCCCTATAGTG | |
| TagI-BIO6-TagA fragment | 9367 | TATTCACGTAGACGGATAGGTATAGCCAGACATCAGCAGCATACTTCGGGAACCGTAGGCTATTACTCTCCTAAACTGAGCACAAGTTTC |
| 3847 | ACTATATGTGAAGGCATGGCTATGG | |
| TagA-pMBP1-Amp-2u-URA3-TagI fragment | 9368 | GTGCCTATTGATGATCTGGCGGAATGTCTGCCGTGCCATAGCCATGCCTTCACATATAGTCGCGCTTGGCGTAATCATGGTC |
| 9366 | GCCTACGGTTCCCGAAGTATGCTGCTGATGTCTGGCTATACCTATCCGTCTACGTGAATACGGTACCCAATTCGCCCTATAGTG | |
| TagA-BIO1-TagB fragment | 4691 | CACCTTTCGAGAGGACGATG |
| 3275 | GTGCCTATTGATGATCTGGCGGAATG | |
| TagB-pMB1-Amp-2u-URA3-TagA fragment | 9370 | ACTATATGTGAAGGCATGGCTATGGCACGGCAGACATTCCGCCAGATCATCAATAGGCACCGGTACCCAATTCGCCCTATAGTG |
| 9369 | GTTGAACATTCTTAGGCTGGTCGAATCATTTAGACACGGGCATCGTCCTCTCGAAAGGTGCGCGCTTGGCGTAATCATGGTC | |
| Southern blot probe | ||
| Amplification of probe for Southern blotting (BIO1) | 2594 | GCTAGGGTTCGCAATATGTCCTGG |
| 2595 | CCACCACCTCATAAAGTTTACTGG | |
| Knockout fragments | ||
| KanMX with TPO1 overhangs for tpo1:kanMX | 523 | CAACTGCTACGGAGGGCAATGGTGGTGCAGATTTAGCGATTCAAAGAACGCGCGCTAAGACAATTCATCACAGCTGAAGCTTCGTACGC |
| 524 | CCTCATGAAAGTGTTTGCTGCGACGGCAGAAGCTGCCAACAATAGATACGCTTCAGTGGCTACGATGAGTGCATAGGCCACTAGTGGATCTG | |
| KanMX with PDR12 overhangs for pdr12::kanMX | 3582 | TCCCAGTTACTAATTTTCACTTAAAAAAAAGGTTTACAGATTTATTGTTATTGTTCTTATCAGCTGAAGCTTCGTACGC |
| 3583 | AAAATTTGTGAAAAAAAATTGAAAATAAAAATTGTGTGTTAAACCACGAAATACAAATATGCATAGGCCACTAGTGGATCTG | |
| Hygromycin cassette with PDR12 overhangs for pdr12::hph | 10447 | TAATTTTCACTTAAAAAAAAGGTTTACAGATTTATTGTTATTGTTCTTATTAATAAAAAATAGGTCTAGAGATCTGTTTAGC |
| 10448 | AAAATTGAAAATAAAAATTGTGTGTTAAACCACGAAATACAAATATATTTGCTTGCTTGTTCGAGAGCTCGTTAAAGCCTTC | |
| qPCR primers | ||
| BIO1 qPCR | 8250 | CCTTTACCATGCCGCAAGTG |
| 8251 | AAGGTCTCCAGTGGCATGTC | |
| BIO2 qPCR | 8383 | GTCTCGGTGAAAGCGAAGAC |
| 8384 | CAGCCATTGGAGTCCCTTTG | |
| BIO3 qPCR | 8254 | GCATCAGTCCTTCCGATCAG |
| 8255 | GCCACCAGTAGGGCTATTTG | |
| BIO4 qPCR | 8318 | AGAGTGGAGCGCAGAGAATC |
| 8319 | TGGCCGCTAGTCTCAATCAG | |
| BIO6 qPCR | 8320 | TTGACGGCCGGATATTTGAC |
| 8321 | AGGTTTGTCCGTGCATGAAG | |
| ACT1 qPCR | 313 | GGCTTCTTTGACTACCTTCCA |
| 314 | AGAAACACTTGTGGTGAACGA |
Plasmid construction.
Promoter fragments of highly expressed yeast genes and promoterless BIO genes were PCR amplified from genomic DNA of S. cerevisiae CEN.PK113-7D. The amplified BIO gene sequences included 0.5-kb terminator sequences. The 3′ and 5′ primers for amplification of promoter and terminator fragments, respectively, contained 60-bp synthetic extensions designed for efficient in vivo assembly of DNA fragments (56). Promoters and coding regions were fused by fusion PCR (57) and subsequently assembled into pJET1.2/blunt vectors with a CloneJet PCR cloning kit (Thermo Scientific), resulting in the vector constructs pUD416 and pUD418 (Table 5). Yeast expression plasmids were assembled in vivo by amplifying promoter-gene cassettes from pUD416 and pUD418 and vector fragments amplified from p426GPD (58). When necessary, oligonucleotide tags were changed to enable in vivo assembly with vector fragments. This assembly yielded the p426GPD-based plasmids pUDE446, pUDE448, and pUDE450 (Table 5).
TABLE 5.
Plasmids used in this study
| Name | Relevant characteristics | Reference |
|---|---|---|
| pUD416 | ampR pJET1.2Blunt TagB-pPYK-BIO1-tBIO1-TagA | This study |
| pUD418 | ampR pJET1.2Blunt TagA-pPGK1-BIO6-tBIO6-5′FlankSGA1 | This study |
| pUDE446 | 2μ URA3 ampR p426-GPD pPYK-BIO1 pPGK1-BIO6 | This study |
| pUDE448 | 2μ URA3 ampR p426-GPD pPGK1-BIO6 | This study |
| pUDE450 | 2μ URA3 ampR p426-GPD pPYK-BIO1 | This study |
Strain construction.
S. cerevisiae strains with increased copy numbers of endogenous BIO genes (IME327, IME329, IME331, and IME334) (Table 1) were constructed by transforming CEN.PK113-5D (ura3) with multicopy expression plasmids (pUDE446, pUDE448, pUDE450, and p426GPD [URA3]). Geneticin (G418) resistance cassettes, PCR amplified from pUG6 (59), were used to delete TPO1 and PDR12 in CEN.PK113-5D, resulting in strains IMK129 and IMK163, respectively. IMK129 was used to construct a TPO1 and PDR12 double-deletion strain by transformation with a hygromycin resistance cassette (hphNT1) amplified from pMEL12 (32), resulting in IMK773. Deletion strains were subsequently transformed with pUDE450 (BIO1 URA3) or with the empty reference vector p426GPD (URA3), resulting in IMZ694, IMZ695, IMZ701, IMZ702, IMZ704, and IMZ705 (Table 1).
Shake flask and plate growth experiments.
Thawed aliquots of frozen stock cultures (1 ml) were inoculated in SMD shake flask cultures and incubated for 12 h. This starter culture was used to inoculate a second shake flask culture on SMD that, after another 12 h, was used to inoculate a third culture on SMD, at an initial optical density at 660 nm (OD660) of 0.05, 0.1, or 0.2. For biotin-free growth studies, all three cultures were grown on SMD without biotin. The OD660 of the third culture was monitored with a Libra S11 spectrophotometer (Biochrom, Cambridge, United Kingdom). Specific growth rates were calculated from at least four time points in the exponential growth phase of each culture. Strain CEN.PK113-7D, which consistently failed to grow on biotin-free SMD in the third culture, was used as a negative control in all growth experiments. Cell numbers were estimated from calibration curves of OD660 versus cell counts in an Accuri flow cytometer (Becton Dickinson B.V., Breda, The Netherlands) generated with exponentially growing shake flask cultures of strain CEN.PK113-7D on SMD medium. For plate assays, precultures were grown on SMD medium. Spot assays on SMD agar (pH 4.5) containing either 50 μM or 2 mM MgSO4 were performed as described previously (60). SMD agar plates were supplemented with either 0.05, 0.15, 0.5, 1, 1.5, 2, or 3 mM pimelic acid; 6 or 15 mM spermidine; or 0.5 mM potassium sorbate (see Fig. S1 in the supplemental material). Liquid cultures of CEN.PK113-7D containing different concentrations of pimelic acid were prepared similarly to shake flask cultivations but were carried out in 50-ml Cellreactor filter top tubes with 25 ml of SMD-urea (pH 4.5) with an inoculation OD660 of 0.1. Filter-sterilized pimelic acid solution (1.25 M, pH 4.5) was added to duplicate cultures to a final concentration of 10, 12.5, 25, 50, 70, 80, 90, or 100 mM.
Laboratory evolution.
Laboratory evolution of S. cerevisiae CEN.PK113-7D for biotin prototrophy was performed in accelerostats and in sequential bioreactor batch cultures. Accelerostat evolution was preceded by serial transfers in 3 parallel shake flask experiments on biotin-free SMD, with an initial OD660 of 0.05 after each transfer. After 15 transfers (in 54 days), a specific growth rate of ca. 0.1 h−1 was reached. Fifteen milliliters of each evolution culture was then used to inoculate separate 450-ml Multifors 2 parallel bioreactors (Infors Benelux, Doetinchem, The Netherlands) with a working volume of 100 ml. These bioreactors were subsequently operated as accelerostats, which are continuous cultures in which the dilution rate is increased over time (27). During the initial batch phase, cells were grown on SM without biotin, supplemented with 20 g · liter−1 glucose and 0.3 g · liter−1 antifoam Pluronic PE 6100 (BASF, Ludwigshafen, Germany). When a decrease in CO2 production indicated glucose depletion (CO2 sensor; Bluesens, Herten, Germany), continuous cultivation was initiated at a dilution rate of 0.10 h−1 on SM without biotin, 7.5 g · liter−1 glucose, and 0.15 g · liter−1 antifoam Pluronic PE 6100. Cultures were grown at 30°C, while a culture pH of 5 was maintained by automated addition of 2 M KOH. The cultures were sparged with air at a flow rate of 50 ml · min−1 and stirred at 1,200 rpm. Dissolved oxygen concentrations remained above 40% of air saturation throughout the experiments. The dilution rate of the accelerostats was controlled by Iris Parallel Bioprocess Control software (Infors Benelux) based on a manually set threshold for the ratio between the CO2 concentration in the outlet gas and the feed rate of the medium supply pump. If this ratio remained above the set point for at least 4 h, the medium feed rate was increased by 5%. When it remained below the set point for the same period, the feed rate was decreased by 4.5%. This setup ensured a steady increase in the dilution rate as the culture evolved to reach higher specific growth rates while preventing culture washout. During evolution experiments, the threshold ratio of CO2 output and feed rate was manually adjusted to compensate for changes in growth stoichiometry caused by a reduced relative contribution of maintenance energy metabolism at higher specific growth rates (61). Accelerostat cultures were terminated when, within 3 months of accelerostat selection, dilution rates of 0.24 to 0.28 h−1 were reached.
Laboratory evolution in sequential batch reactors was preceded by shake flask cultivation of strain CEN.PK113-7D in biotin-free SMD, using a frozen stock culture as the inoculum. After 2 days, 0.1 ml of this culture was transferred to a fresh culture to deplete biotin stores and to generate a preculture from which, after ca. 12 h of incubation, a bioreactor culture was inoculated at an initial OD660 of 0.05. Sequential batch cultivation was performed in 450-ml Multifors 2 parallel bioreactors (Infors Benelux) with a working volume of 100 ml. Aeration, pH, temperature, and dissolved oxygen concentration thresholds were the same as in the accelerostat cultures. Growth was monitored based on the CO2 concentration in the off gas. When, after first having reached the CO2 production peak, the CO2 percentage in the off gas decreased below 0.02%, a computer-controlled peristaltic pump automatically removed approximately 95% of the culture volume, leaving approximately 5% as an inoculum for the next batch. The experiment was stopped when, after 11 batch cultivation cycles (47 days), no further increase in the growth rate was observed over the following six consecutive batches and, moreover, cells with a multicellular clumping phenotype were observed in the culture. Clumping phenotypes, which facilitate sedimentation during empty-refill cycles and thereby enable mutants to escape selection for faster growth in sequential batch reactors (29), were not observed in the accelerostat cultures. Single-colony isolates from accelerostats and sequential batch reactors were obtained by plating on biotin-free SMD. To facilitate analysis of single cell lines and subsequent genetic analysis, nonclumping single cell lines were selected from the sequential batch cultures by microscopic inspection of colonies after plating on biotin-free SMD.
qPCR experiments.
qPCR experiments were performed with duplicate cultures pregrown on SMD with or without biotin. RNA extraction was performed following the method of Schmitt et al. (62), while the sampling procedure was done as described previously (63). cDNA was synthesized using a QuantiTect reverse transcription kit (Qiagen, Düsseldorf, Germany), and concentrations were determined using a Qubit fluorometer (Life Technologies). qPCR experiments with cDNA from duplicate cultures were performed in technical triplicates on three dilutions of each sample, using a QuantiTect SYBR green PCR kit (Qiagen) with a primer concentration of 0.5 μM in a total volume of 20 μl in the Rotor-Gene Q (Qiagen). The qPCR primers are listed in Table 4. ACT1 expression levels determined from the same culture were used as an internal standard. Expression levels of BIO genes relative to those of ACT1 were determined using the ΔΔCT method (64). Briefly, normalized expression levels of the gene of interest (GOI) were calculated by subtracting the average CT value obtained from technical triplicate measurements of the reference gene, ACT1, from the similarly averaged CT values of the GOI (ΔCTsample = average CTGOIsample − average CTACT1). The normalized expression level of the GOI was represented as 2−ΔCT. Data are represented as averages and standard errors of the mean (SEM) of expression levels calculated from independent duplicate cultures (Fig. 3; see Table S2 in the supplemental material).
CHEF electrophoresis and Southern blotting.
Agarose plugs for all the strains were prepared using a contour-clamped homogeneous electric field (CHEF) genomic plug kit following the manufacturer's recommendations (Bio-Rad). One-third of each agarose plug was used per well of a 1% megabase agarose gel (Bio-Rad) buffered with 0.5× TBE (5.4 g Tris base, 2.75 g boric acid, 2 ml 0.5 M EDTA, pH 8, in 1 liter demineralized water). Chromosomes were separated in a CHEF-DR II system (Bio-Rad) for 28 h with a switch time of 60 s, an angle of 120°, and 5 V · cm−2, followed by 16 h with a switch time of 90 s. DNA was stained with ethidium bromide (3 μg · ml−1 in 0.5× TBE buffer) and destained in 0.5× TBE buffer for 20 min with gentle agitation. Imaging was done using an InGenius LHR Gel Imaging System (Syngene, Bangalore, India). Southern blot probes (ca. 1 kb) were amplified from genomic DNA of CEN.PK113-7D and prepared with an AlkPhos direct-labeling and detection system (Amersham Biosciences, Piscataway, NJ) according to the manufacturer's protocol. Separated chromosomes were transferred to Hybond-N+ nylon membranes (GE Healthcare, Diegem, Belgium) using vacuum transfer with a vacuum blotter (Bio-Rad) and fixed to the nylon membrane by UV exposure for 2 min. Signal generation and detection were performed using an Amersham Gene Images AlkPhos direct-labeling and detection system, together with CDP-Star chemiluminescent detection (Amersham Biosciences) and exposure to an Amersham Hyperfilm ECL (Amersham Biosciences), following the manufacturer's recommendations.
DNA sequencing.
Genomic DNA of strains IMS0478, IMS0480, IMS0481, and IMS0496 was isolated with a Qiagen Blood and Cell Culture DNA kit with 100/G genomic tips (Qiagen, Valencia, California) according to the manufacturer's protocol. Paired-end sequencing (126-bp reads) was performed on 350-bp insert libraries with an Illumina HiSeq 2500 sequencer (Baseclear BV, Leiden, The Netherlands) with a minimum sample size of 550 Mb, accounting for a coverage of approximately 45 times. Data mapping to the CEN.PK113-7D genome (22), data processing, and chromosome copy number variation determinations were done as described previously (65). Copy numbers of BIO1, BIO2, BIO3, BIO4, and BIO6 were estimated by comparing their read depths to the average read depths of the single-copy reference genes [YAL001C (TFC3), YBL015W (ACH1), YCL040W (GLK1), YDL029W (ARP2), YEL012W (UBC8), YER049W (TPA1), YBR196C (PGI1), YER178W (PDA1), YFL039C (ACT1), and YJL121C (RPE1)], processed with Pilon (66).
Accession number(s).
The sequencing data are available under the Bioproject accession number PRJNA383023.
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert Mans for programming the accelerostats, Erwin Suir for construction of strains IMK129 and IMK163, Sophie van der Horst for assisting with evolutionary engineering, Wijb Dekker and Stéphanie O'Herne for help with CHEF electrophoresis and Southern blotting, and Maarten Verhoeven and Ioannis Papapetridis for fruitful discussions.
This work was supported by the BE-Basic R&D Program, which was granted an FES subsidy from the Dutch Ministry of Economic Affairs, Agriculture and Innovation (EL&I).
J.M.B., J.-M.G.D., A.J.A.V.M., and J.T.P. designed experiments. J.M.B. and J.T.P. wrote the text. J.M.B. and E.D.H. set up the accelerostats resulting in strains IMS0478, IMS0480, and IMS0481. J.M.B. performed the accelerostat selections, constructed all plasmids and yeast strains, characterized yeast strains, performed CHEF electrophoresis and Southern blotting, did spot plate assays, analyzed whole-genome sequencing data, identified and verified SNPs, made Magnolya plots, and determined gene copy numbers. C.C.K. performed qPCR experiments. E.D.H. carried out the SBR selection resulting in strain IMS0496. M.V.D.B. developed methods and wrote scripts for bioinformatics data analysis and interpretation and developed the Pilon-based method to determine gene copy numbers. We all read and approved the final manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00892-17.
REFERENCES
- 1.Gailiusis J, Rinne RW, Benedict C. 1964. Pyruvate- oxaloacetate exchange reaction in baker's yeast. Biochim Biophys Acta 92:595–601. [DOI] [PubMed] [Google Scholar]
- 2.Losada M, Canovas J, Ruiz A. 1964. Oxaloacetate, citramalate and glutamate formation from pyruvate in baker's yeast. Biochem Z 340:60–74. [PubMed] [Google Scholar]
- 3.Wakil SJ, Titchener EB, Gibson DM. 1958. Evidence for the participation of biotin in the enzymic synthesis of fatty acids. Biochim Biophys Acta 29:225–226. doi: 10.1016/0006-3002(58)90177-X. [DOI] [PubMed] [Google Scholar]
- 4.Sumrada RA, Cooper TG. 1982. Urea carboxylase and allophanate hydrolase are components of a multifunctional protein in yeast. J Biol Chem 257:9119–9127. [PubMed] [Google Scholar]
- 5.Kim HS, Hoja U, Stolz J, Sauer G, Schweizer E. 2004. Identification of the tRNA-binding protein Arc1p as a novel target of in vivo biotinylation in Saccharomyces cerevisiae. J Biol Chem 279:42445–42452. doi: 10.1074/jbc.M407137200. [DOI] [PubMed] [Google Scholar]
- 6.Hoja U, Wellein C, Greiner E, Schweizer E. 1998. Pleiotropic phenotype of acetyl-CoA-carboxylase-defective yeast cells. Eur J Biochem 254:520–526. doi: 10.1046/j.1432-1327.1998.2540520.x. [DOI] [PubMed] [Google Scholar]
- 7.Streit W, Entcheva P. 2003. Biotin in microbes, the genes involved in its biosynthesis, its biochemical role and perspectives for biotechnological production. Appl Microbiol Biotechnol 61:21–31. doi: 10.1007/s00253-002-1186-2. [DOI] [PubMed] [Google Scholar]
- 8.Lin S, Hanson RE, Cronan JE. 2010. Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat Chem Biol 6:682–688. doi: 10.1038/nchembio.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kashiwagi K, Igarashi K. 2011. Identification and assays of polyamine transport systems in Escherichia coli and Saccharomyces cerevisiae. Methods Mol Biol 720:295–308. doi: 10.1007/978-1-61779-034-8_18. [DOI] [PubMed] [Google Scholar]
- 10.Lin S, Cronan JE. 2011. Closing in on complete pathways of biotin biosynthesis. Mol Biosyst 7:1811–1821. doi: 10.1039/c1mb05022b. [DOI] [PubMed] [Google Scholar]
- 11.Burkholder PR, McVeigh I, Moyer D. 1944. Studies on some growth factors of yeasts. J Bacteriol 48:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanabe Y, Maruyama J- I, Yamaoka S, Yahagi D, Matsuo I, Tsutsumi N, Kitamoto K. 2011. Peroxisomes are involved in biotin biosynthesis in Aspergillus and Arabidopsis. J Biol Chem 286:30455–30461. doi: 10.1074/jbc.M111.247338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magliano P, Flipphi M, Arpat BA, Delessert S, Poirier Y. 2011. Contributions of the peroxisome and β-oxidation cycle to biotin synthesis in fungi. J Biol Chem 286:42133–42140. doi: 10.1074/jbc.M111.279687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall C, Dietrich FS. 2007. The reacquisition of biotin prototrophy in Saccharomyces cerevisiae involved horizontal gene transfer, gene duplication and gene clustering. Genetics 177:2293–2307. doi: 10.1534/genetics.107.074963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall C, Brachat S, Dietrich FS. 2005. Contribution of horizontal gene transfer to the evolution of Saccharomyces cerevisiae. Eukaryot Cell 4:1102–1115. doi: 10.1128/EC.4.6.1102-1115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Ito K, Shimoi H. 2005. Identification and characterization of a novel biotin biosynthesis gene in Saccharomyces cerevisiae. Appl Environ Microbiol 71:6845–6855. doi: 10.1128/AEM.71.11.6845-6855.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helliwell KE, Wheeler GL, Smith AG. 2013. Widespread decay of vitamin-related pathways: coincidence or consequence? Trends Genet 29:469–478. doi: 10.1016/j.tig.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Azhar A, Booker GW, Polyak SW. 2015. Mechanisms of biotin transport. Biochem Anal Biochem 4:1. [Google Scholar]
- 19.Stolz J, Hoja U, Meier S, Sauer N, Schweizer E. 1999. Identification of the plasma membrane H+-biotin symporter of Saccharomyces cerevisiae by rescue of a fatty acid-auxotrophic mutant. J Biol Chem 274:18741–18746. doi: 10.1074/jbc.274.26.18741. [DOI] [PubMed] [Google Scholar]
- 20.Phalip V, Kuhn I, Lemoine Y, Jeltsch J-M. 1999. Characterization of the biotin biosynthesis pathway in Saccharomyces cerevisiae and evidence for a cluster containing BIO5, a novel gene involved in vitamer uptake. Gene 232:43–51. doi: 10.1016/S0378-1119(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 21.Gasser B, Dragosits M, Mattanovich D. 2010. Engineering of biotin-prototrophy in Pichia pastoris for robust production processes. Metab Eng 12:573–580. doi: 10.1016/j.ymben.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Nijkamp JF, van den Broek M, Datema E, de Kok S, Bosman L, Luttik MA, Daran-Lapujade P, Vongsangnak W, Nielsen J, Heijne WH. 2012. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN. PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 11:36. doi: 10.1186/1475-2859-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Lang N, Yang G, Yang S, Jiang W, Gu Y. 2016. Improving the performance of solventogenic clostridia by reinforcing the biotin synthetic pathway. Metab Eng 35:121–128. doi: 10.1016/j.ymben.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Piper MD, Daran-Lapujade P, Bro C, Regenberg B, Knudsen S, Nielsen J, Pronk JT. 2002. Reproducibility of oligonucleotide microarray transcriptome analyses; an interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J Biol Chem 277:37001–37008. doi: 10.1074/jbc.M204490200. [DOI] [PubMed] [Google Scholar]
- 25.Canelas AB, Harrison N, Fazio A, Zhang J, Pitkänen J-P, Van den Brink J, Bakker BM, Bogner L, Bouwman J, Castrillo JI. 2010. Integrated multilaboratory systems biology reveals differences in protein metabolism between two reference yeast strains. Nat Commun 1:145. doi: 10.1038/ncomms1150. [DOI] [PubMed] [Google Scholar]
- 26.Daran-Lapujade P, Rossell S, van Gulik WM, Luttik MA, de Groot MJ, Slijper M, Heck AJ, Daran J-M, de Winde JH, Westerhoff HV. 2007. The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc Natl Acad Sci U S A 104:15753–15758. doi: 10.1073/pnas.0707476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paalme T, Kahru A, Elken R, Vanatalu K, Tiisma K, Raivo V. 1995. The computer-controlled continuous culture of Escherichia coli with smooth change of dilution rate (A-stat). J Microbiol Methods 24:145–153. doi: 10.1016/0167-7012(95)00064-X. [DOI] [Google Scholar]
- 28.Kuyper M, Toirkens MJ, Diderich JA, Winkler AA, Dijken JP, Pronk JT. 2005. Evolutionary engineering of mixed-sugar utilization by a xylose-fermenting Saccharomyces cerevisiae strain. FEMS Yeast Res 5:925–934. doi: 10.1016/j.femsyr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Oud B, Guadalupe-Medina V, Nijkamp JF, de Ridder D, Pronk JT, van Maris AJ, Daran J-M. 2013. Genome duplication and mutations in ACE2 cause multicellular, fast-sedimenting phenotypes in evolved Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 110:E4223–E4231. doi: 10.1073/pnas.1305949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuyper M, Hartog MM, Toirkens MJ, Almering MJ, Winkler AA, Dijken JP, Pronk JT. 2005. Metabolic engineering of a xylose-isomerase-expressing Saccharomyces cerevisiae strain for rapid anaerobic xylose fermentation. FEMS Yeast Res 5:399–409. doi: 10.1016/j.femsyr.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Pronk JT. 2002. Auxotrophic yeast strains in fundamental and applied research. Appl Environ Microbiol 68:2095–2100. doi: 10.1128/AEM.68.5.2095-2100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mans R, van Rossum HM, Wijsman M, Backx A, Kuijpers NG, van den Broek M, Daran-Lapujade P, Pronk JT, van Maris AJ, Daran J-MG. 2015. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 15:fov004. doi: 10.1093/femsyr/fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hazelwood LA, Tai SL, Boer VM, De Winde JH, Pronk JT, Daran JM. 2006. A new physiological role for Pdr12p in Saccharomyces cerevisiae: export of aromatic and branched-chain organic acids produced in amino acid catabolism. FEMS Yeast Res 6:937–945. doi: 10.1111/j.1567-1364.2006.00094.x. [DOI] [PubMed] [Google Scholar]
- 34.Albertsen M, Bellahn I, Krämer R, Waffenschmidt S. 2003. Localization and function of the yeast multidrug transporter Tpo1p. J Biol Chem 278:12820–12825. doi: 10.1074/jbc.M210715200. [DOI] [PubMed] [Google Scholar]
- 35.Kren A, Mamnun YM, Bauer BE, Schüller C, Wolfger H, Hatzixanthis K, Mollapour M, Gregori C, Piper P, Kuchler K. 2003. War1p, a novel transcription factor controlling weak acid stress response in yeast. Mol Cell Biol 23:1775–1785. doi: 10.1128/MCB.23.5.1775-1785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piper P, Mahé Y, Thompson S, Pandjaitan R, Holyoak C, Egner R, Mühlbauer M, Coote P, Kuchler K. 1998. The Pdr12 ABC transporter is required for the development of weak organic acid resistance in yeast. EMBO J 17:4257–4265. doi: 10.1093/emboj/17.15.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holyoak CD, Bracey D, Piper PW, Kuchler K, Coote PJ. 1999. The Saccharomyces cerevisiae weak-acid-inducible ABC transporter Pdr12 transports fluorescein and preservative anions from the cytosol by an energy-dependent mechanism. J Bacteriol 181:4644–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Entian K-D, Kötter P. 2007. 25 yeast genetic strain and plasmid collections. Methods Microbiol 36:629–666. doi: 10.1016/S0580-9517(06)36025-4. [DOI] [Google Scholar]
- 39.Wolfe KH, Shields DC. 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387:708–712. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- 40.de Kok S, Nijkamp JF, Oud B, Roque FC, Ridder D, Daran JM, Pronk JT, Maris AJ. 2012. Laboratory evolution of new lactate transporter genes in a jen1Δ mutant of Saccharomyces cerevisiae and their identification as ADY2 alleles by whole-genome resequencing and transcriptome analysis. FEMS Yeast Res 12:359–374. doi: 10.1111/j.1567-1364.2011.00787.x. [DOI] [PubMed] [Google Scholar]
- 41.Torigata K, Akiyama Y. 1968. Tests of sake brewing by yeasts after cultured with ventilation. 1. Rising and falling of vitamins contained in sake moromi and preservative tests of yeasts. J Brew Soc Japan 63:60–63. [Google Scholar]
- 42.Schüller C, Mamnun YM, Mollapour M, Krapf G, Schuster M, Bauer BE, Piper PW, Kuchler K. 2004. Global phenotypic analysis and transcriptional profiling defines the weak acid stress response regulon in Saccharomyces cerevisiae. Mol Biol Cell 15:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomitori H, Kashiwagi K, Sakata K, Kakinuma Y, Igarashi K. 1999. Identification of a gene for a polyamine transport protein in yeast. J Biol Chem 274:3265–3267. doi: 10.1074/jbc.274.6.3265. [DOI] [PubMed] [Google Scholar]
- 44.Chattopadhyay MK, Chen W, Poy G, Cam M, Stiles D, Tabor H. 2009. Microarray studies on the genes responsive to the addition of spermidine or spermine to a Saccharomyces cerevisiae spermidine synthase mutant. Yeast 26:531–544. doi: 10.1002/yea.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uemura T, Tachihara K, Tomitori H, Kashiwagi K, Igarashi K. 2005. Characteristics of the polyamine transporter TPO1 and regulation of its activity and cellular localization by phosphorylation. J Biol Chem 280:9646–9652. doi: 10.1074/jbc.M410274200. [DOI] [PubMed] [Google Scholar]
- 46.Kim SK, Jin YS, Choi IG, Park YC, Seo JH. 2015. Enhanced tolerance of Saccharomyces cerevisiae to multiple lignocellulose-derived inhibitors through modulation of spermidine contents. Metab Eng 29:46–55. doi: 10.1016/j.ymben.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Krüger A, Vowinckel J, Mülleder M, Grote P, Capuano F, Bluemlein K, Ralser M. 2013. Tpo1-mediated spermine and spermidine export controls cell cycle delay and times antioxidant protein expression during the oxidative stress response. EMBO rep 14:1113–1119. doi: 10.1038/embor.2013.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wodicka L, Dong H, Mittmann M, Ho M-H, Lockhart DJ. 1997. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol 15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 49.Madsen CT, Sylvestersen KB, Young C, Larsen SC, Poulsen JW, Andersen MA, Palmqvist EA, Hey-Mogensen M, Jensen PB, Treebak JT. 2015. Biotin starvation causes mitochondrial protein hyperacetylation and partial rescue by the SIRT3-like deacetylase Hst4p. Nat Commun 6:7726. doi: 10.1038/ncomms8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SK, Jo JH, Jin YS, Seo JH. 2017. Enhanced ethanol fermentation by engineered Saccharomyces cerevisiae strains with high spermidine contents. Bioprocess Biosyst Eng 40:683–691. doi: 10.1007/s00449-016-1733-3. [DOI] [PubMed] [Google Scholar]
- 51.Mortimer RK, Johnston JR. 1986. Genealogy of principal strains of the yeast genetic stock center. Genetics 113:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solis-Escalante D, Kuijpers NG, Bongaerts N, Bolat I, Bosman L, Pronk JT, Daran JM, Daran-Lapujade P. 2013. amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res 13:126–139. doi: 10.1111/1567-1364.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lõoke M, Kristjuhan K, Kristjuhan A. 2011. Extraction of genomic DNA from yeasts for PCR-based applications. Biotechniques 50:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gietz RD, Woods RA. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96. doi: 10.1016/S0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 55.Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28. doi: 10.1016/0378-1119(90)90336-P. [DOI] [PubMed] [Google Scholar]
- 56.Kuijpers N, Solis-Escalante D, Bosman L, van den Broek M, Pronk JT, Daran J-M, Daran-Lapujade P. 2013. A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb Cell Fact 12:47. doi: 10.1186/1475-2859-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S. 2004. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res 32:e19–. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mumberg D, Müller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 59.Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Rossum HM, Kozak BU, Niemeijer MS, Dykstra JC, Luttik MA, Daran J-MG, van Maris AJ, Pronk JT. 2016. Requirements for carnitine shuttle-mediated translocation of mitochondrial acetyl moieties to the yeast cytosol. mBio 7:e00520–. doi: 10.1128/mBio.00520-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verduyn C, Postma E, Scheffers WA, Van Dijken JP. 1990. Energetics of Saccharomyces cerevisiae in anaerobic glucose-limited chemostat cultures. Microbiology 136:405–412. [DOI] [PubMed] [Google Scholar]
- 62.Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 18:3091. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huisjes EH, Luttik MA, Almering MJ, Bisschops MM, Dang DH, Kleerebezem M, Siezen R, van Maris AJ, Pronk JT. 2012. Toward pectin fermentation by Saccharomyces cerevisiae: expression of the first two steps of a bacterial pathway for D-galacturonate metabolism. J Biotechnol 162:303–310. doi: 10.1016/j.jbiotec.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 64.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 65.Verhoeven MD, Lee M, Kamoen L, van den Broek M, Janssen DB, Daran J-MG, van Maris AJ, Pronk JT. 2017. Mutations in PMR1 stimulate xylose isomerase activity and anaerobic growth on xylose of engineered Saccharomyces cerevisiae by influencing manganese homeostasis. Sci Rep 7:46155. doi: 10.1038/srep46155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nijkamp JF, van den Broek MA, Geertman J-MA, Reinders MJ, Daran J-MG, de Ridder D. 2012. De novo detection of copy number variation by co-assembly. Bioinformatics 28:3195–3202. doi: 10.1093/bioinformatics/bts601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.