

ABSTRACT
With recent advances in synthetic biology, CO2 could be utilized as a carbon feedstock by native or engineered organisms, assuming the availability of electrons. Two key enzymes used in autotrophic CO2 fixation are the CO dehydrogenase (CODH) and acetyl coenzyme A (acetyl-CoA) synthase (ACS), which form a bifunctional heterotetrameric complex. The CODH/ACS complex can reversibly catalyze CO2 to CO, effectively enabling a biological water-gas shift reaction at ambient temperatures and pressures. The CODH/ACS complex is part of the Wood-Ljungdahl pathway (WLP) used by acetogens to fix CO2, and it has been well characterized in native hosts. So far, only a few recombinant CODH/ACS complexes have been expressed in heterologous hosts, none of which demonstrated in vivo CO2 reduction. Here, functional expression of the Clostridium carboxidivorans CODH/ACS complex is demonstrated in the solventogen Clostridium acetobutylicum, which was engineered to express CODH alone or together with the ACS. Both strains exhibited CO2 reduction and CO oxidation activities. The CODH reactions were interrogated using isotopic labeling, thus verifying that CO was a direct product of CO2 reduction, and vice versa. CODH apparently uses a native C. acetobutylicum ferredoxin as an electron carrier for CO2 reduction. Heterologous CODH activity depended on actively growing cells and required the addition of nickel, which is inserted into CODH without the need to express the native Ni insertase protein. Increasing CO concentrations in the gas phase inhibited CODH activity and altered the metabolite profile of the CODH-expressing cells. This work provides the foundation for engineering a complete and functional WLP in nonnative host organisms.
IMPORTANCE Functional expression of CO dehydrogenase (CODH) from Clostridium carboxidivorans was demonstrated in C. acetobutylicum, which is natively incapable of CO2 fixation. The expression of CODH, alone or together with the C. carboxidivorans acetyl-CoA synthase (ACS), enabled C. acetobutylicum to catalyze both CO2 reduction and CO oxidation. Importantly, CODH exhibited activity in both the presence and absence of ACS. 13C-tracer studies confirmed that the engineered C. acetobutylicum strains can reduce CO2 to CO and oxidize CO during growth on glucose.
KEYWORDS: CO2 fixation, CO2 reduction, CO oxidation, Wood-Ljungdahl pathway, acetogenesis, carbon dioxide fixation, carbon dioxide reduction, metabolic engineering, synthetic biology, acetogens
INTRODUCTION
The Wood-Ljungdahl pathway (WLP) is the most efficient pathway for CO2 fixation, whereby two molecules of CO2 are reduced to form one molecule of acetyl coenzyme A (acetyl-CoA), a primordial biological building block for cellular components (1). The WLP is composed of two linear branches: the so-called eastern, or methyl, branch reduces CO2 to formate via formate dehydrogenase and transfers the carbon molecule to the C1 carrier tetrahydrofolate, while the second branch, the western, or carbonyl, branch, reduces CO2 to CO (2). The final carbonylation step for acetyl-CoA synthesis combines CO with the activated methyl group from the eastern branch and the CoA moiety and is catalyzed by the bifunctional enzyme complex of carbon monoxide dehydrogenase (CODH)-acetyl-CoA synthase (ACS). The native CODH/ACS complex used by acetogens is characterized by an α2β2 tetrameric structure containing a central core of two CODH subunits that are associated on either side with two ACS subunits (3). Each subunit contains an active site; the β site is responsible for CO oxidation and CO2 reduction (C-cluster), and the α site is responsible for acetyl-CoA synthesis (A-cluster). CODH reversibly oxidizes CO to CO2 and allows many bacteria to grow on CO as a sole carbon source (3–6). In addition, the reduction of CO2 allows acetogens to grow on CO2 and H2 as well. In the chemical industry, the interconversion of CO and CO2 is an important process known as the water-gas shift reaction for the generation of H2, in which CO is oxidized under high temperature and pressure. In contrast, the biological reaction occurs at atmospheric pressure and temperatures between 37 and 60°C.
The first study to examine the biological oxidation of CO to CO2 was performed with methanogenic bacteria (7) and later with Clostridium formicoaceticum and Moorella thermoacetica (formerly Clostridium thermoaceticum) (8). All strains were able to oxidize CO to CO2 and grow on CO2 and H2, and they have been shown to contain a WLP and the necessary CODH/ACS proteins/genes. The most extensively studied bifunctional CODH/ACS is from M. thermoacetica, which has been purified (9) and characterized (10) and its crystal structure elucidated (11). There exists evidence that the two subunits are connected to enable substrate channeling, thus enhancing the substrate flux between the two active sites, thereby lowering the internal thermodynamic barrier for CO2 reduction (12–16). CO2 is reduced to CO at the active site of the CODH (C-cluster) and can migrate to the active site of the ACS, the A-cluster, without leaving the hydrophobic pockets of the enzyme. Even though these enzymes have been well studied, much remains to be understood regarding the exact catalytic mechanisms and redox states for the multifaceted process of fixing CO2. Traditionally, CO oxidation is monitored in vitro with an artificial electron carrier, such as methyl viologen; in other cases, ferredoxin isolated from anaerobic bacteria is used (10). The in vitro assay for the ACS involves the carbon exchange reaction between CO and the carbonyl group of acetyl-CoA, monitored using 14C-radioactive tracers. Although in vitro analysis is a convenient measure of enzyme activity, it cannot simulate the physiological conditions involved in in vivo CO2 reduction.
CO2 is the most oxidized form of carbon and is therefore a stable and highly inert compound. Since CO2 reduction and subsequent acetyl-CoA synthesis provide acetogens the sole means for growth on gases alone, reduction of CO2 occurs under physiological conditions. However, under standard conditions, the reduction of CO2 to CO has a ΔG of +32 kJ/mol (2). Clearly, acetogenic bacteria create an intracellular environment and/or low redox potential that allows this reaction to proceed to reduce CO2 to form CO. The CODH/ACS subunits from M. thermoacetica have been expressed in Escherichia coli JM109 but did not exhibit the native tetrameric structure and were thus found inactive (17). More recently, however, Loke et al. demonstrated that expression of the two subunits in the same E. coli host produced a protein complex that migrated as a single band through nondenaturing electrophoretic gels (18). The purified complex exhibited CO oxidation activity in vitro, but the in vitro acetyl-CoA carbon exchange reaction required incubation with nickel after purification.
We hypothesized that expression of the bifunctional CODH/ACS complex would benefit from a host more closely related to native acetogens. E. coli is a Gram-negative prokaryote and does not contain electron carriers, such as ferredoxin, the necessary electron carrier for acetogenic CODHs (10, 19). In contrast, the Gram-positive Clostridium acetobutylicum is an excellent host for expressing the CODH/ACS complex, since it is more closely related phylogenetically to acetogens (20), is strictly anaerobic, and contains the cofactor (ferredoxin) required for in vivo function of the CODH/ACS complex (10). In this study, we sought to express and demonstrate in vivo activities of the CODH/ACS proteins from the acetogen Clostridium carboxidivorans, aiming to engineer CO2 reduction in C. acetobutylicum. With the ultimate goal of engineering a functional WLP in C. acetobutylicum, here, we sought to demonstrate the functional expression of the CODH/ACS complex, an essential first goal, hitherto not accomplished. We demonstrate functional CODH activity expressed in tandem with ACS and alone, and we show that the reduction of CO2 does not require the ACS subunit. In vivo CO2 reduction by the engineered C. acetobutylicum suggests that C. acetobutylicum possesses electron carriers that have a low enough redox potential to overcome the thermodynamic challenge of CO2 reduction. This was achieved in a nonnative host. While there is no known in vivo functional assay for the ACS activity, we also attempted to establish one that would be analogous to the aforementioned carbon exchange reaction between CO and the carbonyl group of acetyl-CoA, but using 13C-tracers.
RESULTS
Expression and purification of recombinant CODH/ACS demonstrates correct assembly of the heterotetrameric protein complex.
To test the functional expression of the CODH/ACS complex in the heterologous host C. acetobutylicum, we expressed both proteins, CODH and ACS, from the acetogen C. carboxidivorans (21) and investigated their expression as a proper tetrameric structure through His tag purification and Western blot identification (Fig. 1). The plasmid-borne construct contained the genes for the two subunits of the complex, CODH and ACS, and a His tag following the ACS coding region prior to the stop codon (Fig. 1C). The operon was under the control of the strong native C. acetobutylicum Pptb promoter (22). Recombinant C. acetobutylicum cells harboring the plasmid vector for expressing the CODH/ACS complex were grown under anaerobic conditions to mid-exponential phase. Harvested cells were lysed, and the supernatant was purified using a nickel-nitrilotriacetic acid (Ni-NTA) affinity column under native conditions so that the heterotetrameric structure is preserved and the four subunits stay tightly associated. After purification, the eluent was subjected to a denaturing gel to verify the presence of the CODH and ACS subunits. Western blots utilizing polyclonal antibodies raised in rabbits against each subunit were used to visualize the two components of the tetramer. The data show that the eluent contained both the ACS and CODH subunits, which moved independently on the denaturing SDS-PAGE gel according to their sizes (Fig. 1A and B). Next, we applied whole-cell lysates to a native PAGE gel and observed the movement of a large protein complex of approximate size of 300 kDa not found in the control cells (Fig. 1D). The expected mass of the C. carboxidivorans heterotetramer is 289 kDa, and it is a little larger with the His tag. Western blotting using antibodies against the His tag confirmed that the large protein complex contained the ACS subunit tightly bound to the CODH subunit (Fig. 1E). We reached this conclusion because the native complex, upon purification, contained the CODH and ACS units (Fig. 1A and B). Taken together, these data suggest a correct assembly of the heterologously expressed CODH/ACS heterotetrameric protein complex in C. acetobutylicum. We noted that the CODH/ACS complex is in the soluble fraction of the cell extracts, as little or none was detected in the insoluble fraction.
FIG 1.

Protein expression analysis of the heterologously expressed CODH/ACS enzyme complex in C. acetobutylicum using His tag purification, SDS-PAGE, and native PAGE. (A and B) Cell lysates were purified on a Ni-NTA column under native conditions. The purified fraction was loaded onto an SDS-PAGE gel and transferred onto a nitrocellulose membrane. Two blots were probed against the ACS subunit (∼77.5 kDa) (A) or against the CODH subunit (∼67.6 kDa) (B) using polyclonal antibodies. I, whole-cell lysates from C. carboxidivorans (positive control); II, protein ladder; III, His tag-purified CODH/ACS protein from C. acetobutylicum under native conditions. (C) Schematic representation of the CODH/ACS expression cassette. Both subunits are under the control of the Pptb promoter, and the ACS subunit contains a His tag prior to the stop codon. (D) Native PAGE gel of wild-type (WT) C. acetobutylicum (control) and C. acetobutylicum expressing CODH/ACS. a, whole-cell extract control; b, insoluble cell extract fraction of control; c, soluble cell extract fraction of control; d, whole-cell extract of strain expressing CODH/ACS; e, insoluble cell extract fraction of strain expressing CODH/ACS; f and g, soluble cell extract fraction of strain expressing CODH/ACS (early and late stage of growth, respectively); h, protein ladder. (E) Western blot of native gel shown in panel D (d to g), whereby the blot was probed with anti-6× His tag and Alexa Fluor 647 antibodies to show that the high-molecular-weight band seen on native PAGE gel is that of the CODH/ACS tetrameric complex.
In vivo reduction of CO2 by C. acetobutylicum expressing CODH/ACS complex.
To test the activity of the CODH/ACS protein complex, the C. acetobutylicum strain expressing CODH/ACS was tested for its ability to reduce CO2. The strain was grown on 40 g/liter glucose in defined medium in sealed serum bottles with 10 lb/in2 CO2 and H2 (20:80) in the headspace. CO formation over 24 h was confirmed using gas chromatography (Fig. 2A). To verify that the CO was a direct product of CO2 reduction, we added 30 mM [13C]sodium bicarbonate to actively growing cells to achieve ∼18% 13C-labeling in CO2, as the headspace already contained 12CO2 from decarboxylation reactions due to the glucose fermentation. The percentage of 13C-label in CO, as measured by gas chromatography coupled to mass spectrometry (GC-MS) (Fig. 2B), increased with time. This confirmed that the CO is produced due to reduction of CO2 in the headspace. Control cells carrying an empty plasmid failed in all experiments to produce CO, that is, no CO could be detected on the chromatogram (Fig. 2C). Since the CODH natively employs reduced ferredoxin (Fdred) to reduce CO2 to CO (Fig. 2D) (9, 10), it is clear that the recombinant CODH used C. acetobutylicum-native Fdred to carry out this reaction.
FIG 2.

In vivo reduction of CO2 by C. acetobutylicum expressing the CODH/ACS complex. CO is a product of CO2 reduction, as shown by 13C-tracing. C. acetobutylicum expressing the CODH/ACS enzyme complex was grown on glucose in defined medium in sealed serum bottles with 10 lb/in2 CO2 and H2 (20/80) in the headspace. (A) CO concentration in headspace at 0, 12, and 24 h. Empty vector (EV) control never produced any CO. (B) Percentage of 13CO in CO after addition of [13C]bicarbonate to actively growing and CO-producing cells. (C) GC-MS spectra of headspace gas samples from C. acetobutylicum expressing CODH/ACS (left) and empty vector control (right). The CODH/ACS strain exhibits a CO peak at 0.98 min after 20 h. Gas analysis from the control strain (EV) shows only the N2 peak at 0.9 min but no CO peak. (D) Schematic depicting the interaction of ferredoxin with the hydrogenase and the CODH. (E) C. acetobutylicum expressing CODH/ACS enzyme grown on glucose in defined medium in sealed serum bottles with 20% CO2 and H2 or He balance in the headspace (10 lb/in2). More CO is produced in the presence of hydrogen than in the absence of hydrogen. The amount of CO (in nanomoles) produced per culture after 24 h was statistically significant (**, P < 0.005) compared to that produced in the presence of He. Error bars indicate the standard error of at least two biological replicates, and statistical significance was tested with a two-sample t test.
Externally added H2 enhances the CODH-catalyzed CO formation from CO2 reduction.
Here, we wanted to further support the logical conclusion that a C. acetobutylicum native ferredoxin is involved in the CODH reaction in this host. Hydrogenases of C. acetobutylicum utilize Fdred as the electron donor (23). Cells can dispose of excess electrons through the production of H2 (Fig. 2E) or can produce reduced products, such as butanol and ethanol (24). Exogenously added H2 to fermentations of C. acetobutylicum has been shown to result in higher concentrations of reduced metabolites (25), suggesting a larger internal Fdred pool that can be used to increase the NADH pool and enable the production of more reduced metabolites (23, 24, 26). Furthermore, it has been shown that during the solventogenic fermentation phase, where the cells produce alcohols, cultures of C. acetobutylicum can consume H2 (27), which would produce more Fdred to be used again for alcohol formation through increased NADH concentrations. Based on these data, we hypothesized that in the presence of H2, the hydrogenase reaction to form H2 will be operating at reduced rates, thus oxidizing the Fdred (produced by the pyruvate decarboxylation reaction) at reduced rates and resulting in a higher intracellular Fdred pool. This in turn could drive the reduction of CO2 more efficiently. The C. acetobutylicum strain expressing CODH/ACS was tested for CO2 reduction in the presence or absence of H2, where helium (He) was used in place of H2. Cells were grown in the presence of 20% CO2 and either 80% H2 or 80% He. CO formation was measured after 24 h (Fig. 2D). Cells grown in the presence of H2 produced significantly more (P < 0.005) CO than the cells grown in the presence of He.
To further pursue the importance of the ferredoxin intracellular pool, we tested the formation of CO under both growing conditions and nongrowing (stationary) conditions. Cells were washed and used to inoculate medium with glucose to an initial optical density at 600 nm (OD600) of ∼0.1 or without glucose to an initial OD600 of ∼3. Both conditions were subject to a headspace composition of 20% CO2–80% H2. Incubation of the cells without glucose failed to produce CO (data not shown). Fdred is produced during glycolysis, namely, by the decarboxylation of pyruvate to acetyl-CoA (23). When glucose is absent, oxidized ferredoxin is not reduced, resulting in a low intracellular Fdred pool. H2 cannot replenish the Fdred pool in the absence of glycolytic activity. This is likely due to the low cellular energy charge that prevents the maintenance of the cellular membrane potential and ΔpH, which are essential for these complex electron exchange reactions. Taken together, these data further support the conclusion that the recombinant CODH interacts with a native C. acetobutylicum ferredoxin as it would in native acetogens.
CO2 is reduced to CO by C. acetobutylicum expressing the CODH subunit alone.
The CODH/ACS protein complex in acetogens couples the reduction of CO2 to CO with the subsequent condensation with a methyl group to form acetyl-CoA (28). The CO which is generated at the C-cluster of the CODH travels through the internal channel to the ACS subunit and catalytic active site, relying on a tight association between the two subunits. Monomeric CODHs, such as those from Carboxydothermus hydrogenoformans and Rhodospirillum rubrum, have been characterized but have been shown to operate only in the energetically favored direction of CO oxidation, and CO2 reduction has not been reported. We pondered whether the C. carboxidivorans CODH alone (i.e., without the presence of its cognate ACS partner) could reduce CO2 to CO. To test this possibility, we expressed the CODH subunit alone in C. acetobutylicum, and as before, we grew the cells on glucose in the presence of 20% CO2–80% H2, and CO production was monitored using gas chromatography. We also added 50 mM [13C]sodium bicarbonate to label the CO2 pool, resulting in roughly ∼20% initial 13C-labeling. CO was produced by the C. acetobutylicum strains expressing the CODH only or both CODH and ACS (Fig. 3A). The 13C-labeling of CO was significantly higher than the natural abundance (P < 0.0001) in both the CODH- and the CODH/ACS-expressing strains compared to the control (Fig. 3B). These results are somewhat surprising in that the CODH enzyme is found to be functioning on its own to reduce CO2. In addition, there seems to be no statistically significant difference between the two strains, indicating the ACS subunit does not play a role in the CO2 reduction to CO. This is a novel and hitherto-unexpected conclusion.
FIG 3.
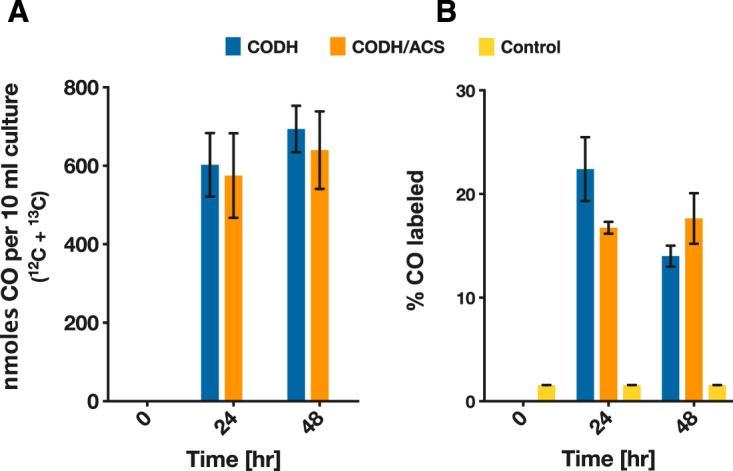
In vivo reduction of CO2 by C. acetobutylicum expressing the CODH/ACS complex or the CODH enzyme alone. Cells of C. acetobutylicum expressing the CODH/ACS complex or the CODH enzyme alone were grown on glucose (40 g/liter) in defined medium in sealed serum bottles with 10 lb/in2 CO2 and H2 (20/80) in the headspace. A concentration of 50 mM [13C]bicarbonate was added to the sealed bottles. (A) Total nanomoles CO (12CO and 13CO) in each serum bottle (10 ml of liquid culture). (B) Percentage of 13CO in produced CO. There was no statistically significant difference in either the amount of CO produced or the percentage of 13C-labeling in CO (n = 4). Error bars indicate the standard error of four biological replicates.
Reversibility shown in oxidation of CO by C. acetobutylicum expressing either the CODH/ACS complex or the CODH subunit alone.
CODH reversibly catalyzes the oxidation of CO to CO2 in native acetogens. Here, we set out to examine if this is the case when expressed in C. acetobutylicum by characterizing the in vivo function of the enzyme in the direction of CO oxidation. C. acetobutylicum expressing the genes for the CODH/ACS complex or the gene for the CODH enzyme by itself was grown in the presence of 5% 13CO, with the balance being helium. Only the two strains expressing the CODH enzyme were able to oxidize CO, whereas the empty vector control was unable to oxidize any CO (Fig. 4A). The decrease in 13CO was monitored throughout the fermentation, and it was inversely proportional to the increase in 13CO2. The increase in 13CO2 resulted in higher-than-natural abundance (∼1%) labeling in the CO2 peak on the chromatogram (Fig. 4B). The CO peak on the chromatogram spectrum did not decrease for the empty vector control (data not shown) but did completely disappear for the CODH/ACS strain and the CODH strain after about 72 h (Fig. 4C). These results show the CODH is capable of both CO oxidation and CO2 reduction.
FIG 4.

In vivo CO oxidation by C. acetobutylicum expressing either the CODH/ACS complex or the CODH subunit alone. (A) C. acetobutylicum strains grown on defined medium in the presence of 5% 13CO (balance N2) in the headspace (5 lb/in2). CO concentration was measured for the CODH/ACS, the CODH, and an empty vector strain (n = 2). The empty vector control strain did not oxidize CO during the 72 h of monitoring. (B) 13C-labeling in CO2 as a function of time. (C) GC-MS analysis of the headspace demonstrating CO oxidization by the C. acetobutylicum strain expressing CODH/ACS. Error bars indicate the standard error of two biological replicates.
CO concentration affects CO oxidation and metabolite distribution in cultures of C. acetobutylicum expressing the CODH subunit.
We then set out to investigate the impact of initial CO concentration on CO oxidation. Because CO is known to affect the growth rate of C. acetobutylicum (29–32), we resorted to a stationary (no-growth) experiment with a high initial cell density (optical density at 600 nm [OD600], ∼3.5) while varying the amount of 13CO (balance was He) in the headspace with 20 lb/in2 total pressure. Small amounts of glucose (ca. 5 g/liter [30 mM]) were added to maintain low metabolic activity. There were small amounts of metabolites present at the setup, all below 4 mM, except for acetate (ca. 43 mM), which is used as pH buffer, and ethanol (ca. 49 mM), which is used to dissolve the antibiotic for plasmid maintenance. Cultures were sampled 24 h after setup. The specific rate of CO2 formation by the CODH was calculated by the change in 13CO2 per min per gram of dry cell weight. As the concentration of CO increased, the rate of CO oxidation decreased (Fig. 5A). At >10% 13CO, we observed a drastic decrease in the production of 13CO2, indicating inhibition of CO oxidation and thus inhibition of the CODH. At 5% CO, the highest rate of 13CO2 formation (which is equal to the rate of 13CO oxidation) was an average of 2.4 μmol per min per mg of dry cell weight (Fig. 5A). Literature values for CO oxidation have been reported as 0.2 to 0.4 μmol per min per mg of wet cell weight (8). For all CO concentrations, glucose was completely consumed in 24 h in the cultures of the CODH expressing C. acetobutylicum. In contrast, in two cultures of the plasmid control strain at 19% CO, glucose was not utilized, and only small amounts of butyric acid were taken up, thus indicating severe inhibition of cell metabolism at this high CO concentration.
FIG 5.

CO concentration affects CO oxidation and metabolite distribution. C. acetobutylicum expressing the CODH enzyme was grown on defined medium in the presence of various amounts of 13CO (balance He) in the headspace (20 lb/in2 total) during high-cell-density low-glucose-concentration experiments (5 g/liter glucose). (A) Concentration of 13CO2 (in micromoles) formed per gram dry weight (gDW) of cells (n = 2). The large error bar for the experiments with 5% CO is largely due to the difficulty of precisely charging the gas phase with low partial pressures of CO, combined with the gas mixing and solubility and the biological variability. (B) Metabolite profile (shown as change in metabolite concentration [ΔConc.] from 0 to 24 h) of cultures in the presence of various CO concentrations. The ethanol data have been corrected for the evaporation of ethanol upon charging the culture with gas. The initial ethanol was ca. 49 mM, since ethanol is used to dissolve the antibiotic for plasmid maintenance. Error bars indicate the standard error of two biological replicates.
CO inhibition of C. acetobutylicum growth derives from the inhibition of the hydrogenase, which contains an active site (H-cluster) containing a novel [4Fe4S] subcluster (33), which can bind CO and inhibit function, similar to the inhibition of hemoglobin by CO. As a result, H2 formation is severely inhibited or totally abolished (31, 32), and cell metabolism, in batch systems, shifts toward metabolites (ethanol and butanol) (30, 31) whose formation requires electrons. This is further accompanied by reduced acetone, butyrate, and acetate formation. In one report from our lab, where CO was introduced in continuous culture of C. acetobutylicum, lactate and acetoin were also induced by CO gassing (32). While the changes in metabolite concentrations were low due to low glucose concentrations (29 mM at t = 0; at such low glucose, wild-type C. acetobutylicum produces acids only), the patterns of metabolite formation were quite informative (Fig. 5B). Under all conditions tested, the cells exposed to CO produced less to no acetone and drastically lower butyrate concentrations than cells not exposed to CO (Fig. 5B). Acetate was consumed at 14% and 19% CO, whereas acetate was produced under the other conditions tested. Butanol and ethanol formation was enhanced by the presence of CO, as previously reported. Lactate and acetoin were produced under all conditions with CO, but virtually none was formed in the absence of CO (Fig. 5B). Overall, the changes in metabolite formation due to CO were not predictable or proportional. For example, lactate production increased as CO concentration increased, but butanol and acetoin production remained largely unchanged after an initial increase by the addition of CO. We note that lactate and acetoin production in response to CO presence has been reported only by us for continuous cultures (32) but not for batch systems. The mechanism by which the inability to produce H2 alters the cell's biosynthetic machinery to produce metabolites requiring electrons is not well understood. It likely involves sensing of the altered redox environment (due to accumulation of reduced ferredoxin, NADH, and other electron carriers) by the redox sensor Rex, which regulates the expression of several product formation genes and pathways (34, 35). As stated above, inhibition of the H2-producing hydrogenase would increase the Fdred pool and NADH pool derived from Fdred. The cells apparently use two mechanisms to deal with the increased NADH and/or Fdred pools. First, they use NADH to produce products (butanol and ethanol) that utilize NADH and suppress the formation of products (acetone, butyrate, and acetate) that use less or no NADH (24). They also apparently slow down the production of Fdred by reducing the rate of pyruvate decarboxylation that produces Fdred; instead, cells direct the carbon flux from the pyruvate pool to acetoin. Lactate production serves both mechanisms: increased NADH utilization by the lactate dehydrogenase, and reduction of Fdred formation by reducing pyruvate decarboxylation. Taken together, these data, compared to the plasmid control data and prior literature, suggest that CODH modulates the response of the cells to CO presence.
Nickel enhances the activity of the recombinant CODH expressed in C. acetobutylicum.
Nickel is an important element found in the active site of oxygen-sensitive acetogenic CODHs. It was previously shown that cells of C. formicoaceticum grown in the absence of nickel had reduced CODH activity, but adding 1 μM NiCl2 to the medium during growth restored CODH activity (36). We assume that this would be the case for CODHs in other acetogens. Given that the intracellular environment of C. acetobutylicum is different than that of C. carboxidivorans, we wanted to test if nickel had an effect on the activity of the recombinant CODH expressed in C. acetobutylicum. To do so, the CODH/ACS-expressing strain was grown in the absence of nickel in a defined medium to mid-exponential phase. Cells were concentrated and used to inoculate serum bottles containing various amounts of nickel. The activity was almost completely absent without nickel present, but CO production increased with increasing nickel concentration (Fig. 6A). This indicates that the nickel is inserted in vivo into the active site of the CODH expressed in C. acetobutylicum, despite the absence of a known Ni-inserting accessory protein. This is pursued further in the next section.
FIG 6.

Impact of added Ni on CO production by C. acetobutylicum expressing the CODH/ACS complex without or with coexpression of the Ni-inserting CooC protein. (A) CO concentration was measured in the headspace for concentrated C. acetobutylicum expressing the CODH/ACS complex alone (blue circles/line) or with the accessory protein CooC (orange squares/line) after 24 h of growth in defined medium with various amounts of Ni. (B) Total CO produced in the absence of nickel for the CODH/ACS-expressing strain (blue) and the CODH/ACS–CooC-expressing strain (orange). There is a significant difference at the 90% confidence level between the two strains (*, P < 0.1; n = 4), indicating that the accessory protein can facilitate nickel insertion into the active site in the presence of trace levels (residual) of Ni. Error bars indicate the standard error of at least two biological replicates.
Ni-inserting accessory protein CooC enhances in vivo CODH activity at smaller nickel amounts.
The CODH/ACS gene cluster of native acetogens contains two small subunits, CooC and AcsF, which are believed to be involved in active-site maturation; namely, they are responsible for nickel insertion (37). Tests in Rhodospirillum rubrum strains known to contain a Ni-CODH bearing CooC mutations required ca. 1,000-fold higher nickel concentrations to sustain CO-dependent growth and restore wild-type levels of growth on CO (38). Our data (Fig. 6A) show that nickel supplementation is sufficient for the assembly of the active nickel-containing C-cluster of the recombinant CODH, thus suggesting that Ni insertion takes place without the presence of a Ni insertase. Nevertheless, we wanted to test the hypothesis that coexpression of the CODH maturation protein CooC (encoded by Ccar_18840) found in the WLP gene cluster of C. carboxidivorans would decrease the amount of nickel required for CO production and result in higher CODH activity. To this effect, we constructed a plasmid bearing the CODH/ACS genes followed by the genes coding for the CooC maturation protein, and we repeated the experiment mentioned in the previous section with various Ni concentrations in the medium. CooC coexpression did not increase CO production significantly over the range of nickel concentrations tested. However, in the absence of added nickel, the accessory protein increased the production of CO, even though the significance was at the 90% confidence level (n = 4, P < 0.1) (Fig. 6B). This might indicate that the accessory protein can facilitate nickel insertion into the active site at trace (residual) Ni concentrations in the medium (from impurities of salts used in medium preparation) and therefore requires less added Ni for higher activity.
An in vivo assay for the ACS exchange activity?
The ACS protein has been studied extensively (15, 16, 39), and its structure and active site have been determined (11). As discussed above, ACS activity is tested in vitro by the carbon exchange reaction between CO and the carbonyl group of acetyl-CoA, using 14C-radioactive tracers. This reaction has been reported only for purified ACS and has been shown to be feasible with the ACS subunit alone (15, 16). We set out to investigate if this exchange activity could be explored in the in vivo setting using 13C-tracers and whole-cell catalysts. In native acetogens, CO release by the cells has not been reported. We hypothesized that 13C-labeling of the acetate carbonyl group could label the carbonyl group of the intracellular acetyl-CoA pool. The ACS enzyme could then bind the acetyl-CoA and exchange the carbon from CO with the carbon from the carbonyl group of acetyl-CoA. If functioning as in in vitro experiments, ACS could bind to [13C]acetyl-CoA and exchange the 13C with the 12C from CO. An increase in the percentage of labeling of CO could then be observed. To test this, C. acetobutylicum strains expressing either the CODH/ACS complex or CODH or ACS alone were grown in the presence of Ni in a defined medium with the addition of 30 mM sodium [1-13C]acetate. The strain expressing the CODH/ACS enzymes showed an increase in the labeling of CO, but so did the strain containing only CODH, indicating the increase was not due to the exchange reaction of ACS. We concluded that through the production of acetone, some of the label from acetate is released as CO2, which resulted in ∼3% labeling in CO2. Conversion of labeled CO2 into labeled CO due to the activity of the CODH could explain the slightly higher labeling in CO in those strains containing the CODH (see Fig. S1 in the supplemental material). To confirm this, we grew the C. acetobutylicum strain containing the ACS protein alone (incapable of reducing the partially labeled CO2 to CO, but still capable of the exchange reaction, if such reaction can take place in vivo) and showed that this strain did not exhibit an increase in the labeling in CO, which showed the labeling of the 13C natural abundance (∼1%). Since the active site of the ACS, the A-cluster, also requires a special Ni-containing center, we set out to investigate the necessity of a Ni accessory protein, AcsF, found in the WLP gene cluster (Ccar_18805). Recently, the Ni insertion accessory protein AcsF from Carboxydothermus hydrogenoformans was shown to be necessary for the ACS active site to fully develop (40). The C. acetobutylicum strain expressing the ACS and the AcsF from C. carboxidivorans did not increase the labeling in CO either. Lastly, a positive control consisting of C. carboxidivorans cells used under the same experimental setup did not lead to an increase in labeling of CO. Taken together, the data from these experiments suggest that the in vitro exchange activity may not function under in vivo conditions, at least for the strains of this study.
DISCUSSION
This study reports a recombinant engineered system capable of reducing CO2, utilizing an acetogenic CODH/ACS expressed in a nonacetogenic host. The CODH/ACS complex has been studied in several acetogens, such as M. thermoacetica, C. formicoaceticum, and Acetobacterium woodii, as well as Desulfovibrio vulgaris and Carboxydothermus hydrogenoformans (9, 28, 36, 41, 42). In these organisms, a reduced anaerobic environment provides the conditions for CO2 fixation (43). The CODH/ACS enzyme has been expressed in a nonnative host (E. coli) before; however, the enzyme was shown to be active only in the direction of CO oxidation in vitro (18). Never before has this enzyme been shown to function in vivo in a nonnative host. To reduce CO2, a suitable low-potential electron carrier is required. The CO2-CO redox couple has a standard redox potential of −520 mV (standard redox potential calculated at 1 M for all reactants at pH 7 by Schuchmann and Müller (44)), and only reduced ferredoxin has a low enough redox potential to provide electrons for this reaction. The CODH/ACS enzymes of M. thermoacetica and A. woodii have been shown to interact in vitro with a ferredoxin derived from Clostridium pasteurianum (10, 45). C. acetobutylicum contains several native ferredoxins, which serve as electron carriers in several reactions, and notably in conserving high energy electrons from the pyruvate decarboxylation reaction to form acetyl-CoA, as well as in the reactions leading to butyryl-CoA synthesis. Reduced ferredoxin(s) serves as an electron donor for H2 formation but also NADH generation used for alcohol production. Here, our data support the hypothesis that a native C. acetobutylicum Fdred apparently interacts with the heterologous CODH/ACS complex. Indeed, we showed that CO2 reduction was affected by H2 pressure and exhibited dependence on growth-associated glucose utilization. Since ferredoxin is produced in the cells during active glycolysis from the conversion of pyruvate to acetyl-CoA or the formation of butyryl-CoA, growing cells contain higher concentrations of Fdred.
Surprisingly, CO2 reduction did not depend on the presence of the ACS protein subunit of the CODH/ACS enzyme complex, and functionality in both oxidation and reduction directions was enabled by the CODH subunit alone. Although the function of the ACS subunit has been studied in vitro (15) and in vivo (46), the CODH subunit from the bifunctional CODH/ACS complex has not been studied by itself before, and it was not known if it can reduce CO2 to CO on its own.
CO oxidation by an engineered C. acetobutylicum strain is the first step toward growth on CO, which could be a source of both carbon and electrons. CO is quite toxic, and only a few anaerobes grow in the presence of 100% (vol/vol) CO (47). Previous studies have engineered the archaeon Pyrococcus furiosus to heterologously express a CODH operon from Thermococcus onnurineus, making the engineered organism capable of CO oxidation and able to generate energy via the generation of an ion gradient using the electrons from CO oxidation (48). Here, we have shown that at gaseous concentrations of 5% (vol/vol) CO, the engineered C. acetobutylicum strain is capable of complete CO oxidation using the acetogenic bifunctional CODH/ACS complex. Developing tolerance to greater amounts of CO by engineering a superior CODH exhibiting higher activity could provide a source of additional electrons from CO and thereby alter the fermentation behavior. Previous studies of heterologous CODH expression reported that incubation with nickel after purification was necessary to restore activity (12, 18, 39), and that chelating agents, such as 1,10-phenanthroline, abolish CODH activity, indicating the need for Ni in the active site (49). Here, we found that the culture medium required nickel supplementation (ca. 50 to 100 μM) to maximize CODH activity (Fig. 6). Many CODH loci code for a nickel-inserting accessory protein, which is believed to facilitate the insertion of nickel into the CODH active site (40). Here, expression of the nickel-inserting accessory protein CooC from C. carboxidivorans did increase activity, but only in the case of no nickel supplementation. Interestingly, the highest activity achieved required nickel at much higher concentrations than what is required in media of acetogens (∼100 μM versus 1.2 μM in DSMZ141 medium). This might suggest that Ni insertion proteins are essential for CODH activity in low-Ni media.
As stated above, in all studies examining the ACS activity, highly purified amounts were needed to achieve a carbon exchange reaction between CO and the carbonyl-carbon from acetyl-CoA (18, 19, 40, 50). The in vitro ACS activity based on this carbon exchange reaction is typically much slower than the in vivo reaction that supports the growth of acetogens. This might indicate that the in vitro reaction uses a different mechanism than the in vivo reaction. The in vitro experiment requires hyperanoxic conditions, extensive ACS purification, and high protein concentrations. Furthermore, it is necessary to insert nickel through in vitro incubation of the purified enzyme with nickel chloride to restore activity and achieve nickel insertion into the active site. Recently, Gregg et al. showed the necessity of a nickel accessory protein, AcsF, to facilitate the insertion of nickel into the active site in Carboxydothermus hydrogenoformans (40). Even though we tested our system with additional expression of the AcsF accessory protein, the in vivo carbon exchange reaction could not be detected. The native acetogen C. carboxidivorans was also incapable of demonstrating the exchange activity in vivo, but this might be due to the presence of a functional WLP that prevents CO accumulation.
The tetrameric CODH/ACS complex plays an important role as the acetyl-CoA synthase in the WLP, in which the reaction of CO2 reduction is coupled with the formation of acetyl-CoA from a methyl group and CO. The first step of CO2 reduction is an important step for achieving synthetic CO2 fixation in an organism that does not fix CO2. We have shown that the reduction of CO2 is feasible without any additional codon optimization or protein engineering. The enzymes were expressed and active in a closely related Clostridium host. C. acetobutylicum presents a great platform organism due to its native production of butanol and acetone, but also because of the availability of an extensive set of metabolic engineering tools (23, 51). Here, we have shown that the organism can be engineered to reduce CO2. Further engineering efforts toward reduction and incorporation of CO2 hold great promise to achieve synthetic CO2 fixation in C. acetobutylicum using a functional WLP.
MATERIALS AND METHODS
Chemicals.
Stable isotope gases and all chemicals were purchased from Sigma-Aldrich.
Strains, culture conditions, and medium.
C. acetobutylicum ATCC 824 was grown anaerobically at 37°C in either 2× YTG medium (16 g/liter Bacto tryptone, 10 g/liter yeast extract, 4 g/liter NaCl, and 5 g/liter glucose [pH 5.8]) or defined clostridial growth medium (dCGM; 0.75 g/liter K2HPO4, 0.75 g/liter KH2PO4, 0.7 g/liter MgSO4·7H2O, 0.01 g/liter MnSO4·H2O, 0.01 g/liter FeSO4·7H2O, 1 g/liter NaCl, 0.004 g/liter p-aminobenzoic acid, 10 μg/liter biotin, 3.3 g/liter ammonium acetate and 40 g/liter glucose [pH 6.8]). Recombinant strains were grown on solid 2× YTG medium supplemented with 40 μg/ml erythromycin, and single colonies were picked and grown anaerobically in 10 ml of dCGM as preculture. In vivo experiments were conducted as described below. Transformations into C. acetobutylicum were performed as previously described (52). All molecular cloning steps were completed in E. coli, and strains were grown aerobically at 37°C and 220 rpm in liquid LB medium with shaking at 220 rpm, or on LB with 1.5% agar supplemented with 50 μg/ml ampicillin. C. carboxidivorans DSM 15243 was grown anaerobically in American Type Culture Collection (ATCC) medium 1754 with 10 g/liter glucose at 37°C.
Vector and strain construction.
All plasmid vectors were constructed in either E. coli (NEB Turbo) or E. coli (NEB 5α). All primers are listed in Table 1. Plasmid vector p94_CODH/ACS was designed for expression of the CODH/ACS enzyme complex in C. acetobutylicum. It contained both the codh (Ccar_18845) and the acs (Ccar_18785) genes from C. carboxidivorans in an operon arrangement under the control of the strong Pptb promoter native to C. acetobutylicum (22). The codh gene was amplified from C. carboxidivorans genomic DNA (gDNA) using primers CODH_F and CODH_R. Similarly, the acs gene was amplified from the same gDNA using primers ACS_F and ACS_R. Vector pSOS94_MCS was digested with BamHI, and the gene encoding CODH and the acs gene were cloned into the vector using Gibson Assembly (New England BioLabs), resulting in p94_CODH/ACS. The His tag was added using p94_CODH/ACS digested with SbfI and KspI and primers p94_sbfI_f and CODH/ACS_his_R. The CODH subunit by itself was constructed using primers that flanked the acs gene to be deleted (CODHonly_f and CODHonly_r) using p94_CODH/ACS as the template. The Q5 site-directed mutagenesis kit from NEB BioLabs was used for easy plasmid isolation. The resulting vector (p94_CODH) has the same promoter configuration as p94_CODH/ACS (described above). The strain containing the codh, cooC (Ccar_18840), and acs genes was constructed by amplifying two fragments from C. carboxidivorans gDNA using primers p94_CODH/CooC_f and p94_CODH/CooC _r, and p94_ACS_f and p94_ACS_r. The pSOS94_MCS vector was digested with BamHI and XbaI, and the three fragments were assembled using Gibson Assembly (New England BioLabs). For the strain containing the acs gene alone, the gene was amplified from C. carboxidivorans gDNA using primers (ACS_BamHI_f and ACS_XbaI_r) and inserted into the digested pSOS95_MCS vector (BamHI and XbaI) using Gibson Assembly. The resulting vector p95_ACS contained the acs gene under the control of the thiolase promoter (Pthl). The gene coding for the AcsF accessory protein (Ccar_18805) was amplified using primers p95_AcsF_f and p95_AcsF_r using C. carboxidivorans gDNA as the template and ligated into the digested p95_ACS (digested with SphI and MluI) using Gibson Assembly.
TABLE 1.
Primers for construction of strains used in this work
| Primer | Sequence (5′ → 3′) |
|---|---|
| CODH_F | TCATTTAACATAGATAATTGGATCCGTAATTCAGCAAGGAGGG |
| CODH_R | ATTTATATAAAATTTATATTCCTAATTTTTTACGTTTTTCATTG |
| ACS_F | TAGGAATATAAATTTTATATAAATTTTAATTTTAGGGAGGG |
| ACS_R | GCGGATTCTAGATTGGATTACATTATTGGATCCATAGTTAATG |
| CODH/ACS_his_R | ACCGCGGATTCTAGATTGGATTAATGATGATGATGATGATGCATTATTGGATCCATAGT |
| p94_sbfI_f | AAAGCTCCTGCAGGTCGACTGTGGATGG |
| CODHonly_f | GTAAAAGAGATTGTTTCTAGCTC |
| CODHonly_r | TAAAATTTATATTCCTAATTTTTTACGTTTTTC |
| ACS_BamHI_f | GGTTTATCTGTTACCCCGTAGGATCCTAGGGAGGGGCAAAAAAA |
| ACS_XbaI_r | GATGCATGCTACCGCGGATTCTAGATGTCGTATTATAAGTTTTATTACATTATTG |
| p95_AcsF_f | CTAGAATCCGCGGTAGCATGCTGCATGACAATAGGAGGAG |
| p95_AcsF_r | ATCATTAAGTGGCGCCTTTACGCGTAGGAAGAACACTTTCTCC |
| p94_CODH/CooC_f | GTTAATCATTTAACATAGATAATTGGATCCAGGAGGGAATTTATTAAAATGG |
| p94_CODH/CooC _r | GTTAATCATTTAACATAGATAATTGGATCCAGGAGGGAATTTATTAAAATGG |
| p94_ACS_f | GGGGGTATTTTAAAGGGAGGGGCAAAAAAATG |
| p94_ACS_r | GGGGGTATTTTAAAGGGAGGGGCAAAAAAATG |
Vector transformations.
Vectors were transformed into E. coli (NEB Turbo) and isolated using the QIAprep spin kit (Qiagen). Before transformation into C. acetobutylicum, vectors were transformed into electrocompetent E. coli ER2275(pAN3) (53) for the in vivo methylation by the Φ3T I methyl transferase contained on the pAN3 vector, as described previously (54). Methylated vectors were isolated using the QIAprep spin kit (Qiagen) and transformed into C. acetobutylicum ATCC 824 using a well-established protocol (52).
Culture conditions and harvesting of recombinant C. acetobutylicum.
A recombinant C. acetobutylicum strain expressing the CODH/ACS enzyme complex was grown in 2× YTG to an OD600 of around 0.7. The cultures were harvested by centrifugation at 4°C for 10 min at 7,000 × g, and cell pellets were frozen at −82°C. Cells of the C. acetobutylicum empty vector control strain and wild-type C. carboxidivorans were collected in the same manner.
Native His tag purification.
The His-tagged proteins were purified with a Ni-NTA spin column kit (Qiagen), according to the protocol for purification under native conditions. First, the frozen cell pellets were thawed, washed once, and resuspended in lysis buffer, followed by sonication for 5 min at 50% power with intervals of 15 s of sonication and 15 s of rest using a Fisher Scientific sonic dismembrator and a cup horn. The samples were clarified by centrifugation for 10 min at 16,000 rpm and 4°C, and the remainder of the protocol was performed as per the manufacturer's recommendations. Eluates were quantified using a protein assay (Bio-Rad RC DC protein assay).
SDS-PAGE.
The cell pellets were resuspended in 500 μl of 100 mM Tris-HCl (pH 7.0) and sonicated as described above. The samples were clarified by centrifugation for 10 min at 16,000 rpm and 4°C. Supernatants were collected, and the cell debris was resuspended in the same amount of Tris-HCl buffer. Samples were mixed in a 1:5 dilution with 5× loading buffer (0.2 M Tris-HCl [pH 6.8], 10% SDS, 0.05% bromophenol blue, 50% glycerol) and boiled at 95°C for 10 min. Aliquots of 30 to 50 μl were loaded onto an SDS-PAGE gel (4 to 20% gradient, ExpressPlus; GenScript), and proteins were separated at 120 V for 2 h. The gels were either stained with Coomassie blue or transferred onto 0.2-μm-pore-size nitrocellulose membranes (Bio-Rad) using the Bio-Rad Mini Trans-Blot. Purified polyclonal antibodies against the CODH and ACS subunits were custom produced by Proteintech (Rosemont, IL) and obtained from rabbit serum. The CODH subunit was raised against the peptide sequence PEVVDILTNKMEDWVGAKFF and the ACS subunit against the peptide sequence VVAFLEEKGHPALTMDPIM. Polyclonal antibodies were used at a 1:10,000 dilution. An alkaline phosphatase (AP) colorimetric kit (Bio-Rad) was used to visualize the bands on the Western blot.
Nondenaturing PAGE.
Cell pellets were resuspended and sonicated as described above, except that the samples were mixed in a 1:5 dilution with 5× nondenaturing loading buffer and loaded onto an PAGE gel (4 to 20% gradient, ExpressPlus; GenScript), according to the manufacturer's protocol for native gels. All subsequent steps, including Western blotting, were performed as described earlier. The tetrameric band was visualized on the Western blot using 6×His tag monoclonal antibody (Invitrogen) and Alexa Fluor 647 anti-mouse antibody on a Typhoon FLA 9000 laser scanner.
GC-MS.
All gas composition measurements were done with a Shimadzu QP2010 Ultra gas chromatograph-mass spectrometer. Headspace samples of 50 μl were manually collected and injected using an airtight syringe. Gas separation was performed on a Carboxen-1006 porous layer open tubular (PLOT) column (30 m by 0.32 mm; 15-μm pore size; Sigma-Aldrich) with an injector temperature of 150°C, split ratio of 5, and carrier gas flow of 15.4 lb/in2 He (∼3 ml/min). The oven temperature was initially held at 30°C for 2.5 min, with ramping at 40°C/min to 120°C. CO separated from N2 on the column around 0.9 min. The mass spectrometer was set at a scan speed of ∼450 and scanning from m/z 4.00 to 60.00. Additional single-ion monitoring (SIM) was performed for N2 (m/z 28.00 and 29) and CO2 (m/z 44.00 and 45.00).
Detection of metabolites.
High-performance liquid chromatography (HPLC) was used to measure metabolite concentrations in fermentation medium. Metabolites were detected using an Agilent HPLC instrument with a Bio-Rad Aminex HPX 87H column using a 5 mM H2SO4 mobile phase and a flow rate of 0.5 ml/min (55).
CO2 reduction using growing cells.
C. acetobutylicum cells expressing either the CODH/ACS complex or CODH and an empty vector control were grown to mid-exponential phase and collected by centrifugation. Cell pellets were resuspended in fresh dCGM with 40 g/liter glucose and 200 μM NiCl2. The final OD600 of the cultures was 0.1. Ten milliliters of the cell suspension was added to 26-ml serum bottles (Wheaton) sealed with aluminum crimp caps. The headspace was pressurized to ∼10 pounds per square inch gauge (psig) using a 20:80 mixture of CO2-H2. The headspace was monitored with GC-MS, as previously described. For the Ni experiments, cells were grown in the absence of Ni to mid-exponential phase and collected by centrifugation. The cells were resuspended in fresh medium (dCGM with 40 g/liter glucose), and various amounts of Ni were added. Ten milliliters was transferred and grown in 110-ml serum bottles (Wheaton). The headspace was monitored using GC-MS.
CO oxidation using growing cells.
All experiments were performed in 26-ml serum bottles (Wheaton). C. acetobutylicum cells expressing either the CODH/ACS complex or CODH and an empty vector control were grown to mid-exponential phase and collected by centrifugation. Cell pellets were resuspended in fresh dCGM with 40 g/liter glucose. A concentration of 200 μM NiCl2 and 5 ml of the resuspended cells was dispensed into the bottles secured with a thick butyl rubber stopper and crimped with aluminum seals. The headspace was pressurized to 3 psig using a 20:80 mixture of CO2 and H2. One hundred percent 13CO was added into the headspace via a syringe. The headspace was monitored using GC. Metabolites were detected using HPLC.
CO oxidation using resting cells.
All experiments were performed in 26-ml serum bottles (Wheaton). C. acetobutylicum cells expressing the CODH and an empty vector control were grown to mid-exponential phase and collected by centrifugation. Cell pellets were concentrated in fresh dCGM with 5 g/liter glucose supplemented with 200 mM NiCl2 to an OD of ∼3.5. Five milliliters of cell suspensions was dispensed into 26-ml bottles secured with a thick butyl rubber stopper and crimped with aluminum seals. The headspace was pressurized with various amounts of 13CO (the balance was He) to a total pressure of 20 psig. The headspace gas composition was monitored using GC-MS. Metabolites were detected using HPLC.
ACS exchange activity.
All experiments were performed in 26-ml serum bottles (Wheaton). C. acetobutylicum cells expressing either the CODH/ACS complex or CODH and an empty vector control were grown to mid-exponential phase and collected by centrifugation. Cell pellets were resuspended in fresh dCGM prepared with [13C]sodium acetate, 40 g/liter glucose, and 200 μM NiCl2. Five milliliters was dispensed into each bottle, and each bottle was secured with a thick butyl rubber stopper and crimped with aluminum seals. The headspace was pressurized to 3 psig using a 20:80 mixture of CO2 and H2. A small amount (1 ml) of 100% CO was added into the headspace. The headspace was monitored using GC for the increase in the 13CO peak using SIM for m/z 28.00 and 29.00.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Science Foundation (USA) under award number CBET-1511660.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00829-17.
REFERENCES
- 1.Fast AG, Papoutsakis ET. 2012. Stoichiometric and energetic analyses of non-photosynthetic CO2-fixation pathways to support synthetic biology strategies for production of fuels and chemicals. Curr Opin Chem Eng 1:380–395. doi: 10.1016/j.coche.2012.07.005. [DOI] [Google Scholar]
- 2.Bar-Even A, Noor E, Milo R. 2012. A survey of carbon fixation pathways through a quantitative lens. J Exp Bot 63:2325–2342. doi: 10.1093/jxb/err417. [DOI] [PubMed] [Google Scholar]
- 3.Can M, Armstrong FA, Ragsdale SW. 2014. Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase. Chem Rev 114:4149–4174. doi: 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abrini J, Naveau H, Nyns EJ. 1994. Clostridium autoethanogenum, sp. nov., an anaerobic bacterium that produces ethanol from carbon monoxide. Arch Microbiol 161:345–351. doi: 10.1007/BF00303591. [DOI] [Google Scholar]
- 5.Genthner BRS, Bryant MP. 1987. Additional characteristics of one-carbon-compound utilization by Eubacterium limosum and Acetobacterium woodii. Appl Environ Microbiol 53:471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Köpke M, Mihalcea C, Bromley JC, Simpson SD. 2011. Fermentative production of ethanol from carbon monoxide. Curr Opin Biotechnol 22:320–325. doi: 10.1016/j.copbio.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Daniels L, Fuchs G, Thauer RK, Zeikus JG. 1977. Carbon monoxide oxidation by methanogenic bacteria. J Bacteriol 132:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diekert GB, Thauer RK. 1978. Carbon monoxide oxidation by Clostridium thermoaceticum and Clostridium formicoaceticum. J Bacteriol 136:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drake HL, Hu SI, Wood HG. 1980. Purification of carbon monoxide dehydrogenase, a nickel enzyme from Clostridium thermoaceticum. J Biol Chem 255:7174–7180. [PubMed] [Google Scholar]
- 10.Shanmugasundaram T, Wood HG. 1992. Interaction of ferredoxin with carbon monoxide dehydrogenase from Clostridium thermoaceticum. J Biol Chem 267:897–900. [PubMed] [Google Scholar]
- 11.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. 2002. A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science 298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 12.Maynard EL, Lindahl PA. 2001. Catalytic coupling of the active sites in acetyl-CoA synthase, a bifunctional CO-channeling enzyme. Biochemistry 40:13262–13267. doi: 10.1021/bi015604+. [DOI] [PubMed] [Google Scholar]
- 13.Volbeda A, Fontecilla-Camps J. 2004. Crystallographic evidence for a CO/CO2 tunnel gating mechanism in the bifunctional carbon monoxide dehydrogenase/acetyl coenzyme A synthase from Moorella thermoacetica. J Biol Inorg Chem 9:525–532. doi: 10.1007/s00775-004-0565-9. [DOI] [PubMed] [Google Scholar]
- 14.Maynard EL, Lindahl PA. 1999. Evidence of a molecular tunnel connecting the active sites for CO2 reduction and acetyl-CoA synthesis in acetyl-CoA synthase from Clostridium thermoaceticum. J Am Chem Soc 121:9221–9222. doi: 10.1021/ja992120g. [DOI] [Google Scholar]
- 15.Tan X, Lindahl PA. 2008. Tunnel mutagenesis and Ni-dependent reduction and methylation of the alpha subunit of acetyl coenzyme A synthase/carbon monoxide dehydrogenase. J Biol Inorg Chem 13:771–778. doi: 10.1007/s00775-008-0363-x. [DOI] [PubMed] [Google Scholar]
- 16.Tan X, Loke H-K, Fitch S, Lindahl PA. 2005. The tunnel of acetyl-coenzyme a synthase/carbon monoxide dehydrogenase regulates delivery of CO to the active site. J Am Chem Soc 127:5833–5839. doi: 10.1021/ja043701v. [DOI] [PubMed] [Google Scholar]
- 17.Roberts DL, James-Hagstrom JE, Garvin DK, Gorst CM, Runquist JA, Baur JR, Haase FC, Ragsdale SW. 1989. Cloning and expression of the gene cluster encoding key proteins involved in acetyl-CoA synthesis in Clostridium thermoaceticum: CO dehydrogenase, the corrinoid/Fe-S protein, and methyltransferase. Proc Natl Acad Sci U S A 86:32–36. doi: 10.1073/pnas.86.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loke HK, Bennett GN, Lindahl PA. 2000. Active acetyl-CoA synthase from Clostridium thermoaceticum obtained by cloning and heterologous expression of acsAB in Escherichia coli. Proc Natl Acad Sci U S A 97:12530–12535. doi: 10.1073/pnas.220404397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ragsdale SW, Clark JE, Ljungdahl LG, Lundie LL, Drake HL. 1983. Properties of purified carbon monoxide dehydrogenase from Clostridium thermoaceticum, a nickel, iron-sulfur protein. J Biol Chem 258:2364–2369. [PubMed] [Google Scholar]
- 20.Bruant G, Lévesque M-J, Peter C, Guiot SR, Masson L. 2010. Genomic analysis of carbon monoxide utilization and butanol production by Clostridium carboxidivorans strain P7. PLoS One 5:e13033. doi: 10.1371/journal.pone.0013033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liou JS-C, Balkwill DL, Drake GR, Tanner RS. 2005. Clostridium carboxidivorans sp. nov., a solvent-producing clostridium isolated from an agricultural settling lagoon, and reclassification of the acetogen Clostridium scatologenes strain SL1 as Clostridium drakei sp. nov. Int J Syst Evol Microbiol 55:2085–2091. doi: 10.1099/ijs.0.63482-0. [DOI] [PubMed] [Google Scholar]
- 22.Tummala SB, Welker NE, Papoutsakis ET. 1999. Development and characterization of a gene expression reporter system for Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 65:3793–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papoutsakis ET. 2008. Engineering solventogenic clostridia. Curr Opin Biotechnol 19:420–429. doi: 10.1016/j.copbio.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Papoutsakis ET. 1984. Equations and calculations for fermentations of butyric acid bacteria. Biotechnol Bioeng 26:174–187. doi: 10.1002/bit.260260210. [DOI] [PubMed] [Google Scholar]
- 25.Yerushalmi L, Volesky B, Szczesny T. 1985. Effect of increased hydrogen partial pressure on the acetone-butanol fermentation by Clostridium acetobutylicum. Appl Microbiol Biotechnol 22:103–107. doi: 10.1007/BF00250028. [DOI] [Google Scholar]
- 26.Meyer CL, Papoutsakis ET. 1989. Increased levels of ATP and NADH are associated with increased solvent production in continuous cultures of Clostridium acetobutylicum. Appl Microbiol Biotechnol 30:450–459. doi: 10.1007/BF00263849. [DOI] [Google Scholar]
- 27.Kim BH, Zeikus GJ. 1992. Hydrogen metabolism in Clostridium acetobutylicum fermentation. J Microbiol Biotechnol 2:248–254. [Google Scholar]
- 28.Hadj-Saïd J, Pandelia ME, Léger C, Fourmond V, Dementin S. 2015. The carbon monoxide dehydrogenase from Desulfovibrio vulgaris. Biochim Biophys Acta 1847:1574–1583. doi: 10.1016/j.bbabio.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Datta R, Zeikus JG. 1985. Modulation of acetone-butanol-ethanol fermentation by carbon monoxide and organic acids. Appl Environ Microbiol 49:522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim B, Bellows P, Datta R, Zeikus J. 1984. Control of carbon and electron flow in Clostridium acetobutylicum fermentations: utilization of carbon monoxide to inhibit hydrogen production and to enhance butanol yields. Appl Environ Microbiol 48:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer CL, Papoutsakis ET, McLaughlin JK. 1985. The effect of CO on growth and product formation in batch cultures of Clostridium acetobutylicum. Biotechnol Lett 7:37–42. doi: 10.1007/BF01032417. [DOI] [Google Scholar]
- 32.Meyer CL, Roos JW, Papoutsakis ET. 1986. Carbon monoxide gasing leads to alcohol production and butyrate uptake without acetone formation in continuous cultures of Clostridium acetobutylicum. Appl Microbiol Biotechnol 24:159–167. doi: 10.1007/BF01982561. [DOI] [Google Scholar]
- 33.Cornish AJ, Gärtner K, Yang H, Peters JW, Hegg EL. 2011. Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum. J Biol Chem 286:38341–38347. doi: 10.1074/jbc.M111.254664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q, Venkataramanan KP, Huang H, Papoutsakis ET, Wu CH. 2013. Transcription factors and genetic circuits orchestrating the complex, multilayered response of Clostridium acetobutylicum to butanol and butyrate stress. BMC Syst Biol 7:120. doi: 10.1186/1752-0509-7-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wietzke M, Bahl H. 2012. The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Environ Microbiol 96:749–761. doi: 10.1007/s00253-012-4112-2. [DOI] [PubMed] [Google Scholar]
- 36.Diekert G, Thauer RK. 1980. The effect of nickel on carbon monoxide dehydrogenase formation in Clostridium thermoaceticum and Clostridium formicoaceticum. FEMS Microbiol Lett 7:187–189. doi: 10.1111/j.1574-6941.1980.tb01622.x. [DOI] [Google Scholar]
- 37.Bender G, Pierce E, Hill JA, Darty JE, Ragsdale SW. 2011. Metal centers in the anaerobic microbial metabolism of CO and CO2. Metallomics 3:797–815. doi: 10.1039/c1mt00042j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerby RL, Ludden PW, Roberts GP. 1997. In vivo nickel insertion into the carbon monoxide dehydrogenase of Rhodospirillum rubrum: molecular and physiological characterization of cooCTJ. J Bacteriol 179:2259–2266. doi: 10.1128/jb.179.7.2259-2266.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loke HK, Tan X, Lindahl PA. 2002. Genetic construction of truncated and chimeric metalloproteins derived from the alpha subunit of acetyl-CoA synthase from Clostridium thermoaceticum. J Am Chem Soc 124:8667–8672. doi: 10.1021/ja025924w. [DOI] [PubMed] [Google Scholar]
- 40.Gregg CM, Goetzl S, Jeoung JH, Dobbek H. 2016. AcsF catalyzes the ATP-dependent insertion of nickel into the Ni, Ni-[4Fe4S] cluster of acetyl-CoA synthase. J Biol Chem 291:18129–18138. doi: 10.1074/jbc.M116.731638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertsch J, Müller V. 2015. CO metabolism in the acetogen Acetobacterium woodii. Appl Environ Microbiol 81:5949–5956. doi: 10.1128/AEM.01772-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang VC-C, Ragsdale SW, Armstrong FA. 2013. Investigations of two bidirectional carbon monoxide dehydrogenases from Carboxydothermus hydrogenoformans by protein film electrochemistry. Chembiochem 14:1845–1851. doi: 10.1002/cbic.201300270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Müller V. 2003. Energy conservation in acetogenic bacteria. Appl Environ Microbiol 69:6345–6353. doi: 10.1128/AEM.69.11.6345-6353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schuchmann K, Müller V. 2014. Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat Rev Microbiol 12:809–821. doi: 10.1038/nrmicro3365. [DOI] [PubMed] [Google Scholar]
- 45.Schuchmann K, Müller V. 2013. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 342:1382–1385. doi: 10.1126/science.1244758. [DOI] [PubMed] [Google Scholar]
- 46.Liew F, Henstra AM, Winzer K, Köpke M, Simpson SD, Minton NP. 2016. Insights into CO2 fixation pathway of Clostridium autoethanogenum by targeted mutagensis. mBio 7(3):e00427-16. doi: 10.1128/mBio.00427-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mörsdorf G, Frunzke K, Gadkari D, Meyer O. 1992. Microbial growth on carbon monoxide. Biodegradation 3:61–82. [Google Scholar]
- 48.Schut GJ, Lipscomb GL, Nguyen DMN, Kelly RM, Adams MWW. 2016. Heterologous production of an energy-conserving carbon monoxide dehydrogenase complex in the hyperthermophile Pyrococcus furiosus. Front Microbiol 7:29. doi: 10.3389/fmicb.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin W, Lindahl PA. 1992. Function and carbon monoxide binding properties of the nickel-iron complex in carbon monoxide dehydrogenase from Clostridium thermoaceticum. Biochemistry 31:12870–12875. doi: 10.1021/bi00166a023. [DOI] [PubMed] [Google Scholar]
- 50.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Romer P, Huber R, Meyer O. 2004. A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc Natl Acad Sci U S A 101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tracy BP, Jones SW, Fast AG, Indurthi DC, Papoutsakis ET. 2012. Clostridia: the importance of their exceptional substrate and metabolite diversity for biofuel and biorefinery applications. Curr Opin Biotechnol 23:364–381. doi: 10.1016/j.copbio.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 52.Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET. 1992. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Biotechnology 10:190–195. doi: 10.1038/nbt0292-190. [DOI] [PubMed] [Google Scholar]
- 53.Al-Hinai MA, Fast AG, Papoutsakis ET. 2012. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl Environ Microbiol 78:8112–8121. doi: 10.1128/AEM.02214-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mermelstein LD, Papoutsakis ET. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 59:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones SW, Fast AG, Carlson ED, Wiedel CA, Au J, Antoniewicz MR, Papoutsakis ET, Tracy BP. 2016. CO2 fixation by anaerobic non-photosynthetic mixotrophy for improved carbon conversion. Nat Commun 7:12800. doi: 10.1038/ncomms12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.