

Abstract
Edema factor (EF), a key anthrax exotoxin, has an anthrax protective antigen-binding domain (PABD) and a calmodulin (CaM)-activated adenylyl cyclase domain. Here, we report the crystal structures of CaM-bound EF, revealing the architecture of EF PABD. CaM has N- and C-terminal domains and each domain can bind two calcium ions. Calcium binding induces the conformational change of CaM from closed to open. Structures of the EF–CaM complex show how EF locks the N-terminal domain of CaM into a closed conformation regardless of its calcium-loading state. This represents a mechanism of how CaM effector alters the calcium affinity of CaM and uncouples the conformational change of CaM from calcium loading. Furthermore, structures of EF–CaM complexed with nucleotides show that EF uses two-metal–ion catalysis, a prevalent mechanism in DNA and RNA polymerases. A histidine (H351) further facilitates the catalysis of EF by activating a water to deprotonate 3′OH of ATP. Mammalian adenylyl cyclases share no structural similarity with EF and they also use two-metal–ion catalysis, suggesting the catalytic mechanism-driven convergent evolution of two structurally diverse adenylyl cyclases.
Keywords: adenylyl cyclase, anthrax, calmodulin, edema factor, two-metal–ion catalysis
Introduction
Anthrax bacteria secrete three key virulence factors: edema factor (EF), lethal factor (LF), and protective antigen (PA) (Collier and Young, 2003). EF has two functional domains: an N-terminal PA-binding domain (PABD) followed by a calmodulin (CaM)-activated adenylyl cyclase domain (ACD) (Drum et al, 2002). The EF PABD shares 35% sequence identity with the PABD of LF, a metalloprotease that can inactivate mitogen-activated protein kinase kinase (Collier and Young, 2003). PA serves as a transporter to deliver EF and LF into host cells. Inside the cells, the adenylyl cyclase activity of EF is activated by CaM, which leads to the rise of intracellular cyclic AMP (cAMP) to pathological levels (Shen et al, 2002). EF can impair host innate and adaptive immunity by altering the phagocytic activity of macrophages, cytokine production by monocytes and macrophages, and antigen presentation of T cells (Rossi Paccani et al, 2005). Consequently, the disruption of the EF gene results in reduced survival and lethality of anthrax bacteria (Brossier et al, 2000).
CaM, a highly evolutionary conserved calcium sensor, controls structurally and functionally diverse effectors to modulate a plethora of fundamental processes. CaM has N- and C-terminal globular domains (N-CaM, C-CaM) that are connected by a flexible, central α-helix. Each globular domain of CaM has two calcium-binding motifs (Fallon and Quiocho, 2003). The calcium binding to CaM induces conformational changes from a mainly hydrophilic closed state to an open conformation, exposing a large, hydrophobic binding pocket (Hoeflich and Ikura, 2002). CaM uses these hydrophobic binding pockets along with other interacting surfaces from both N and C lobes of CaM to bind diverse structures (Bhattacharya et al, 2004).
The binding of CaM can significantly alter the biochemical properties of its target effectors. The binding of CaM can remove the inhibitory domain of its effectors from the catalytic site to achieve catalytic activation. CaM can also trigger the dimerization of small-conductance potassium (SK) channels to increase ion conductivity and induce the reorganization of the catalytic site of EF (Schumacher et al, 2001; Drum et al, 2002). Conversely, the binding of CaM effectors can affect the conformational state and calcium loading of CaM. The interaction of CaM with its effectors typically facilitates the calcium loading and open state of CaM. However, growing examples have shown that CaM effectors can also trigger the opposite effects on CaM. CaM is constitutively associated with SK channel and their interaction locks the C-terminal domain of CaM in a calcium-free, semi-open state (Schumacher et al, 2001). EF binds the closed conformation of N-CaM to initiate the interaction with CaM (Shen et al, 2002; Ulmer et al, 2003). This relatively weak interaction (2 mM affinity) triggers the insertion of the open conformation of C-CaM into EF to achieve the much tighter binding of EF with CaM (20 nM affinity). Interestingly, the binding of EF significantly reduces the affinity of calcium to N-CaM.
Sequence analysis reveals the existence of five classes of adenylyl cyclase, four of which are found only in bacteria (class I, II, IV, and V), and the fifth family (class III) exists in many prokaryotes and eukaryotes (Linder and Schultz, 2003; Baker and Kelly, 2004). Class II adenylyl cyclases such as EF are secreted by pathogenic bacteria and require host cellular factors for their catalytic activation (Shen et al, 2002). This class also includes CyaA from Bordetella pertussis, the causative agent of whooping cough, and ExoY from Pseudomonas aeruginosa, bacteria responsible for various nosocomial infections (Yahr et al, 1998; Ladant and Ullmann, 1999). Two related Yersinia species, Y. pestis (plague) and Y. pseudotuberculosis (gastrointestinal symptoms), may also secrete adenylyl cyclase toxins (Shen et al, 2002). The structural comparison of the catalytic domain of EF with those of class III enzymes reveals no structural similarity between two classes (Tesmer et al, 1999; Bieger and Essen, 2001; Drum et al, 2002).
EF has a catalytic rate of approximately 1–2 m/s, substantially faster than those of class III enzymes (Tang et al, 1991; Shen et al, 2002). Based on the structure of the complex of EF-ACD and CaM, it has been proposed that EF uses one catalytic metal to stabilize the transition state and to facilitate the departure of pyrophosphate (PPi). In addition, EF uses a histidine as a catalytic base to deprotonate the 3′OH of ATP (Drum et al, 2002). Such a mechanism differs from that of class III adenylyl cyclase, which uses two-metal–ion catalysis, a prevalent mechanism for the phosphoryl transfer reaction of many DNA and RNA polymerases (Steitz, 1998; Tesmer et al, 1999). A major difference of two-metal–ion catalysis from the proposed mechanism of EF is the presence of an additional metal ion, instead of histidine, to facilitate the deprotonation of the 3′OH. One key feature for enzymes that use two-metal–ion catalysis utilizes two closely spaced aspartates to coordinate the catalytic metal ions. Interestingly, EF also has the same setup, raising the possibility that EF may also use two-metal–ion catalysis.
Here, we describe six crystal structures of EF in complex with CaM. Three of these structures were determined in the presence of a substrate analog (3′-deoxy-ATP (3′dATP)), a reaction product, either cAMP or PPi, while the other three were with three different calcium concentrations. In combination with biochemical analyses, we have analyzed the structural basis for the calcium-independent interaction of EF with the N-CaM and the catalytic mechanism of EF.
Results
Structure of the complex of EF and CaM
The 30 kDa EF PABD is absent from the structures of the EF-ACD alone or the EF-ACD–CaM complex. To obtain the structure of EF PABD and address its interaction with EF-ACD, we optimized the expression and purification of EF to obtain 20–30 mg of >95% pure EF from each liter of Escherichia coli culture (Soelaiman et al, 2003). Crystals of the EF–CaM complex that diffracted to 3.2–3.6 Å were then grown, and structures of the EF–CaM complex were solved (Table I, Supplementary Figure 1 and Table 1S). The EF–CaM complex contains the PABD followed by the EF-ACD, which can be subdivided into the catalytic core (CA and CB) and helical domains, and the extended form of CaM. The model of the EF–CaM complex includes amino acids 64–798 of EF and 3–147 of CaM. No visible electron density can be observed for the N-terminal amino acids 33–63 of EF, likely because the N-terminal end of EF is folded in a way that interfere with the ideal protein packing. Thus, we next generated an EF truncation mutant (EF-Δ64), aiming to improve resolution. However, crystals of EF-Δ64–CaM complex diffracted no better than those of EF–CaM (Table I) and the structure of the EF-Δ64–CaM complex is near identical to that of the EF–CaM complex.
Table 1.
Data collection of EF–CaM complexes
| Crystal name | EF–CaM–Ca2+ (1 μM) | EF–CaM–Ca2+ (1 mM) | EF–CaM–Ca2+ (10 mM) | EF–CaM–3′dATP | EF–CaM–cAMP | EF–Δ64–CaM | EF–CaM–PPia |
| Beamline | APS, 14-BM-C | APS, 19-ID-B | APS, 14-ID-B | APS, 19-ID-B | APS, 19-ID-B | APS, 19-ID-B | APS, 19-ID-B |
| Space group | C2 | C2 | C2 | C2 | C2 | C2 | C2 |
| Unit cell (Å) | |||||||
| a | 315.62 | 183.26 | 318.30 | 184.60 | 320.50 | 317.56 | 317.51 |
| b | 182.04 | 316.47 | 183.76 | 319.28 | 185.04 | 183.11 | 183.35 |
| c | 141.02 | 140.80 | 141.52 | 142.04 | 142.45 | 141.12 | 141.81 |
| β | 89.93 | 89.91 | 90.02 | 90.22 | 90.22 | 90.05 | 90.05 |
| Resolution (Å) | 30-3.25 | 50-3.25 | 30-3.2 | 50-3.35 | 30-3.3 | 50-3.3 | 30-3.6 |
| Unique reflections | 118 338 (11 456)b | 128 284 (12 913)b | 132 318 (12 758)b | 114 975 (11 643)b | 120 342 (12 066)b | 120 891 (11 990)b | 97 720 (9 746)b |
| Completeness (%) | 97.9 (95.2)b | 99.1 (100)b | 98.9 (95.3)b | 98.5 (100)b | 99.1 (100)b | 99.8 (100)b | 99.9 (100)b |
| Redundancyc | 3.4 (2.7)b | 3.8 (3.7)b | 3.7 (2.7)b | 3.8 (3.7)b | 3.8 (3.7)b | 3.8 (3.8)b | 3.8 (3.7)b |
| Rsym(%)d | 5.4 (49.5)b | 7.6 (47.4)b | 3.8 (33.4)b | 8.1 (53.0)b | 9.4 (55.5)b | 5.2 (46.3)b | 6.3 (55.5)b |
| I/σ | 19.2 (2.3)b | 17.3 (2.3)b | 31.0 (2.6)b | 17 (2.6)b | 14 (2.4)b | 26 (2.7)b | 19.4 (2.1)b |
| Refinement | |||||||
| Rcryst (%)e | 26.9 | 25.8 | 26.2 | 26.3 | 26.6 | 28.3 | 28.6 |
| Rfree (%)f | 28.9 | 27.0 | 27.8 | 28.2 | 28.3 | 30.0 | 30.7 |
| R.m.s.bond(Å) | 0.011 | 0.010 | 0.012 | 0.011 | 0.011 | 0.011 | 0.011 |
| R.m.s.angle(deg) | 1.8 | 1.7 | 1.8 | 1.8 | 1.7 | 1.7 | 1.7 |
| Number of | |||||||
| protein atoms | 42 834 | 42 834 | 42 834 | 42 834 | 42 834 | 42 834 | 42 834 |
| ligand atoms | 0 | 0 | 0 | 180 | 132 | 0 | 54 |
| solvent molecules | 0 | 0 | 0 | 6 | 6 | 0 | 0 |
| metal atoms | 18 | 24 | 30 | 30 | 24 | 24 | 24 |
| aEF–CaM crystal was soaked with 5 mM Rp-ATPαS. | |||||||
| bThe outer resolution shell. | |||||||
| cNobs/Nunique. | |||||||
| dRsym=∑j ∣〈I〉−Ij∣/∑〈I〉 where Ij is the intensity of the jth reflection and 〈I〉 is the average intensity. | |||||||
| eRcryst=∑hkl∣Fobs−Fcalc∣/∑hklFobs. | |||||||
| fRfree, calculated the same as for Rcryst but on the 5% data excluded from the refinement calculation. | |||||||
Although structures of EF-ACD and CaM in the EF–CaM complex are similar to the previously determined structure of EF-ACD–CaM, there are two major differences. The first is the contact region between N-CaM and the helical domain of EF as well as their adjacent segments (aa 6–81 of N-CaM; aa 672–695, aa 737–745, and aa 784–793 of EF) (Supplementary Figure 2A). This reveals how the helical domain of EF locks the N-CaM in the closed conformation regardless of the calcium-loading states of CaM (see later Discussion). The second difference involves a 12° rigid body rotation of CB relative to CA (Supplementary Figure 2B). This substantially alters the catalytic pocket formed between these two domains, particularly the interaction of EF with the triphosphate of ATP.
EF PABD can be further divided into two subdomains. The N-terminal domain (blue) consists of an N-terminal of three layers, α/β sandwich domain (four β-sheets, β1–β4, sandwiched by four α-helices, α1–α4) and a C-terminal five-helix domain (purple) (Figure 1A and C). The PABDs of EF and LF share ∼35% sequence identity and similar structural architecture (Figure 1A) (Pannifer et al, 2001). However, the overall structure of these two domains differs significantly (r.m.s.d.=3.85 Å for a total of 224 Cα atoms). The major differences lie in five joining loops, L1–L5, where L5 contributes the key residues to bind PA (Figure 1B; Supplementary Figure 2C) (Lacy et al, 2002).
Figure 1.
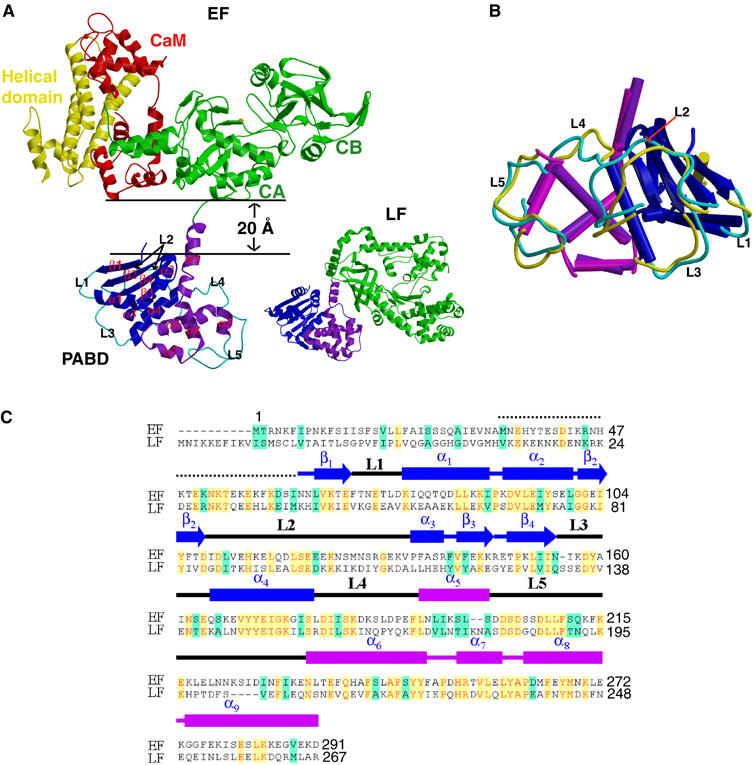
Structure of EF–CaM complex. (A) Ribbon diagram of EF in complex with CaM and that of LF. Catalytic core domain, helical domain, N-terminal PABD, and C-terminal PABD of EF are colored in green, yellow, blue, and purple, respectively, and CaM in red. N-terminal PABD, C-terminal PABD, and protease domain of LF are in blue, purple and green, respectively. (B) Comparison of PABDs of EF and LF. The similar secondary structures of the N-terminal α/β sandwich of PABDs of EF and LF are depicted in blue and dark blue, respectively, and those of the C-terminal five-helix domain of PABD of EF and LF are colored in purple and magenta, respectively. Five loops, L1–L5, which have significant differences between EF-PABD and LF-PABD, are colored in cyan and yellow, respectively. (C) Sequence alignment of PABD of EF and LF. Identical sequences are colored in yellow and similar sequences are in green.
The comparison of EF and LF structures reveals a striking difference in the interaction of the catalytic domains of EF and LF with their PABDs (Figure 1A). The PABD of LF interacts with the protease domain with a buried solvent-accessible surface area of 492 Å2. In contrast, EF PABD is 20 Å away from EF-ACD and does not make contact with CaM or EF-ACD (Figure 1A). We postulate that the detachment of EF-PABD from EF-ACD only occurs after the association with CaM, therefore safeguarding EF from proteolytic degradation prior to its entry into host cells and binding with CaM. A limited trypsin digestion of EF in the absence and presence of CaM showed that the EF–CaM complex is indeed more sensitive to trypsin digestion (Supplementary Figure 3). This suggests that EF PABD makes significant contacts with the EF-ACD in the absence of CaM. The binding of CaM then triggers the detachment of EF PABD from EF-ACD.
Biochemical analyses have shown that one PA heptamer can bind up to three EF and/or LF, and both PABDs of EF and LF have comparably high affinity (∼1 nM) to the PA heptamer (Elliott et al, 2000). The surface-exposed residues in L5, α6, α7, and the joining loop between α7 and α8 at the C-terminal five-helix domain of LF-PABD and EF-PABD have been mapped by mutational analysis to make the primary contacts with PA (Lacy et al, 2002). A comparison of the PA-binding surfaces of EF–PABD with those of LF–PABD reveals that both surfaces are richly negatively charged, which could promote interaction with the positively charged residues at EF/LF interacting surface of PA (Cunningham et al, 2002) (Supplementary Figure 4). However, there are substantial differences in the surface charge distribution and noticeable differences in the relative positions for critical residues that can alter the affinity of EF or LF to PA when mutated (Supplementary Figure 4). This suggests that the interaction of EF and LF with PA may be significantly different. How PA interacts with EF and LF awaits the future structural analysis.
Molecular basis of how EF locks N-CaM in its closed state
Structural and mutational analyses revealed that at least two steps are required for CaM to bind and activate EF (Drum et al, 2002; Ulmer et al, 2003). The calcium-free, closed conformation of N-CaM initiates the contact with the exposed helical domain of EF. This allows calcium-loaded C-CaM in its open conformation to insert itself between the catalytic core and helical domain. The structure of CaM-bound EF-ACD provides a molecular basis illustrating how the calcium-loaded C-CaM makes extensive interactions with both helical domain and the catalytic core for the high-affinity interaction (Drum et al, 2002). However, due to the poor electron density map at these regions, the structure of CaM-bound EF-ACD fails to offer the molecular detail needed to determine how the helical domain of EF interacts with the N-CaM, and why such interactions drastically reduce the affinity of N-CaM for calcium (Drum et al, 2002). Fortunately, the electron density map of the N-CaM and helical domain of EF are greatly improved in the structure of the EF–CaM complex, enabling us to address the interactions between the helical domain of EF and N-CaM. We have determined structures of EF–CaM complex at three different calcium concentrations (1 μM, 1 mM, and 10 mM), and find that the conformations of the N-CaM are similar to each other and adopt the closed conformation (Figure 2A). Helices I and II of N-CaM form hydrogen bonds and a salt bridge with helices L and M of the helical domain of EF, with a buried solvent-accessible surface area of 433 Å2 (Figure 2B and C).
Figure 2.
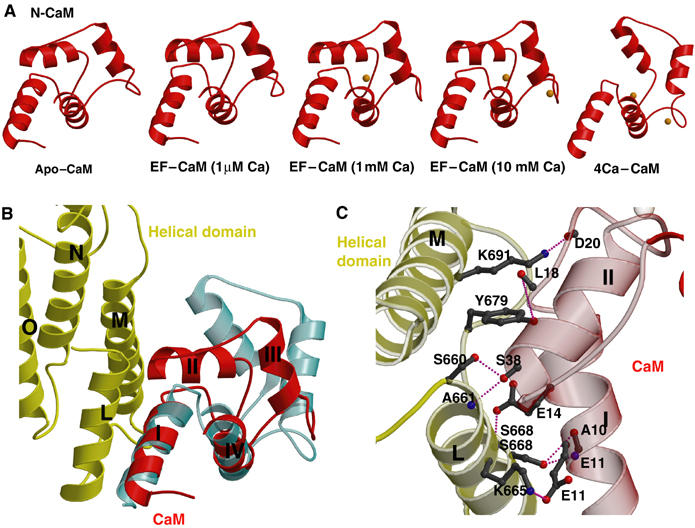
Structures of N-CaM and its interaction with EF. (A) Structures of N-CaM (red) in EF–CaM complex at 1 μM calcium, 1 mM calcium, 10 mM calcium concentrations in comparison with the calcium-free N-CaM structure (left, PDB code: 1CFD) and the crystal structure of four calcium-loaded CaM (right, PDB code: 1CLL). Calcium ions are colored in orange. (B) The interaction between N-CaM and the helical domain of EF. The helical domains of EF and N-CaM of the EF–CaM complex at 10 mM calcium concentration are colored in yellow and red, respectively. For comparison, four calcium-loaded CaM is shown in cyan. (C) Detailed hydrogen bonding and salt bridge formed at the interface between helices I and II of N-CaM and helices L and M of EF.
N-CaM has two calcium-binding sites (sites 1 and 2) and the loading of calcium in these sites can trigger the conformational change of N-CaM from a closed state to an open state. The interaction with the helical domain of EF might lock N-CaM in the closed state by precluding the calcium binding. Alternatively, such an interaction could prevent the conformational switch of N-CaM. At estimated 1 μM calcium concentration, no electron density corresponding to a calcium ion was observed in either site 1 or 2. Interestingly, a calcium ion can be seen in calcium-binding site 1 at 1 mM calcium concentration, whereas both calcium-binding sites 1 and 2 are loaded with calcium when the calcium concentration is raised to 10 mM (Figures 2A and 3). This is consistent with the fact that both calcium-binding sites are still accessible to solvent, and thus are capable of binding calcium ions. However, the interaction of the helical domain of EF with helices I and II of N-CaM restricts the movement of helix III and IV of CaM, abolishing the cooperative movement of these two calcium-binding helix–turn–helix motifs of N-CaM.
Figure 3.
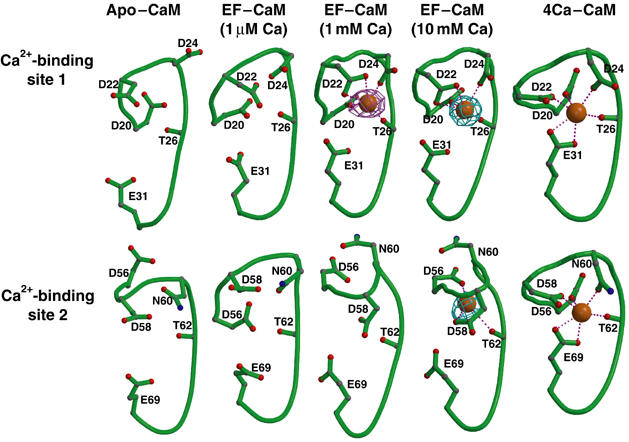
Conformations of calcium-binding sites 1 and 2 of N-CaM. Calcium-binding sites 1 and 2 of N-CaM in the calcium-free NMR average solution structure, structures of EF–CaM in the presence of 1 μM, 1 mM, and 10 mM CaCl2, and four calcium-loaded CaM. Oxygen, carbon, nitrogen, backbone, and calcium are in red, gray, blue, green, and orange. The simulated annealing omit map of calcium ion (cyan and pink) are contoured at the 5.0σ level.
Although our EF–CaM structures have a relatively low resolution (3.2–3.3 Å), they reveal the likely molecular basis of how the binding of EF could significantly decrease the binding affinity of N-CaM to calcium (Ulmer et al, 2003). In calcium-binding sites 1 and 2 of N-CaM, the calcium ion is coordinated by the side chains of five critical conserved residues (Figure 3). By comparing the side chains that are involved in calcium binding at calcium-binding site 1 in the presence and absence of EF, the most apparent difference is that glutamate 31 is too far to effectively coordinate the calcium ion (Figure 3). This leaves four side chains (Asp 20, Asp 22, Asp 24, and Thr 26) to coordinate the calcium ion at calcium-binding site 1. Similar analysis of calcium-binding site 2 reveals the loss of both asparagine 60 and glutamate 69 to coordinate calcium, leaving only three side chains (Asp 56, Asp 58, and Thr 62) to coordinate a calcium ion (Figure 3).
Two metal ions are coordinated by the catalytic site of EF and ATP
The structure of EF-ACD in complex with CaM and the noncyclizable ATP analog, 3′dATP, was determined in the presence of a lanthanide ion (ytterbium) which serves as a vital additive to facilitate crystal formation (Drum et al, 2001). This reveals that a ytterbium ion occupies the catalytic site of EF to coordinate both α and β phosphates of 3′dATP (Figure 4), which has led to the hypothesis that only one catalytic metal is used for the catalysis of EF. However, the lanthanide ion is capable of altering the metal-binding state of E. coli DNA polymerase from two metal ions to one, raising doubt as to the validity of the proposed one-metal–ion catalysis for EF (Brautigam et al, 1999). Crystals of the EF–CaM complex were grown in the presence of the physiological catalytic metal ion, magnesium (200 mM). Notably, these crystals are highly active, based on the fact that the EF–CaM crystal can effectively convert ATP to cAMP (Supplementary Figure 5A). In addition, only a reaction product, PPi, can be observed at the catalytic site of the EF–CaM complex when they are soaked with Rp-ATPαS, a substrate at least 10 000 less reactive than ATP (Table I and Supplementary Figure 5B) (Guo et al, 2004). Thus, structures of EF–CaM in complex with nucleotides allow us to address how magnesium ion and substrate analog bind to the catalytic site of EF.
Figure 4.
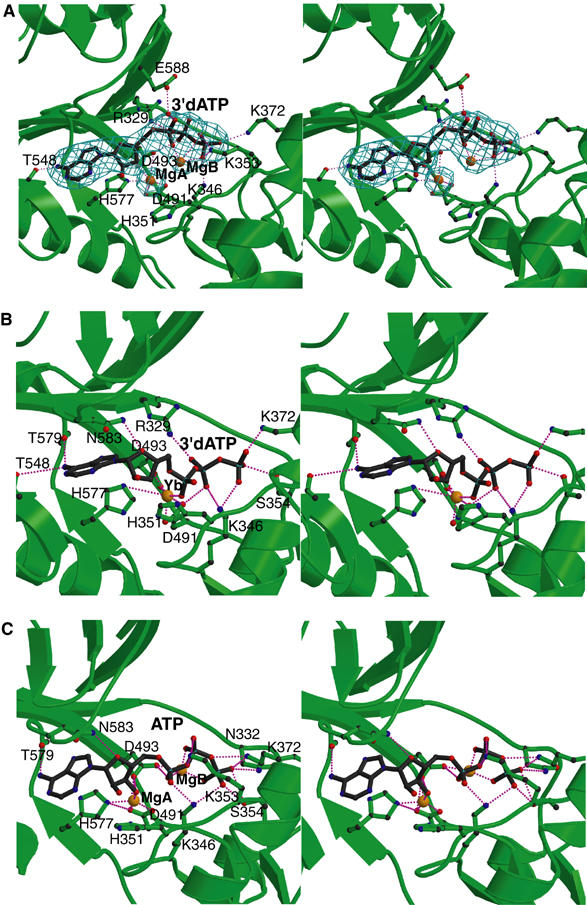
The active site of EF. The comparison of active sites of EF–CaM in complex with 3′dATP (A) and the complex of EF-ACD and CaM with 3′dATP (B). The simulated annealing omit map was contoured at 3.0σ level. Oxygen, nitrogen, carbon, and metal atoms are in red, blue, black, and orange, respectively. Secondary structures of EF and ligands are in green and black, respectively. (C) The catalytic site of the simulated model based on the EF–CaM–3′dATP structure. ATP conformation and coordination of Mg2+ ions obtained from a representative snapshot along the MD trajectory of the EF–CaM–ATP complex in aqueous solution. Hydrogen atoms and water molecules are not shown.
In the structure of EF–CaM–3′dATP complex, 3′dATP is clearly visible at the active site of EF based on a simulated annealing omit map contoured at 3.0σ level (Figure 4A). In comparison with the structure of EF-ACD in complex with CaM and 3′dATP, the positions of adenosine and the corresponding interacting residues of EF do not change significantly (Figure 4A and B). However, the positions of α, β, and γ phosphates in the structure of the EF–CaM complex are moved toward switch B (aa 582–590), a catalytic loop that is stabilized by the binding of CaM. Several residues that interact with the triphosphate of ATP, including K346, K353, and K372, are also moved upward, in part due to the rigid-body rotation between CA and CB. A magnesium ion coordinated by the aspartate pairs, D491 and D493, and a histidine, H577, are clearly visible in the EF–CaM structure, which is similar to the position of the ytterbium ion in the structure of EF-ACD in complex with CaM and 3′dATP (Figure 4A and B). An additional magnesium ion coordinated by the nonesterified oxygens of α, β, and γ phosphates is also visible. These two magnesium ions are about 4.5 Å away from each other.
To determine which of the two ATP-binding states in the catalytic site of EF is more likely the starting point for the enzymatic reaction, we modeled the 3′OH group into the substrates present in crystal structures of EF–CaM and EF-ACD–CaM, and calculated the structure and stability of the resulting complexes in an aqueous solution. Separate 2 ns molecular dynamics (MD) trajectories were initiated from all six noncrystallographic symmetric EF–CaM molecules and all three EF-ACD–CaM molecules. Simulation of each EF–CaM molecule yielded stable two-metal active site configurations, with the distance between the metal atoms fluctuating in the 4.5–5.3 Å range. The calculated free energies of ATP binding in the active sites of the EF–CaM and EF-ACD–CaM were −5.7 and −6.0 kcal/mol, respectively, representing the calculated Kd values of 60 and 40 μM for ATP binding (Supplementary Table 2S). This is in agreement with the experimental Kd value (130 μM) for the binding of 3′dATP to the complex of EF-ACD and CaM (Labruyère et al, 1990). These energies indicate that neither the single- nor two-metal structure has a clear advantage for substrate-binding energetics.
Further analysis of the active site geometries showed that the trajectories of EF–CaM complex are more likely to sample the configuration space in which the nucleophilic oxygen is in the immediate vicinity of the Pα atom. More specifically, if we define the pre-attack configurations as geometries having the distance between O3′ (nucleophile) and Pα atoms of ATP shorter than 3.5 Å, we can see that these configurations are present in about 10% of the sampled EF–CaM configurations (Figure 4C and Supplementary movie), but in no configurations from the trajectories of EF-ACD–CaM. These pre-attack configurations are exclusively associated with the 3′-endo conformation of the ribose and with the direct coordination of the O3′ atom by the catalytic metal (Figure 4C). This geometry appears to be ideal for the initiation of nucleophilic attack of the 3′-oxygen on the α-phosphorus.
The role of histidine 351 in the adenylyl cyclase reaction of EF
The structure of the complex of EF-ACD and CaM suggests that histidine 351 could serve as a catalytic base to deprotonate the 3′OH of ATP (Drum et al, 2002). However, the structure of EF–CaM reveals that histidine 351 is about 6 Å away from the putative 3′OH group of 3′dATP. Such a large distance makes it difficult for histidine 351 to act as a general base that accepts a proton from the 3′-OH group in the initial step of the cyclization reaction. Therefore, we have further investigated the role of histidine 351 in the EF catalysis by steady-state kinetic analysis and computer simulations of the EF–ATP complex in aqueous solution.
We made EF-H351K, EF-H351A, and EF-H351R mutants, where histidine 351 was substituted with lysine, alanine, and arginine, respectively. All three mutants could be purified effectively and had the profiles of circular dichroism similar to wild-type EF, suggesting that no global change in protein structure was caused by these mutations (Supplementary Figure 6A and B). We found that the substitution of histidine 351 by alanine or arginine caused about a 200-fold reduction in the catalytic activity without altering the EC50 values for CaM, confirming the critical role of histidine 351 (Figure 5A). However, histidine 351 could be effectively substituted by lysine (Figure 5A). Most importantly, the mutation of histidine 351 by lysine did not alter the pH optimum of EF (Figure 5B). These data are consistent with the hypothesis that histidine 351 does not serve as a catalytic base.
Figure 5.
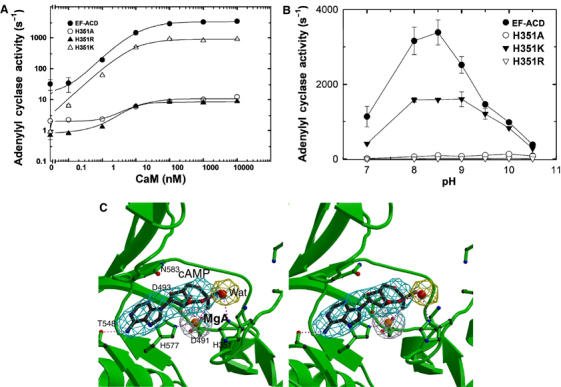
The catalytic mechanism of EF. (A) The adenylyl cyclase activity of wild-type EF-ACD and its mutants, H351A, H351K, and H351R (0.8 nM each) in response to the activation of CaM. The assay was performed at pH 7.2 in the presence of 10 mM MgCl2, 1 mM EDTA, 1 μM free CaCl2, 10 μM CaM, and 10 mM ATP. (B) The adenylyl cyclase activity of wild-type EF-ACD and its mutants H351A, H351K, and H351R in response to the pH changes. Adenylyl cyclase activity was measured in the presence of 10 mM ATP, 10 μM CaM, 1.2 μM free CaCl2, 500 μM BAPTA, and 10 mM MgCl2. Mean±s.e. are representative of at least two experiments. (C) The active site of EF in complex with CaM and cAMP. The simulated annealing omit map was contoured at the 3.0σ level. Oxygen, nitrogen, carbon, and metal atoms are in red, blue, black, and orange, respectively. Secondary structures of EF and ligands are in green and black, respectively.
The catalytic rate of EF to convert ATP to cAMP and PPi (1–2 m/s) is approximately the same as that to convert cAMP and PPi back to ATP (Guo et al, 2004). Thus, we soaked the crystal of the EF–CaM complex with cAMP and PPi (5 mM each) to address how the catalytic site of EF binds its reaction products, cAMP and PPi, for the reverse reaction. From the simulated annealing omit map contoured at the 3.0σ level, cAMP and a metal ion, presumably magnesium, are clearly visible (Figure 5C), but PPi is not. The binding of cAMP and a metal ion to the catalytic site of EF is similar to that revealed from the structure of EF–ACD in complex with CaM (Supplementary Figure 7). Interestingly, the 3′O atom of cAMP is about 6.0 Å away from histidine 351, and it is clear that a water molecule lies between them in the structure of EF–CaM complex (Figure 5C).
The static view from the crystal structures discussed above is consistent with the motions of histidine 351 observed in our MD simulations (Supplementary movie). The calculated trajectory of the imidazole moiety of histidine 351 shows fast and large amplitude motions of this moiety in the area on the protein–solvent boundary. The imidazole ring is found with nearly equal probability between 4 Å and 8 Å from the ribose, and in many configurations between these extremes. No direct contacts between histidine 351 and ribose lead to the formation of the O3′-H…N hydrogen bond that could facilitate the direct abstraction of the proton from the nucleophile. These results, together with our mutational and crystallographic results, suggest that histidine 351 does not serve as an acceptor of 3′OH proton (general base), but rather it facilitates an increase in the concentration of OH− ions in the vicinity of the 3′OH group.
The role of the residues in CyaA and ExoY corresponding to EF H351
Adenylyl cyclase toxins from five species of pathogenic bacteria have similar ∼37 kDa adenylyl cyclase core domain (Figure 6A). Outside the adenylyl cyclase core domain, they are quite diverse. Sequence comparison of adenylyl cyclase core domains of these toxins reveals three conserved segments (Figure 6A and B). Segment A contains conserved residues that interact with the triphosphate of ATP (R329, K346, K353, and K372 of EF). Segment B has two conserved aspartates (D491 and D493 of EF) that bind the catalytic metal ions. Segment C consists of a catalytic loop (aa 579–590 of EF) that is stabilized upon CaM binding. This loop has a conserved asparagine (N583 of EF) to hold ribose in place. Segment C also contains a conserved histidine (H577 of EF) to coordinate a catalytic metal ion.
Figure 6.
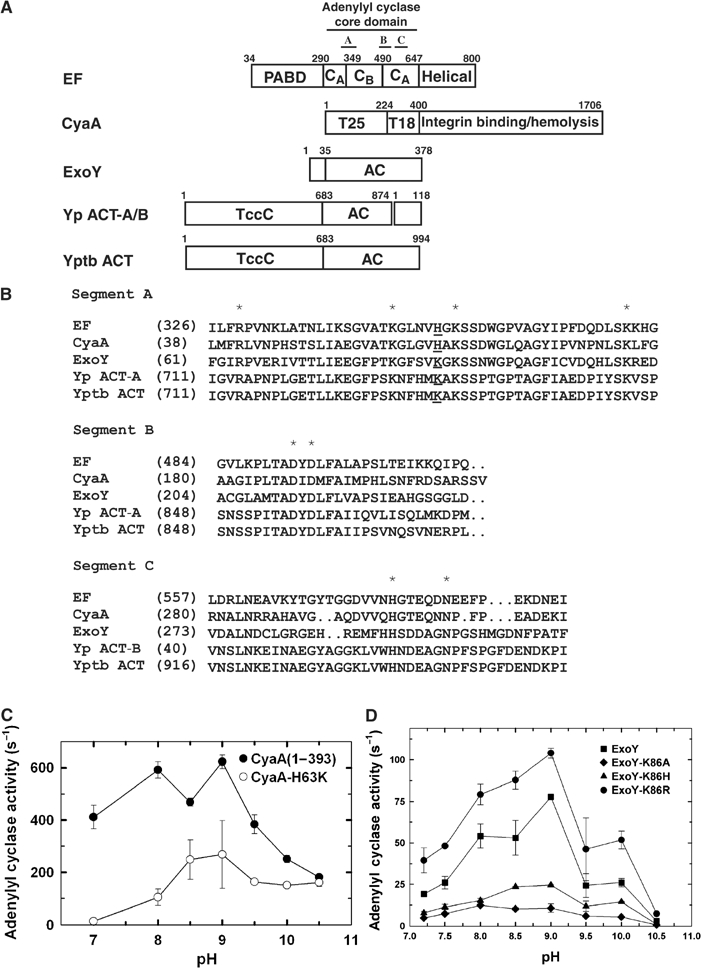
The comparison of class II adenylyl cyclase toxins. (A) Schematic diagram of domain organization of five adenylyl cyclase toxins. The adenylyl cyclase core domains and the conserved segments, A, B, and C are indicated. Yp ACT-A/B and Yptb ACT refer to the adenylyl cyclase toxin from Yersinia pestis and Yersinia pseudotuberculosis, respectively. Accession numbers for EF, CyaA, ExoY, Yp ACT-A, Yp ACT-B, and Yptb ACT are P40126, P15318, AAC78299, NP993432, NP993433, and YP070748, respectively. Yp ACT consists of two polypeptide chains due to the frameshift mutation. Yp ACT-B has sequence homology with bacterial DNA gyrase and oligopeptide ATP transporter. The C-terminal 145 kDa domain of CyaA can bind αMβ2 integrin and has hemolytic activity (El-Azami-El-Idrissi et al, 2003). The N-terminal domains of Yptb and Yp ACT are homologous to the C locus of insecticidal toxin complex c (TccC) (Bowen et al, 1998). (B) Sequence alignment of three conserved segments of class II adenylyl cyclases. The highly conserved residues crucial for catalysis are highlighted by asterisk. EF H351 and the corresponding residues are underlined. The adenylyl cyclase activity of CyaA (C), ExoY (D) and their mutants in response to the pH changes. Adenylyl cyclase activity was measured in the presence of 10 mM ATP, 1.2 μM free CaCl2, 500 μM BAPTA, 10 mM MgCl2, 1 μM CaM (for CyaA) or 10 μg spleen lysate (for ExoY).
The corresponding residue of histidine 351 of EF is not conserved among adenylyl cyclase toxins (Figure 6B). While this residue in CyaA is histidine (H63), the corresponding residue in the remaining three adenylyl cyclase toxins is lysine. To examine the role of histidine 63 in CyaA, we constructed CyaA mutants with this residue mutated to lysine, arginine, and alanine. Similar to EF, the mutation of histidine 63 to lysine (H63K) caused a marginal reduction in adenylyl cyclase activity, with no obvious alteration in the pH optimum of CyaA (Figure 6C). The mutation of histidine 63 of CyaA-ACD to arginine caused 6–10-fold reduced adenylyl cyclase activity and the pH optimum shifted slightly toward alkali pH (Supplementary Figure 9), which is consistent with the previous report (Munier et al, 1992). Furthermore, the mutation of histidine 63 to alanine drastically reduced adenylyl cyclase activity (>100 fold). Surprisingly, CyaA-ACD H63A mutant also had the shift in its pH optimum toward alkali pH (Supplementary Figure 9). At present, the mechanism for the shift in the pH optimum of CyaA H63R and H63A remains elusive.
The residue corresponding to EF H351 and CyaA H63 is lysine 86 in ExoY, which we subsequently mutated to alanine, arginine, and histidine. The circular dichroism spectra showed that these ExoY mutants had a profile similar to the wild-type enzyme, suggesting no obvious structural alteration induced by the mutation (Supplementary Figure 10). Interestingly, the mutation of lysine 86 to arginine slightly increased the catalytic activity of ExoY, while that to histidine or alanine caused significant reduction in activity (Figure 6D). No significant shift in their pH optimum was observed in all three ExoY mutants.
Discussion
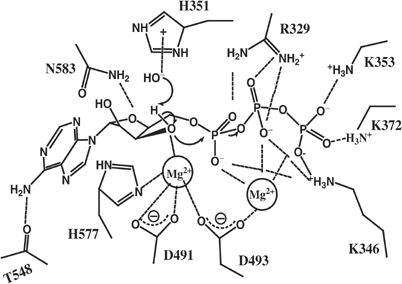
Our structural, mutational, and computational analyses form the basis for our current model for the catalytic mechanism of EF (Figure 7). Similar to the cyclization reaction of mammalian adenylyl cyclase, or the nucleotidyl transfer reaction of DNA and RNA polymerase, the catalytic reaction of EF is mediated by the nucleophilic attack of the 3′ oxygen atom on the α-phosphate (Steitz, 1998; Tesmer et al, 1999; Guo et al, 2004). To promote this reaction, EF needs to bind ATP, facilitate the deprotonation of 3′OH, stabilize pentacoordinated phosphorane intermediate, and effectively release cAMP and PPi. We propose that EF uses asparagine 583 to restrain the rotational freedom of ribose, thus holding 3′OH in place for its nucleophilic attack during the catalysis (Figure 7). The protonated histidine 351 is used to stabilize the OH− ion near the 3′OH group. A variant of this mechanism is that EF uses neutral histidine 351 to deprotonate a neutral water molecule. This OH− in turn facilitates the deprotonation of 3′OH of ATP.
Figure 7.
The proposed two-metal–ion catalytic mechanism of EF.
A suitable catalytic metal ion, MgA, near the 3′OH of ATP is coordinated by two aspartates (491 and 493) and a histidine (577). This metal can also facilitate the deprotonation of 3′OH by stabilizing the negative charge on the resulting 3′-oxyanion. The action of MgA ion and histidine 351 can be additive. Using the energy term, the free energy cost associated with the proton transfer of 3′OH is proportional to the difference between pKa of the 3′OH group and local pH of the solution, ΔG∼pKa−pH. The catalytic role of histidine 351 is to increase local pH, whereas the role of the catalytic metal is to decrease the pKa component to make ΔG small or even negative. In addition, this metal ion, MgA, may also stabilize the reaction intermediate by moving towards the nonbridging oxygen of α-phosphate during the nucleophilic attack step of the catalytic reaction. A second metal ion, MgB, is found to be coordinated by D493 and nonbridging oxygens of all three phosphates of ATP. This metal ion can facilitate the bond breakage between α- and β-phosphates by stabilizing the developing negative charges. Several positive residues (R329, K353, K372 and K346) are also involved in stabilizing the reaction intermediate and the departure of PPi.
Based on our structural and biochemical data, we propose that EF uses a two-metal-ion catalysis. This mechanism is also employed by many structurally diverse enzymes, including mammalian adenylyl cyclase, DNA and RNA polymerase, alkaline phosphatase, as well as ribozymes such as self-splicing introns (Steitz, 1998; Horton and Perona, 2001; Fedor, 2002). Interestingly, pol I and pol β in DNA polymerase families have quite different structural architectures, but they share the same two-metal–ion catalytic mechanism (Pelletier et al, 1996; Kiefer et al, 1997; Brautigam and Steitz, 1998). Such catalytic mechanism-driven convergent evolution has also been seen in many other families of enzymes such as phosphotyrosine protein phosphatase, polysaccharide lyase, and cytosine deaminase (Barford et al, 1998; Charnock et al, 2002; Ko et al, 2003).
Based on the sequence comparison, the residues that are involved in the metal binding and catalysis of EF are also conserved in the other four members of class II adenylyl cyclase (Figure 6B). Thus, it is tempting to hypothesize that all adenylyl cyclase toxins use two-metal–ion catalytic mechanism. One major difference among these toxins is the role of H351 of EF and its corresponding residues. Our structural data and computer simulations suggest that H351 serves to activate the surrounding water molecule, which, in turn, facilitates the deprotonation of 3′OH. Depending upon their distance to 3′OH, we envision that the role of corresponding residues in other adenylyl cyclase toxins may vary. They serve as the catalytic base directly or, similar to EF, indirectly facilitate the deprotonation of 3′OH. The answers to these hypotheses await the future structural analysis of these toxins in complex with substrate analogs.
Our structures provide the first molecular basis of how CaM effectors can uncouple the calcium binding of CaM from its conformational change. CaM acts both as a calcium sensor and a signal transducer (Eldik and Watterson, 1998). To do so, CaM tightly couples its calcium loading with its role as a conformational switch, so that the calcium-loaded CaM undergoes the transformation from having mostly hydrophilic surface (closed state) to make hydrophobic surfaces accessible (open state). However, binding of CaM to its target proteins can profoundly alter the calcium binding as well as the coordinated conformational changes of CaM (Jurado et al, 1999; Schumacher et al, 2001; Ulmer et al, 2003). Our structures show that the helical domain of EF interacts with the hydrophilic surface of the first calcium-binding, helix–loop–helix motif of CaM to effectively reduce the calcium-binding affinity of this loop. This interaction also locks CaM in the closed conformation, which, in turn, drastically decreases the affinity of the second calcium-binding, helix–loop–helix of CaM to calcium. Furthermore, calcium binding to N-CaM cannot unlock N-CaM from its closed state. The interaction of N-CaM with EF differs significantly from the binding of C-CaM to the CaM-binding domain of the calcium-activated potassium channel, which locks C-CaM into a calcium-free, partially open state, and completely eliminates the ability of C-CaM to bind calcium ions (Schumacher et al, 2004). As more structures of CaM complexed with its targets are solved, we expect to observe even greater structural diversity in how CaM targets can bind and affect the calcium-binding ability and conformation of CaM.
Materials and methods
Expression and purification of EF, CyaA-ACD, and ExoY
Mutations of EF, EF-ACD, CyaA-ACD, and ExoY were generated using the QuickChange mutagenesis kit (Biocrest Manufacturing L P). The expression and purification of EF-ACD, EF mutants, CyaA-ACD, CaM, and selenomethionyl CaM were performed as described (Shen et al, 2002; Soelaiman et al, 2003). The expression and purification of ExoY was performed as described (Yahr et al, 1998) and ExoY was then further purified with an additional Q-sepharose and gel filtration columns to achieve greater than 95% pure protein.
Crystallization of EF–CaM
The complex of EF with CaM and calcium was formed by mixing proteins in a 2:3:6 molar ratios and isolated by a superdex-200 column. Crystals of EF–CaM were grown by the hanging drop method. EF–CaM (1 μl of 30 mg/ml protein in 20 mM Tris–HCl (pH 8.6) and 20 mM NaCl) was mixed with 1 μl of well solution (100 mM Tris–HCl (pH 8.5), 28% PEG400, and 200 mM MgCl2). Crystals were formed within 1-week incubation at 4°C and were frozen after 5 min equilibration in the cryoprotectant solution (100 mM Tris–HCl (pH 8.5), 31% PEG400, 200 mM MgCl2). EF–CaM crystals were soaked with 5 mM cAMP plus 5 mM pyrophosphates, 5 mM 3′dATP, and 5 mM Rp-ATPαS in cryoprotectant for 2 days at 4°C for the structure determination of EF–CaM-cAMP, EF–CaM-3′dATP, and EF–CaM-PPi, respectively. A similar procedure was applied for the structure determination of EF–CaM with 1 μM, 1 and 10 mM CaCl2. Crystals of EF-Δ64-CaM complex were grown with slightly different concentrations of precipitant and additive (20% PEG400, 220 mM MgCl2). Data were collected at APS Biocars 14-BM-C, 14-ID-B, and SBC 19-ID, and processed with HKL2000 program (Otwinowski and Minor, 1997).
Structure determination
The initial phases of EF–CaM with 1 μM CaCl2 were obtained by molecular replacement with the structure of EF-ACD from the complex of EF-ACD–CaM as a search model using AMoRe (Navaza, 2001), resulting in six EF molecules in one asymmetric unit of C2 space group. The molecular replacement solution was refined by the rigid-body and positional refinements with noncrystallographic symmetry (NCS) restraints using CNS program (Brunger et al, 1998). The phases were further improved based on the location of 48 selenomethionines in CaM using the data of EF in complex with Se-Met-CaM and programs of CNS and Shake-and-Bake (Weeks and Miller, 1999). CaM molecules were then modeled into the complex. The sigma-A weighted Fourier maps (2Fo−Fc and Fo−Fc) and Program O were then used to build the model of EF PABD (Jones et al, 1991), followed by annealing refinement with the NCS restraints using CNS program. Iterative cycles of positional refinement and model building were carried out until free R factor was converged. Structures of EF–CaM–cAMP, EF–CaM with 10 mM Ca2+, EF–CaM–PPi and EF–Δ64–CaM were determined by Sigma-A weighted 2Fo−Fc and Fo−Fc maps using the template of EF–CaM complex with 1 μM Ca2+. Structures of EF–CaM-3′dATP and EF–CaM with 1 mM Ca2+ were obtained by molecular replacement using EF–CaM complex with 1 μM Ca2+ as a search model. The simulated annealing omit maps were calculated to reveal additional density to model cAMP, 3′dATP, PPi and calcium ions. All six structures were refined following the similar strategy for the structure refinement of EF–CaM with 1 μM Ca2+ as described above. Final PDB files have been deposited into the PDB bank (PDB code: 1XFY, 1XFZ, 1XFX, 1XFV, 1XFW, 1XFU, and 1Y0V for EF–CaM with 1 μM Ca2+, EF–CaM with 1 mM Ca2+, EF–CaM with 10 mM Ca2+, EF–CaM–3′dATP, EF–CaM–cAMP, EF–Δ64–CaM, and EF–CaM–PPi, respectively).
In vitro adenylyl cyclase assay
Adenylyl cyclase activities were measured after 10 min at 30°C in the presence of 20 mM Hepes (pH 7.2), 5 mM ATP with trace amount of 32P-ATP, 1 mM EDTA, 0.1 M KCl, 10 mM MgCl2, and ATP and cAMP were separated as described (Drum et al, 2002). In pH-titration experiment, 500 μM BAPTA instead of EDTA was used to buffer the free calcium to avoid the pH-dependent change in the buffering capacity of the calcium chelator. Following isolation, mouse spleens were homogenized with a motor-driven Teflon homogenizer and centrifuged at 150 000 g for 30 min. The high-speed supernatant of mouse spleen homogenate was used to activate ExoY.
Computer simulations
Starting structures for classical MD simulations were generated from the crystal structure of EF–CaM (PDB code: 1XFV) and from the structure of EF-ACD-CaM (PDB code: 1K90) (Drum et al, 2002) after adding 3′O atom to the substrate. All crystallographic water molecules and CaM were removed. The selection of the charged residues was consistent with Glu, Asp, Lys, Arg, and phosphate groups having charges based on their pKa constants in water (see Table 2S in Supplementary data). All residues farther than 18 Å from the Pα atom of ATP were kept in their electroneutral forms. New water molecules were added by immersing the simulation sphere into the sphere of bulk water molecules. Those water molecules that were not sterically overlapping with the atoms present in the crystal structure were retained in the starting structure for the MD simulations.
MD trajectories were calculated in 23 Å simulation sphere centered on the Pα atom using the program Q (Marelius et al, 1999). Amber 95 force field (Cornell et al, 1995) was used for standard protein and DNA residues. The force field parameters used for ATP and Mg2+ ions have been reported previously (Florián et al, 2003). Positions of atoms lying beyond the 23 Å sphere were fixed at their crystallographic positions, and their nonbonded interactions with the atoms within the simulation sphere were turned off. Following a 200 ps equilibration period, MD simulations were carried out at 298 K using the step-size of 2 fs and the ‘shake' algorithm (Ryckaert et al, 1977). The nonbonded interactions were evaluated explicitly for distances shorter than 10 Å. The local-reaction field method was used to include long-range electrostatic interactions for distances beyond a 10 Å cutoff (Lee and Warshel, 1992). The trajectory analysis was carried out using program VMD 1.8.2 (Humphrey et al, 1996). In all, 11 separate 2 ns production trajectories, which differed in the starting geometry and charges of some ionizable residues (see Table 2S in Supplementary data), were analyzed. The binding free energy of ATP was calculated using the linear interaction energy (LIE) approximation (Aqvist et al, 1994). LIE coefficients α=0.56 and β=0, which were found optimal in our previous studies of nucleotide triphosphate binding to DNA polymerases (Florián et al, 2002a, 2002b), were used.
Supplementary Material
Supplementary Figures and Tables
Supplementary Movie Legend
Supplementary Movie (RealPlayer or QuickTime recommended for viewing)
Acknowledgments
We are grateful to Fritz Eckstein for (Rp)-ATPαS, Xiaojing Yang for her computer support and insightful advises, to Pengyun Li and staff of APS SBC 19-ID, APS BioCars 14-BMC, and APS BioCars 14-IDB for their help in data collection, to Sandriyana Soelaiman for her superb technical assistance, to Dara Frank (Medical College of Wisconsin) for the plasmid to express recombinant ExoY, to John Crispino, James Bowen, and Pamela Bergson for critical reading of manuscript. This research was supported by the National Institute of Health GM53459, GM62548, and GLRCE pilot grant to W-J Tang. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Basic Energy Sciences, under contract no. W-31-109-ENG-38.
References
- Aqvist J, Medina C, Samuelson JE (1994) A new model for predicting binding affinity in computer-aided drug design. Protein Eng 7: 385–391 [DOI] [PubMed] [Google Scholar]
- Baker DA, Kelly JM (2004) Structure, function and evolution of microbial adenylyl and guanylyl cyclases. Mol Microbiol 52: 1229–1242 [DOI] [PubMed] [Google Scholar]
- Barford D, Das AK, Egloff MP (1998) The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 27: 133–164 [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Bunick CG, Chazin WJ (2004) Target selectivity in EF-hand calcium binding proteins. Biochim Biophys Acta 1742: 69–79 [DOI] [PubMed] [Google Scholar]
- Bieger B, Essen LO (2001) Structural analysis of adenylate cyclases from Trypanosoma brucei in their monomeric state. EMBO J 20: 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen D, Rocheleau TA, Blackburn M, Andreev O, Golubeva E, Bhartia R, Ffrench-Constant RH (1998) Insecticidal toxins from the bacterium Photorhabdus luminescens. Science 280: 2129–2132 [DOI] [PubMed] [Google Scholar]
- Brautigam CA, Aschheim K, Steitz TA (1999) Structural elucidation of the binding and inhibitory properties of lanthanide (III) ions at the 3′–5′ exonucleolytic active site of the Klenow fragment. Chem Biol 6: 901–908 [DOI] [PubMed] [Google Scholar]
- Brautigam CA, Steitz TA (1998) Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr Opin Struct Biol 8: 54–63 [DOI] [PubMed] [Google Scholar]
- Brossier F, Weber-Levy M, Mock M, Sirard JC (2000) Role of toxin functional domains in anthrax pathogenesis. Infect Immun 68: 1781–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Charnock SJ, Brown IE, Turkenburg JP, Black GW, Davies GJ (2002) Convergent evolution sheds light on the anti-beta-elimination mechanism common to family 1 and 10 polysaccharide lyases. Proc Natl Acad Sci USA 99: 12067–12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier RJ, Young JA (2003) Anthrax Toxin. Annu Rev Cell Dev Biol 19: 45–70 [DOI] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz JKM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Cunningham K, Lacy DB, Mogridge J, Collier RJ (2002) Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci USA 99: 7049–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drum CL, Shen Y, Rice PA, Bohm A, Tang WJ (2001) Crystallization and preliminary X-ray study of the edema factor exotoxin adenylyl cyclase domain from Bacillus anthracis in the presence of its activator, calmodulin. Acta Crystallogr D 57: 1881–1884 [DOI] [PubMed] [Google Scholar]
- Drum CL, Yan SZ, Bard J, Shen Y, Lu D, Soelaiman S, Grabarek Z, Bohm A, Tang W-J (2002) Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415: 396–402 [DOI] [PubMed] [Google Scholar]
- El-Azami-El-Idrissi M, Bauche C, Loucka J, Osicka R, Sebo P, Ladant D, Leclerc C (2003) Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: role of toxin acylation and identification of the main integrin interaction domain. J Biol Chem 278: 38514–38521 [DOI] [PubMed] [Google Scholar]
- Eldik LV, Watterson DM (1998) Calmodulin and Signal Transduction. New York: Academic Press [Google Scholar]
- Elliott JL, Morgridge J, Collier RJ (2000) A quantitative study of the interactions of Bacillus anthracis edema factor and lethal factor with activated protective antigen. Biochemistry 39: 6706–6713 [DOI] [PubMed] [Google Scholar]
- Fallon JL, Quiocho FA (2003) A closed compact structure of native Ca2+-calmodulin. Structure (Camb) 11: 1303–1307 [DOI] [PubMed] [Google Scholar]
- Fedor MJ (2002) The role of metal ions in RNA catalysis. Curr Opin Struct Biol 12: 289–295 [DOI] [PubMed] [Google Scholar]
- Florián J, Goodman MF, Warshel A (2002a) Theoretical investigation of the binding free energies and key substrate-recognition components of the replication fidelity of human DNA polymerase β. J Phys Chem B106: 5739–5753 [Google Scholar]
- Florián J, Goodman MF, Warshel A (2003) Computer simulation of the chemical catalysis of DNA polymerases: discriminating between alternative nucleotide insertion mechanism for T7 DNA polymerase. J Am Chem Soc 125: 8163–8177 [DOI] [PubMed] [Google Scholar]
- Florián J, Warshel A, Goodman MF (2002b) Molecular dynamics free-energy simulations of the binding contribution to the fidelity of T7 DNA polymerase. J Phys Chem B106: 5754–5760 [Google Scholar]
- Guo Q, Shen Y, Zhukovskaya NL, Florian J, Tang W-J (2004) Structural and kinetic analyses of the interaction of anthrax adenylyl cyclase toxin with reaction products, cAMP and pyrophosphate. J Biol Chem 279: 29427–29435 [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Ikura M (2002) Calmodulin in action: diversity in target recognition and activation mechanisms. Cell 108: 739–742 [DOI] [PubMed] [Google Scholar]
- Horton NC, Perona JJ (2001) Making the most of metal ions. Nat Struct Biol 8: 290–293 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD – Visual Molecular Dynamics. J Mol Graphics 14: 33–38 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Jurado LA, Chockalingam PS, Jarrett HW (1999) Apocalmodulin. Physiol Rev 79: 661–682 [DOI] [PubMed] [Google Scholar]
- Kiefer JR, Mao C, Hansen CJ, Basehore SL, Hogrefe HH, Braman JC, Beese LS (1997) Crystal structure of a thermostable Bacillus DNA polymerase I large fragment at 2.1 Å resolution. Structure (Camb) 5: 95–108 [DOI] [PubMed] [Google Scholar]
- Ko TP, Lin JJ, Hu CY, Hsu YH, Wang AH, Liaw SH (2003) Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J Biol Chem 278: 19111–19117 [DOI] [PubMed] [Google Scholar]
- Labruyère E, Mock M, Ladant D, Michelson S, Gilles AM, Laoide B, Barzu O (1990) Characterization of ATP and calmodulin-binding properties of a truncated form of Bacillus anthracis adenylate cyclase. Biochemistry 29: 4922–4928 [DOI] [PubMed] [Google Scholar]
- Lacy DB, Mourez M, Fouassier A, Collier RJ (2002) Mapping the anthrax protective antigen binding site on the lethal and edema factors. J Biol Chem 277: 3006–3010 [DOI] [PubMed] [Google Scholar]
- Ladant D, Ullmann A (1999) Bordatella pertussis adenylate cyclase: a toxin with multiple talents. Trends Microbiol 7: 172–176 [DOI] [PubMed] [Google Scholar]
- Lee FS, Warshel A (1992) A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J Chem Phys 97: 3100–3107 [Google Scholar]
- Linder JU, Schultz JE (2003) The class III adenylyl cyclase: multi-purpose signalling modules. Cell Signal 15: 1081–1089 [DOI] [PubMed] [Google Scholar]
- Marelius J, Kolmodin K, Feierberg I, Aqvist J (1999) Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J Mol Graphics Model 16: 213–225 [DOI] [PubMed] [Google Scholar]
- Munier H, Bouhss A, Krin E, Danchin A, Gilles AM, Glaser P, Barzu O (1992) The role of histidine 63 in the catalytic mechanism of Bordetella pertussis adenylate cyclase. J Biol Chem 267: 9816–9820 [PubMed] [Google Scholar]
- Navaza J (2001) Implementation of molecular replacement in AMoRe. Acta Crystallogr D Biol Crystallogr 57: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pannifer A, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S, Leppla SH, Hanna PC, Liddington RC (2001) Crystal structure of the anthrax lethal factor. Nature 414: 229–233 [DOI] [PubMed] [Google Scholar]
- Pelletier H, Sawaya MR, Wolfle W, Wilson SH, Kraut J (1996) Crystal structure of human DNA polymerase β complexed with DNA: implications for catalytic mechanism, processivity and fidelity. Biochemistry 35: 12742–12761 [DOI] [PubMed] [Google Scholar]
- Rossi Paccani S, Tonello F, Ghittoni R, Natale M, Muraro L, D'elios MM, Tang W-J, Montecucco C, Baldari CT (2005) Anthrax toxins suppress T-lymphocyte activation by disrupting antigen receptor signaling. J Exp Med 201, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23: 327–341 [Google Scholar]
- Schumacher MA, Crum M, Miller CM (2004) Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure (Camb) 12: 849–860 [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Rivard AF, Bachinger HP, Adelman JP (2001) Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410: 1120–1124 [DOI] [PubMed] [Google Scholar]
- Shen YQ, Lee Y-S, Soelaiman S, Bergson P, Lu D, Chen A, Beckingham K, Grabarek Z, Mrksich M, Tang W-J (2002) Physiological calcium concentrations regulate calmodulin binding and catalysis of adenylyl cyclase exotoxins. EMBO J 21: 6721–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soelaiman S, Wei BQ, Bergson P, Lee Y-S, Shen Y-Q, Mrksich M, Shoichet BK, Tang W-J (2003) Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J Biol Chem 278: 25990–25997 [DOI] [PubMed] [Google Scholar]
- Steitz TA (1998) A mechanism for all polymerases. Nature 391: 231–232 [DOI] [PubMed] [Google Scholar]
- Tang WJ, Krupinski J, Gilman AG (1991) Expression and characterization of calmodulin-activated (type I) adenylylcyclase. J Biol Chem 266: 8595–8603 [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, Sprang SR (1999) Two-metal-Ion catalysis in adenylyl cyclase. Science 285: 756–760 [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Soelaiman S, Li S, Klee CB, Tang WJ, Bax A (2003) Calcium dependence of the interaction between calmodulin and anthrax edema factor. J Biol Chem 278: 29261–29266 [DOI] [PubMed] [Google Scholar]
- Weeks CM, Miller R (1999) The design and implementation of SnB version 2.0. J Appl Crystallogr 32: 120–124 [Google Scholar]
- Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW (1998) ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci USA 95: 13899–13904 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables
Supplementary Movie Legend
Supplementary Movie (RealPlayer or QuickTime recommended for viewing)