

SUMMARY
Diarrhea is a host response to enteric pathogens, but its impact on pathogenesis remains poorly defined. By infecting mice with the attaching and effacing bacteria Citrobacter rodentium, we defined the mechanisms and contributions of diarrhea and intestinal barrier loss to host defense. Increased permeability occurred within 2 days of infection and coincided with IL-22-dependent upregulation of the epithelial tight junction protein claudin-2. Permeability increases were limited to small molecules, as expected for the paracellular water and Na+ channel formed by claudin-2. Relative to wildtype, claudin-2-deficient mice experienced severe disease, including increased mucosal colonization by C. rodentium, prolonged pathogen shedding, exaggerated cytokine responses, and greater tissue injury. Conversely, transgenic claudin-2 overexpression reduced disease severity. Chemically-induced osmotic diarrhea reduced colitis severity and C. rodentium burden in claudin-2 deficient, but not transgenic, mice, demonstrating that claudin-2-mediated protection is the result of enhanced water efflux. Thus, IL-22-induced claudin-2 upregulation drives diarrhea and pathogen clearance.
Keywords: Tight junction, bacterial infection, innate defense, diarrhea, permeability, enteric infection, colitis
Graphical Abstract
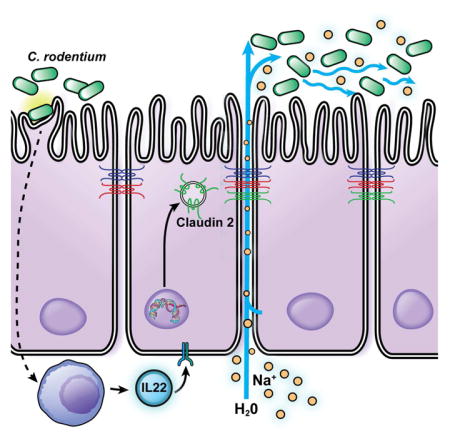
Diarrhea is common in enteric infection, but whether this reflects disease progression or host defense is unknown. Using the C. rodentium model, Tsai et al. show that diarrhea is critical to pathogen clearance and demonstrate that diarrhea development requires claudin-2 upregulation that increases tight junction permeability to Na+ and water.
INTRODUCTION
The intestinal barrier is often compromised during enteric infection. In the case of invasive and toxigenic organisms, this typically involves direct epithelial damage. In contrast, non-invasive, attaching and effacing (A/E) pathogens, such as the human and mouse pathogens enteropathogenic E. coli and Citrobacter rodentium, respectively, use a type III secretion system to inject proteins into host cells (Deng et al., 2010). These effector proteins promote cytoskeletal reorganization and increase intestinal permeability (Guttman et al., 2006). However, neither the mechanisms that underlie these permeability increases nor their significance to pathogenesis are well understood.
Intercellular tight junctions define the epithelial paracellular barrier, but do not form an absolute seal, as paracellular permeability is necessary for ion, water, nutrient, and waste transport. Flux across tight junctions occur by two distinct routes, defined as the pore and leak pathways (Fig. 1A). The pore pathway is a high capacity, charge- and size-selective route with a maximum diameter of 6 A (Anderson and Van Itallie, 2009; Turner, 2009). In contrast, the low conductance leak pathway is relatively nonselective and accommodates molecules with diameters up to ~100 A. Finally, materials can cross the barrier via the unrestricted pathway (Fig. 1A), which allows free passage of ions, water, macromolecules, bacteria, and viruses as a result of epithelial damage. The unrestricted pathway is therefore tight junction-independent.
Figure 1.

Pore pathway permeability and claudin-2 expression are increased early in the course of C. rodentium infection. (A) Pore, leak, and unrestricted pathway permeabilities were assessed using creatinine, 4kD dextran, and 70kD dextran, respectively. (B) Creatinine, 4kD dextran, and 70kD dextran fluxes increased 2, 4, and 6 days after infection, respectively (n=6). (C) Specific pore and leak pathway permeabilities increased shortly after C. rodentium infection but were undetectable by day 6 due to epithelial damage. Creatinine and 4kD dextran flux at these times therefore reflects increased unrestricted pathway, not tight junction, permeability. (D) Quantitative RT-PCR analysis shows that only claudin-2 mRNA is significantly increased at day 2 of infection. (E) Western blots and densitometry of isolated colonic epithelial cells demonstrate increased claudin-2 (CLDN2), but not claudin-15 (CLDN15) or E-cadherin (Ecad) expression. (F) Immunofluorescence microscopy shows claudin-2 (red), F-actin (green), and DNA (blue) during infection. Brackets indicate zone of claudin-2 expression. Bar = 50 μm.
Barrier loss in disease is frequently attributed to tight junction dysregulation, but the tools used in most in vivo analyses cannot discriminate between increased tight junction permeability and epithelial damage. As a result, the relative contributions of the pore, leak, and unrestricted pathways to intestinal barrier loss, disease progression, and host responses during enteric infection have not been defined. We developed an in vivo assay that allowed independent analysis of each permeability pathway (Fig. 1A) during C. rodentium colitis. This made it possible to identify an early increase in pore pathway permeability resulting from IL-22-dependent claudin-2 upregulation. Genetic and pharmacological interventions showed that this claudin-2-mediated increase in pore pathway permeability increased water efflux and enhanced C. rodentium clearance. These results identify an unrecognized mechanism of IL-22-dependent host defense and elucidate the essential roles of tight junction regulation and diarrhea in enteric pathogen clearance.
RESULTS
C. rodentium infection disrupts the intestinal barrier in three distinct phases
C. rodentium infection disrupts the intestinal barrier, but the evolution of barrier loss and its role in disease pathogenesis have not been determined. Most studies have relied on a single probe, FITC-4kD dextran, which cannot distinguish between leak and unrestricted pathways (Fig 1A). We characterized barrier loss during disease progression using three probes: creatinine, FITC-4kD dextran, and rhodamine-70kD dextran, which have hydrodynamic diameters of 6A, 28A, and 120A, and can be used to probe pore, leak, and unrestricted pathways, respectively (Fig. 1A).
Intestinal permeability to creatinine was elevated within 2 days of C. rodentium infection and continued to increase thereafter (Fig. 1B). 4kD dextran permeability increased 1 day later, but 70kD dextran flux did not increase until day 6 of infection. To specifically asses pore and leak pathways, creatinine or 4kD dextran fluxes were normalized to 70kD dextran permeability. These ratiometric analyses demonstrated specific increases in pore and leak pathways at 2 and 3 days post-infection, respectively (Fig. 1C). The increased 70kD dextran permeability that occurred as a result of epithelial damage and increased unrestricted pathway flux at day 6 caused pore and leak pathway ratios to return to baseline values (Fig. 1C). The increased creatinine and 4kD dextran flux at 6 days after C. rodentium infection and beyond is therefore a function of unrestricted pathway permeability rather than specific upregulation of tight junction pore and leak pathways.
Increased pore pathway permeability is accompanied by increased epithelial claudin-2 expression
Pore pathway permeability is primarily regulated by claudin protein expression and function. Quantitative RT-PCR analyses of isolated colonocytes showed that claudin-2 mRNA was uniquely upregulated within 2 days of C. rodentium infection (Fig. 1D). Limited increases in mRNA encoding Cldn15, which complements claudin-2 functionally (Wada et al., 2013), were only significant at day 11. mRNA encoding other tight junction proteins was unchanged in the first week of infection (Fig. 1D). Immunoblots confirmed increased claudin-2 protein expression within 2 days of C. rodentium infection (Fig. 1E).
Immunofluorescence microscopy demonstrated expansion of the zone of claudin-2 expression within 2 days of infection (Fig. 1F), well before histologically-detectable inflammatory changes or epithelial damage were present (Fig. S1). Claudin-2 expression within transit-amplifying cells peaked by day 11 and, consistent with qRT-PCR and western blot data, regressed by day 15 of infection (Fig. 1E, F). Although marked mucosal hyperplasia does occur during C. rodentium infection, the absence of increased claudin-15 expression demonstrates that claudin-2 is specifically upregulated and not merely a consequence of increased cell numbers (Fig. 1D, E). Given the well-established role of claudin-2 as a mediator of pore pathway permeability, these data suggest that claudin-2 upregulation drives the increased pore pathway permeability detected within 2 days of infection.
C. rodentium upregulates claudin-2 via IL-22 signaling
Cytokine-mediated claudin-2 transcriptional regulation has been described in vitro and in vivo (Mankertz et al., 2009; Suzuki et al., 2011; Weber et al., 2010). Analyses of mucosal cytokines during infection showed that only IL-22 was significantly increased by day 2 (Fig. 2A). IL-22 has not, however, been reported to regulate claudin-2 transcription. Cytokines that have been linked to claudin-2 upregulation were increased only at later times (IL-6, TNF, IL-17A) or were unchanged (IL-13).
Figure 2.

IL-22 induces claudin-2 expression during C. rodentium infection. (A) Cytokine expression at indicated times during C. rodentium infection (n=6). (B, C, D) IL-22 upregulates claudin-2 mRNA and protein expression in mouse organoids, as shown by qRT-PCR, western blots, and densitometry (n=3). (E) Immunofluorescence microscopy of IL-22-treated organoids demonstrates increased claudin-2 (red) expression at tight junctions and lateral membranes. F-actin (green), DNA (blue). Bar = 20 μm. (F) Western blots and densitometry of claudin-2 (CLDN2) and E-cadherin (Ecad) expression in isolated colonic epithelial cells after in vivo IL-22 treatment (n=3). (G) Claudin-2 (red) expression after IL-22 treatment in vivo. F-actin (green), DNA (blue). Brackets indicate zone of claudin-2 expression. Bar = 20 μm. (H) Western blots and densitometry of claudin-2 (CLDN2) and E-cadherin (Ecad) expression in isolated colonic epithelial cells from mice pre-treated with control IgG or anti-IL-22 after 2 days of infection (n=3). (I) Immunofluorescence microscopy of claudin-2 (red) expression after C. rodentium infection in mice pre-treated with control IgG or anti-IL-22 at day 2 of infection. Uninfected mice are shown for reference. E-cadherin (green), DNA (blue). Brackets indicate zone of claudin-2 expression. Bar = 20 μm.
Despite many advantages, the presence of numerous cell types and potential for complex intercellular signaling limit utility of in vivo models to determine whether IL-22 signals directly to intestinal epithelia. We used intestinal epithelial organoid cultures to overcome this limitation. In organoid cultures IL-22 significantly upregulated claudin-2, but not claudin-15, mRNA and protein expression (Fig. 2B, C, D). Induced claudin-2 was localized to tight junctions and lateral membranes (Fig. 2E). IL-22 is therefore a direct, potent, and selective inducer of epithelial claudin-2.
To determine if IL-22 could induce claudin-2 upregulation in vivo, wildtype mice were treated with recombinant IL-22. This increased colonic epithelial claudin-2 expression within 24 hours (Fig. 2F). Similar to upregulation during infection, both the magnitude of claudin-2 expression within individual cells and the number of cells expressing claudin-2 were increased (Fig. 2G). IL-22 is therefore sufficient to induce colonic epithelial claudin-2 expression in vivo. We cannot, however, exclude contributions from other cytokines at later times during C. rodentium infection, as IL-22 expression peaks at 6 days after infection, but claudin-2 upregulation progresses until day 11.
The essential contributions of IL-22 to host defense (Zheng et al., 2008) constrain use of IL-22-deficient mice to further probe the role of IL-22 in claudin-2 upregulation during advanced C. rodentium infection. However, such comparisons are possible at 2 days after C. rodentium infection, when claudin-2 upregulation first occurs, as no differences between wildtype and either IL-22-deficient or anti-IL-22-treated mice are apparent at this early time. Treatment with anti-IL-22 prior to infection prevented C. rodentium-induced increases in claudin-2 expression (Fig. 2H, I). In contrast, robust claudin-2 upregulation was apparent in mice treated with a control IgG (Fig. 2H, I). These data show that IL-22 is essential for claudin-2 induction during early stages of C. rodentium infection. Claudin-2 upregulation may therefore be an unrecognized mechanism by which IL-22 activates innate host defense.
Transgenic claudin-2 expression increases fecal Na+ and water content
To define the impact of intestinal epithelial claudin-2 expression on C. rodentium infection, transgenic mice with intestinal epithelial specific expression of EGFP-claudin-2, which we have shown to be functional (Raleigh et al., 2011), were developed. Analyses of isolated colonic epithelial cells from uninfected claudin-2 transgenic, wildtype, and claudin-2 knockout (Wada et al., 2013) mice showed a 12-fold increase in claudin-2 mRNA in transgenic mice relative to wildtype littermates (Fig. 3A). Endogenous claudin-2 protein was expressed at similar levels in wildtype and transgenic mice, but EGFP-claudin-2 expression was 8-fold greater than endogenous claudin-2 (Fig. 3B). No claudin-2 transcripts or protein were detected in knockout mice (Fig. 3A, B). Neither claudin-2 overexpression nor claudin-2 deficiency affected mRNA or protein expression of claudin-15 or any other tight junction components (Fig. 3A, B).
Figure 3.

Transgenic claudin-2 expression increases fecal Na+ and water. (A) Tight junction protein mRNA expression in wildtype (WT), claudin-2 transgenic (Vil-Cldn2Tg), and claudin-2 knockout (Cldn2KO) mice (n=5). Only claudin-2 expression differed between genotypes. (B) Immunoblots of claudin-2 (21kD) and EGFP-claudin-2 (48kD), claudin-15 (21kD), E-cadherin (80kD), and β-actin (42kD). (C) Histology of wildtype, claudin-2 transgenic, and claudin-2 knockout mice. Bar = 50 μm. (D) Immunofluorescent microscopy of claudin-2 and EGFP-claudin-2 (red), F-actin (green), and DNA (blue). Brackets indicate zone of claudin-2 expression. Bar = 50 μm. (E) Fecal Na+ content of wildtype, claudin-2 transgenic, and claudin-2 knockout mice (n≥21). (F) Fecal water content of wildtype, claudin-2 transgenic, and claudin-2 knockout mice (n=15).
Neither transgenic claudin-2 expression nor claudin-2 knockout affected colonic histology (Fig. 3C). Nevertheless, fluorescence microscopy demonstrated that EGFP-claudin-2 was expressed within an expanded population of epithelial cells (Fig. 3D). Thus, both the magnitude and distribution of total claudin-2 expression in uninfected claudin-2 transgenic mice are similar to that in C. rodentium-infected wildtype mice. Claudin-2 transgenic mice can, thus be used to represent maximal claudin-2 expression. Conversely, claudin-2 knockout mice can be used to model the low endogenous claudin-2 expression seen in adult mice (Holmes et al., 2006; Wada et al., 2013). Claudin-2 transgenic and knockout mice can therefore serve as stable reference points for the low and high claudin-2 expression, respectively, seen in wildtype mice before and during infection.
Uninfected claudin-2 transgenic mice demonstrated significantly increased fecal Na+ relative to wildtype or claudin-2 knockout mice (Fig. 3E), thereby demonstrating that the transgenically-expressed EGFP-claudin-2 did function as a cation channel. Further, fecal water, defined as the fraction of total stool mass lost upon desiccation, of claudin-2 transgenic mice was significantly greater than that of wildtype or claudin-2 knockout mice (Fig. 3F). These data, which confirm that EGFP-claudin-2 is functional, are consistent with the ability of claudin-2 to conduct Na+ and water across the tight junction (Rosenthal et al., 2016). As expected given the low claudin-2 expression in adult mice, fecal water content was similar in wildtype and claudin-2 knockout mice (Fig. 3F).
Claudin-2 promotes C. rodentium clearance and limits mucosal immune activation
C. rodentium infection induced significant weight loss in claudin-2 knockout mice (Fig. 4A). In contrast, neither wildtype nor claudin-2 transgenic mice lost weight, as is typical of C. rodentium colitis. In addition to weight loss, fecal C. rodentium shedding in claudin-2 knockout mice was increased 10-fold on day 9 and 100-fold on days 15 and 18 of infection relative to wildtype or transgenic mice (Fig. 4B). On day 11 of infection, which represents both the nadir of body weight in claudin-2 knockout mice (Fig. 4A) and the peak of claudin-2 expression in wildtype mice (Fig. 1D–F), wildtype and claudin-2 transgenic mice had 8- and 40-fold fewer mucosa-associated C. rodentium than claudin-2 knockout mice, respectively (Fig. 4C). These differences in mucosal colonization were evident morphologically, as bacteria penetrated into colonic crypts of claudin-2 knockout mice, yet were restricted to the surface in wildtype and transgenic mice (Fig. 4D, arrows). Although this could reflect an effect of claudin-2 on C. rodentium colonization, the observation that claudin-2 knockout, claudin-2 transgenic, and wildtype mice shed similar numbers of fecal C. rodentium until day 9 suggests that overall colonization was not affected by claudin-2 expression.
Figure 4.

Claudin-2 promotes C. rodentium clearance and limits mucosal immune activation. (A) Weights of wildtype (WT), claudin-2 transgenic (Vil-Cldn2Tg), and claudin-2 knockout (Cldn2KO) mice during C. rodentium colitis (n=7). (B) Fecal C. rodentium in wildtype, claudin-2 transgenic, and claudin-2 knockout mice at indicated times after infection (n=5). (C) Mucosa-associated C. rodentium in wildtype, claudin-2 transgenic, and claudin-2 knockout mice 11 days after infection (n=5 per condition). (D) Immunofluorescent microscopy of C. rodentium (red), F-actin (green), DNA (blue) 11 days after infection. Crypt spaces are only colonized in claudin-2 knockout mice (P < 0.05). Bar = 50 μm. (E) Mucosal cytokine content in wildtype, claudin-2 transgenic, and claudin-2 knockout mice before and 11 days after C. rodentium infection (n=5).
Increased mucosal C. rodentium numbers might be expected to result in greater immune activation in claudin-2 knockout mice. Consistent with this, infection-associated IL-1β, IL-6, IL-22, and TNF expression were significantly greater in knockout, relative to wildtype or transgenic, mice (Fig. 4E). Conversely, TNF expression was significantly reduced in claudin-2 transgenic mice. These differences were not due to pre-existing alterations, as mucosal IL-1β, IL-6, IL-17, IL-22, TNF, and IFN-γ expression were similar in uninfected wildtype, claudin-2 transgenic, and claudin-2 knockout mice (Fig. 4E). The markedly increased IL-22 expression in infected claudin-2 knockout mice further suggests that claudin-2 may participate in a negative feedback loop that, by reducing IL-22 production, limits the degree of claudin-2 upregulation.
Claudin-2 upregulation limits C. rodentium-induced tissue damage
Transgenic expression or knockout of claudin-2 had no effect on intestinal epithelial proliferation or apoptosis in healthy mice (Fig. 5A). C. rodentium infection markedly increased epithelial proliferation and apoptosis in all mice, but these changes were far greater in claudin-2 knockout, relative to wild type or transgenic, mice (Fig. 5A). The greater epithelial damage and proliferative responses of claudin-2 knockout mice to infection were accompanied by significantly more severe colonic histopathology. While isolated intraepithelial neutrophils were readily identified in wildtype and claudin-2 transgenic mice, neutrophil aggregates were associated with surface epithelial erosions in claudin-2 knockout mice (Fig. 5B, arrows).
Figure 5.

Claudin-2 upregulation limits C. rodentium-induced tissue damage. (A) Epithelial proliferative and apoptotic responses at day 11 of C. rodentium infection. Ki67 (red), cleaved caspase-3 (CC3, red), DNA (blue), (n=5). (B) Colonic histopathology at day 11 of C. rodentium infection (n=7). (C) Creatinine, 4kD dextran, and 70kD dextran permeabilities on day 11 of infection. Data shown are normalized to means of infected wildtype mice (n=5). (D) Ratiometric analyses of specific pore (creatinine/70kD dextran) and leak (40kD dextran/70kD dextran) pathway permeabilities (n=5). (E) Fecal water content during C. rodentium infection (n=5).
Consistent with the presence of surface erosions and more frequent apoptotic epithelial cells, increases in creatinine, 4kD dextran, and 70kD dextran permeability were exaggerated in C. rodentium-infected claudin-2 knockout mice at day 11 of infection (Fig. 5C). However, the increases for each probe were proportional, such that the ratios of creatinine and 4kD dextran permeabilities to 70kD dextran permeability were unchanged (Fig. 5D). This indicates that the increased absolute permeabilities observed reflect increased flux across the unrestricted, i.e. epithelial damage, pathway. The greater permeability increases in claudin-2 knockout mice are therefore indicative of severe mucosal damage, consistent with the analyses of histopathology, apoptosis, and compensatory proliferation.
As shown in Figure 3, fecal water content of uninfected claudin-2 transgenic mice was greater than that of uninfected wildtype or claudin-2 knockout mice (Fig. 5E). However, fecal water content of claudin-2 transgenic and wildtype mice was similar beginning at day of infection, consistent with the kinetics of claudin-2 upregulation in wildtype mice. More strikingly, fecal water content in claudin-2 knockout mice increased dramatically at day 11 and remained significantly greater than that of both wildtype and claudin-2 transgenic mice until day 21 (Fig. 5E). The increase in fecal water coincided with the peak of disease in claudin-2 knockout mice and correlated with the marked greater tissue damage experienced by these mice at that time. As a whole, the analyses of apoptosis, histopathology, permeability, and fecal water content therefore indicate that increased fecal water from days 11 to 18 in claudin-2 knockout mice was secondary to marked increases in unrestricted pathway permeability. These results therefore indicate that increased intestinal epithelial claudin-2 expression is an important host defense response that limits C. rodentium-induced intestinal pathology. The data further suggest that, in the absence of pore pathway water efflux, increased unrestricted pathway permeability allows diarrhea to develop. The temporal correlation between increased fecal water and bacterial shedding in claudin-2 knockout mice further suggests that diarrhea resulting from water efflux is critical to pathogen clearance.
Osmotically-driven water efflux rescues claudin-2 knockout mice from severe infectious colitis
As noted above, claudin-2 forms a paracellular channel that conducts Na+ and water. We therefore hypothesized that the protection against C. rodentium colitis observed in wildtype and claudin-2 transgenic, relative to claudin-2 knockout, mice might simply be due to increased paracellular water efflux. To test this idea without expressing claudin-2, mice received polyethylene glycol (PEG) in their drinking water. PEG is a nontoxic, nonabsorbable molecule that, due to its osmotic activity within the lumen, draws fluid across the paracellular space and induces diarrhea. This approach is used routinely in patients in order to cleanse the bowel prior to colonoscopy.
The bowel-preparation solutions used to induce diarrhea in patients generally contain low molecular mass, 3.35kD PEG, at 6% (w/v). Preliminary studies showed that administration of low molecular mass PEG at a much lower dose (2%) was insufficient to cause diarrhea in mice, but did increase fecal water content to a level similar to that in uninfected claudin-2 transgenic mice when given continuously for 7 days (Fig. S2). Importantly, PEG treatment did not alter expression of claudin-2 or other tight junction proteins (Fig. S3).
Treatment with PEG for 7 days, beginning at day 4 of infection (Fig. 6A) significantly reduced numbers of mucosa-associated C. rodentium in wildtype and claudin-2 knockout mice (Fig. 6B). In contrast, PEG did not reduce mucosa-associated C. rodentium numbers in claudin-2 transgenic mice (Fig. 6B). This was expected, as water efflux is increased in claudin-2 transgenic mice even in the absence of PEG treatment. The data, therefore, support the conclusion that the PEG-induced water efflux maximizes pathogen clearance in wildtype and claudin-2 knockout mice. These results also suggest that pathogen clearance was maximized by transgenic claudin-2 expression and could not be enhanced further. Consistent with this, PEG treatment eliminated genotype-dependent differences in fecal C. rodentium shedding (Fig. 6C).
Figure 6.

Osmotic diarrhea reduces bacterial colonization and mucosal immune responses induced by C. rodentium infection. (A) Schematic of infection and PEG treatment. (B) Mucosa-associated C. rodentium numbers in wildtype (WT), claudin-2 transgenic (Vil-Cldn2Tg), and claudin-2 knockout (Cldn2KO) mice at day 11 (n=5). (C) Fecal C. rodentium at day 11 after infection (n=5). (D) Immunofluorescent microscopy of C. rodentium (red) and DNA (blue) 11 days after infection. Colonization of crypt spaces in claudin-2 knockout mice is blocked by PEG treatment (P < 0.05). Bar = 50 μm. (E) Mucosal cytokines on day 11 of infection (n=5).
Although a previous study reported that 5% PEG reduced C. rodentium attachment to cultured colonic epithelial monolayers by downregulating β1-integrin (Qi et al., 2011), PEG did not affect intestinal epithelial β1-integrin expression in vivo (Fig. S4). Moreover, as treatment was not initiated until 4 days after infection, PEG would not be expected to affect initial bacterial adherence in the experiments shown here. We also considered the possibility that PEG might interfere with C. rodentium growth, but this was excluded by in vitro analyses of bacterial growth (Fig. S5) as well as the failure of PEG to reduce fecal C. rodentium numbers in wildtype or claudin-2 transgenic mice (Fig. 6C).
Morphologically, PEG prevented C. rodentium from colonizing colonic crypts in claudin-2 knockout mice, although surface colonization persisted (Fig. 6D, arrows). Finally, PEG significantly diminished mucosal IL-1β, IL-6, TNF, IFN-γ, and IL-22 mucosal cytokine content in claudin-2 knockout, but not wildtype or claudin-2 transgenic, mice (Fig. 6E). In contrast, PEG had no effect on mucosal cytokine expression in uninfected mice (Fig. S6). These data suggest that, by increasing water efflux and promoting pathogen clearance, claudin-2 or PEG provide negative feedback that limits IL-22 production. PEG-induced water efflux therefore promotes C. rodentium clearance and limits mucosal immune activation in a manner similar to claudin-2 upregulation.
Osmotic diarrhea limits barrier loss, epithelial apoptosis, and tissue damage
PEG treatment of uninfected mice had no effect on creatinine, 4kD dextran, or 70kD dextran permeabilities (Fig. S7). In contrast, PEG markedly reduced infection-associated increases in creatinine (Fig. 7A), 4kD dextran (Fig. 7B), and 70kD dextran (Fig. 7C) permeabilities in claudin-2 knockout mice and had more modest effects on wildtype mice. The magnitudes to which PEG limited infection-induced permeability increases in wildtype and claudin-2 knockout mice were proportional for all probes, including 70kD dextran. This effect of PEG must therefore reflect reduced tissue damage, i.e. decreased unrestricted pathway flux, as that is the only route that accommodates 70kD dextran. Consistent with their increased basal fecal water content, claudin-2 transgenic mice did not benefit from PEG treatment (Fig. 7A, B, C). Creatinine, 4kD dextran, and 70kD dextran permeabilities were therefore similar in C. rodentium-infected, PEG-treated mice regardless of genotype.
Figure 7.

Osmotic diarrhea protects claudin-2 knockout mice from severe C. rodentium colitis. (A, B, C) Creatinine, 4kD dextran, and 70kD dextran permeabilities in wildtype (WT), claudin-2 transgenic (VilCldn2Tg), and claudin-2 knockout (Cldn2KO) mice on day 11 after infection (n=5). (D) Epithelial apoptosis as detected by cleaved caspase-3 (CC3) staining on day 11 after infection (n=5). (E) Epithelial proliferation as detected by Ki67 staining on day 11 after infection (n=5). (F) Fecal water on day 11 after infection (n=5). (G, H) Histopathology in C. rodentium infection (n=5). Bar = 50 μm.
Epithelial apoptosis is characteristic of C. rodentium infection and is further increased in claudin-2 knockout mice (Fig. 4B). PEG treatment reduced apoptotic frequency in both wildtype and claudin-2 knockout mice (Fig. 7D) and was mirrored by reductions in epithelial proliferation (Fig. 7E). As with C. rodentium colonization, cytokine production, and probe permeability, PEG reduced fecal water content in claudin-2 knockout and wildtype mice at day 11 (Fig. 7F), when water efflux is primarily mediated by the unrestricted pathway. Consistent with these benefits, PEG treatment reduced histopathology in claudin-2 knockout mice (Fig. 7G, H). Taken together, these results demonstrate that the impaired C. rodentium clearance and more severe disease observed in claudin-2 knockout mice are due to reduced paracellular water transport and can be largely reversed by induction of an osmotic diarrhea that draws water into the gut lumen by claudin-2-independent mechanisms.
DISCUSSION
The hypothesis that diarrhea clears enteric pathogens has been debated for centuries (Davison, 1922). Data supporting this idea are limited to the observation that antimotility agents, which can lessen diarrhea severity, can also delay pathogen clearance (DuPont and Hornick, 1973). Nevertheless, support for the idea that diarrhea is beneficial is weak, and, despite limited evidence of efficacy in most infections, efforts to develop therapeutic ion channel inhibitors that may prevent diarrhea continue. We sought to define the role of diarrhea in pathogen clearance using the model A/E pathogen C. rodentium.
Infection by A/E pathogens results in intestinal barrier loss due to tight junction dysregulation and epithelial damage. Here, we developed an assay that allows size-specific analysis of intestinal permeability and dissection of three distinct modes of barrier loss during C. rodentium infection. The results indicate that, within 2 days of infection, before morphological features of inflammation or tissue damage have developed, IL-22-dependent signaling promotes colonic epithelial claudin-2 expression that enhances permeability of small probes, e.g. creatinine, but not more commonly-used larger probes such as 4kD dextran. The increased fecal water induced by transgenic claudin-2 expression in uninfected mice provides further evidence that inflammation and epithelial damage are not required for the permeability increases or diarrhea that develop during infection. Claudin-2-dependent water efflux does, however, promote pathogen clearance and limit disease severity. Whether these claudin-2-dependent increases in water efflux simply wash away adherent C. rodentium or, alternatively, have some other effects on the microbiome has not been addressed here but could be an interesting question. Regardless of the mechanism, the claudin-2-dependent pathogen clearance described here is distinct from IL-13-dependent helminth expulsion, where claudin-2 upregulation occurs but does not contribute significantly to host defense (Sun et al., 2016).
The data presented here show that IL-22 is required for claudin-2 upregulation in vivo. In vitro studies indicate that claudin-2 upregulation depends on STAT3 and STAT6 signaling (Rosen et al., 2013; Weber et al., 2010). Separate studies have shown that epithelial STAT3 signaling is essential for effects of IL-22 on epithelial homeostasis and repair during acute colitis (Pickert et al., 2009). Available data therefore indicate that IL-22 upregulates epithelial STAT signaling in vivo and that STAT signaling is required for epithelial claudin-2 upregulation in vitro. By demonstrating that IL-22 signaling is necessary and sufficient to drive epithelial claudin-2 upregulation in C. rodentium infection, these data link the previous observations and lead to the conclusion that IL-22-mediated claudin-2 expression occurs via a STAT-dependent process. Although IL-22 has not been previously recognized as a regulator of claudin-2 expression, other effects of IL-22-dependent epithelial STAT3 signaling, such as inhibition of enteric viral infection, might also reflect claudin-2 upregulation (Xue et al., 2017). Thus, claudin-2-mediated water efflux may be a widely used mechanism of innate defense.
Our data indicate that diarrhea, whether driven by claudin-2-mediated paracellular Na+ and water flux or an osmotic agent, promotes pathogen clearance. Although not tested in vivo, in vitro studies suggest that increased water flux driven by other means, such as chloride secretion induced by activation of apical chloride channels, could have similar effects (Keely et al., 2012). Water efflux might also occur via transcellular, rather than paracellular, transport. Consistent with this idea, epithelial aquaporin-4 expression has been reported to be enhanced by IL-22 signaling (Pham et al., 2014). However, most studies have failed to identify a role for aquaporins in intestinal water transport, and, recent data suggest that intestinal epithelial aquaporins are more important for transport of bioactive signaling molecules, such as H2O2, than water (Thiagarajah et al., 2017; Yang et al., 2005). Thus, despite the potential involvement of many processes, including several activated by IL-22, in triggering the water efflux that enhances bacterial clearance, claudin-2 upregulation is the primary mechanism employed in C. rodentium colitis.
In addition to promoting pathogen clearance, the diarrhea driven by claudin-2 expression limited epithelial apoptosis and the associated repair response. Epithelial proliferation and apoptosis were not, however, affected by claudin-2 knockout or transgene expression in the absence of challenge. This contrasts with the increased colonic epithelial proliferation reported in a different claudin-2 transgenic mouse (Ahmad et al., 2014). Given the absence of mucosal hypertrophy in that mouse, one can conclude that the previously-reported claudin-2 transgenic mouse suffered from chronic, low-grade epithelial damage. The absence of this phenotype in the claudin-2 transgenic mouse reported here indicates that the damage is not a direct result of increased claudin-2 expression. Further, our data suggest that increased fecal water content and resulting DSS dilution may be a simple explanation for the DSS-resistant phenotype of the previously-reported claudin-2 transgenic mice (Ahmad et al., 2014). Given the increased claudin-2 expression that normally occurs during DSS colitis, this hypothesis is also consistent with the enhanced DSS colitis severity described in claudin-2 knockout mice (Nishida et al., 2013).
Multiple immune cell subsets produce IL-22 during C. rodentium infection (Basu et al., 2012; Rankin et al., 2016). This IL-22 production requires epithelial NFκB signaling (Giacomin et al., 2015), thereby explaining how a noninvasive pathogen can activate subepithelial immune cells. Previous studies have shown that intestinal epithelial IL-22RA signaling contributes to host defense during C. rodentium infection, and that intestinal epithelial IL-22RA knockout results in exaggerated mucosal IL-22 production (Pham et al., 2014). This is remarkably similar to the effects of claudin-2 knockout and supports the hypothesis that IL-22-dependent claudin-2 upregulation feeds back to limit IL-22 production.
In sum, the results presented here indicate that innate immune activation and IL-22-dependent increases in claudin-2 expression and pore pathway paracellular permeability are essential to host defense. This suggests that, while inhibition of permeability increases might limit diarrhea in early stages of infection, they may also delay pathogen clearance and prolong disease. Thus, while therapeutic claudin-2 inhibition has been proposed, it may be contraindicated in infectious enterocolitis. As a whole, our studies provide direct evidence that increased tight junction permeability and the diarrhea that follows are critical to enteric pathogen clearance and define these as a previously unrecognized contribution of IL-22 to host defense.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Jerrold R Turner, MD, PhD (jrturner@bwh.harvard.edu).
Experimental Model and Subject Details
Mice
C57BL/6 mice (Stock# 000664) were purchased from The Jackson Laboratory. Claudin-2 knockout mice on a C57BL/6 background were obtained from Dr. Sachiko Tsukita (Wada et al., 2013). A mammalian expression construct with the 9kB villin promoter (Pinto et al., 1999) followed by EGFP and mouse claudin-2 was injected into C57BL/6 embryos by the Transgenic Mouse and Embryonic Stem Cell Facility at the University of Chicago. Claudin-2 transgenic pups were born at expected Mendelian proportions, grew and gained weight identically to their littermates, and did not develop spontaneous disease. Preliminary experiments demonstrated that EGFP-claudin-2 was expressed, trafficked to the tight junction, and was functional. EGFP-claudin-2 expression was not detected in the kidney. Individual experiments were segregated, but all studies were performed in both sexes. Mice were bred under specific pathogen-free conditions and used at 6 – 8 weeks of age. Littermates or co-housed mice were used for all experiments. All studies were approved by the Animal Care and Use Committees of the University of Chicago, Brigham and Women’s Hospital, and Boston Children’s Hospital.
Method Details
Epithelial cell isolation
Colonic epithelium were isolated using Cell Recovery Solution (Corning) as described (Nik and Carlsson, 2013). Briefly, colons were removed and flushed several times with ice-cold PBS. The tissue was inverted by inserting a gavage needle, securing the needle end with suture, and pulling the needle back. Following inversion, the other end of the inverted colon was secured with a suture. The gavage needle was then attached to a 1 ml syringe and the tissue was submerged in at 3 ml ice-cold Cell Recovery Solution in a FACS tube. The tissue was deflated and re-inflated with the plunger every 5 min for 30 min to recover epithelial sheets.
Primary cell culture and cytokine stimulation
For stem cell culture, intestinal epithelial cells were harvested from 8–10-week-old C57BL/6 mice and cultured in Matrigel (Corning) and fed with media containing epidermal growth factor, Noggin, and R547 spondin, as previously described (Sato et al., 2011). Two days after splitting, organoid cultures were treated with recombinant murine IL-1β (10 ng/ml), IL-6 (50 ng/ml), IL-17 (10 ng/ml), IL-22 (10 ng/ml), TNF (10 ng/ml), or IFN-γ (10 ng/ml) for 48 hours. Cells were harvested from Matrigel using Cell Recovery Solution.
Citrobacter rodentium infection
C. rodentium strain DBS100 (ATCC 51459; American Type Culture Collection) was grown in LB broth overnight with shaking at 37°C. Mice were fasted for 4 hours before oral inoculation of 2 × 109 colony554 forming unit (CFU) C. rodentium in a volume of 0.2ml sterile phosphate-buffered saline (PBS). Bacterial concentration was measured as absorbance at OD600 and confirmed independently for each experiment by serial dilution and growth on MacConkey agar.
For analysis of the effect of low molecular weight polyethylene glycol (PEG) on infection, filter-sterilized 2% PEG (MW 3,350) was added to the drinking water beginning on day 4 of C. rodentium infection.
Citrobacter rodentium quantification
For analysis of C. rodentium shedding, feces were collected, weighed, homogenized in sterile PBS using with 0.1mm diameter zirconia-silica beads and a beadbeater (Biospec) for 20 s at room temperature. Mucosa-associated bacteria were assessed in 0.5 cm segments of distal colon that had been flushed with ice-cold sterile PBS to removal luminal contents and loosely adherent bacteria. Colonic segments were then weighed and homogenized identically to fecal samples. Serial dilutions of fecal and colonic homogenates were plated on MacConkey agar. C. rodentium were identified as pink colonies after 20 hours of growth at 37°C.
For analysis of the effect of PEG on bacterial growth, stationary phase cultures were diluted 1:100 into fresh LB media with or without 2% PEG and OD600 was measured at indicated intervals.
Cytokine and antibody treatment
Mice were injected i.p with 5 μg recombinant murine IL-22 (PeproTech). Colonic epithelia were harvested after 24 hrs. Mice were injected i.p with 100 μg anti-IL-22 neutralizing or isotype-matched control monoclonal antibodies 48 hours prior to infection.
Intestinal permeability assay
Mice were denied access to food but allowed water for 3 hours prior to gavage with 0.2 ml saline containing 12 mg fluorescein isothiocyanate-4kD dextran, 8 mg rhodamine B isothiocyanate-70kD dextran, and 20 mg creatinine (Sigma). Serum was harvested after 5 hours. Creatinine was measured as described previously (Clayburgh et al., 2005). Recovery of creatinine and fluorescent probes was measured in a Synergy HT plate reader (BioTek) using freshly-prepared standards. Fluorescence of fluorescein and rhodamine B were measured using excitation wavelengths of 495nm and 555nm and emission wavelengths of 525nm and 585nm, respectively.
Fecal Na+ and water determination
Fresh stools were collected into pre-tared 1.5ml tubes and immediately sealed. After weighing, tubes were uncapped and completely desiccated by incubation in a dry oven at 60°C for 24 hours. Tubes were then re-weighed, and fecal water was determined as the fraction of total mass lost upon desiccation. In order to provide sufficient sample for the Na+ assay, dried stools were rehydrated with 5× the initial water volume and homogenized using a beadbeater, as above. After brief centrifugation to pellet insoluble debris, Na+ content of the supernatant was measured using a B-722 LAQUAtwin Compact Sodium Ion Meter (Horiba). Fecal Na+ mass was normalized to dry fecal mass.
RNA isolation and quantitative real-time RT-PCR
For qRT-PCR, total RNA was extracted using the RNeasy Mini Kit and on-column DNAse I digestion (Qiagen). cDNA synthesis was performed using the iScript cDNA kit (Bio-Rad). Real-time PCR used primers indicated (Supplemental Table I), and reactions were run using the SsoFast EvaGreen supermixes on a CFX96 thermocycler (Bio-Rad). Raw data were analyzed by the ΔCt method using the epithelial-specific gene keratin 8 for normalization. Changes were calculated by the ΔΔCt method.
ELISA
Portions of distal colon (0.5 cm) were washed using Bio-Plex wash buffer (Bio-Rad). and then homogenized in 0.5 ml Bio-Plex cell lysis buffer with 2mM PMSF using a sterile, 7 cm polypropylene pestle (Kimble). After freeze-thaw, samples were sonicated on ice, centrifuged for 5 min at 4,500 × g, and total protein within the supernatant was quantified using a Bradford protein assay (Bio-Rad). Supernatant (0.25 mg protein in 0.1 ml) was assayed for cytokines using Ready-SET-Go! ELISA kits (eBioscience).
Western blots
Isolated epithelial cell and organoid lysates were separated by SDS-PAGE, transferred to PVDF membranes, and blotted using primary antibodies to claudin-2 (1:2,000 dilution, Abcam), claudin-15 (1:2,000 dilution, ThermoFisher), E-Cadherin (1:5,000 dilution, Cell Signaling Technologies), or β-actin (1:5,000 dilution, Sigma-Aldrich) overnight at 4°C. Membranes were then incubated with horseradish peroxidase (HRP)-conjugated or infrared dye-conjugated secondary antibodies for 1 hr at room temperature. Protein was detected using HyBlotCL film and quantified using ImageJ (HRP conjugates) or a LI-COR Odyssex Fc imager and quantified using ImageStudio (infrared conjugates).
Immunofluorescence staining and microscopy
Distal colons were snap-frozen as described previously (Clayburgh et al., 2005). Snap-frozen distal colons were incubated in 1% PFA in PBS for 10 minutes after cutting 5μm sections. Sections were permeabilized with 0.5% NP-40 and blocked with 10% goat serum for 1 hour at room temperature. Sections were incubated with primary antibody diluted in PBS with 5% goat serum. Primary antibodies consisted of the same antibodies used for western blots or antibodies to C. rodentium (1:250 dilution, Abcam), Ki67 (1:250 dilution, Abcam), or cleaved caspase-3 (1:250 dilution, Cell Signaling Technologies). After washing, sections were incubated with secondary Alexa 594 goat anti-rabbit IgG, Alexa 488-conjugated phalloidin, and Hoechst 33342 (Key Resources Table). In some tissues, mouse anti-E-cadherin was included with the primary antibodies and Alexa 488 goat anti-mouse IgG was included with the secondary antibodies in place of Alexa 488-conjugated phalloidin. Images of tissue sections were collected as stacks at 0.2 μm intervals using an Axioplan 2 (Zeiss), Chroma single channel ET filter sets, 63X NA 1.4 Plan-Apochromat oil immersion objective, and Coolsnap HQ camera. Images were deconvoluted using Autoquant X3 (MediaCybernetics). Images of organoids were collected using a DMI6000 (Leica) with a 63X NA1.3 Plan-Apo glycerol immersion objective, CSUX Yokogawa spinning disc (Andor), Borealis illumination system (Andor), and Zyla Plus camera (Andor). Both microscopes were controlled by MetaMorph 7.8 (Molecular Devices). Images of H&E-stained sections were acquired using a DMLB microscope (Leica) with a 40X NA 0.65 FL-Plan objective, and Micropublisher 3 camera (QImaging) controlled by QCapPro 7 (QImaging).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-claudin-2 (ab53032) | Abcam | RRID:AB_869174 |
| Rabbit polyclonal anti-claudin-15 (38-9200) | ThermoFisher | RRID:AB_2533391 |
| Rabbit polyclonal anti-Citrobacter koseri (ab37056) | Abcam | RRID:AB_726896 |
| Rabbit polyclonal anti-Ki67 (ab15580) | Abcam | RRID:AB_443209 |
| Rabbit monoclonal anti-cleaved caspase-3 (9664) | Cell Signaling Technology | RRID:AB_2070042 |
| Rabbit monoclonal anti-E-cadherin (3195) | Cell Signaling Technology | RRID:AB_2291471 |
| Mouse monoclonal anti-E-cadherin (76055) | Abcam | RRID:AB_1310159 |
| Mouse monoclonal anti-b actin (A1978) | Sigma-Aldrich | RRID:AB_476692 |
| Goat anti-rabbit IgG (H+L), HRP conjugate (7074) | Cell Signaling Technology | RRID:AB_2099233 |
| Horse anti-mouse IgG (H+L), HRP conjugate (7076) | Cell Signaling Technology | RRID:AB_330924 |
| Donkey anti-rabbit IgG (H+L), Alexa 594 conjugate, highly cross-adsorbed F(ab’)2 fragments (711-586-152) | Jackson ImmunoResearch | RRID:AB_2340622 |
| Donkey anti-mouse IgG (H+L), Alexa 488 conjugate, highly cross-adsorbed F(ab’) 2 fragments (715-546-151) | Jackson ImmunoResearch | RRID:AB_2340850 |
| IRDye 800CW Goat anti-rabbit IgG (H+L) (925-32211) | LI-COR Biosciences | RRID:AB_2651127 |
| IRDye 680RD Goat anti-Mouse IgG (H+L) (925-68070) | LI-COR Biosciences | RRID:AB_2651128 |
| Monoclonal anti-IL-22 neutralizing antibody (IgG1, clone 8E11.9) | Genentech | RRID:AB_2651129 |
| Mouse anti-OVA control antibody (IgG1, clone F2-3.58) | BioXCell | RRID:AB_2651130 |
| Bacterial and Virus Strains | ||
| Citrobacter rodentium (strain DBS100) | ATCC | Cat#ATCC51459 |
| Biological Samples | ||
| Organoids were derived from isolated intestinal epithelium of 8–10 week old C57BL/6J mice | N/A | |
| Epithelial cells isolated from distal colon of 8–10 week old C57BL/6J, claudin-2 knockout/C57BL/6J, or claudin-2 transgenic/C57BL/6J mice; all were bred on-site as cage-or litter-mates | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant murine IL-1b (211-11B) | PeproTech | N/A |
| Recombinant murine IL-6 (216-16) | PeproTech | N/A |
| Recombinant murine IL-17 (210-17) | PeproTech | N/A |
| Recombinant murine IL-22 (210-22) | PeproTech | N/A |
| Recombinant murine TNF (315-01A) | PeproTech | N/A |
| Recombinant murine IFN-g (315-05) | PeproTech | N/A |
| Matrigel (356234) | Corning | N/A |
| Cell Recovery Solution (354253) | Corning | N/A |
| Epidermal growth factor (315-09) | PeproTech | N/A |
| Noggin (250-38) | PeproTech | N/A |
| R-spondin (315-32) | PeproTech | N/A |
| Fluorescein isothiocyanate-4kD dextran (46944) | Sigma-Aldrich | N/A |
| Rhodamine B isothiocyanate-70kD dextran (R9379) | Sigma-Aldrich | N/A |
| Creatinine (C4255) | Sigma-Aldrich | N/A |
| MacConkey agar (M7408) | Sigma-Aldrich | N/A |
| Polyethylene glycol, MW 3,350 (P4338) | Sigma-Aldrich | N/A |
| Zirconia-silica beads, 0.1mm dia (11079101z) | Biospec | N/A |
| Alexa Fluor 647 Phalloidin (A22287) | Invitrogen | N/A |
| Alexa Fluor 594 Phalloidin (A12381) | Invitrogen | N/A |
| Hoechst 33342 (H3570) | Invitrogen | N/A |
| Bio-Plex Cell Lysis Kit (171-304011) | Bio-Rad | N/A |
| Critical Commercial Assays | ||
| RNeasy Mini Kit (74106) | Qiagen | N/A |
| RNase-Free Dnase Set (79254) | Qiagen | N/A |
| iScript cDN/A kit (1725038) | Bio-Rad | N/A |
| SsoFast EvaGreen supermixes (1725202) | Bio-Rad | N/A |
| Protein Assay Kit II (5000002) | Bio-Rad | N/A |
| Mouse IL-1b ELISA Ready-SET-Go! (88-7013-86) | eBioscience | N/A |
| Mouse IL-6 ELISA Ready-SET-Go! (88-7064-86) | eBioscience | N/A |
| Mouse IL-17 ELISA Ready-SET-Go! (88-7371-86) | eBioscience | N/A |
| Mouse IL-22 ELISA Ready-SET-Go! (88-7422-86) | eBioscience | N/A |
| Mouse TNF ELISA Ready-SET-Go! (88-7324-86) | eBioscience | N/A |
| Mouse IFN-g ELISA Ready-SET-Go! (88-7314-88) | eBioscience | N/A |
| Mouse IL-10 ELISA Ready-SET-Go! (88-7137-86) | eBioscience | N/A |
| Mouse IL-13 ELISA Ready-SET-Go! (88-7105-86) | eBioscience | N/A |
| Oligonucleotides | ||
| qPCR primers, see Supplemental Table I | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J (000664) | The Jackson Laboratory | N/A |
| Mouse: claudin-2 knockout | Wada et al., 2013 | N/A |
| Mouse: claudin-2 transgenic | This paper | N/A |
| Software and Algorithms | ||
| MetaMorph 7.8 | Molecular Devices | N/A |
| Autoquant X3 | MediaCybernetics | N/A |
| ImageJ | NIH | N/A |
| GraphPad Prism 6.0 | GraphPad Software | N/A |
| ImageStudio 5.0 | LI-COR Biosciences | N/A |
| Microsoft Excel | Microsoft | N/A |
Histopathological scoring
Histopathological analysis of colitis was performed by a pathologist blinded to the experimental conditions. Colon tissues were scored on a scale of 0–3 for eight parameters, yielding a maximum score of 24. Scoring parameters were goblet cell depletion, mucosal hyperplasia, crypt cell apoptosis, erosion, lymphocytic infiltrate, polymorphonuclear (PMN) leukocyte infiltrate, crypt architectural distortion, and involvement of the submucosa.
Quantification and Statistical Analysis
All data are representative of at least 3 independent experiments with 3–10 mice per group. Specific numbers of mice per group is annotated in corresponding figure legends. Data are presented as mean ± SEM. Statistical significance was determined by two-tailed Student’s t test, two-tailed Mann-Whitney U test, or Kaplan-Meier log-rank test. P values are indicated in the figures (*P <0.05, ** P < 0.01, *** P < 0.001).
Supplementary Material
IL-22 induced by enteric infection upregulates the tight junction protein claudin-2
Intestinal epithelial claudin-2 expression promotes paracellular Na+ and water efflux
Na+ and water efflux results in diarrhea that facilitates pathogen clearance
Claudin-2-mediated diarrhea is an innate mechanism of host defense
Acknowledgments
We are grateful to Natalie Ronaghan, Tiffany S. DaVanzo, Edda Fiebiger, and Richard S. Blumberg for their conceptual and technical contributions. This work was supported by National Institute of Health grants F30DK103511 (M.A.O.); T32HD007009 (M.A.O.); R01DK61931 (J.R.T.); R01DK68271 (J.R.T.); R24DK099803 (J.R.T.); Crohn’s and Colitis Foundation of America (J.R.T.); and the State Scholarship Fund of China 201208110294 (B.K.Z.).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization: PYT, BZ, YXF, JRT; Methodology: PYT, BZ, MAW, LS, YXF, JRT; Investigation: PYT, BZ, WQH, JMZ, MAW, GS, AS, SY, WTK, JRT; Resources: AT, YXF, ST; Writing – original draft preparation: PYT, JRT; Writing – review and editing: PYT, JRT; Supervision: JRT; Funding acquisition: JRT, BKZ, MAO.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad R, Chaturvedi R, Olivares-Villagomez D, Habib T, Asim M, Shivesh P, Polk DB, Wilson KT, Washington MK, Van Kaer L, et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014;7:1340–1353. doi: 10.1038/mi.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, O’Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison WC. A Bacteriological and Clinical Consideration of Bacillary Dysentery in Adults and Children. Medicine. 1922;1:389–510. [Google Scholar]
- Deng W, de Hoog CL, Yu HB, Li Y, Croxen MA, Thomas NA, Puente JL, Foster LJ, Finlay BB. A comprehensive proteomic analysis of the type III secretome of Citrobacter rodentium. J Biol Chem. 2010;285:6790–6800. doi: 10.1074/jbc.M109.086603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont HL, Hornick RB. Adverse effect of lomotil therapy in shigellosis. JAMA. 1973;226:1525–1528. [PubMed] [Google Scholar]
- Giacomin PR, Moy RH, Noti M, Osborne LC, Siracusa MC, Alenghat T, Liu B, McCorkell KA, Troy AE, Rak GD, et al. Epithelial-intrinsic IKKalpha expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. 2015;212:1513–1528. doi: 10.1084/jem.20141831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell Microbiol. 2006;8:634–645. doi: 10.1111/j.1462-5822.2005.00656.x. [DOI] [PubMed] [Google Scholar]
- Holmes JL, Van Itallie CM, Rasmussen JE, Anderson JM. Claudin profiling in the mouse during postnatal intestinal development and along the gastrointestinal tract reveals complex expression patterns. Gene Expr Patterns. 2006;6:581–588. doi: 10.1016/j.modgep.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Keely S, Kelly CJ, Weissmueller T, Burgess A, Wagner BD, Robertson CE, Harris JK, Colgan SP. Activated fluid transport regulates bacterial-epithelial interactions and significantly shifts the murine colonic microbiome. Gut microbes. 2012;3:250–260. doi: 10.4161/gmic.20529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankertz J, Amasheh M, Krug SM, Fromm A, Amasheh S, Hillenbrand B, Tavalali S, Fromm M, Schulzke JD. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009;336:67–77. doi: 10.1007/s00441-009-0751-8. [DOI] [PubMed] [Google Scholar]
- Nik AM, Carlsson P. Separation of intact intestinal epithelium from mesenchyme. BioTechniques. 2013;55:42–44. doi: 10.2144/000114055. [DOI] [PubMed] [Google Scholar]
- Nishida M, Yoshida M, Nishiumi S, Furuse M, Azuma T. Claudin-2 regulates colorectal inflammation via myosin light chain kinase-dependent signaling. Dig Dis Sci. 2013;58:1546–1559. doi: 10.1007/s10620-012-2535-3. [DOI] [PubMed] [Google Scholar]
- Pham TA, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, Keane JA, Page AJ, Kumasaka N, Kane L, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. 2014;16:504–516. doi: 10.1016/j.chom.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Joshi S, Weber CR, Wali RK, Roy HK, Savkovic SD. Polyethylene glycol diminishes pathological effects of Citrobacter rodentium infection by blocking bacterial attachment to the colonic epithelia. Gut microbes. 2011;2:267–273. doi: 10.4161/gmic.2.5.18256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, Wang Y, Wu L, Schneeberger EE, Shen L, et al. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193:565–582. doi: 10.1083/jcb.201010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, Wieduwild E, Putoczki T, Mondot S, Lantz O, et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat Immunol. 2016;17:179–186. doi: 10.1038/ni.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MJ, Chaturvedi R, Washington MK, Kuhnhein LA, Moore PD, Coggeshall SS, McDonough EM, Weitkamp JH, Singh AB, Coburn LA, et al. STAT6 Deficiency Ameliorates Severity of Oxazolone Colitis by Decreasing Expression of Claudin-2 and Th2-Inducing Cytokines. J Immunol. 2013 doi: 10.4049/jimmunol.1201373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Gunzel D, Krug SM, Schulzke JD, Fromm M, Yu AS. Claudin-2-mediated cation and water transport share a common pore. Acta Physiol (Oxf) 2016 doi: 10.1111/apha.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterol. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- Sun R, Urban JF, Jr, Notari L, Vanuytsel T, Madden KB, Bohl JA, Ramalingam TR, Wynn TA, Zhao A, Shea-Donohue T. Interleukin-13 Receptor alpha1-Dependent Responses in the Intestine Are Critical to Parasite Clearance. Infect Immun. 2016;84:1032–1044. doi: 10.1128/IAI.00990-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem. 2011;286:31263–31271. doi: 10.1074/jbc.M111.238147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajah JR, Chang J, Goettel JA, Verkman AS, Lencer WI. Aquaporin-3 mediates hydrogen peroxide-dependent responses to environmental stress in colonic epithelia. Proc Natl Acad Sci USA. 2017;114:568–573. doi: 10.1073/pnas.1612921114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- Wada M, Tamura A, Takahashi N, Tsukita S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterol. 2013;144:369–380. doi: 10.1053/j.gastro.2012.10.035. [DOI] [PubMed] [Google Scholar]
- Weber CR, Raleigh DR, Su L, Shen L, Sullivan EA, Wang Y, Turner JR. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–12046. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Zhao J, Ying L, Fu F, Li L, Ma Y, Shi H, Zhang J, Feng L, Liu P. IL-22 suppresses the infection of porcine enteric coronaviruses and rotavirus by activating STAT3 signal pathway. Antiviral Res. 2017;142:68–75. doi: 10.1016/j.antiviral.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Song Y, Zhao D, Verkman AS. Phenotype analysis of aquaporin-8 null mice. Am J Physiol - Cell Physiol. 2005;288:C1161–1170. doi: 10.1152/ajpcell.00564.2004. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.