

Abstract
Colorectal cancer (CRC) remains the leading cause of cancer-related death in the world. Aspirin (ASA) and curcumin (CUR) are widely investigated chemopreventive candidates for CRC. However, the precise mechanisms of their action and their combinatorial effects have not been evaluated. The purpose of the present study was to determine the effect of ASA, CUR, and their combination in azoxymethane/dextran sulfate sodium (AOM/DSS)-induced colitis-accelerated colorectal cancer (CAC). We also aimed to characterize the differential gene expression profiles in AOM/DSS-induced tumors as well as in tumors modulated by ASA and CUR using RNA-seq. Diets supplemented with 0.02% ASA, 2% CUR or 0.01% ASA+1% CUR were given to mice from 1 week prior to the AOM injection until the experiment was terminated 22 weeks after AOM initiation. Our results showed that CUR had a superior inhibitory effect in colon tumorigenesis compared to that of ASA. The combination of ASA and CUR at a lower dose exhibited similar efficacy to that of a higher dose of CUR at 2%. RNA isolated from colonic tissue from the control group and from tumor samples from the experimental groups was subjected to RNA-seq. Transcriptomic analysis suggested that the low-dose combination of ASA and CUR modulated larger gene sets than the single treatment. These differentially expressed genes were situated in several canonical pathways important in the inflammatory network and liver metastasis in CAC. We identified a small subset of genes as potential molecular targets involved in the preventive action of the combination of ASA and CUR. Taken together, the current results provide the first evidence in support of the chemopreventive effect of a low-dose combination of ASA and CUR in CAC. Moreover, the transcriptional profile obtained in our study may provide a framework for identifying the mechanisms underlying the carcinogenesis process from normal colonic tissue to tumor development as well as the cancer inhibitory effects and potential molecular targets of ASA and CUR.
Keywords: Colitis-associated colorectal cancer, aspirin, curcumin, RNA-seq
Graphical abstract
1. Introduction
Colorectal cancer (CRC) is the third most common malignancy and the fourth leading cause of cancer-related deaths worldwide, which is estimated to account for more than 49,190 deaths in 2016 in the United States (1). Chronic inflammation is one of the hallmarks of cancer (2) and has been linked to the pathogenesis of tumors in multiple human cancers, including CRC (3). Colitis-accelerated colon cancer (CAC) is a subtype of CRC with a high mortality that is closely associated with inflammatory bowel disease (IBD) (4). As one of the top high-risk conditions for CRC, epidemiological studies show that patients with long-standing IBD have a significantly higher risk of developing CRC (5). The azoxymethane (AOM)-initiated and dextran sulfate sodium (DSS)-promoted mouse model has been widely used to simulate the pathogenesis observed in patients with CAC (6). Specifically, the multistep carcinogenesis process is induced by an AOM injection [an initiation factor that induces aberrant crypt foci (ACF) by causing DNA damage] and DSS in the drinking water (a promotion factor that induces colitis by imposing inflammatory damage in the epithelial lining of the colon) in rodents.
Although the efficacy of CRC treatment has improved in recent years, the side effects of these treatment strategies, such as surgery, radiation therapy, chemotherapy, and targeted therapy, cannot be neglected. CRC is highly associated with environmental and lifestyle factors and usually undergoes a relatively long precancerous stage that provides individuals with opportunities to interfere before adenomas progress into malignancies. Hence, chemoprevention, the intake of agents with a relatively low toxicity to prevent the progression of cancer at a premalignant stage, has gained increasing attention in the management of CRC as a cost-effective alternative to CRC treatment (7). Among proposed chemopreventive interventions, aspirin (ASA, acetylsalicylic acid) is perhaps the agent with the most extensive evidence that long-term and regular use lowers the risk of different types of CRC (8), including sporadic CRC (9), hereditary CRC (10), and CAC (11). In addition, recent studies from our laboratory demonstrated that dietary administration of ASA (0.2% w/w, equivalent to a dose of approximately 110 mg/day in humans) for 20 weeks effectively prevented carcinogenesis in AOM/DSS-induced CF-1 mice (12). Curcumin (CUR), the main component of turmeric (also called curry powder), is another widely studied chemopreventive candidate for CRC with a promising effect in suppressing inflammation and colon cancer cell growth (13,14) with no reported adverse effects. In a phase IIa clinical trial, CUR at a dose of 4 g/day for 30 days significantly reduced ACF formation (15). Although CUR has been shown to effectively inhibit tumor growth in AOM-induced rats (16,17), its effect in suppressing AOM/DSS-induced CAC has not yet been determined.
The potential side effects of gastrointestinal bleeding from chronic use of high-dose ASA have limited its use in the general population for the prevention of CRC. With regards to CUR, although up to 12 g/day was well-tolerated in humans (18), chronic administration of CUR at a very high dose may lead to poor patient compliance. Co-administration of two or more chemopreventive agents with different molecular mechanisms at a lower dosage may act as a promising strategy to maximize efficacies and minimize toxicities. For example, a synergistic effect has been observed in the combination of green tea polyphenols and atorvastatin in the inhibition of lung tumorigenesis (19), atorvastatin and celecoxib in the suppression of prostate tumors (20), and metformin and ASA in the inhibition of pancreatic cancer (21). In addition, data from two different randomized clinical trials suggested a synergistic interaction between calcium supplementation and the use of ASA in the reduction of the risk of advanced colorectal adenomas; however, the combination of calcium and ASA failed to exert a synergistic action in the suppression of ACF formation in AOM-induced rodent models (22). Regarding the combinational action of ASA and CUR, Thakkar et al. reported a synergistic effect of ASA, CUR, and sulforaphane in the reduction of pancreatic cancer cell viability (23). Furthermore, Perkins et al. found that ASA and CUR exerted an optimal adenoma-retarding effect at different stages in Apcmin/+ mice, although synergism was not achieved when CUR and ASA were administered sequentially (24). In the present study, we aimed to investigate the combinatorial effect of concomitant administration of ASA and CUR at half the dose of their single treatment in the prevention of AOM/DSS-induced CAC.
Previous studies have indicated that both ASA and CUR are multi-target chemopreventive agents that impact various signaling pathways and molecules involved in inflammation, tumor initiation, and tumor progression (8,25). However, an overview of the genes and signaling pathways associated with the chemopreventive actions of ASA and CUR in AOM/DSS-induced CAC remains relatively understudied. In particular, the molecular targets influenced by the combination of these two agents have not yet been investigated. RNA sequencing (RNA-seq) is a recently developed deep-sequencing approach in transcriptome profiling that offers a relatively unbiased and more precise measurement of the levels of transcripts and their isoforms (26). Because RNA-seq technology provides several key advantages over hybridization-based microarrays for transcriptome profiling, it is rapidly becoming an attractive tool to identify the differentially expressed genes in multiple experimental conditions. To depict a comprehensive picture of the molecular mechanisms underlying the chemopreventive effect of ASA and CUR, especially their combination at a lower dose, the present study utilized RNA-seq to analyze the differential gene expression and pathways in tumors induced by AOM/DSS with or without treatment with ASA and CUR, alone or in combination, in a rodent CAC model.
2. Materials and methods
2.1. Animals and diets
Animal experiments were conducted under an animal protocol (01-016) approved by the Institutional Animal Care and Use Committee (IACUC) of Rutgers University. C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA) at the age of 4 weeks old. Upon arrival, the animals were maintained at a controlled temperature (20–22°C), controlled relative humidity (45–55%), and 12-h light and 12-h dark cycles at the Rutgers Animal Care Facility. After 1 week of acclimatization, mice at the age of 5 weeks were randomly assigned to 5 experimental groups (n = 14–15) and fed with AIN-93M rodent diet (Research Diet Inc. New Brunswick, NJ, USA) or special diet (Research Diet Inc.) supplemented with ASA, CUR, or their combination ad libitum. ASA was purchased from Sigma-Aldrich (St Louis, MO, USA) and blended into AIN-93M rodent diet at a final concentration of 0.02% as previously described (12). Curcumin C3 Complex® was a kind gift from Sabinsa Corporation (East Windsor, NJ, USA) and mixed into the AIN-93M rodent diet at a final concentration of 2%. The diet containing the combination of ASA and CUR was prepared by simultaneously mixing ASA and CUR into AIN-93M diet at a final concentration of 0.01% and 1%, respectively.
2.2. Experimental procedure
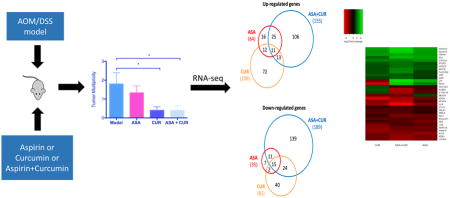
The AOM/DSS model was carried out as previously described (12,27) and is summarized in Figure 1. Briefly, 6-week-old mice were injected with AOM (Sigma-Aldrich) subcutaneously in the lower flank at a dose of 10 mg/kg body weight. The mice in the control group instead received an injection of saline (Thermo Fisher Scientific, Waltham, MA, USA). One week later, the drinking water for the mice, other than the control group, was replaced with DSS (MP Biomedicals, Solon, OH, USA) at a concentration of 1.2% (w/v) for 7 days. The disease activity index (DAI) was calculated daily during the administration of DSS to monitor the symptoms of acute colitis using the scoring system published previously (12). The body weight and the consumption of food and fluid were recorded weekly during the entire experiment. Twenty-two weeks after the AOM injection, all mice were sacrificed by CO2 asphyxiation. Colons were removed, cleaned, and opened longitudinally followed by careful examination of the tumors. The left portion of the colon was saved for histological analysis. After removing the proximal end and palpable tumors, the remaining right portion of the colon was snap frozen and stored at -80°C for molecular assays.
Figure 1. The experimental protocol for a chemoprevention study with ASA and CUR, alone or in combination, in AOM/DSS-induced C57/BL6 mice.

Mice at 5 weeks of age were fed the AIN-93M diet or this diet supplemented with 0.02% ASA, 2% CUR, or 0.01% ASA+1% CUR until the end of the experiment. Mice in groups other than the control group received a subcutaneous injection of AOM at 10 mg/kg at the age of 6 weeks, followed by the administration of water containing DSS at 1.2% (w/v) for 7 consecutive days. Twenty-two weeks after AOM initiation, the mice were sacrificed for further analysis.
2.3. Histopathological analysis
The histopathological analysis was performed as described previously (12). The left portion of the colon was fixed in 10% buffered formalin (Thermo Fisher Scientific) for 24 h, serially dehydrated, embedded in paraffin (Thermo Fisher Scientific), and stored at 4°C. The tissue blocks were then serially sectioned at 4 μm and mounted on glass slides. Histopathological abnormalities in the colon were examined by hematoxylin and eosin staining and evaluated by the histopathologist, Dr. Guangxun Li.
2.4. RNA extraction, library preparation, and next-generation sequencing
Total RNA was isolated from snap-frozen colonic tissue from the control group and tumor samples from the experimental groups (model, ASA, CUR, and ASA+CUR) using the AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA, USA). The quality and quantity of the extracted RNA samples were determined with an Agilent 2100 Bioanalyzer and NanoDrop, respectively. A total of 10 RNA samples [2 samples per group × 5 groups (control, model, ASA, CUR, and ASA+CUR)] were sent to RUCDR for library preparation and sequencing. Briefly, the library was constructed using the Illumina TruSeq RNA preparation kit (Illumina, San Diego, CA, USA) according to the manufacturer’s manual. Samples were sequenced on the Illumina NextSeq 500 instrument with 50–75 bp paired-end reads, to a minimum depth of 30 million reads per sample.
2.5. Computational analyses of RNA-seq data
The reads were aligned to the mouse genome (mm10) with TopHat v2.0.9 (28). Reference gene annotations from UCSC were supplied to TopHat (-G genes.gtf); otherwise, default parameters were used. The Cufflinks v2.2.1 (29) program cuffdiff was used to calculate expression levels, using the UCSC gene annotations and default parameters.
2.6. Ingenuity Pathway Analysis (IPA)
Isoforms that exhibited a log2 fold change greater than 1 and a false detection rate (FDR) less than 0.05 were subjected to Ingenuity Pathway Analysis (IPA 4.0, Ingenuity Systems, www.Ingenuity.com). The input isoforms were mapped to IPA’s knowledge bases, and the relevant biological functions, networks, and pathways related to the treatment of ASA, CUR, and their combination were identified.
2.7. Quantitative polymerase chain reaction (qPCR)
qPCR was used to validate selected differentially expressed genes observed in RNA-seq. After synthesis of first-strand cDNA from 500 ng of RNA using TaqMan® Reverse Transcription reagents (Applied Biosystems, Carlsbad, CA, USA), qPCR was performed using a QuantStudio® 5 Real-Time PCR System (Applied Biosystems) with SYBR Green PCR Master Mix (Applied Biosystems). The results were normalized to the expression of Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) using the 2^ΔΔCT method. All of the primers were designed and ordered from Integrated DNA Technologies (IDT, Coralville, Iowa, USA).
2.8. Statistical analysis
The data are presented as the mean ± SD. Comparisons among multiple groups were analyzed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. The DAI data were analyzed with the repeated measure ANOVA method. A P-value less than 0.05 was considered statistically significant.
3. Results
3.1. Effects of ASA, CUR, and their combination in the prevention of colon tumorigenesis
The clinical symptoms of colitis were evaluated by recording the DAI during DSS treatment. It is widely accepted that the DAI is associated with colitis severity as well as erosions and inflammation (30). As shown in Figure 2A, the DAI score increased gradually in the model group, indicating elevated clinical colitis symptoms in the mice that received AOM/DSS treatment. Dietary administration of ASA (0.02%) slightly attenuated the increase of the DAI starting at day 5, whereas treatment of CUR (2%) and the combination (ASA 0.01% + CUR 1%) started to exhibit effective suppression of the DAI at day 4. Notably, the inhibitory effect of the combination treatment, although at only half the dose of the single compound, was statistically significant by repeated measure ANOVA.
Figure 2. The effect of dietary administration of ASA, CUR, and their combination in AOM/DSS-induced CAC. (A).

The suppression of the DAI by 0.02% ASA, 2% CUR, and 0.01% ASA+1% CUR. (B) The effect of ASA, CUR, and their low-dose combination on tumor incidence in AOM/DSS-induced CAC. The tumor incidence (%) of each group was calculated from the number of mice with tumor growth over the number of mice examined. (C) The effect of ASA and CUR, alone or in combination, in decreasing tumor multiplicity in AOM/DSS-induced CAC. Tumor multiplicity was calculated from the total number of tumors in each group divided by the number of mice in each group. (D–I) Histological observation of the control group (D), the model group with AOM/DSS administration (E, F), mice treated with 0.02% ASA (G), mice treated with 2% CUR (H), and mice treated with 0.01% ASA+1% CUR (I). * P < 0.05 versus the model group.
We did not observe any noticeable body weight loss in mice fed the diet supplemented with ASA, CUR, or the combination compared with mice fed the control diet. Twenty-two weeks after the AOM injection, the tumors in the colon were carefully examined and recorded. As shown in Figure 2B and 2C, 8 of the 15 mice in the model group developed tumors in the colon with a tumor multiplicity of 1.80 ± 0.60 tumors per animal, which is comparable to a previous publication with a similar experimental protocol in C57BL/6 mice (27). Although mice fed the diet supplemented with ASA at the level of 0.02% developed slightly fewer tumors (1.33 ± 0.6 tumors per mouse), the tumor incidence was not decreased. Compared to the model group, dietary administration of CUR at 2% resulted in a lower tumor incidence as well as significantly decreased tumor multiplicity (0.40 ± 0.19 tumors per animal). The combination treatment with only half of the dose compared to the single treatment exhibited the lowest tumor incidence among the four experimental groups and resulted in significantly lower tumor multiplicity (0.41 ± 0.24 tumors in each mouse).
Hematoxylin and eosin staining showed that AOM/DSS-treated mice had severe colonic damage including crypt dysplasia, adenomas, and adenocarcinoma. Inflammation, an increased nucleus to cytoplasm ratio, nuclear crowding, mitosis, and nuclear hyperchromasia were observed in the adenomas (Figure 2E). Adenocarcinomas were associated with severe inflammation, infiltration of leukocytes into the lumen, the composition of cribriform glands, the loss of nuclear polarity to the basement membrane, nuclear hyperchromasia, and mitosis (Figure 2F). Although ASA (0.02%) treatment attenuated inflammation, hyperplasia and adenomas were observed (Figure 2G). CUR (2%) treatment and the combination treatment (ASA 0.01% + CUR 1%) suppressed inflammation severity and cancer lesions, and treated animals exhibited normal colon morphology (Figure 2H–I).
Taken together, these results demonstrate that the dietary administration of CUR at 2% showed a superior inhibitory effect in colitis and colon tumorigenesis over ASA at 0.02%. The combination of ASA and CUR at only half of the dose effectively and significantly suppressed acute colitis and tumor growth in AOM/DSS-induced mice without affecting body weight. One of the limitations of our present study is that we did not include a group of animals administered with 1% CUR alone, thus it is possible that 1% CUR is more effective than 2% CUR. Several studies have indicated a dose-dependent effect of CUR in suppressing AOM-induced tumors. For example, 2% CUR exhibited superior effect in inhibiting AOM-induced ACFs than 0.2% CUR (31) and 2% CUR showed better inhibition of AOM-induced adenomas and adenocarcinomas than 0.5% CUR (32). In addition, Pereira et al. reported that AOM-induced animals developed less adenoma upon treatment with 1.6% CUR than 0.8% CUR (16). Therefore, we postulate that 1% CUR may not be as effective as 2% CUR and ASA increased the efficacy of CUR such that combination treatment with only half of the dose was effective as 2% CUR alone. Thus, combining these two compounds at a lower dosage may provide a promising effect in colon cancer prevention.
3.2. Top differentially expressed genes and canonical pathways affected in the model group compared to the control group
To understand the mechanisms underlying the carcinogenicesis process from normal colonic tissue to tumor tissue, we compared the gene expression profiles of tumors induced by AOM/DSS in the model group to those of age-matched colonic tissue in the control group. We found that a total of 1,291 differentially expressed genes showed a log2 fold change greater than 1 and an FDR less than 0.05. The top 10 down-regulated and up-regulated genes under this comparison are presented in Table 1. The dramatic fold change of these genes (as low as 0.021-fold or as high as 389.9-fold) could be due to the different nature and cell populations of the tumor mass compared to those of normal colon tissue or to alterations triggered by AOM/DSS treatment. To understand the possible biological functions associated with these differentially expressed genes observed in the model group versus the control group, canonical pathway analysis in IPA was used. Based on the ratio of the number of differentially expressed genes in our dataset to the total number of reference genes in the corresponding pathways in the IPA knowledge bases, IPA utilized Fisher’s exact test to determine the significant canonical pathways associated with differentially expressed genes observed from RNA-seq. Using a cutoff P-value less than 0.05, a total of 378 canonical pathways were identified as being significantly correlated with the alterations of gene expression in AOM/DSS-induced tumors compared to normal colonic tissue. Table 2 displays the 10 most significant pathways, their −log (P-value), the ratio of affected genes over total genes in that particular pathway, and the details of significant expressed genes in our dataset contained in that specific pathway. These results suggested that AOM/DSS-induced tumors displayed substantially differentially expressed genes with dramatic mRNA expression changes compared to age-matched normal colon tissue.
Table 1.
Top 10 down-regulated and up-regulated genes in tumors from mice in Model group compared to Control group. Fold change larger than 1 indicates higher expression in tumors in Model group. Fold change smaller than 1 indicates lower expression in tumors in Model group.
| Gene ID | Gene name | Fold Change | FDR |
|---|---|---|---|
| Down-regulated | |||
| RETNLB | resistin like beta | 0.021 | 2.12E-03 |
| CA3 | carbonic anhydrase III | 0.028 | 3.89E-03 |
| NOS1 | nitric oxide synthase 1 (neuronal) | 0.031 | 1.45E-02 |
| SYCN | syncollin | 0.031 | 2.12E-03 |
| ZCCHC12 | zinc finger, CCHC domain containing 12 | 0.032 | 3.28E-02 |
| CHRNA3 | cholinergic receptor, nicotinic, alpha 3 (neuronal) | 0.036 | 1.34E-02 |
| SST | somatostatin | 0.037 | 2.12E-03 |
| NAALADL 1 | N-acetylated alpha-linked acidic dipeptidase-like 1 | 0.041 | 2.12E-03 |
| Pln | phospholamban | 0.042 | 9.63E-03 |
| STMN3 | stathmin-like 3 | 0.044 | 5.44E-03 |
| Up-regulated | |||
| MMP7 | matrix metallopeptidase 7 | 389.911 | 2.12E-03 |
| LYZ | lysozyme | 328.329 | 2.12E-03 |
| CXCL6 | chemokine (C-X-C motif) ligand 6 | 181.900 | 3.63E-02 |
| WIF1 | WNT inhibitory factor 1 | 167.266 | 2.12E-03 |
| PNLIPRP 1 | pancreatic lipase-related protein 1 | 154.236 | 2.12E-03 |
| SLC30A2 | solute carrier family 30 (zinc transporter), member 2 | 124.673 | 6.89E-03 |
| CLCA4 | chloride channel accessory 4 | 114.246 | 2.12E-03 |
| ALOX15 | arachidonate 15-lipoxygenase | 92.411 | 6.89E-03 |
| MMP10 | matrix metallopeptidase 10 | 91.456 | 2.12E-03 |
| ALB | albumin | 87.913 | 2.12E-03 |
Table 2.
The 10 most significant canonical pathways regulated by AOM/DSS-induced tumors compared to normal colonic tissue in Control group. Genes in bold are down-regulated in tumors.
| Canonical Pathways | −log (p value) | Ratio | Target genes |
|---|---|---|---|
| Hepatic Fibrosis / Hepatic Stellate Cell Activation | 19.8 | 50/187 (0.267) | CCR5,COL9A3,MMP13,COL8A1,IL1R2,TGFB1,LA MA1,SERPINE1,PDGFRB,TNFRSF11B,IL4R,COL4 A1,MYH14,MMP2,IGFBP5,PDGFB,MYL7,MYL9,A CTA2,IL10RA,COL6A5,TNF,CCR7,COL7A1,IGFBP 4,ICAM1,FN1,IL1RL1,EGF,CCL5,COL4A2,MYH11, PDGFC,COL15A1,PGF,COL1A2,NGFR,PDGFRA,L BP,TNFRSF1B,COL18A1,VCAM1,COL5A2,COL12 A1,FGF1,COL1A1,COL5A3,IL1B,EDNRA,MMP9 |
| Atheroscleros is Signaling | 13.9 | 34/125 (0.272) | APOE,ICAM1,APOB,MMP3,PLA2G10,CMA1,MMP 13,ALOX12,PLA2G7,PDGFC,PRDX6,COL1A2,LYZ ,Pla2g2a,TGFB1,COL18A1,PLA2G12A,ALOX15,V CAM1,CXCR4,PLA2G3,F3,PDGFB,TPSAB1/TPSB 2,SELPLG,COL1A1,ITGB2,COL5A3,ALB,SELP,IL1 B,TNF,MMP9,CLU |
| Granulocyte Adhesion and Diapedesis | 13.1 | 40/179 (0.223) | MMP7,ICAM1,MMP3,CLDN15,IL1RL1,MMP14,MM P13,CCL22,CCL5,Cxcl9,IL1R2,CLDN4,SDC2,CCL 28,NGFR,CXCL14,MMP11,TNFRSF1B,MMP12,Ccl 6,MMP17,TNFRSF11B,VCAM1,CXCR4,PF4,MMP1 0,THY1,MMP2,CXCL6,SELPLG,ITGB2,CXCL16,IT GAM,SELP,CDH5,CLDN8,IL1B,TNF,MMP9,HSPB1 |
| LPS/IL-1 Mediated Inhibition of RXR Function | 12.9 | 45/224 (0.201) | GAL3ST2,APOE,CYP3A7,CHST4,GSTM5,IL1RL1, ABCC2,CYP2C9,ABCG1,HMGCS2,SOD3,ABCA1, CHST2,IL1R2,GSTT2/GSTT2B,MAOB,ALDH1A1, ACSBG1,ALDH1A3,Gstm3,NGFR,PPARGC1B,CH ST11,FABP5,LBP,TNFRSF1B,ALDH6A1,TNFRSF 11B,GSTA3,GSTM1,ALDH1B1,ABCB1,MGST1,SU LT1C2,CHST12,Sult1d1,SULT2B1,HS3ST3B1,FA BP2,ALDH1A2,IL1B,ABCC3,TNF,SULT1B1,MAO A |
| Agranulocyte Adhesion and Diapedesis | 11.5 | 39/190 (0.205) | MMP7,ICAM1,FN1,MMP3,CLDN15,MMP14,MMP1 3,CCL22,MYH11,CCL5,Cxcl9,CLDN4,CCL28,CXC L14,MMP11,ACTG2,MMP12,Ccl6,MMP17,ACTA1, VCAM1,CXCR4,PF4,MYH14,MMP10,MMP2,CXCL 6,SELPLG,MYL7,MYL9,ITGB2,CXCL16,SELP,CDH 5,CLDN8,ACTA2,IL1B,TNF,MMP9 |
| Xenobiotic Metabolism Signaling | 7.73 | 41/274 (0.15) | GAL3ST2,CYP3A7,CHST4,GSTM5,CAMK1D,ABC C2,CYP2C9,SOD3,CHST2,HMOX1,GSTT2/GSTT2 B,MAOB,CES1,ALDH1A1,ALDH1A3,Gstm3,Ugt1a 7c,CHST11,PRKCE,NOS2,PRKD1,ALDH6A1,GST A3,GSTM1,ALDH1B1,ABCB1,MGST1,SULT1C2,U GT2B10,CHST12,Sult1d1,SULT2B1,Ces1e,HS3S T3B1,PRKCD,ALDH1A2,IL1B,ABCC3,TNF,SULT1 B1,MAOA |
| Leukocyte Extravasation Signaling | 7.69 | 34/204 (0.167) | RAC2,MMP7,ICAM1,MMP3,CLDN15,MMP14,MMP 13,CLDN4,CYBA,CYBB,PRKCE,MMP11,ACTG2,M MP12,PRKD1,MMP17,ACTA1,VCAM1,CXCR4,TH Y1,MMP10,PLCG1,MMP2,NCF4,SELPLG,ITGB2,N CF1,ITGAM,EDIL3,CDH5,CLDN8,ACTA2,PRKCD, MMP9 |
| Role of Macrophages , Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 7.43 | 43/302 (0.142) | FZD10,SOCS3,TCF4,FN1,ICAM1,MMP3,IL1RL1,L TB,MMP13,WNT6,CCL5,PDGFC,FCGR1A,CCND1, PGF,PLCD1,IL1R2,WIF1,PLCE1,TGFB1,DKK3,NG FR,TLR7,DKK2,PRKCE,TNF RSF1B,NOS2,FCGR3A/FCGR3B,PRKD1,TNFRSF 11B,ADAMTS4,VCAM1,PLCG1,CREB3L4,PDGFB, TLR2,PRKCD,Tlr13,IL1B,LEF1,TNF,Tcf7,WNT5A |
| Complement System | 7 | 13/38 (0.342) | C1R,C4A/C4B,ITGB2,SERPING1,ITGAM,C3,C1QA ,C1QC,CFH,C1QB,C3AR1,C2,ITGAX |
| Axonal Guidance Signaling | 6.9 | 54/440 (0.123) | RAC2,ADAMTS8,BMP4,MMP13,WNT6,ADAMTS2, ADAM8,PLCE1,CFL2,PTCH2,PRKD1,ADAMTS4,T UBB3,PAPPA,MMP2,RAC3,PDGFB,MYL7,MYL9,A DAMTS6,ADAM12,PRKCD,EPHA2,FZD10,ADAMT S7,MMP7,BMP3,EGF,PDGFC,ROBO1,SEMA4C,P GF,TUBB2B,PLCD1,SDC2,NGFR,PRKCE,PLXNB1 ,MMP11,TUBB4A,SEMA4A,SEMA3F,BMP1,GNG4, PLXNC1,CXCR4,MMP10,PLCG1,PLXND1,SEMA4 G,BMP7,MMP9,SEMA7A,WNT5A |
3.3. Overview of differentially expressed genes regulated by the treatment of ASA, CUR, and their combination compared to the model group
To determine how ASA, CUR, and their combination exerted a preventive effect in AOM/DSS-induced CAC, we compared the global gene expression profiles of AOM/DSS-induced tumors to those treated by ASA, CUR, or their combination. A cut-off value of a log2 fold change greater than 1 and an FDR less than 0.05 were used to extract the differentially expressed genes. We identified 99 differentially expressed genes in the comparison of tumors from ASA 0.02% treated mice versus tumors from the model group (64 genes were up-regulated by ASA treatment, while 35 genes were down-regulated by ASA treatment). We observed 189 genes with differential expression in comparison with tumors from CUR 2% treated mice versus tumors from the model group (108 genes were up-regulated by CUR treatment, while 81 genes were down-regulated by CUR treatment). The combination of ASA and CUR at only half the dose of the single compound was found to modulate more genes than ASA or CUR alone when compared to the model group. A total of 344 genes that showed significantly differential expression levels were identified (155 genes were up-regulated while 189 genes were down-regulated by combination treatment compared to the model group). This result indicated that dietary administration of CUR at 2% alone modulated a larger number of genes than ASA at 0.02%, whereas the combined treatment was able to regulate an even broader gene set. Of the genes increased by ASA, 56% (36/64) were also up-regulated by the combination of ASA and CUR (Figure 3A), whereas 74% (26/35) of the down-regulated genes in tumors from ASA-treated mice were also decreased in the tumors from combination-treated animals (Figure 3B). However, the differentially expressed genes regulated by CUR treatment showed less commonality with those regulated by the combination treatment. As shown in Figure 3A and B, only 22% (24/108) and 48% (36/81) of the up-regulated and down-regulated genes, respectively, also appeared in the subset of the differentially expressed genes regulated by the combination treatment.
Figure 3. Overview of the genes regulated by ASA and CUR, alone or in combination, compared to the model group. (A, B).

Venn diagrams comparing the number of up-regulated genes (A) and down-regulated genes (B) in tumors from mice treated with 0.02% ASA, 2% CUR, or 0.01% ASA+1% CUR compared to tumors from mice treated with AOM/DSS alone. Genes with log2 fold changes greater than 1 and an FDR less than 0.05 were counted. (C) Heat map of 32 genes with differential expression that appeared in all three treatment groups (ASA, CUR, and ASA+CUR) compared to the model group.
In addition, we identified a total of 32 genes that appeared in all three treatment groups (ASA, CUR, and their combination) compared to the model group. Of the differentially expressed genes in these three groups, 81% (26/32) showed the same direction of regulation (either up-regulated or down-regulated). As shown in the heat map (Figure 3C), the color of the combination group was overall slightly brighter than that of the ASA and CUR groups, suggesting that combined treatment with ASA and CUR may regulate the expression of these shared genes at a higher fold change than treatment with the single compound. We randomly selected 6 genes from this set of shared genes and validated their expression in tumor samples from the model, ASA, CUR, and ASA+CUR groups using qPCR analysis. As shown in Figure 4, the fold change determined by qPCR (bar with black color) was consistent with that observed in RNA-seq (bar with white color) for all 6 selected genes, confirming the quantitative properties of the RNA-seq analysis used in this study. Furthermore, it was found that ASA at 0.02% and CUR at 2% could down-regulate the expression of Alb and Mfap4, and the combination treatment (ASA 0.01%+CUR 1%) resulted in the lowest expression of Alb and Mfap4 among the four groups. The mRNA expression of Krt36, Tacstd2, Hoxd10, and Hoxd13 was up-regulated by treatment with ASA, CUR, or their combination compared with their levels in tumors in the model group, whereas the combination treatment was able to induce this mRNA expression to a slightly higher level than was induced by the single treatment.
Figure 4. Validation of the mRNA expression of selected genes regulated by ASA, CUR, or their combination compared to the model group.

mRNA isolated from tumors in mice from the model, 0.02% ASA, 2% CUR, and 0.01% ASA+1% CUR groups was subjected to qPCR analysis. Black bar: the qPCR results are presented as the fold change compared with the model group using Gapdh as the endogenous control. White bar: fold change from the RNA-seq analysis. The data are presented as the mean ± SD (n = 2).
Altogether, these data showed that combination treatment with ASA and CUR at half of the dose of the single compound had an impact on more gene targets than ASA alone or CUR alone. In addition, for the gene sets regulated by all three treatments (ASA, CUR, and their combination), the combination treatment resulted in a slightly higher fold change than the single compound.
3.4. Top differentially expressed genes and canonical pathways modulated by ASA, CUR, and their combination
Upon further examination of the significant differentially expressed gene profiles of tumors treated by ASA, CUR, or their combination compared to those in the model group, we listed the top 10 down-regulated or up-regulated genes (ranked by fold change) for these three treatments, as shown in Table 3 and Table 4. A fold change lower than 1 indicated that the expression of the gene was decreased by this particular treatment compared to the expression in the model group, whereas a fold change higher than 1 suggested an elevated mRNA expression in tumors receiving that treatment compared to the expression with AOM/DSS alone. Interestingly, Alb was among the top 10 down-regulated genes in all the three comparisons (Table 3), and its relative expression was only 0.185, 0.134, and 0.032 in tumors treated by ASA, CUR, and ASA+CUR, respectively (expression of Alb in AOM/DSS alone was set as 1). As shown in Figure 4A, the expression of Alb was validated in tumor samples by qPCR analysis, confirming that its expression was decreased by these treatments and that the combination was able to decrease its expression to a higher magnitude. Similarly, Hoxd13 was among the top 10 up-regulated genes in all three comparisons (Table 4), and its expression was 9.044-, 8.497-, and 18.126-fold higher in tumors treated by ASA, CUR, and the combination, respectively, compared to the expression in the model group, and this trend was also confirmed by qPCR analysis (Figure 4F).
Table 3.
Top 10 down-regulated genes in tumors from mice treated by ASA, CUR, and their combination compared to Model group. The expression of these genes in Model group was set to 1.
| Gene ID | Gene name | Fold Change | FDR |
|---|---|---|---|
| ASA | |||
| IGFBP2 | insulin-like growth factor binding protein 2 | 0.078 | 4.94E-02 |
| Krt10 | keratin 10 | 0.141 | 2.12E-03 |
| JCHAIN | joining chain of multimeric IgA and IgM | 0.184 | 2.12E-03 |
| ALB | albumin | 0.185 | 2.12E-03 |
| MZB1 | marginal zone B and B1 cell-specific protein | 0.206 | 2.12E-03 |
| DES | desmin | 0.207 | 2.12E-03 |
| HSPA1A | heat shock 70kDa protein 1A | 0.217 | 3.89E-03 |
| FMOD | fibromodulin | 0.225 | 3.37E-02 |
| NOV | nephroblastoma overexpressed | 0.230 | 2.12E-03 |
| HSPA1B | heat shock protein 1B | 0.242 | 2.12E-03 |
| CUR | |||
| Defa3 | defensin, alpha, 3 | 0.048 | 6.89E-03 |
| FMOD | fibromodulin | 0.051 | 2.12E-03 |
| PSCA | prostate stem cell antigen | 0.069 | 4.86E-02 |
| GPC3 | glypican 3 | 0.071 | 2.12E-03 |
| AFM | afamin | 0.095 | 3.18E-02 |
| ANGPT4 | angiopoietin 4 | 0.095 | 1.68E-02 |
| ALB | albumin | 0.134 | 2.12E-03 |
| Hspa1b | heat shock protein 1B | 0.139 | 2.12E-03 |
| HSPA1A | heat shock 70kDa protein 1A | 0.159 | 2.12E-03 |
| MFAP4 | microfibrillar-associated protein 4 | 0.164 | 2.12E-03 |
| ASA+CUR | |||
| B3GNT6 | UDP-GlcNAc:betaGal beta-1,3-Nacetylglucosaminyltransferase 6 | 0.032 | 8.28E-03 |
| ALB | albumin | 0.032 | 2.12E-03 |
| GPC3 | glypican 3 | 0.046 | 2.12E-03 |
| TMIGD1 | transmembrane and immunoglobulin domain containing 1 | 0.046 | 5.44E-03 |
| Apol7e | apolipoprotein L 7e | 0.050 | 2.12E-03 |
| KRT1 | keratin 1, type II | 0.070 | 4.39E-02 |
| B4GALNT | beta-1,4-N-acetyl-galactosaminyl transferase 2 | 0.072 | 2.12E-03 |
| ACTA1 | actin, alpha 1, skeletal muscle | 0.073 | 2.12E-03 |
| MFAP4 | microfibrillar-associated protein 4 | 0.077 | 2.12E-03 |
| FABP2 | fatty acid binding protein 2, intestinal | 0.090 | 2.12E-03 |
Table 4.
Top 10 up-regulated genes in tumors from mice treated by ASA, CUR, and their combination compared to Model group. The expression of these genes in Model group was set to 1.
| Gene ID | Gene name | Fold Change | FDR |
|---|---|---|---|
| ASA | |||
| LTF | lactotransferrin | 11.096 | 2.12E-03 |
| LIX1 | limb and CNS expressed 1 | 10.541 | 1.68E-02 |
| KRT5 | keratin 5, type II | 9.044 | 2.12E-03 |
| HOXD13 | homeobox D13 | 9.044 | 2.12E-03 |
| ISL1 | ISL LIM homeobox 1 | 8.982 | 2.12E-03 |
| HOXD10 | homeobox D10 | 8.938 | 1.45E-02 |
| TRPV6 | transient receptor potential cation channel, subfamily V, member 6 | 8.179 | 2.11E-02 |
| KRT84 | keratin 84, type II | 7.890 | 2.12E-03 |
| ATP12A | ATPase, H+/K+ transporting, nongastric, alpha polypeptide | 7.600 | 2.12E-03 |
| GBP2 | Guanylate binding protein 2 | 7.321 | 2.12E-03 |
| CUR | |||
| HOXD12 | homeobox D12 | 12.951 | 4.62E-02 |
| HOXD13 | homeobox D13 | 8.497 | 2.12E-03 |
| TRPV6 | transient receptor potential cation channel, subfamily V, member 6 | 7.989 | 2.11E-02 |
| IDO1 | indoleamine 2,3-dioxygenase 1 | 7.989 | 2.12E-03 |
| SLFN12L | schlafen family member 12-like | 7.765 | 2.12E-03 |
| HOXD10 | homeobox D10 | 7.738 | 1.68E-02 |
| GABRA4 | gamma-aminobutyric acid (GABA) A receptor, alpha 4 | 7.280 | 1.45E-02 |
| BTNL2 | butyrophilin-like 2 | 7.190 | 3.28E-02 |
| Tgtp1 | T cell specific GTPase 1 | 6.723 | 2.12E-03 |
| Cxcl9 | chemokine (C-X-C motif) ligand 9 | 6.498 | 2.12E-03 |
| ASA+CUR | |||
| HOXD12 | homeobox D12 | 48.068 | 6.89E-03 |
| KLK15 | kallikrein-related peptidase 15 | 45.192 | 2.21E-02 |
| LTF | lactotransferrin | 21.511 | 2.12E-03 |
| CNTN3 | contactin 3 (plasmacytoma associated) | 21.511 | 1.79E-02 |
| KRT5 | keratin 5, type II | 20.649 | 2.12E-03 |
| SHH | sonic hedgehog | 20.224 | 8.28E-03 |
| HOXD13 | homeobox D13 | 19.685 | 2.12E-03 |
| HOXD10 | homeobox D10 | 18.126 | 5.44E-03 |
| Xlr3c | X-linked lymphocyte-regulated 3C | 16.656 | 1.22E-02 |
| PSCA | prostate stem cell antigen | 16.427 | 2.12E-03 |
Among the top 10 down-regulated genes in the ASA+CUR treated group, the expression of genes such as B3gnt6, Alb, Gpc3, Tmigd1, and Apol7e was more than 20-fold lower than that in tumors from the AOM/DSS group without any treatment. Similarly, the combined treatment with ASA and CUR increased the mRNA expression of Hoxd12, Klk15, Ltf, Cntn3, Krt5, and Shh by more than 20-fold compared to treatment with AOM/DSS alone. Because the combination treatment with ASA and CUR at half the dose of the single treatment effectively prevented colitis and colon carcinogenesis in our study, modulated broader targets, and induced/suppressed genes at higher fold change, we continued to investigate the possible biological function and pathways that were influenced by the combination regimen. Similar to the canonical pathway analysis we performed using the contrast of the control versus AOM/DSS-induced tumors, we were also interested in finding the significant pathways associated with the alterations of gene expression in ASA+CUR-treated tumors versus those treated with AOM/DSS alone. We identified a total of 235 canonical pathways significantly associated with the differentially expressed genes in the combination compared to the model group, with a P-value less than 0.05. The top 10 most significant pathways are displayed in Table 5. Interestingly, 6 of the top 10 pathways modulated by combination treatment compared to AOM/DSS alone, including hepatic fibrosis/hepatic stellate cell activation; agranulocyte adhesion and diapedesis; granulocyte adhesion and diapedesis; atherosclerosis signaling; LPS/IL-1 mediated inhibition of RXR function; and the role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis, were also recognized as the top 10 pathways associated with differentially expressed genes in AOM/DSS-induced tumors compared to normal colonic tissue. This finding may suggest that these pathways not only play an important role in the carcinogenesis process induced by AOM/DSS but also contain the molecular targets potentially modulated by the combination treatment. Notably, some of the molecules in these pathways showed the opposite direction of change when contrasting the combination versus the model and the model versus the control group. For example, Mmp9 was significantly up-regulated in AOM/DSS-induced tumors compared to its expression in normal tissue, whereas its expression was down-regulated in tumors from the combination treatment compared to that in AOM/DSS-induced tumors (Table 2 and Table 5).
Table 5.
The 10 most significant canonical pathways regulated by ASA 0.01%+CUR 1% compared to tumors in Model group. Genes in bold are down-regulated in the combination treatment.
| Canonical Pathways | −log (p value) | Ratio | Target genes |
|---|---|---|---|
| Hepatic Fibrosis / Hepatic Stellate Cell Activation | 6.75 | 15/18 7 (0.08) | IL1RL1,BAMBI,MMP13,MYH11,CCL5,MYL 7,MYL9,CXCL3,NGFR,HGF,SERPI,NE1,C OL9A2,MMP9,COL7A1,TNFRSF11B |
| Agranulocyte Adhesion and Diapedesis | 5.9 | 14/19 0 (0.07) | CLDN15,Ppbp,ITGA6,MMP13,MYH11,CCL 5,MYL7,MYL9,CXCL3, Cxcl3,Ccl6,ACTA1,MMP9,MMP19 |
| Granulocyte Adhesion and Diapedesis | 5.45 | 13/17 9 (0.07) | CLDN15,IL1RL1,Ppbp,ITGA6,MMP13,CCL 5,CXCL3,NGFR,Cxcl3,Ccl6,MMP9,MMP19 ,TNFRSF11B |
| LXR/RXR Activation | 4.59 | 10/12 8 (0.08) | ALB,APOB,IL1RL1,NGFR,SAA1,LPL,PTG S2,NOS2,MMP9,TNFRSF11B |
| Atherosclerosis Signaling | 3.92 | 9/125 (0.07) | ALOX15,ALB,APOB,LPL,MMP13,ALOX12 ,F3,TPSAB1/TPSB2,MMP9 |
| Glucocorticoid Receptor Signaling | 3.43 | 13/28 1 (0.05) | Hspa1b,HSPA1A/HSPA1B,TAT,PCK1,CC L5,KRT36,CD3G,CXCL3,ANXA1,PTGS2,N RIP1,NOS2,SERPINE1 |
| Role of IL-17A in Arthritis | 2.81 | 5/56 (0.09) | CXCL3,MMP13,CCL5,PTGS2,NOS2 |
| LPS/IL-1 Mediated Inhibition of RXR Function | 2.65 | 10/22 4 (0.04) | ALDH1B1,HS3ST3B1,FABP2,ALDH1A3,IL 1RL1,NGFR,FABP4,CYP2C9,HMGCS2,T NFRSF11B |
| Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 2.64 | 12/30 2 (0.04) | WNT10A,IL1RL1,NGFR,DKK2,LTB,MMP1 3,FZD9,SFRP1,CCL5,NOS2,FZD2,TNFRS F11B |
| Role of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis | 2.63 | 10/22 5 (0.04) | WNT10A,IL1RL1,NGFR,DKK2,MMP13,FZ D9,SFRP1,FZD2,IL11,TNFRSF11B |
3.5. The subset of genes modified by AOM/DSS-induced tumors was also influenced by ASA+CUR
In light of the finding that some molecules in the shared pathways changed in the opposite direction, we further compared the significant differentially expressed genes in the following two data sets: the model versus the control and the combination versus the model groups. A total of 54 genes were found to be regulated by AOM/DSS and the combination treatment in the opposite direction (Figure 5). Specifically, 13 genes that were significantly down-regulated by AOM/DSS alone (the expression in the control colon tissue was set to 1) were further up-regulated by the combination treatment when we set the expression in the AOM/DSS group to 1. Forty-one significantly up-regulated genes in tumors induced with AOM/DSS alone were decreased by the combined treatment of ASA+CUR. Thus, these 54 genes might represent a set of molecular targets that underlies the preventive action of the combination of ASA+CUR in the AOM/DSS-induced model.
Figure 5. The list of 54 genes that showed regulation in the opposite direction when comparing the model versus the control and the combination versus the model groups.

There were 13 genes that were down-regulated in the model group compared to the control group, and their expression was up-regulated by the combination of ASA and CUR. There were 41 genes that were up-regulated in the model group compared to control group, and their expression was down-regulated by the combination of ASA and CUR.
Given the potential functional role of the set of genes presented in Figure 5, we randomly selected 14 genes and validated their expression using qPCR analysis. As shown in Figure 6, the trend of fold change determined by qPCR (black bar) was overall in accordance with the fold change observed by RNA-seq (white bar). Among these 14 genes, the relative mRNA expression of Hoxb13 and Ngfr was suppressed in AOM/DSS-induced tumors, whereas ASA+CUR treatment could alleviate this suppression. Furthermore, the mRNA expression of Ccl5, Esam, Fmod, Ggt1, Ltb, Mmp9, Alb, Mfap4, Sox18, Slc38a4, Gpc3, and Nos2 were all increased in AOM/DSS-induced tumors, although combined treatment with ASA+CUR could ameliorate this induction.
Figure 6. Validation of the mRNA expression of selected genes that showed regulation in the opposite direction when comparing AOM/DSS alone and the combination of ASA and CUR.

mRNA isolated from the colonic tissues of mice from the control group and tumors from mice receiving 0.01%ASA+1% CUR was subjected to qPCR analysis. Black bar: the qPCR results are presented as the fold change compared with the control group using Gapdh as the endogenous control. White bar: fold change from the RNA-seq analysis. The data are presented as the mean ± SD (n = 2).
4. Discussion
In the present study, we investigated the preventive effect of dietary administration of ASA and CUR in AOM/DSS-induced CAC in C57/BL6 mice. Although oral feeding of CUR at a dose of 2% has been shown to prevent tumor formation in AOM-induced mice and Apcmin/+ mice previously (14,32), our study provides the first evidence that dietary administration of CUR (2%) effectively suppresses tumor multiplicity and tumor incidence in inflammation-related CRC induced by AOM/DSS (Figure 2B–C). However, to our surprise, ASA treatment in the current study in C57BL/6 mice was not as effective as in our recent study in AOM/DSS-induced CF-1 mice (12). In addition, another recent study found that ASA at a similar dose failed to significantly inhibit the tumor number in AOM/DSS-induced Balb/c mice (33). The discrepancy in the effect of ASA in these two previous studies and our current study could possibly be explained by the presence of strain differences in the susceptibility to AOM/DSS-induced colonic tumorigenesis (34), suggesting that the chemopreventive effect of ASA in AOM/DSS-induced CAC might be strain specific. Further studies comparing the preventive effect of ASA against CRC in different strains of animals and different ethnicities of patients should be considered.
The highlight of the current study is that we investigated the effect of concomitant administration of ASA and CUR in an AOM/DSS-induced CAC model for the first time. Our results indicate that combined treatment with ASA and CUR is effective at reducing tumor incidence and tumor multiplicity (Figure 2B–C). Although mice in the combination group received a low dose of ASA (0.01%) and CUR (1%), the tumor incidence and multiplicity were similar to those for CUR alone at a higher dose (2%) and much lower than those for ASA alone at 0.02%. Notably, the equivalent human dose of the combinational treatment in the present study is approximately 55 mg/day ASA + 5.5 g/day CUR, which is more feasible and will possibly lead to less adverse effects from chronic use of ASA in humans compared to single treatment with ASA at 110 mg/day or CUR at 11 g/day. With the help of RNA-seq, we established, for the first time, a global transcriptome profile associated with ASA, CUR, and their combination in AOM/DSS-induced tumors. When comparing the gene expression profiles of tumors from these three treatment groups (ASA, CUR, and both) to those from AOM/DSS induction alone without any intervention, we found that the combination treatment, even at a lower dose, had an impact on a larger gene set (344 differentially expressed genes) than for ASA alone (99 differentially expressed genes) or CUR alone (189 differentially expressed genes) (Figure 3A–B). In addition, when we looked at a smaller set of significant differentially expressed genes modulated by all three treatment groups (ASA, CUR, and their combination), the combined treatment with ASA+CUR resulted in a slightly higher fold change than ASA alone or CUR alone (Figure 3C and 4). These results suggest that co-administration of ASA and CUR at a lower dose may provide a promising preventive regimen against CAC, possibility by targeting more molecular targets and magnifying the fold change of certain genes. Interestingly, other than the impact at the molecular level, as we showed in the current study by combination of ASA and CUR, a previous study reported that the stability of CUR could be improved in the presence of ASA, whereas the stability of ASA was not affected by the presence of CUR (35). In addition, the cellular uptake of CUR and the cytotoxicity of CUR in HCT116 cells could be enhanced when incubated with ASA (35). Moreover, it was postulated that the acidic properties and antioxidant potential of ASA could provide favorable conditions for stabilizing CUR and prevent the degradation of CUR (36). Therefore, further studies should be carried out to determine if co-administration of ASA and CUR could enhance the absorption and half-life of CUR in humans.
In addition to investigating the effect of the chemopreventive agents ASA, CUR, and their combination in the AOM/DSS-induced CAC model, the present study also aimed to identify the global profile of gene expression changes related to AOM/DSS-induced tumors. The top-ranked genes with decreased or increased expression that are listed in Table 1 may provide novel insight to facilitate the discovery of critical genes driving the carcinogenesis process in AOM/DSS-induced CAC as well as potential therapeutic targets and biomarkers for the prevention of CAC. For example, the expression levels of two matrix metalloproteinases [MMP7 (fold change = 389.9) and MMP10 (fold change = 91.5)] were dramatically up-regulated in AOM/DSS-induced tumors compared to the levels in normal colonic tissue (Table 1). MMPs comprise a large family of zinc-dependent endopeptidases that are involved in the physiological and pathological remodeling of the extracellular matrix in proliferation, angiogenesis, tumor invasion, and metastasis (37). However, their function in inflammation-associated colorectal cancer remains largely unknown. Our observation of increased expression of MMP7 in tumors from AOM/DSS-treated mice was in accordance with various previous studies showing that MMP7 is overexpressed in advanced stages of CRC (38) and is a potential prognostic marker (39,40). On the contrary, the function of MMP10 in CRC is more ambiguous. Although the overexpression of MMP10 in the serum of CRC patients is considered to be a prognostic marker (40) and the expression of MMP10 is up-regulated in DSS-induced colitis in mice (41), MMP10 knockout mice develop more severe colitis after DSS exposure, suggesting that MMP10 may play a beneficial role in favor of colitis resolution (41). Our results show for the first time that MMP10 is dramatically elevated in AOM/DSS-induced tumors; however, the mechanisms underlying the role of MMP10 in tumorigenesis and metastasis require further investigation. In addition, chemokine (C-X-C motif) ligand 6 (CXCL6) is another target that showed significantly higher expression in tumors than in normal tissue (fold change = 181.9, Table 1). CXCL6 belongs to the family of ELR+ CXC chemokines that play important roles in the activation and recruitment of neutrophils at sites of inflammation (42), and CXCL6 has been shown to be overexpressed in the inflamed tissue of IBD patients (43). Nevertheless, Rubie et al. found that although other members of the ELR+ CXC chemokine family (CXCL1 and CXCL5) were up-regulated in colorectal adenoma and carcinoma tissue specimens, the expression of CXCL6 was not significantly altered (44). Thus, it is possible that CXCL6 only plays a pivotal role in CRC associated with IBD instead of hereditary CRC, but this hypothesis requires further investigation. Furthermore, a member of the carbonic anhydrases, CA III, was shown for the first time to be down-regulated in AOM/DSS-induced tumors (fold change = 0.028, Table 1). CA isozymes have been considered to be important players in maintaining the pH homeostasis in tumors and thereby modulate the behavior of cancer cells. Among these isozymes, CA I, II, IV, VII, and XIII were implicated as potential tumor suppressors in CRC with down-regulated expression in CRC specimens compared to normal tissue (45–47). Specifically, it was found that promoter hypermethylation may contribute to the silence of CA IV in CRC, where its tumor suppressor action involves the inhibition of the Wnt signaling pathway (46). The expression of CA III may be associated with the invasiveness and metastasis of liver cancer (48); however, its role in CRC has not yet been investigated. Based on our current observation that the expression of CA III was down-regulated in AOM/DSS-induced tumors, the tumor suppressive potential of CA III and the mechanism leading to the inactivation of CA III in CAC should be explored in human specimens.
Our canonical pathway analysis highlighted “Hepatic Fibrosis/Hepatic Stellate Cell Activation” as the most significantly regulated pathway influenced by AOM/DSS-induced tumors (Table 2). Interestingly, this pathway was also recognized as the most significant pathway containing the differentially expressed genes regulated by ASA+CUR compared to the model group (Table 5). Hepatic stellate cells (HSCs) are considered to be critical players in colon cancer-induced liver metastasis. It was found that colonic tumor-derived factors lead to the activation of HSCs in the liver, and in turn, activated HSCs promote hepatic fibrosis and produce cytokines, chemokines, and matrix-degrading MMPs to enhance metastatic growth in the liver (49). Our results show that up-regulation of cytokines (such as CCL5, IL1B, and TNF), growth factors (such as PGF and TGFB1), and MMPs (such as MMP2, MMP9, and MMP13) in colonic tumors may be involved in the activation of HSCs in the liver in AOM/DSS-induced CAC. In addition, concomitant administration of ASA and CUR significantly down-regulated the expression of CCL5 and MMP9, which showed elevated expression in AOM/DSS-induced colonic tumors in the hepatic fibrosis/HSC activation pathway. However, our current study did not examine the effects of ASA, CUR, or their combination on the inhibition of colon cancer-induced liver metastasis, which may be worth investigating in the future. Other canonical pathways worth noting are the “Agranulocyte Adhesion and Diapedesis” and “Granulocyte Adhesion and Diapedesis” pathways. These two pathways are associated with the migration of leukocytes and immune cells from the vascular system to sites of inflammation. Our results suggested that these two pathways were the top significant pathways involved in AOM/DSS-induced colonic tumorigenesis and contain the molecular targets that underlie the preventive action of the combined treatment of ASA and CUR (Table 2 and 5). Further research is warranted to understand the pivotal role of agranulocytes, granulocytes, and their mediators in the constitution of the tumor microenvironment during the progression of CAC and how the combination of ASA and CUR suppresses CAC by modulating the infiltration of these inflammatory cells.
Additionally, we identified a subset of 54 differentially expressed genes as the potential molecular targets underlying the protective action of the concomitant administration of ASA and CUR in AOM/DSS-induced CAC. Among these genes, the overexpression of 41 genes was found in AOM/DSS-induced tumors, while their expression was down-regulated by ASA+CUR (Figure 5). Some of these genes, such as REG3A (50), MMP9 (51), NOS2 (12), CCL5 (52), LTB (53), DUOXA2 (54), and BST2 (55), were also found to be overexpressed in AOM/DSS-induced CAC, CRC specimens, or CRC cell lines in previous reports, and inhibition of these targets has been implicated as a promising therapeutic strategy in CRC. On the contrary, 13 genes were identified with a decreased expression in tumors from the model group, and their expression was restored by the combination of ASA and CUR (Figure 5). Some of these genes, such as HOXB13 (56) and NGFR (57), have been suggested to be tumor suppressors with diminished expression in CRC in previous studies. The modulation of the genes listed in Figure 5, except for MMP9 and NOS2 (12), following treatment with ASA, CUR, or the combination of ASA and CUR has not been investigated previously. However, we also noticed that the alterations of several genes in our list were not in accordance with previous reports. For example, ANO1, a gene with decreased expression in AOM/DSS-induced tumors in our study, was reported to be overexpressed in CRC cell lines (58). In addition, GPX3 expression was increased in tumors from the model group, whereas Barrett et al. showed that GPX3 was down-regulated in AOM/DSS-treated mice (59). Although further detailed mechanistic studies are needed, our current study provides a novel list of genes that may be responsible for the preventive effect of concomitant administration of ASA and CUR in AOM/DSS-induced CAC.
Unlike the 54 genes regulated by AOM/DSS versus the ASA/CUR combination treatment displayed opposite direction, we observed 104 genes showed same direction of regulation by AOM/DSS and the combination treatment (data not shown). Interestingly, several tumor suppressor genes were in this set of genes and their expression was down-regulated by AOM/DSS and concomitant administration of ASA and CUR could further decrease their expression in tumors. For instance, the relative expression of CDX2 (a widely known tumor suppressor gene (60), with decreased expression in ~30% human CRC (61)) was 0.48 in AOM/DSS-induced tumors and 0.05 in tumors from combination group (the expression of CDX2 in control group was set as 1). The inhibition of these tumor suppressor genes by the combination treatment may be one of the reasons for the tumor growth in the presence of these chemopreventive agents ASA/CUR. In addition, the expression of several membrane transporters in tumors was altered by the combination of ASA and CUR, which could change the influx or efflux of the chemopreventive agents in tumor cells. The altered expression of transporters could also influence the uptake of essential nutrients for tumor growth and survival, therefore making the tumors resistant to ASA and CUR and leading to the tumor growth in the presence of chemopreventive agents.
In summary, our study is the first to show that concomitant administration of ASA 0.01% + CUR 1% effectively attenuates tumor growth in the AOM/DSS-induced CAC model. Our results provide a quantitative gene expression profile of AOM/DSS-induced tumors as well as tumors from mice treated with ASA, CUR, and their combination at half of the dose. Furthermore, a small set of genes was postulated as potential molecular targets involved in the action of ASA+CUR in the prevention of AOM/DSS-induced CAC. These findings provide novel insights that further the understanding of the carcinogenesis of inflammatory CRC as well as the mechanisms underlying the preventive effect of ASA and CUR in CAC.
Acknowledgments
Funding
This work was supported by R01AT007065 from the National Center for Complementary and Alternative Medicines (NCCAM) and the Office of Dietary Supplements (ODS).
We thank Dr. Guangxun Li for his assistance in the histology evaluation. We also thank Drs. Mou-Tuan Huang and Tin Oo Khor for their help in the animal study. The authors are grateful to Sabinsa Corporation for providing Curcumin C3 Complex®. We thank all of the members of Dr. Kong’s laboratory for their helpful discussions and the preparation of this manuscript.
Footnotes
Conflict of Interest Statement
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Howlader NNA, Krapcho M. Institute, N.C, editor . SEER Cancer Statistics Review. 2012. [Google Scholar]
- 2.Hanahan D, et al. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Lasry A, et al. Inflammatory networks underlying colorectal cancer. Nat Immunol. 2016;17:230–40. doi: 10.1038/ni.3384. [DOI] [PubMed] [Google Scholar]
- 4.Feagins LA, et al. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- 5.Soderlund S, et al. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology. 2009;136:1561–7. doi: 10.1053/j.gastro.2009.01.064. quiz 1818–9. [DOI] [PubMed] [Google Scholar]
- 6.De Robertis M, et al. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sporn MB, et al. Chemoprevention: an essential approach to controlling cancer. Nat Rev Cancer. 2002;2:537–43. doi: 10.1038/nrc844. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Albeniz X, et al. Aspirin for the prevention of colorectal cancer. Best Pract Res Clin Gastroenterol. 2011;25:461–72. doi: 10.1016/j.bpg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Algra AM, et al. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13:518–27. doi: 10.1016/S1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- 10.Burn J, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081–7. doi: 10.1016/S0140-6736(11)61049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velayos FS, et al. Predictive and protective factors associated with colorectal cancer in ulcerative colitis: A case-control study. Gastroenterology. 2006;130:1941–9. doi: 10.1053/j.gastro.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 12.Guo Y, et al. The epigenetic effects of aspirin: the modification of histone H3 lysine 27 acetylation in the prevention of colon carcinogenesis in azoxymethane- and dextran sulfate sodium-treated CF-1 mice. Carcinogenesis. 2016;37:616–24. doi: 10.1093/carcin/bgw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Y, et al. Curcumin inhibits anchorage-independent growth of HT29 human colon cancer cells by targeting epigenetic restoration of the tumor suppressor gene DLEC1. Biochem Pharmacol. 2015;94:69–78. doi: 10.1016/j.bcp.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy EA, et al. Curcumin’s effect on intestinal inflammation and tumorigenesis in the ApcMin/+ mouse. J Interferon Cytokine Res. 2011;31:219–26. doi: 10.1089/jir.2010.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll RE, et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res (Phila) 2011;4:354–64. doi: 10.1158/1940-6207.CAPR-10-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pereira MA, et al. Effects of the phytochemicals, curcumin and quercetin, upon azoxymethane-induced colon cancer and 7,12-dimethylbenz[a]anthracene-induced mammary cancer in rats. Carcinogenesis. 1996;17:1305–11. doi: 10.1093/carcin/17.6.1305. [DOI] [PubMed] [Google Scholar]
- 17.Rao CV, et al. Inhibition by dietary curcumin of azoxymethane-induced ornithine decarboxylase, tyrosine protein kinase, arachidonic acid metabolism and aberrant crypt foci formation in the rat colon. Carcinogenesis. 1993;14:2219–25. doi: 10.1093/carcin/14.11.2219. [DOI] [PubMed] [Google Scholar]
- 18.Vareed SK, et al. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol Biomarkers Prev. 2008;17:1411–7. doi: 10.1158/1055-9965.EPI-07-2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu G, et al. Synergistic inhibition of lung tumorigenesis by a combination of green tea polyphenols and atorvastatin. Clin Cancer Res. 2008;14:4981–8. doi: 10.1158/1078-0432.CCR-07-1860. [DOI] [PubMed] [Google Scholar]
- 20.Zheng X, et al. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent LNCaP xenograft prostate tumors to androgen independence. Cancer Prev Res (Phila) 2010;3:114–24. doi: 10.1158/1940-6207.CAPR-09-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue W, et al. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget. 2015;6:21208–24. doi: 10.18632/oncotarget.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, et al. Effects of combination of calcium and aspirin on azoxymethane-induced aberrant crypt foci formation in the colons of mice and rats. Nutr Cancer. 2008;60:660–5. doi: 10.1080/01635580802290215. [DOI] [PubMed] [Google Scholar]
- 23.Thakkar A, et al. The molecular mechanism of action of aspirin, curcumin and sulforaphane combinations in the chemoprevention of pancreatic cancer. Oncol Rep. 2013;29:1671–7. doi: 10.3892/or.2013.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perkins S, et al. Age-related difference in susceptibility of Apc(Min/+) mice towards the chemopreventive efficacy of dietary aspirin and curcumin. Br J Cancer. 2003;88:1480–3. doi: 10.1038/sj.bjc.6600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park W, et al. New perspectives of curcumin in cancer prevention. Cancer Prev Res (Phila) 2013;6:387–400. doi: 10.1158/1940-6207.CAPR-12-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung KL, et al. Differential in vivo mechanism of chemoprevention of tumor formation in azoxymethane/dextran sodium sulfate mice by PEITC and DBM. Carcinogenesis. 2010;31:880–5. doi: 10.1093/carcin/bgp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murthy SN, et al. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38:1722–34. doi: 10.1007/BF01303184. [DOI] [PubMed] [Google Scholar]
- 31.Kubota M, et al. Preventive effects of curcumin on the development of azoxymethaneinduced colonic preneoplastic lesions in male C57BL/KsJ-db/db obese mice. Nutr Cancer. 2012;64:72–9. doi: 10.1080/01635581.2012.630554. [DOI] [PubMed] [Google Scholar]
- 32.Huang MT, et al. Inhibitory effects of dietary curcumin on forestomach, duodenal, and colon carcinogenesis in mice. Cancer Res. 1994;54:5841–7. [PubMed] [Google Scholar]
- 33.Tian Y, et al. Aspirin promotes apoptosis in a murine model of colorectal cancer by mechanisms involving downregulation of IL-6-STAT3 signaling pathway. Int J Colorectal Dis. 2011;26:13–22. doi: 10.1007/s00384-010-1060-0. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki R, et al. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis. 2006;27:162–9. doi: 10.1093/carcin/bgi205. [DOI] [PubMed] [Google Scholar]
- 35.Choi HA, et al. Interaction of over-the-counter drugs with curcumin: influence on stability and bioactivities in intestinal cells. J Agric Food Chem. 2012;60:10578–84. doi: 10.1021/jf303534e. [DOI] [PubMed] [Google Scholar]
- 36.Dinis TC, et al. Action of phenolic derivatives (acetaminophen, salicylate, and 5-aminosalicylate) as inhibitors of membrane lipid peroxidation and as peroxyl radical scavengers. Arch Biochem Biophys. 1994;315:161–9. doi: 10.1006/abbi.1994.1485. [DOI] [PubMed] [Google Scholar]
- 37.Gialeli C, et al. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 38.Polistena A, et al. MMP7 expression in colorectal tumours of different stages. In Vivo. 2014;28:105–10. [PubMed] [Google Scholar]
- 39.Koskensalo S, et al. MMP-7 as a prognostic marker in colorectal cancer. Tumour Biol. 2011;32:259–64. doi: 10.1007/s13277-010-0080-2. [DOI] [PubMed] [Google Scholar]
- 40.Klupp F, et al. Serum MMP7, MMP10 and MMP12 level as negative prognostic markers in colon cancer patients. BMC Cancer. 2016;16:494. doi: 10.1186/s12885-016-2515-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koller FL, et al. Lack of MMP10 exacerbates experimental colitis and promotes development of inflammation-associated colonic dysplasia. Lab Invest. 2012;92:1749–59. doi: 10.1038/labinvest.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D, et al. The role of chemokines in intestinal inflammation and cancer. Curr Opin Pharmacol. 2009;9:688–96. doi: 10.1016/j.coph.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gijsbers K, et al. CXCR1-binding chemokines in inflammatory bowel diseases: downregulated IL-8/CXCL8 production by leukocytes in Crohn’s disease and selective GCP-2/CXCL6 expression in inflamed intestinal tissue. Eur J Immunol. 2004;34:1992–2000. doi: 10.1002/eji.200324807. [DOI] [PubMed] [Google Scholar]
- 44.Rubie C, et al. ELR+ CXC chemokine expression in benign and malignant colorectal conditions. BMC Cancer. 2008;8:178. doi: 10.1186/1471-2407-8-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kummola L, et al. Expression of a novel carbonic anhydrase, CA XIII, in normal and neoplastic colorectal mucosa. BMC Cancer. 2005;5:41. doi: 10.1186/1471-2407-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, et al. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut. 2016;65:1482–93. doi: 10.1136/gutjnl-2014-308614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang GZ, et al. Prognostic value of carbonic anhydrase VII expression in colorectal carcinoma. BMC Cancer. 2015;15:209. doi: 10.1186/s12885-015-1216-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai HY, et al. Carbonic anhydrase III promotes transformation and invasion capability in hepatoma cells through FAK signaling pathway. Mol Carcinog. 2008;47:956–63. doi: 10.1002/mc.20448. [DOI] [PubMed] [Google Scholar]
- 49.Kang N, et al. Hepatic stellate cells: partners in crime for liver metastases? Hepatology. 2011;54:707–13. doi: 10.1002/hep.24384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye Y, et al. Up-regulation of REG3A in colorectal cancer cells confers proliferation and correlates with colorectal cancer risk. Oncotarget. 2016;7:3921–33. doi: 10.18632/oncotarget.6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garg P, et al. Matrix metalloproteinase-9 functions as a tumor suppressor in colitisassociated cancer. Cancer Res. 2010;70:792–801. doi: 10.1158/0008-5472.CAN-09-3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang LY, et al. Tumor-derived chemokine CCL5 enhances TGF-beta-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res. 2012;72:1092–102. doi: 10.1158/0008-5472.CAN-11-2493. [DOI] [PubMed] [Google Scholar]
- 53.Ihara A, et al. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J Pharmacol Sci. 2007;103:24–32. doi: 10.1254/jphs.fp0060651. [DOI] [PubMed] [Google Scholar]
- 54.MacFie TS, et al. DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5-aminosalicylic acid. Inflamm Bowel Dis. 2014;20:514–24. doi: 10.1097/01.MIB.0000442012.45038.0e. [DOI] [PubMed] [Google Scholar]
- 55.Mukai S, et al. Overexpression of Transmembrane Protein BST2 is Associated with Poor Survival of Patients with Esophageal, Gastric, or Colorectal Cancer. Ann Surg Oncol. 2016 doi: 10.1245/s10434-016-5100-z. [DOI] [PubMed] [Google Scholar]
- 56.Jung C, et al. HOXB13 is downregulated in colorectal cancer to confer TCF4-mediated transactivation. Br J Cancer. 2005;92:2233–9. doi: 10.1038/sj.bjc.6602631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Z, et al. Epigenetic inactivation and tumor-suppressor behavior of NGFR in human colorectal cancer. Mol Cancer Res. 2015;13:107–19. doi: 10.1158/1541-7786.MCR-13-0247. [DOI] [PubMed] [Google Scholar]
- 58.Sui Y, et al. Inhibition of TMEM16A expression suppresses growth and invasion in human colorectal cancer cells. PLoS One. 2014;9:e115443. doi: 10.1371/journal.pone.0115443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barrett CW, et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 2013;73:1245–55. doi: 10.1158/0008-5472.CAN-12-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hryniuk A, et al. Cdx1 and Cdx2 function as tumor suppressors. J Biol Chem. 2014;289:33343–54. doi: 10.1074/jbc.M114.583823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baba Y, et al. Relationship of CDX2 loss with molecular features and prognosis in colorectal cancer. Clin Cancer Res. 2009;15:4665–73. doi: 10.1158/1078-0432.CCR-09-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]