

Abstract
ClC-7 is a chloride channel of late endosomes and lysosomes. In osteoclasts, it may cooperate with H+-ATPases in acidifying the resorption lacuna. In mice and man, loss of ClC-7 or the H+-ATPase a3 subunit causes osteopetrosis, a disease characterized by defective bone resorption. We show that ClC-7 knockout mice additionally display neurodegeneration and severe lysosomal storage disease despite unchanged lysosomal pH in cultured neurons. Rescuing their bone phenotype by transgenic expression of ClC-7 in osteoclasts moderately increased their lifespan and revealed a further progression of the central nervous system pathology. Histological analysis demonstrated an accumulation of electron-dense material in neurons, autofluorescent structures, microglial activation and astrogliosis. Like in human neuronal ceroid lipofuscinosis, there was a strong accumulation of subunit c of the mitochondrial ATP synthase and increased amounts of lysosomal enzymes. Such alterations were minor or absent in ClC-3 knockout mice, despite a massive neurodegeneration. Osteopetrotic oc/oc mice, lacking a functional H+-ATPase a3 subunit, showed no comparable retinal or neuronal degeneration. There are important medical implications as defects in the H+-ATPase and ClC-7 can underlie human osteopetrosis.
Keywords: channelopathy, NCL/TCIRG1, transgenic rescue
Introduction
Several chloride channels of the CLC family play important roles in regulating electrical excitability and transepithelial transport. Over the past few years, however, it emerged that many CLC proteins are expressed in membranes of the endocytotic pathway (Jentsch et al, 2002). A common denominator of their function may be the electrical neutralization of protons that are actively pumped into the lumen of intracellular vesicles. The importance of luminal acidification is illustrated by the severe consequences entailed by the mutational inactivation of vesicular CLC channels in mice and men. The disruption of the endosomal ClC-5 channel led to defective renal endocytosis (Piwon et al, 2000), resulting in the complex phenotype of a kidney stone disorder (Lloyd et al, 1996). Disrupting ClC-3, a channel expressed in endosomes and synaptic vesicles, resulted in a severe degeneration of the hippocampus and in blindness (Stobrawa et al, 2001). In both cases, the acidification of specific vesicle populations was significantly reduced (Piwon et al, 2000; Stobrawa et al, 2001; Yoshikawa et al, 2002; Günther et al, 2003; Hara-Chikuma et al, 2005).
Immunocytochemistry and cell fractionation revealed that the ubiquitously expressed ClC-7 channel (Brandt and Jentsch, 1995) resides in late endosomes and lysosomes (Kornak et al, 2001). Mice with a disruption of the corresponding gene Clcn7 showed severe osteopetrosis, retinal degeneration, and died within 7 weeks (Kornak et al, 2001). Their osteopetrosis resulted from defective osteoclasts that failed to acidify the resorption lacuna and hence could not degrade bone. In WT osteoclasts, ClC-7 is inserted together with the V-type H+-ATPase into the ruffled border, a specialized, acid-secreting plasma membrane domain. The impaired osteoclast function in Clcn7−/− mice may result from a failure to electrically balance proton transport by passive Cl− currents (Kornak et al, 2001).
The osteopetrosis of Clcn7−/− mice led to the identification of CLCN7 mutations also in humans. Homozygous mutations lead to malignant infantile osteopetrosis (Kornak et al, 2001), whereas patients heterozygous for dominant mutations suffer from a less severe form (Cleiren et al, 2001; Frattini et al, 2003). Homozygous mutations in the a3 subunit of the H+-ATPase also cause severe osteopetrosis both in mice (Li et al, 1999; Scimeca et al, 2000) and men (Frattini et al, 2000; Kornak et al, 2000). These genetic findings further support the notion that both the proton pump and the ClC-7 channel are crucial for the acidification of the resorption lacuna.
Besides osteopetrosis, mice lacking ClC-7 display severe retinal degeneration (Kornak et al, 2001). Visual impairment is frequently observed in patients with severe infantile osteopetrosis (Steward, 2003) and has often been ascribed to a compression of the optic nerve by the osteopetrotic process. There are a few reports on central nervous system (CNS) degeneration of patients suffering from malignant infantile osteopetrosis (Steward (2003) and references therein), prompting us to investigate whether the loss of ClC-7 might be associated with a more general degeneration in the nervous system.
We now show that ClC-7 disruption leads to a widespread degeneration of the CNS with typical features of neuronal ceroid lipofuscinosis (NCL), a subtype of human lysosomal storage disease. This suggests that the recently observed neurological abnormalities in several patients with CLCN7 osteopetrosis (Frattini et al, 2003) are caused by an NCL-like phenotype directly linked to the lack of ClC-7. Although the disruption of ClC-3 also caused severe retinal and CNS degeneration that led to an almost total loss of the hippocampus, we show that this is not associated with the typical signs of lysosomal storage disease in our mouse model (Stobrawa et al, 2001). We neither observed retinal nor CNS degeneration in osteopetrotic oc/oc mice that carry a mutation in the a3 subunit of the V-type H+-ATPase (Scimeca et al, 2000). These findings suggest that patients with malignant infantile osteopetrosis carrying mutations in the proton pump may be cured by bone marrow transplantation, whereas patients with a total loss of ClC-7 are likely to develop blindness and severe CNS degeneration even if the osteopetrosis is successfully treated by such an intervention.
Results
ClC-7 resides on neuronal lysosomes
Previous work had shown that ClC-7 is broadly expressed in the mouse embryo, with particularly high levels in the central and peripheral nervous system (Kornak et al, 2001). We now determined the expression of ClC-7 in the adult brain using mice expressing a lacZ fusion protein from the endogenous Clcn7 locus (Kornak et al, 2001). LacZ staining revealed ClC-7 expression in virtually every region of the CNS, including the hippocampus, cerebellum and cortex (Figure 1A–C). As exemplified by the strong labeling of the pyramidal cell layer in the hippocampus (Figure 1A) and of Purkinje and granule cells in the cerebellum (Figure 1B), neurons were stained prominently. Expression was also detected in other cells, including ependymal cells and most likely glia. Astrocytes were clearly immunostained for ClC-7 in cell culture (data not shown).
Figure 1.

Localization of ClC-7 in the CNS. (A–C) X-gal staining of brain cryosections from mice expresssing a ClC-7/lacZ fusion protein. Strong signals were detected in neuronal cell layers of the hippocampus (A), cerebellum (B) and cortex (C). Brains from WT mice were not stained (not shown). Scale bars (A–C): 0.25 mm. (D) Punctate, perinuclear ClC-7 localization in the pyramidal layer of the hippocampal CA1 region as shown by immunofluorescence. Only weak, diffuse staining was present in the ClC-7 KO (not shown). (E) Localization of ClC-7 by immunogold labeling in a cortical neuron. The limiting membrane of a lysosome (arrow) was prominently stained, whereas mitochondria (arrowhead) were devoid of staining. Scale bar: 0.25 μm. (F) Staining of ClC-7 (green) and lamp-1 (red) in a cultured hippocampal neuron, revealing a large degree of co-localization (yellow). Scale bars for (D, F): 10 μm.
Immunostaining of ClC-7 revealed a punctate pattern in neuronal cell bodies that was absent in ClC-7 knockout (KO) tissue (Figure 1D). Immunogold electron microscopy localized ClC-7 to the limiting membrane of large cytoplasmic vesicles that most likely represent lysosomes (Figure 1E). In cultured hippocampal neurons, ClC-7 was distributed in a punctate pattern in cell bodies and proximal neurites. It nearly perfectly co-localized with lamp-1, a marker for late endosomes and lysosomes (Figure 1F). Hence, ClC-7 resides in late endosomal and lysosomal membranes in fibroblasts (Kornak et al, 2001) and neurons.
Neurodegeneration in the CNS of Clcn7−/− mice
Histological analysis revealed a conspicuous degeneration of the hippocampal CA3 region of ClC-7 KO mice (Figure 2). A reduction in cell density became visible at postnatal day 20 (p20) (Figure 2C) and in most animals CA3 pyramidal cells were almost completely lost by p30–p40 (Figure 2B). The apparent absence of cell loss at p12 (Figure 2D) argued against a developmental defect. The degeneration was paralleled by an activation of microglia, as revealed by GSA staining of the hippocampus (Figure 2F and G). Microglia invaded the pyramidal layer of the CA3, but not of the CA1 region. Similarly, there was increased staining for the astrocyte marker GFAP (Figure 2H and I). No neurodegeneration was visible in osteopetrotic oc/oc mice (Figure 2E), which neither showed astrocytosis (not shown). In Clcn7−/− mice close to their death around p40, neuronal loss also occurred in the cortex and the number of cerebellar Purkinje cells seemed decreased. To obtain older animals and exclude effects of osteopetrosis on the nervous system, we generated transgenic mice expressing ClC-7 under the control of the tartrate-resistant acid phosphatase (TRAP) promoter that is nearly exclusively active in osteoclasts and macrophages (Schwartzberg et al, 1997). Introducing this TRAP::ClC-7 transgene into the Clcn7−/− background yielded mice with normal bone density (unpublished data). Their lifespan was prolonged only by 3 weeks (to an age of ∼2 months). They displayed severe CNS degeneration that progressed beyond the stage seen in the total KO. At 2 months of age, there was a dramatic reduction in the size of the hippocampus, with its CA3 region totally lacking neuronal cell bodies (Figure 2J and K). The thickness of the neocortex was reduced, and, in the cerebellum, roughly 50% of Purkinje cells were lost (Figure 2L and M).
Figure 2.
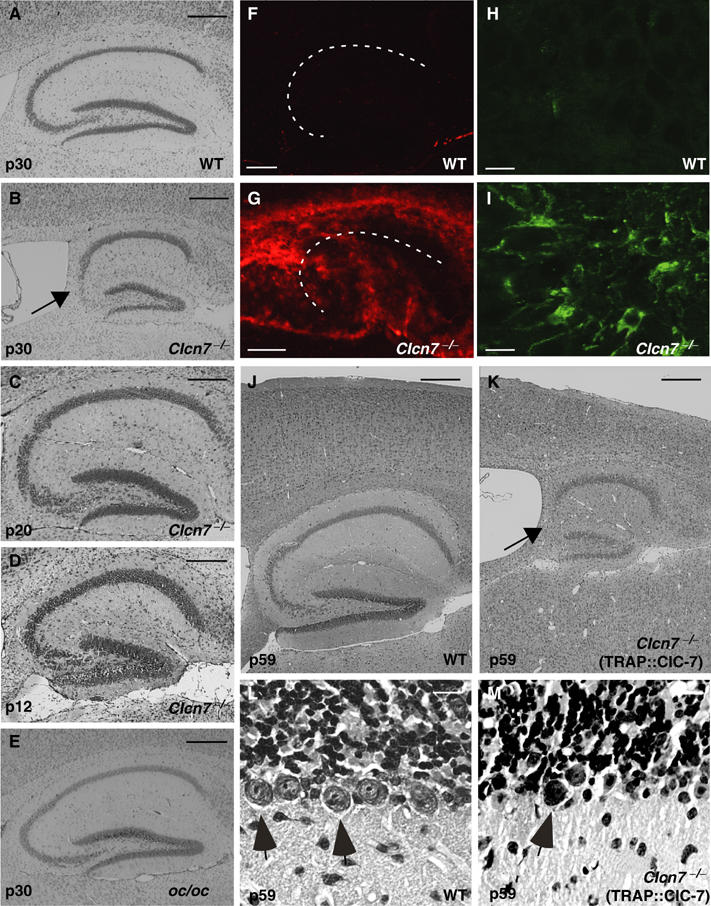
Neurodegeneration in Clcn7−/−, but not in oc/oc mice. (A–E) Neuronal loss in the hippocampal CA3 region (arrow in B) at p30 as revealed by Nissl staining was found in Clcn7−/− mice (B). oc/oc mice (E) were similar to WT controls (A). At p12, hippocampal sections of Clcn7−/− mice appeared normal (D). A reduction of cell density became visible at p20 (C). These pathological changes were observed in all animals that were analyzed at p30 or older (n>8). Neurodegeneration in Clcn7−/− mice was accompanied by increased staining with the microglial marker GSA (F, G; the pyramidal cell layer is indicated by a dashed line) and for the astrocyte marker protein GFAP (H, I). (K) In 2-months-old Clcn7−/− mice carrying a TRAP::ClC-7 transgene to rescue osteopetrosis, a further degeneration of cerebral cortex and hippocampus (arrow, CA3 region) is visible in Nissl staining as compared to WT (J). Severe loss of Purkinje cells (arrows) in the cerebellum of Clcn7−/− TRAP::ClC-7 mice (M) compared to WT (L) at p59. Scale bars: (A–G, J, K), 0.25 mm; (H, I, L, M), 10 μm.
Retinal degeneration does not depend on the osteopetrosis
Human malignant osteopetrosis is often associated with visual impairment (Steward, 2003). The retina of Clcn7−/− mice degenerates within the first 2–4 weeks after birth, leading to an almost complete loss of photoreceptors at p28 (Kornak et al, 2001). We now compared the retinal morphology of Clcn7−/− and oc/oc mice at p30. Unlike the severely degenerated Clcn7−/− retina (Figure 3C), the retina of oc/oc mice appeared normal (Figure 3B).
Figure 3.

Retinal degeneration in Clcn7−/−, but not in oc/oc mice. (A–D) Methylene blue-stained semithin retinal sections from WT (A), oc/oc (B), Clcn7−/− (C) and Clcn7−/− TRAP::ClC-7 (D) mice at p30. Scale bar: 20 μm; RPE, retinal pigment epithelium; OS/IS: photoreceptor outer/inner segments; ONL/INL: outer/inner nuclear layer; GCL: ganglion cell layer. (E–G) Scotopic full-field ERGs at p24–25. The electrical responses to different flash energies (indicated by numbers 1–8: 0.1, 0.4, 1, 4, 10, 100, 1000, 3000 mcds/m2) are superimposed. There is barely any difference in the response between Clcn7+/− (control) (E) and oc/oc mice (F). In Clcn7−/− mice (G), both a- and b-waves were markedly reduced. (H) Comparison of mean a- and b-wave amplitudes between Clcn7−/− and Clcn7+/− mice in response to 3000 mcds/m2 at p17, p24 and p29. At p29, the electrical response of the retina had virtually disappeared. n=7, 5, 3 for +/− and 6, 5, 4 for KO. Error bars, s.e.m. (*P<0.05).
To study the functional consequences of retinal degeneration, scotopic electroretinograms (ERGs) were measured in Clcn7+/− (control), Clcn7−/− and oc/oc mice at p24–25 (Figure 3E–G). The early electronegative response of the retina to a light pulse (a-wave) reflects the function of the photoreceptors, while the b-wave represents the response of the second retinal neuron (mainly bipolar cells). Both ERG components were significantly reduced in Clcn7−/− mice (Figure 3G and H), but not in oc/oc mice at the same age (Figure 3F). As osteopetrosis is equally severe in these mice, we conclude that the retinal degeneration of Clcn7−/− mice is not due to optic nerve compression. This conclusion is strongly supported by unchanged retinal degeneration in Clcn7−/− mice with a transgenic rescue of the osteopetrosis (Figure 3D).
CNS degeneration in Clcn7−/− mice displays features of NCL
Electron microscopy identified electron-dense, osmiophilic material in the perikarya of hippocampal and cortical neurons of p40 Clcn7−/− mice (Figure 4A). Already at p11, storage material was abundant in Clcn7−/− neurons (data not shown) and increased further with age, whereas no deposits were found in the brains of p30 oc/oc mice (Figure 4C). Higher magnification (Figure 4B) revealed a mixed ultrastructure consisting of amorphous, granular or, infrequently, lamellar deposits (Figure 4B, inset). These resembled the avacuolar lipopigments, granular osmiophilic deposits (GROD) and fingerprint profiles observed in variable proportions in a form of human lysosomal storage diseases, the neuronal ceroid lipofuscinoses (NCL) (Goebel and Wisniewski, 2004). Recently, mice disrupted for the homologous Cl− channel ClC-3 were reported to show storage material and to recapitulate some aspects of NCL (Yoshikawa et al, 2002). However, no electron-dense deposits were found in neurons of our Clcn3−/− mouse model (Stobrawa et al, 2001), not even in the regions most severely affected by neurodegeneration (data not shown).
Figure 4.
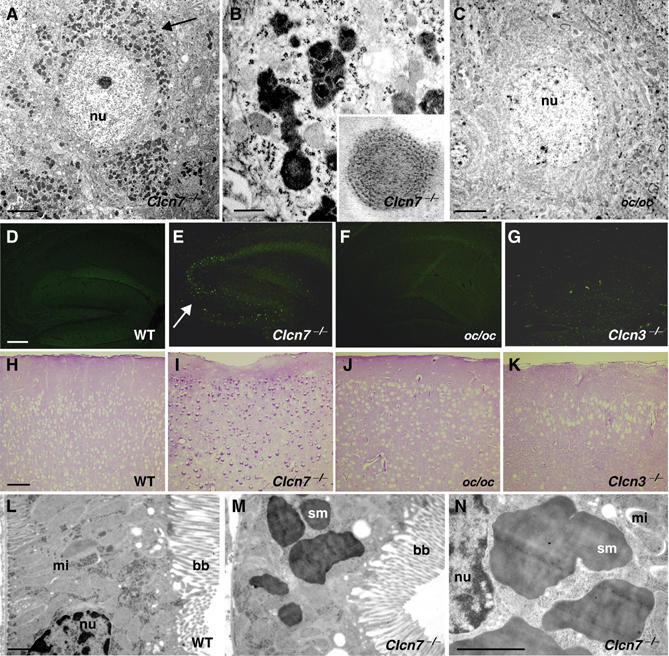
Morphology of lysosomal storage disease in Clcn7−/− mice. (A–C) Electron microscopy of cortex sections of p40 Clcn7−/− (A, B) and p30 oc/oc mice (C). Accumulation of osmiophilic material (arrow) was observed in Clcn7−/−, but not in oc/oc mice. Higher magnification (B) showed avacuolar lipopigments and, less frequently, lamellar profiles (inset). Scale bars: 2 μm for (A, C), 200 nm for (B), inset 4 × . (D–G) Autofluorescence in the hippocampus of Clcn7−/− (E, p33, arrow indicates CA3), but not in WT (D, p37) and oc/oc (F, p33) mice. Faint autofluorescence was visible in p45 Clcn3−/− mice (G). (H–K) PAS staining reveals perinuclear accumulation of carbohydrates in the cortex of p30 Clcn7−/− (I), but not in p30 oc/oc (J) or in 3-months-old Clcn3−/− (K) or WT (H) mice. Scale bars for (D–K): 0.2 mm. (L–N) Electron microscopy of proximal tubular cells from WT (L) and Clcn7−/− (M, N) kidneys at p30. Note the electron-dense storage material (sm) in the KO. (bb, brush border; mi, mitochondrion; nu, nucleus). Scale bars: 1.5 μm.
One of the hallmarks of NCL is the presence of autofluorescent pigments (Goebel and Wisniewski, 2004). Autofluorescence appearing as bright punctae was observed in Clcn7−/− brains as early as p20 and progressed over time. It was found predominantly in the CA3 region of the hippocampus (Figure 4E, arrow), underneath the corpus callosum, in the cerebral cortex and the thalamus. Its preferential localization in regions of intense GSA staining suggested an association with microglia. Fluorescence of brain sections from oc/oc mice (Figure 4F) was not different from WT (Figure 4D), and Clcn3−/− mice displayed only weak fluorescence in their degenerating hippocampus (Figure 4G). Intracellular carbohydrate accumulation in lysosomal storage diseases can be detected by periodic acid Schiff (PAS) staining (Goebel and Wisniewski, 2004). A perinuclear deposition of carbohydrates was seen in cortical neurons of p30 Clcn7−/− mice (Figure 4I) and virtually all other brain regions (data not shown). In contrast, PAS staining was increased neither in oc/oc (Figure 4J) nor in Clcn3−/− (Figure 4K) brain, despite the massive neurodegeneration at that age (3 months) in the latter mice.
As in many forms of human NCL, storage material in Clcn7−/− mice was not restricted to neurons. Electron-dense deposits were prominent in proximal tubule cells of KO, but not WT kidneys (Figure 4L–N). We used immunohistochemistry and postembedding immunogold techniques to further characterize the storage material in Clcn7−/− neurons. In Clcn7−/− hippocampus and cortex, the lysosomal marker lamp-1 was changed from a WT pattern of discrete cytoplasmic punctae (Figure 5A) to a more diffuse and more intense staining (Figure 5B). Similar changes were seen for the late endosomal/lysosomal protein saposin D (Figure 5C and D). At the EM level, electron-dense deposits were positive for cathepsin D (Figure 5E), indicating a lysosomal origin. Storage material in most forms of human NCL contains large amounts of subunit c of the mitochondrial ATP synthase. In the WT, immunogold label for subunit c was confined to the mitochondria, whereas in Clcn7−/− mice, it was also prominently found on electron-dense storage material (Figure 5F). Western blotting revealed an increase of subunit c in brain lysates from Clcn7−/−, but not from Clcn3−/− mice (Figure 6H).
Figure 5.
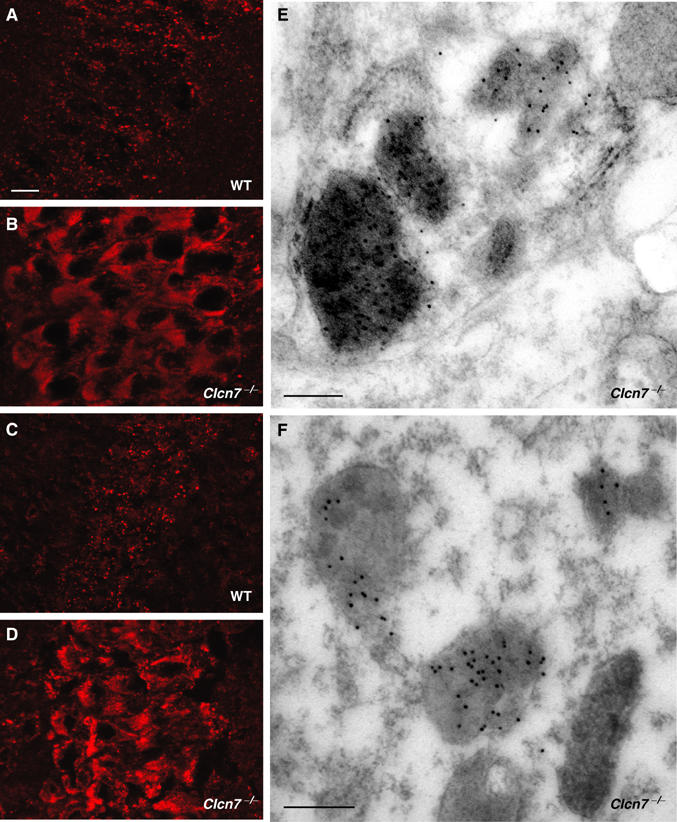
Characterization of lysosomal storage material in Clcn7−/− mice. (A–D) Immunostaining for lamp-1 (A, B) and saposin D (C, D) in the CA1 hippocampal region of p30 WT (A, C) and Clcn7−/− mice (B, D). Scale bars: 10 μm. (E, F) Postembedding immunogold localization of cathepsin D (E) and subunit c of the mitochondrial ATP synthase (F) in a Clcn7−/− neuron. Both proteins are contained within storage deposits characterized by their electron-dense appearance, large size and distorted shape. Scale bars: 0.25 μm.
Figure 6.
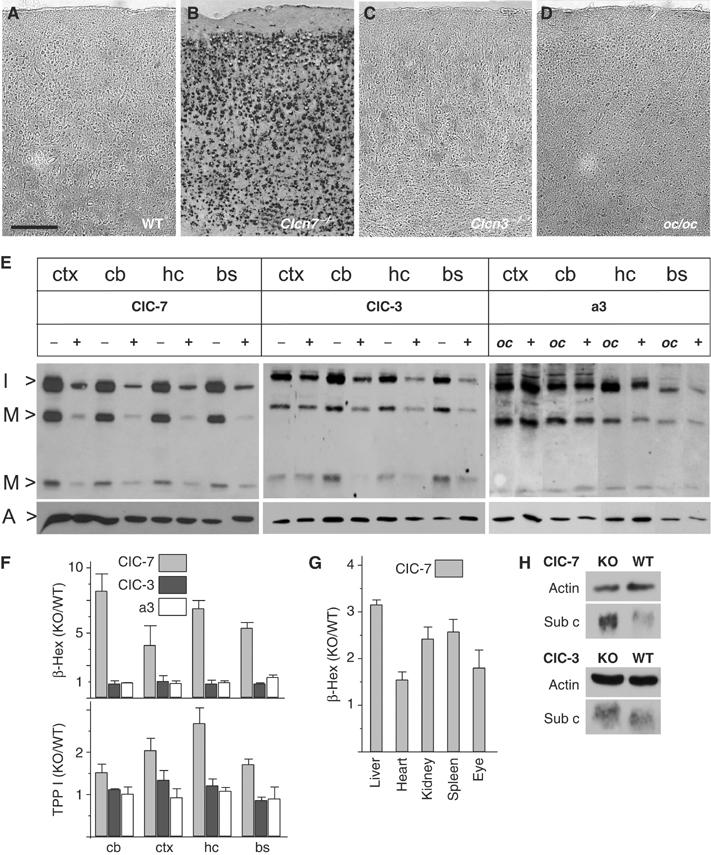
Alteration of lysosomal enzymes in Clcn7−/−, but not in oc/oc or Clcn3−/− mice. (A–D) Lysosomal acid phosphatase activity was visualized in situ as dark staining on cortex sections of p40 WT (A), p40 Clcn7−/− (B), 3 months Clcn3−/− (C) and p29 oc/oc mice (D). Scale bar: 20 μm. (E) Immunoblot analysis of cathepsin D. Extracts of different brain regions (ctx, cortex; cb, cerebellum; hc, hippocampus; bs, brainstem) of p30 Clcn7−/−, Clcn3−/− and oc/oc mice were compared to WT controls. Bands represent the intermediate (I>, 42 kDa) and the mature forms (M>, 32 and 12 kDa) of cathepsin D. Staining for actin (A>) served as a loading control. (F) Enzymatic activity of β-hexosaminidase (β-Hex) and TPP I in brain extracts from Clcn7−/− (gray bars), Clcn3−/− (black bars) and oc/oc mice (white bars) at p30 (shown is the ratio of mutant over WT; error bars, s.e.m.; n=4). (G) Increase of β-hexosaminidase activity in the eye and peripheral organs from Clcn7−/− mice. (H) Western blot analysis of subunit c of the mitochondrial ATP synthase of total brain homogenates from Clcn7−/−, Clcn3−/− and WT mice. Actin served as loading control.
Changes in lysosomal enzymes characteristic for NCL
To further characterize the Clcn7−/− storage phenotype, we measured several lysosomal enzyme activities. Lysosomal acid phosphatase activity was determined in brain cryosections in situ (Figure 6A–D). Clcn7−/− mice showed drastically increased staining, which was predominantly localized to neuronal cell bodies (Figure 6B). By contrast, no difference to WT controls (Figure 6A) was found in oc/oc (Figure 6D) or in Clcn3−/− mice, even when the latter were examined at 3 months of age (Figure 6C). The expression and processing of the lysosomal protease cathepsin D were assessed by Western blot (Figure 6E). Irrespective of the brain region, intermediate and mature forms of cathepsin D were increased approximately six-fold in Clcn7−/− mice. The increase in Clcn3−/− mice was ∼2-fold, whereas no change of cathepsin D was observed in oc/oc mice.
In vitro activities of the lysosomal enzymes β-hexosaminidase and tripeptidyl peptidase I (TPP I) were determined from lysates of different brain regions. Compared to WT, a four- to seven-fold increase in β-hexosaminidase activity was detected in Clcn7−/−, but neither in Clcn3−/− nor in oc/oc mice (Figure 6F). Similarly, in vitro TPP I activity was elevated approximately two-fold in Clcn7−/− brain but not in oc/oc, and only marginally in Clcn3−/− mice. β-Hexosaminidase activity was also increased in Clcn7−/− liver, heart, kidney, spleen and eye (Figure 6G).
Lysosomal function and pH in living cells
A cell-permeable fluorogenic substrate for TPP I was used to measure lysosomal enzyme activity in living cells. Cleavage of the peptide Ala–Ala–Phe–Rhodamine110–Phe– Ala–Ala resulted in intracellular fluorescence that could be analyzed by FACS. No significant difference was observed between WT and Clcn7−/− fibroblasts (Figure 7A). TPP I activity of primary neuronal cell cultures was determined by measuring the fluorescent product that was released into the culture medium. There was no difference between WT and KO either (Figure 7B).
Figure 7.
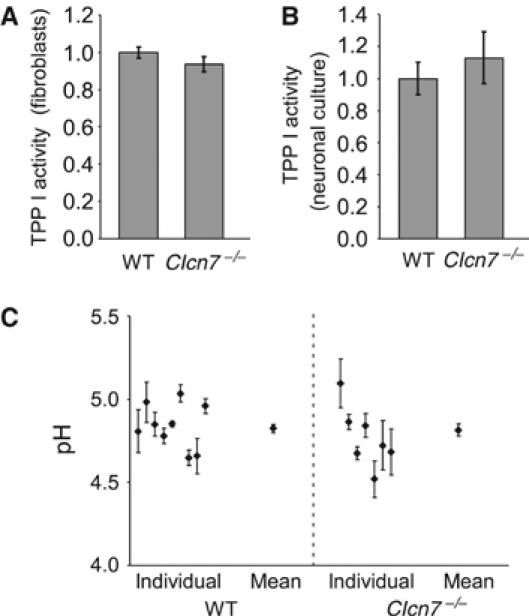
Lysosomal pH and enzyme activity in Clcn7−/− cells. (A, B) Measurement of TPP I activity in living cells using Ala–Ala–Phe–Rhodamine110–Phe– Ala–Ala as a substrate. (A) Intracellular fluorescence generated after a 1 h incubation of fibroblasts with the fluorogenic substrate, as analyzed by FACS. (B) TPP I activity in neuronal cultures, as indicated by fluorescence released into the supernatant and normalized to cell density. (C) Lysosomal pH of WT and Clcn7−/− neurons was determined with dextrane-coupled pH-sensitive dye Oregon green 488 that was loaded by endocytosis. The average pH of WT and KO lysosomes was 4.82±0.03 (s.e.m.) and 4.82±0.04, respectively (n=100 visual fields (containing one to two cells) for WT and 69 for KO; the plot shows pH obtained from individual animals as well as the mean).
Like other vesicular CLC proteins, ClC-7 might provide an electrical shunt for proton pumping (Jentsch et al, 2002). We therefore determined lysosomal pH in cultured neurons by ratiometric imaging. The dextrane-coupled pH indicator Oregon green was loaded into cells by endocytosis and chased into late endosomes and lysosomes. Co-staining for lamp-1 confirmed that the dye reached late endosomal/lysosomal compartments in both WT and KO cells (data not shown). The average pH of WT lysosomes was 4.82±0.03 (s.e.m.) and was unchanged in Clcn7−/− neurons (pH=4.82±0.04) (Figure 7C). We neither observed changes in lysosomal pH nor in the delivery of the dye to late endosomes/lysosomes between KO and WT fibroblasts (data not shown).
Changes in gene expression in the hippocampus of Clcn7−/− mice
Genome-wide expression profiling was used to detect changes in expression levels in the hippocampi of Clcn7−/− mice as compared to WT littermates. It was performed at a time point (p14) before degeneration became obvious. Applying a threshold of 1.5-fold change, 37 transcripts were upregulated and 15 downregulated in the KO. The changes in 25 of the upregulated genes were validated by real-time RT–PCR. In all but one case, this analysis confirmed the qualitative changes observed in the microarray experiments (Table I; for the complete list, see Supplementary data). The genes with increased transcription can be broadly categorized into four classes: genes involved in antigen presentation (MHC class I genes and the proteasome subunit LMP7), complement components, genes with a potential role in microglia/macrophages and often known to be induced by interferons, and finally GFAP, the classical marker of astrocytes. The activation of astrocytes and of components of the immune system in Clcn7−/− brains, as revealed by expression profiling, are compatible with the microglia activation and astrocytosis described above.
Table 1.
Genes upregulated in the Clcn7−/− hippocampus
| Description | FC |
|---|---|
| 2′-5′ Oligoadenylate synthetase-like 2 | 8.51 |
| Interferon, alpha-inducible protein | 6.16 |
| Peptidylprolyl isomerase C-associated protein | 3.56 |
| Proteasome subunit, beta type 8 (LMP7) | 3.12 |
| Interferon-induced protein with tetratricopeptide repeats 3 | 2.66 |
| Beta-2 microglobulin | 2.63 |
| Guanylate nucleotide-binding protein 3 | 2.53 |
| Interferon-induced transmembrane protein 3 | 2.47 |
| Signal transducer and activator of transcription 1 | 2.45 |
| Sex-limited protein | 2.27 |
| Histocompatibility 2, Q region locus 1 | 2.12 |
| Histocompatibility 2, K region | 2.11 |
| Histocompatibility 2, K region | 1.95 |
| Interferon regulatory factor 7 | 1.85 |
| Complement component 1, q subcomponent, beta | 1.82 |
| Histocompatibility 2, D region locus 1 | 1.74 |
| CD52 antigen | 1.72 |
| Nuclear factor I/A | 1.66 |
| TYRO protein tyrosine kinase-binding protein | 1.61 |
| Glial fibrillary acidic protein | 1.59 |
| Complement component 1, q subcomponent, gamma | 1.57 |
| Interferon-induced protein with tetratricopeptide repeats 1 | 1.54 |
| Affymetrix chip analysis of genes upregulated in p14 Clcn7−/− mouse hippocampus in comparison to WT. Quantification is given as ‘fold change' (FC), indicating the increase in KO mice compared to WT. The upregulation of all shown genes was confirmed by real-time RT–PCR. Genes without assigned function and downregulated genes were omitted. The complete list, including RT–PCR data and GeneBank accession, is given in Supplementary data. | |
Discussion
Mice lacking ClC-7 show severe osteopetrosis as a consequence of defective HCl secretion by osteoclasts (Kornak et al, 2001). Here we describe that the absence of ClC-7 also leads to a severe lysosomal storage phenotype that is associated with neurodegeneration. We compare this phenotype to that of another osteopetrosis model, the osteosclerotic (oc) mouse that is deficient in a proton pump subunit, and to that of ClC-3 KO mice which lack a related endosomal Cl− channel and which also show severe neurodegeneration.
ClC-7 KO has a severe CNS phenotype with signs of NCL
Clcn7−/− mice displayed neurological deficits like hindfeet clasping, which became more severe until the time of death at about 6 weeks. Rescuing the osteopetrotic phenotype by expressing ClC-7 in osteoclasts under the control of the TRAP promoter increased the lifespan of the animals by only a few weeks. Old ‘bone rescue' mice showed hypomotility, abnormal postures and a loss of motor control, and lost weight before dying after about 2 months. We assume that severe neurological problems at least contribute strongly to their death.
We focused on the cellular basis of the neurological deficits and observed a severe lysosomal storage disease with many typical features of NCL. NCLs are a genetically heterogeneous group of human progressive encephalopathies that are associated with neurocognitive and physical decline and ultimately lead to premature death (Mitchison et al, 2004). One of their hallmarks is the neuronal accumulation of autofluorescent lipopigment (Goebel and Wisniewski, 2004). An impairment of lysosomal function has been suggested as a common underlying mechanism. Eight types of NCL are classified according to the age of onset, histological features and genetic loci. The underlying genes, two of which are still unknown, are called CLN1–CLN8. None of these NCL variants is associated with osteopetrosis, making it less likely that CLCN7 is a candidate for one of the unknown CLN genes.
Neuronal cell death in the retina and CNS is common to most NCL subtypes. The cortex is affected strongly in all characterized NCL mouse models that frequently display also hippocampal or Purkinje cell loss (Mitchison et al, 2004). In Clcn7−/− mice, hippocampal (CA3) and cortical neurons as well as Purkinje cells degenerated preferentially, while many other cells, although accumulating storage material, were relatively spared. Cell-intrinsic differences in vulnerability might explain the differential degeneration. Neurodegeneration in Clcn7−/− mice was accompanied by microglial activation and astrogliosis, a hallmark of NCL and other CNS pathologies. This was supported by gene expression profiling, which revealed changes similar to those found in mouse models of mucopolysaccharidoses I and IIIB, lysosomal storage diseases associated with neurodegeneration (Ohmi et al, 2003). The activation of microglia is probably a consequence of the neuronal pathology and may serve to eliminate dead cells. However, an inappropriate microglial reaction may also have adverse effects as in other degenerative CNS diseases (Wada et al, 2000).
Regions affected by neurodegeneration showed increased autofluorescence. In neuronal cell bodies, electron microscopy revealed storage material which displayed heterogeneous morphologies, including amorphic deposits, GRODs and lamellar structures resembling fingerprints. All of these are found in varying proportions in different NCL entities (Goebel and Wisniewski, 2004). In most human NCLs, the major protein accumulating in storage bodies is subunit c of the mitochondrial ATP synthase (Palmer et al, 1992; Ezaki and Kominami, 2004). It is believed to be particularly resistant to degradation because of its hydrophobicity. In contrast, saposins are more prominently stored in NCL1 (Tyynelä et al, 1993). Levels of both proteins were elevated in Clcn7−/− brain and immunogold labeling showed the presence of subunit c in storage material.
Lysosomal function in Clcn7−/− mice
Several parameters associated with lysosomal function were altered in Clcn7−/− brains. As typically found in NCL (Prasad and Pullarkat, 1996; Sleat et al, 1998; Tyynelä et al, 2000), the abundance or in vitro activity of lysosomal enzymes was increased. Staining for saposin D, which promotes sphingolipid hydrolysis in late endosomes/lysosomes, and for lamp-1, a structural protein of these compartments, suggested an expansion of lysosome-derived structures. Electron microscopy indicated that this apparent expansion was caused by storage material rather than by an increased number of lysosomes.
Despite histological and biochemical findings that are typical for a lysosomal disorder, an analysis of cultured fibroblasts and neurons from Clcn7−/− mice failed to reveal defects in lysosome function. No change in lysosomal TPP I activity was observed in living cells. There was no measurable difference in lysosomal pH, neither in cultured neurons nor in fibroblasts. This finding was unexpected, as ClC-7, like other vesicular CLCs, is thought to facilitate vesicular acidification by electrically shunting electrogenic proton pumping (Jentsch et al, 2002). A role of ClC-7 in lysosome acidification would also fit to reports that alkalinizing lysosomes by chloroquine caused a neuronal accumulation of lipofuscin-like storage material (Ivy et al, 1984). Although we did not detect differences in lysosomal steady-state pH, we cannot exclude more subtle changes, for example, in the rate of acidification during the travel along the endocytotic–lysosomal pathway. The trafficking along this pathway may depend on its luminal acidification, as evident from the defect in endocytosis in mice lacking the endosomal ClC-5 Cl− channel (Piwon et al, 2000). However, we did not detect an obvious defect in targeting the pH indicator to lysosomes, which was an important control for our pH measurements.
However, ClC-7 should be considered a putative Cl− channel, as its absence from the plasma membrane precluded a biophysical characterization. Other potential roles cannot be excluded. For example, ClC-7 might share the transport property of the bacterial homolog ClC-e1, which functions as an electrogenic 2Cl−/H+ exchanger (Accardi and Miller, 2004). In this case, ClC-7 could still electrically shunt lysosomal proton pumping by mediating Cl− influx, but the lysosomal Cl− accumulation would be directly coupled to the pH gradient. Aside from allowing acidification, little is known about a lysosomal function for chloride. It might drive unknown transport processes across lysosomal membranes or might influence enzymatic activities. For instance, the yeast CLC gef1p is thought to raise luminal Cl− in late Golgi compartments, which allosterically influenced the copper loading of an oxidase (Davis-Kaplan et al, 1998). Finally, like all mammalian CLCs, ClC-7 has large cytoplasmic domains that may interact with other proteins and that contain CBS domains which may bind ATP (Scott et al, 2004). The function of these domains, which carry mutations in several patients with osteopetrosis, is still unknown, but invites speculations.
Consistent with the ubiquitous expression of ClC-7, the lysosomal enzyme β-hexosaminidase was at least moderately increased in every Clcn7−/− tissue examined. Similarly, and like in many human forms of NCL (Goebel and Wisniewski, 2004), storage material was not restricted to neurons. Renal proximal tubular cells showed large amounts of deposits, whereas several other cell types expressing comparable levels of ClC-7 were inconspicuous. A possible explanation is that cells characterized by a very high lysosomal activity or a long lifespan are most severely affected. Neurons are nondividing cells that turn over significant amounts of membrane lipid and protein, and proximal tubular cells are highly active in endocytosis and lysosomal degradation of urinary proteins. In view of the high endocytotic load of cells like those of the proximal tubule, a strong impairment of lysosomal degradation would be expected to cause a much more severe pathology than observed in Clcn7−/− mice. In this respect, the failure to detect changes in lysosomal function in cell culture, disappointing as it might be, fits well to the disease phenotype. We suggest that the lysosomal dysfunction of Clcn7−/− mice is mild, leading to an only slow intracellular accumulation of undigested material in particularly susceptible cells.
Finally, one should address the apparent paradox of impaired lysosomal function (indicated by the accumulation of storage material) in the presence of elevated levels of lysosomal enzymes. This situation is frequently observed in NCL (Ezaki and Kominami, 2004) and is interpreted as an accumulation of enzymes in compartments whose environment is not optimal for their function. Indeed, whereas in vitro TPP I activity in extracts from several brain regions was increased, there was no change of in vivo activity measured with a cleavable, fluorogenic substrate.
Retina degeneration separated from the osteopetrosis phenotype
Cell death in the retina of Clcn7−/− mice was even more dramatic and occurred faster than in the CNS. In electroretinograms of Clcn7−/− mice, the reduction of the a-wave (generated by photoreceptors) was not preceded, but paralleled by a decrease of the b-wave (generated mainly by bipolar cells), consistent with a primary reduction in photoreceptor function. Neither the histological nor the more sensitive electrophysiological analysis indicated a retinal dysfunction in osteopetrotic oc/oc mice at p24/25. Together with the equally severe retinal degeneration in transgenic ‘bone rescue' animals, this proves that this degeneration is retina-intrinsic in Clcn7−/− mice and that the constriction of the optic nerve (Kornak et al, 2001) plays a minor role. Instead, the retinal degeneration is probably closely related to the NCL phenotype. Visual impairment and blindness are observed in most subtypes of NCL (Goebel and Wisniewski, 2004). The increased β-hexosaminidase activity in the retina indicates a lysosomal dysfunction also in this tissue, and we detected minor amounts of electron-dense storage material in Clcn7−/− ganglion cells (data not shown). Retinal cells might degenerate too fast to allow a more pronounced intracellular storage.
Neurodegeneration in ClC-3 KO mice differs from typical NCL
ClC-3 is a Cl− channel of endosomes and synaptic vesicles (Stobrawa et al, 2001) and is important for the acidification of these compartments (Stobrawa et al, 2001; Yoshikawa et al, 2002). Disruption of ClC-3 led to a severe neurodegeneration in which the hippocampus was nearly totally lost after 3 months (Stobrawa et al, 2001; Dickerson et al, 2002; Yoshikawa et al, 2002). Such an extreme and preferential degeneration of the hippocampus has not been described in human NCL. Clcn3−/− mice generated by Yoshikawa et al (2002) were reported to display lipofuscin-like granules in the perikarya of hippocampal neurons and increased levels of subunit c in subcellular fractions enriched for lysosomes. In contrast, electron microscopy of hippocampal neurons from our Clcn3−/− mice detected signs of cytolysis in the absence of storage material (Stobrawa et al, 2001). In these mice, we observed only weak autofluorescence, unchanged total subunit c levels, no change in PAS staining or in lysosomal enzymes, except for a slight increase in cathepsin D. Our electron microscopical analysis of Clcn3−/− and Clcn7−/− mice was performed in C57Bl/6 and mixed C57Bl/6x129SvJ backgrounds. Only minor differences in the amount of neuronal storage material were seen in the Clcn7−/− lines, whereas none of the two different ClC-3 KO lines showed significantly more intracellular electron dense material than was normal for the age (up to 13 months) analyzed. The source of the apparent differences between our Clcn3−/− mice (Stobrawa et al, 2001) and those of Yoshikawa et al (2002) thus remains unclear. The difference in lysosomal pathology between Clcn7−/− and Clcn3−/− mice, however, correlates with the lysosomal localization of ClC-7 and not ClC-3.
Phenotypic comparison of ClC-7 and a3-deficient mice
Mutations in CLCN7 (Kornak et al, 2001) and TCIRG1 (Atp6i; encoding the a3 subunit of the V-type H+-ATPase) (Frattini et al, 2000; Kornak et al, 2000) are common causes of malignant infantile osteopetrosis in humans, supporting the concept that currents of ClC-7 are required for the acidification of the resorption lacuna (Kornak et al, 2001). The gene for the a3 subunit has been disrupted in mice by homologous recombination (Li et al, 1999) and bears a deletion in the spontaneous oc/oc mouse mutant (Scimeca et al, 2000). Both mouse strains develop severe osteopetrosis. Although a3 levels in brain are low, both ClC-7 and a3 are broadly expressed (Nishi and Forgac, 2000; Toyomura et al, 2000). In fibroblasts and in macrophage precursors, the a3 subunit has been located in lysosomes (Toyomura et al, 2003). Importantly, we show that CNS and primary retinal degeneration are present in Clcn7−/− and absent in a3-deficient mice. Thus, a3 can probably be replaced by the homologous a1 or a2 proteins. Both latter subunits are expressed in brain (Nishi and Forgac, 2000; Toyomura et al, 2000), although they are believed to be mainly present in endosomes and synaptic vesicles (Morel et al, 2003; Toyomura et al, 2003).
Absence of CNS and retinal degeneration in oc/oc mice: medical implications
The lack of neuronal and retinal degeneration in oc/oc mice has potentially important medical implications. As osteopetrosis is equally severe in oc/oc and Clcn7−/− mice (unpublished results), our comparison of these mouse models and in particular the ‘bone rescue' Clcn7−/− mice indicate that the blindness and neurodegeneration of Clcn7−/− mice are not secondary consequences of their osteopetrosis. Primary retinal and neurodegeneration or lysosomal storage disease were observed in some patients with malignant infantile osteopetrosis (Steward (2003) and references therein). A recent report describes neurological symptoms and brain atrophy in osteopetrotic patients with CLCN7 mutations (Frattini et al, 2003). We conclude that the brain and retina pathology described here is probably present in human malignant osteopetrosis caused by CLCN7, but not TCIRG1 (H+-ATPase a3) mutations.
Specific mutations in CLCN7, however, cause a less severe recessive intermediate form of osteopetrosis (Campos-Xavier et al, 2003) and autosomal dominant osteopetrosis type II (ADOII) (Cleiren et al, 2001; Frattini et al, 2003). Unlike malignant infantile osteopetrosis, ADOII is not generally associated with visual impairment or blindness, and neurological symptoms and neurodegeneration have not been reported. As CLC channels are dimers, heterozygous dominant-negative mutations can reduce currents to 25% of WT levels (Jentsch et al, 2002). The remaining activity may suffice for lysosomal function in neurons, but not for bone resorption by osteoclasts.
Bone marrow transplantation is the only therapy available for recessive malignant infantile osteopetrosis (Gerritsen et al, 1994). When performed early enough, it can also prevent the loss of vision that is caused by a compression of the optic nerve. As there were no signs of CNS or retinal degeneration in oc/oc mice, a mouse model for a3 H+-ATPase deficiency (Scimeca et al, 2000), bone marrow transplantation may be an effective cure for this subtype of recessive osteopetrosis. Patients with malignant osteopetrosis due to a total loss of ClC-7, however, may develop blindness and severe NCL-like neurodegeneration despite transplantation.
Materials and methods
Animals
The generation of Clcn3−/− and Clcn7−/− mice was described (Kornak et al, 2001; Stobrawa et al, 2001). Transgenic mice expressing ClC-7 under the control of the TRAP promoter will be described in detail elsewhere. Introducing the TRAP::ClC-7 transgene into the Clcn7−/− background normalized their bone density (unpublished data). ClC-7 expression was restricted to osteoclasts and few other tissues (liver and low amounts in kidney) and was undetectable in brain or eye. Analysis was carried out in two independent lines. Osteosclerotic (oc) mice (Scimeca et al, 2000) were obtained from Jackson Laboratory.
Cell culture
Primary fibroblasts of WT and Clcn7−/− mice were from tails of p30 animals. After dissociation with collagenase/dispase, cells were cultured in DMEM containing 10% (w/v) fetal calf serum. Cultures of hippocampal neurons were prepared as described (Fuhrmann et al, 2002), but p0 mice were used and plating density was 120 000 cells/14 mm coverslip.
Histology, immunohistochemistry and electron microscopy
Primary antibodies for immunohistochemistry were: rabbit α-cathepsin D (Oncogene), rabbit α-ClC-7 (Kornak et al, 2001), rat α-lamp-1 (BD PharMingen), mouse α-GFAP (Roche), goat α-saposin D (gift of K Sandhoff, Bonn) and rabbit α-subunit c (gift of E Kominami, Tokyo). Detailed procedures are described in Supplementary data.
Western blot
Tissues were homogenized in PBS with protease inhibitors (Complete®, Roche) and cleared by centrifugation at 1000 g. Equal amounts of protein were separated by SDS–PAGE, blotted onto nitrocellulose, and labeled with rabbit α-cathepsin D (gift of R Pohlmann, Münster), rabbit α-subunit c (gift of E Kominami, Tokyo) or rabbit α-actin (Sigma) antibodies. Secondary antibodies were conjugated to HRP (Chemicon) and detected by chemoluminescence (SuperSignalWest, Pierce).
Biochemical assays
TPP I activity in brain homogenates was measured using a fluorometric assay with Ala–Ala–Phe–7-amido-4-methylcoumarin (Sigma) as substrate (Kleijer et al, 2001). For β-hexosaminidase activity, the substrate was p-nitrophenyl-2-acetamido-2-deoxy-β-D-glucopyranosid (Sigma). Brain homogenate was added to 0.1 M sodium citrate, pH 4.6, containing 0.04% (w/v) NaN3, 0.2% (w/v) BSA, 0.5% (w/v) Triton X-100 and 10 mM substrate. The reaction was stopped with 5 vol of 0.4 M glycine, pH 10.4, and OD405 was measured in a spectrophotometer.
TPP I activity in living cells
The activity of TPP I was determined using the cell-permeable substrate Ala–Ala–Phe–Rhodamine110–Phe– Ala–Ala (Bis-AAF-R110, synthesized by ThermoHybaid, Ulm, Germany). Sequential cleavage of the nonfluorescent bisamide substrate by TPP I results in the highly fluorescent rhodamine110. Detailed procedures are available as Supplementary data.
Determination of lysosomal pH
Lysosomal pH was measured using the dextran-coupled pH-sensitive ratiometric dye Oregon Green 448 (Molecular Probes). The procedures for loading the dye into lysosomes by endocytosis and pH measurements by imaging are described in Supplementary data.
Electroretinogram
Mouse electroretinograms (ERGs) were recorded as described (Ruether et al, 1997). Briefly, the dark adapted and anesthetized mice got a monopolar contact lens electrode and were placed in a dome. A flash series consisting of eight steps started at 10−4 cds/m2 and reached 3.0 cds/m2.
Expression profiling
RNA was extracted from the hippocampi of p14 Clcn7−/− mice and control littermates, converted into doubled-stranded cDNA and hybridized to Affymetrix murine genome U74v2 microarrays. A detailed protocol is available as Supplementary data. The microarray data have been submitted to ArrayExpress, accession number E-MEXP-242.
Supplementary Material
Supplementary Information
Acknowledgments
We thank S Bauer, J Enderich, N Krönke and B Merz for technical assistance, E Becker for work on the rescue construct, and E Kominami (Tokyo), R Pohlmann (Münster) and K Sandhoff (Bonn) for antibodies. This work was supported by grants from the DFG, and the NGFN program of the BMBF. R Planells-Cases was supported by a Marie-Curie fellowship from the European Union and an EMBO short-term fellowship, and M Poët by a Marie-Curie fellowship.
References
- Accardi A, Miller C (2004) Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 427: 803–807 [DOI] [PubMed] [Google Scholar]
- Brandt S, Jentsch TJ (1995) ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett 377: 15–20 [DOI] [PubMed] [Google Scholar]
- Campos-Xavier AB, Saraiva JM, Ribeiro LM, Munnich A, Cormier-Daire V (2003) Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis. Hum Genet 112: 186–189 [DOI] [PubMed] [Google Scholar]
- Cleiren E, Benichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, deVernejoul MC, Van Hul W (2001) Albers–Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet 10: 2861–2867 [DOI] [PubMed] [Google Scholar]
- Davis-Kaplan SR, Askwith CC, Bengtzen AC, Radisky D, Kaplan J (1998) Chloride is an allosteric effector of copper assembly for the yeast multicopper oxidase Fet3p: an unexpected role for intracellular chloride channels. Proc Natl Acad Sci USA 95: 13641–13645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC, Nehrke K, Williamson RA, Lamb FS (2002) Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res 958: 227–250 [DOI] [PubMed] [Google Scholar]
- Ezaki J, Kominami E (2004) The intracellular location and function of proteins of neuronal ceroid lipofuscinoses. Brain Pathol 14: 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A (2000) Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 25: 343–346 [DOI] [PubMed] [Google Scholar]
- Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM, Mihci E, Notarangelo LD, Ramenghi U, Teti A, Van Hove J, Vujic D, Young T, Albertini A, Orchard PJ, Vezzoni P, Villa A (2003) Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res 18: 1740–1747 [DOI] [PubMed] [Google Scholar]
- Fuhrmann JC, Kins S, Rostaing P, El Far O, Kirsch J, Sheng M, Triller A, Betz H, Kneussel M (2002) Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J Neurosci 22: 5393–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritsen EJ, Vossen JM, Fasth A, Friedrich W, Morgan G, Padmos A, Vellodi A, Porras O, O'Meara A, Porta F, Bordigoni P, Cant A, Hermans J, Griscelli C, Fischer A (1994) Bone marrow transplantation for autosomal recessive osteopetrosis. A report from the Working Party on Inborn Errors of the European Bone Marrow Transplantation Group. J Pediatr 125: 896–902 [DOI] [PubMed] [Google Scholar]
- Goebel HH, Wisniewski KE (2004) Current state of clinical and morphological features in human NCL. Brain Pathol 14: 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther W, Piwon N, Jentsch TJ (2003) The ClC-5 chloride channel knock-out mouse—an animal model for Dent's disease. Pflügers Arch 445: 456–462 [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S, Verkman AS (2005) ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J Biol Chem 280: 1241–1247 [DOI] [PubMed] [Google Scholar]
- Ivy GO, Schottler F, Wenzel J, Baudry M, Lynch G (1984) Inhibitors of lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain. Science 226: 985–987 [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA (2002) Molecular structure and physiological function of chloride channels. Physiol Rev 82: 503–568 [DOI] [PubMed] [Google Scholar]
- Kleijer WJ, van Diggelen OP, Keulemans JL, Losekoot M, Garritsen VH, Stroink H, Majoor-Krakauer D, Franken PF, Eurlings MC, Taschner PE, Los FJ, Galjaard RJ (2001) First-trimester diagnosis of late-infantile neuronal ceroid lipofuscinosis (LINCL) by tripeptidyl peptidase I assay and CLN2 mutation analysis. Prenat Diagn 21: 99–101 [DOI] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ (2001) Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104: 205–215 [DOI] [PubMed] [Google Scholar]
- Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C (2000) Mutations in the a3 subunit of the vacuolar H+-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet 9: 2059–2063 [DOI] [PubMed] [Google Scholar]
- Li YP, Chen W, Liang Y, Li E, Stashenko P (1999) Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 23: 447–451 [DOI] [PubMed] [Google Scholar]
- Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, Harding B, Bolino A, Devoto M, Goodyer P, Rigden SP, Wrong O, Jentsch TJ, Craig IW, Thakker RV (1996) A common molecular basis for three inherited kidney stone diseases. Nature 379: 445–449 [DOI] [PubMed] [Google Scholar]
- Mitchison HM, Lim MJ, Cooper JD (2004) Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol 14: 86–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel N, Dedieu JC, Philippe JM (2003) Specific sorting of the a1 isoform of the V-H+ATPase a subunit to nerve terminals where it associates with both synaptic vesicles and the presynaptic plasma membrane. J Cell Sci 116: 4751–4762 [DOI] [PubMed] [Google Scholar]
- Nishi T, Forgac M (2000) Molecular cloning and expression of three isoforms of the 100-kDa a subunit of the mouse vacuolar proton-translocating ATPase. J Biol Chem 275: 6824–6830 [DOI] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF (2003) Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA 100: 1902–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, Jolly RD (1992) Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease). Am J Med Genet 42: 561–567 [DOI] [PubMed] [Google Scholar]
- Piwon N, Günther W, Schwake R, Bösl MR, Jentsch TJ (2000) ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 408: 369–373 [DOI] [PubMed] [Google Scholar]
- Prasad VV, Pullarkat RK (1996) Brain lysosomal hydrolases in neuronal ceroid-lipofuscinoses. Mol Chem Neuropathol 29: 169–179 [DOI] [PubMed] [Google Scholar]
- Ruether K, van de Pol D, Jaissle G, Berger W, Tornow RP, Zrenner E (1997) Retinoschisislike alterations in the mouse eye caused by gene targeting of the Norrie disease gene. Invest Ophthalmol Vis Sci 38: 710–718 [PubMed] [Google Scholar]
- Schwartzberg PL, Xing L, Hoffmann O, Lowell CA, Garrett L, Boyce BF, Varmus HE (1997) Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes Dev 11: 2835–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimeca JC, Franchi A, Trojani C, Parrinello H, Grosgeorge J, Robert C, Jaillon O, Poirier C, Gaudray P, Carle GF (2000) The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 26: 207–213 [DOI] [PubMed] [Google Scholar]
- Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG (2004) CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 113: 274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Sohar I, Pullarkat PS, Lobel P, Pullarkat RK (1998) Specific alterations in levels of mannose 6-phosphorylated glycoproteins in different neuronal ceroid lipofuscinoses. Biochem J 334: 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward CG (2003) Neurological aspects of osteopetrosis. Neuropathol Appl Neurobiol 29: 87–97 [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ (2001) Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29: 185–196 [DOI] [PubMed] [Google Scholar]
- Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M (2003) From lysosomes to the plasma membrane: localization of vacuolar-type H+-ATPase with the a3 isoform during osteoclast differentiation. J Biol Chem 278: 22023–22030 [DOI] [PubMed] [Google Scholar]
- Toyomura T, Oka T, Yamaguchi C, Wada Y, Futai M (2000) Three subunit a isoforms of mouse vacuolar H+-ATPase. Preferential expression of the a3 isoform during osteoclast differentiation. J Biol Chem 275: 8760–8765 [DOI] [PubMed] [Google Scholar]
- Tyynelä J, Palmer DN, Baumann M, Haltia M (1993) Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett 330: 8–12 [DOI] [PubMed] [Google Scholar]
- Tyynelä J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, Baumann M, Haltia M, Lobel P (2000) A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J 19: 2786–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL (2000) Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA 97: 10954–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M, Uchida S, Ezaki J, Rai T, Hayama A, Kobayashi K, Kida Y, Noda M, Koike M, Uchiyama Y, Marumo F, Kominami E, Sasaki S (2002) CLC-3 deficiency leads to phenotypes similar to human neuronal ceroid lipofuscinosis. Genes Cells 7: 597–605 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information