

Abstract
The key molecular mechanism governing the cancer cell state (stem cell-like state vs differentiation state) to control the cancer stem cell (CSC) pool remains elusive. This study provides the first evidence showing that all-trans retinoic acid (ATRA) induces the interaction and chromatin recruitment of a novel RARβ-TET2 complex to epigenetically activate a specific cohort of gene targets, including MiR-200c. TET2-activated miR-200c further targets and suppresses PKCζ, a cell polarity protein that has a pivotal role in directing asymmetric division of mammalian stem cells to sustain the stem cell pool. Our data reveal that pharmacological concentration of ATRA effectively downregulates PKCζ through activation of miR-200c, leading to a decrease of the stem cell-like populations from non-tumorigenic mammary epithelial cells and non-aggressive breast cancer cells. However, aggressive breast cancer cells that manifest TET2-miR-200c dysregulation sustain a CSC pool highly resistant to ATRA, where inhibition of PKCζ directs the resistant CSCs to the luminal cell-like state and sensitization to tamoxifen, resulting in abrogation of mammary tumor growth and progression. Together, these findings elucidate a novel RARβ-TET2-miR-200c-PKCζ signaling pathway that directs cancer cell state changes and also provide previously unidentified therapeutic implications for PKCζ inhibitors in diminishment of breast CSCs to eradicate breast cancer.
Supplementary information
The online version of this article (doi:10.1038/onc.2016.467) contains supplementary material, which is available to authorized users.
Subject terms: Breast cancer
Introduction
During tumorigenesis, the generation of cancer stem cells (CSCs), a subset of the cancer cell population that possesses self-renewal properties associated with normal stem cells, allows the perpetual rise of the bulk of a tumor as the ‘seed’ of the cancer.1, 2, 3, 4 Accumulated evidence shows that the cell fate decision that favors asymmetric division (AD) of stem cells is critical to ensure a self-renewing and sustaining stem cell pool.5, 6, 7 It has been known that atypical protein kinase C (aPKC), the cell polarity protein family that includes PKCζ and PKCι/λ, directs self-renewing AD through phosphorylation and asymmetric segregation of the cell fate determinant NUMB (S276/S295) to only one of the progenies to maintain the stem cell pool.6, 7 In contrast, loss of aPKC expression/activity leads to a uniform distribution of NUMB with the consequent symmetric commitment (SC) of both daughter cells to the differentiation cell state, resulting in decrease of the stem cell pool.5, 6, 7 Nonetheless, the critical mechanism underlying regulation of aPKC that directs the cell fate decision to govern the cancer cell state changes (stem cell-like state vs differentiation state) still remains to be elucidated.
Numerous studies have shown that RA signaling directs stem cell differentiation and is critical for the maintenance of the stemness differentiation homeostasis.8 All-trans retinoic acid (ATRA), a natural derivative of vitamin A, is a potent molecule for inducing cell differentiation and the only example of a clinically useful cytodifferentiating agent in treatment of several solid tumor types, including breast cancer,8, 9, 10, 11, 12 presents an attractive modality of treatment that targets the CSC population and tends to be less toxic and more specific than conventional chemotherapy.8 Even through some of the trial patients receive significant therapeutic benefits, a large proportion of patients with aggressive/relapsed breast cancers exhibit only modest/nonsignificant therapeutic effects.8, 13 Notably, studies have shown that retinoic acid receptors (RARs), a nuclear receptor family that binds to retinoid X receptor (RXRs) as heterodimer (RAR/RXR) to the RAR response element (RAR-RE) in the target gene promoters, undertake conformational changes to recruit transcriptional co-activators or to dissociate co-repressors once the RARs have interacted with the RA ligands, such as ATRA, leading to transcriptional activation of the target genes.13 Among the RARs (RARα, RARβ and RARγ), lost RARβ expression is most significantly correlated with tumor grade and is also associated with RA resistance.13 However, the mechanism that links RARβ to RA response (sensitivity vs resistance) is yet to be established.
This study uncovers a novel RARβ-TET2-miR-200c-PKCζ signaling pathway that directs breast cancer cell state changes in response to ATRA. The resulting findings provide important molecular insights into new strategies to overcome the therapeutic hurdles of aggressive breast cancer.
Results
ATRA induces interaction and nuclear colocalization of RARβ-TET2
To determine the mechanism(s) underlying RARβ-regulated ATRA response, a mass spectrometry analysis was used to identify potential chromatin proteins that specifically interact with RARβ in response to a pharmacological concentration of ATRA treatment (10−6 M)8, 13 in MCF12A, a non-tumorigenic, immortalized mammary epithelial cell line with intact RARβ expression and RA response. Interestingly, we found that a methylcytosine dioxygenase, ten eleven translocation2 (TET2), was associated with RARβ in ATRA-treated MCF12A cells (Supplementary Table S1, TET2 had the highest number of the peptides matched for the protein with the highest sequence coverage among the identified RARβ-interacting proteins), where endogenous RARβ/RXRα can also be reciprocally co-immunoprecipitated with TET2 in MCF12A cells, as well as in T47D cells, a non-invasive breast cancer cell line (Figure 1a, Supplementary Figure S1). GST pull down data also validated a direct physical interaction between RARβ and TET2 proteins in vitro, where their interaction was substantially enhanced in the presence of ATRA (Figure 1b). It is well recognized that the TET protein family has a key role in active DNA demethylation by catalyzing the conversion of the modified genomic base 5-methylcytosine into 5-hydroxymethylcytosine (5 hmC), thereby activating the target gene expression.14 Among the three human TET proteins (TET1–TET3), TET2 is the most predominantly expressed in normal breast tissue and the expression is significantly repressed in aggressive breast cancers.15 To gain functional insight into RARβ–TET2 interaction, we analyzed the interaction of RARβ with TET2 mutant proteins along with deletions of specific functional domains16 (the domains are conserved between human and mouse). The data showed that an intact C-terminal catalytic domain (CD, containing DSBH and cysteine-rich domains) was required for both human and mouse TET2 to have optimal interactions with RARβ (Figure 1c, Supplementary Figure S2). Furthermore, it is known that RAR nuclear translocation can be induced by ATRA;13 however, the regulatory mechanism underlying nuclear localization and chromatin recruitment of TET2, which has no apparent nuclear localization signal sequence or DNA-binding domain, still remains largely unknown. We found that compared with the mock-treated cells, ATRA enhanced the nuclear localization of RARβ; interestingly, ATRA also elevated TET2 expression in the nuclear fraction, whereas knockdown of RARβ blocked ATRA-mediated TET2 nuclear localization and substantially increased TET2 expression in the cytoplasm fraction (Figure 1d). Concordantly, confocal immunofluorescence analysis showed that ATRA significantly increased localization of TET2 to the nucleus, where it colocalized with RARβ (Figures 1e and f). However, TET2 re-localized to the cytoplasm upon knockdown of RARβ using lentivirus-expressed short hairpin RNA (sh-RARβ) in MCF12A cells (Figures 1e and f). These results could be recapitulated in a luminal breast cancer cell line, T47D, where RARβ expression is required for increased TET2 nuclear localization along with an enhanced 5hmc level by ATRA treatment (Supplementary Figure S3).
Figure 1.
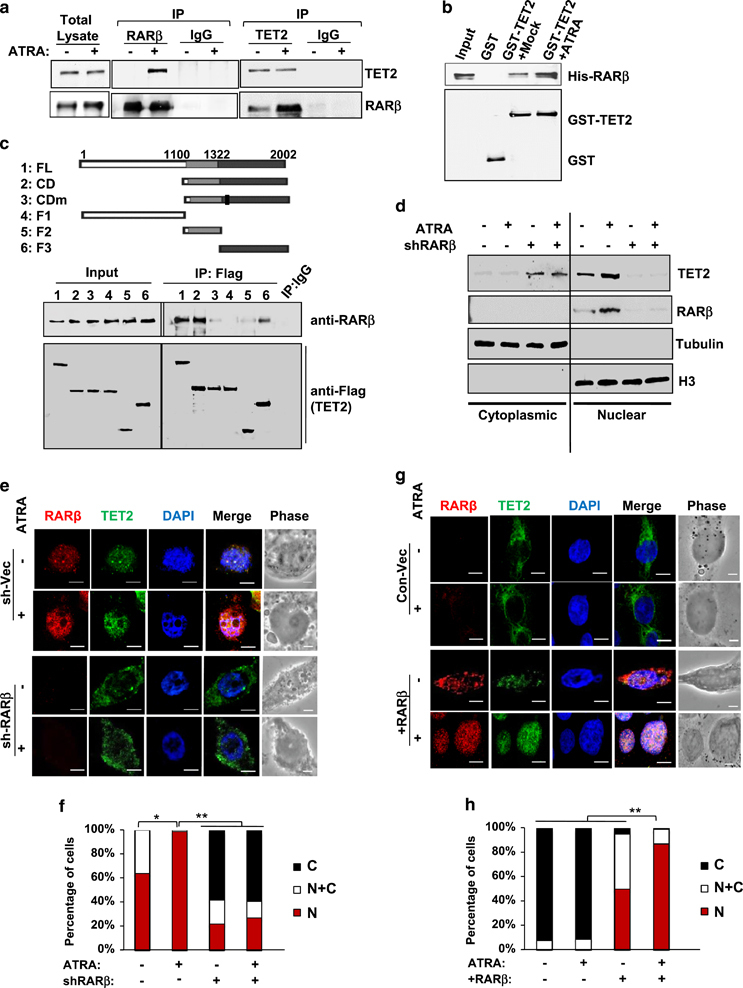
ATRA induces interaction of RARβ-TET2 and nuclear localization of TET2. (a) Reciprocal co-immunoprecipitation assay showing interaction between endogenous RARβ and TET2 in MCF12A cells treated with 10−6M ATRA or vehicle for 6 h (input: 10% total cell lysate). (b) GST pull down assay showing direct association of His-RARβ and GST-TET2 recombinant proteins in vitro. (c) Full-length (FL) and deletion mutants of Flag-tagged human TET2 were expressed and immunoprecipitated with the Flag antibody followed by immunoblotting for RARβ in MCF12A cells treated with 10−6 M ATRA or vehicle for 6 h. The TET2 mutants include the C-terminal catalytic domain (CD, containing DSBH and cysteine-rich domains), CD domain carrying H1382Y/D1384A mutation (CDm), N-terminal domain (NT), cysteine-rich domain (F1) and DSBH (double-strand beta helix) domain (F2). (d) Protein levels in the nuclear and cytoplasmic fractions of MCF12A cell lysates that stably expressed the control vector or sh-RARβ and treated with 10−6 M ATRA or vehicle for 6 h. (e) Confocal immunofluorescence staining images showing subcellular localization of RARβ and TET2 in MCF12A cells that expressed the control vector (sh-Vec) or sh-RARβ and treated with 10−6M ATRA or vehicle for 6 h. (f) Bar graphs showing the percentage of cells with TET2 localized to the nucleus (N), cytoplasm (C) or both (N+C) analyzed from the images in e. (g) Confocal immunofluorescence staining images showing subcellular localization of RARβ and TET2 in MDA-MB-231 cells that expressed the control vector (Con-Vec) or RARβ expression plasmid and treated with 10−6 M ATRA or vehicle for 6 h. (h) Bar graphs showing the percentage of cells with TET2 localized to the nucleus (N), cytoplasm (C) or both (N+C) analyzed from the images in g. In all, 700–1000 cells were counted per sample from three independent experiments, asterisk indicates P<0.05, double asterisk indicates P<0.01. Comparable amount of control vector and vehicle were included as negative control (-) in each group. Error bars denote ±s.d.
In contrast to MCF12A and T47D cells, TET2 was predominantly localized in the cytoplasm in aggressive triple-negative breast cancer (TNBC) cell line MDA-MB-231, which is deficient in endogenous RARβ expression (Figures 1g and h). Re-expression of RARβ in MDA-MB-231 cells re-localized TET2 to the nucleus, and the nuclear TET2 level was further enhanced by ATRA treatment (Figures 1g and h). Next, we ectopically expressed the TET2 full-length (FL) and mutant proteins (NT, N-terminal domain, and CD, catalytic domain, Figure 1c) along with wild-type RARβ in the MDA-MB-231 cells. We found that the FL- and CD-TET2 proteins, which showed strong interaction with RARβ in Figure 1c, were localized to the cytoplasm in the absence of RARβ. FL-and CD-TET2 started to exhibit nuclear localization upon ectopic expression of RARβ, and their nuclear localization was further enhanced by ATRA, along with the increased nuclear RARβ (Supplementary Figure S4). In contrast to FL-and CD-TET2, NT-TET2, which could not interact with RARβ (Figure 1c), remained in the cytoplasm and did not respond to ATRA nor to the ectopic expression of RARβ (Supplementary Figure S4).
ATRA-induced RARβ-TET2 complex co-occupies targeted promoters of the genes involved in cell differentiation
To understand whether and how the interaction of RARβ and TET2 impacts global gene expression, we performed genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) analysis to identify the RARβ and TET2 bound chromatin regions. We found that ATRA most significantly enhanced RARβ-TET2 co-occupancy in promoters of a cohort of genes involved in cell differentiation (Figures 2a and b), such as RUNX1, BMP6, IKZF1 and CAV1, by gene ontology enrichment analysis (Supplementary Table S2, false discovery rate <0.5, P<0.01). Furthermore, RARβ-TET2 also co-occupied promoters of several non-coding RNAs, such as MicroRNA-200c-3p (MiR-200c-3p) and MicroRNA-205-5p (MiR-205-5p; representative MiR-200c peak graph shown in Figure 2c), both of which are known to regulate stemness properties.17, 18, 19, 20 Peak and motif analysis revealed that the majority of RARβ-TET2 peaks were located at promoters/transcription start sites of high CpG (HCP) content, where putative RAR-REs-AGGTCAnnnnnAGGTCA- are located right next to CpG islands that were strongly associated with 5 hmc (Figures 2c–f: the gray bar denotes RAR-RE; the black bar denotes CpG island, P<0.001). Consistently, the expression levels of RARβ-TET2 targeted genes/miRNAs were significantly upregulated by ATRA treatment, among which miR-200c was the most activated (Figure 2g). Furthermore, the gene/miRNA expression levels increased by ATRA were abolished upon knockdown of RARβ or TET2 (sh-RARβ, sh-TET2) in MCF12A cells (Figures 2g and h), where knocking-down RARβ also markedly diminished the nuclear TET2 level (Figure 2h). Together, these data suggest that ATRA induces physical association and chromatin recruitment of the RARβ-TET2 complex to specific promoter regions that contain RAR-REs next to CpG islands, which can be critical for ATRA-mediated activation of the target genes involved in regulation of cell differentiation stemness homeostasis.
Figure 2.
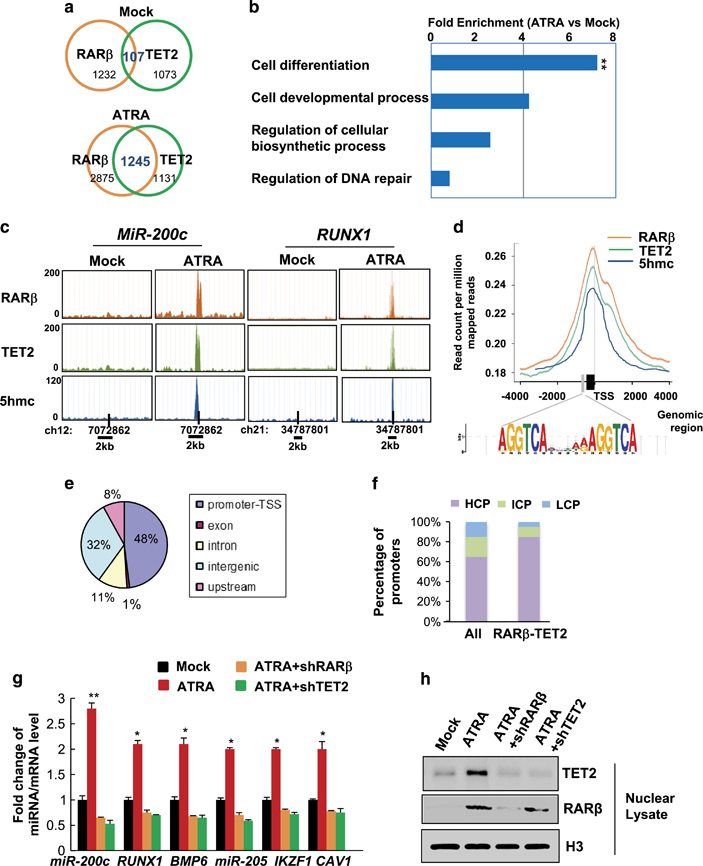
ATRA-induced RARβ-TET2 co-occupancy in targeted promoters of genes involved in cell differentiation. (a–f) ChIP-sequencing analysis results of MCF12A cells treated with 10−6 M ATRA or vehicle for 12 h (n=3 independent experiments, double asterisk indicates P<0.01): (a) Venn diagram showing overlap of RARβ and TET2-bound genes, (b) gene ontology functional annotation of RARβ and TET2-bound genes showing enrichment in specific biological processes, (c) peak histograms showing RARβ and TET2 co-occupied MiR-200c (left) and RUNX1 (right) promoters with enhanced 5hmc (bar positions denote the transcription start site (TSS)), (d) RARβ, TET2, and 5hmc peak distribution around TSS (gray bar denotes RARE motif, black bar denotes CpG island), (e) pie chart showing the percentage of RARβ and TET2 co-occupied genomic regions that belong to promoter-TSS, exon, intron, intergenic or upstream regions, and (f) histograms showing the percentage of gene promoters that are divided into high-, intermediate- or low CpG density promoters (HCP, ICP or LCP) for all genes and for RARβ-TET2 co-occupied genes. (g) Fold change of miRNA/mRNA expression, and (h) nuclear protein expression levels in MCF12A cells that stably expressed sh-RARβ or sh-TET2 and treated with 10−6 M ATRA or vehicle for 12 h (n=3 independent experiments, asterisk indicates P<0.05, double asterisk indicates P<0.01). Comparable amount of control vector and vehicle were included as negative control (-) in each group. Error bars denote ±s.d.
ATRA activates miR-200c expression via recruitment of RARβ-TET2 to a specific MiR-200c promoter region and mediates conversion of 5-methylcytosine to 5 hmc
Following the ChIP-seq data, we analyzed the changes in the global miRNA expression profile in response to the ATRA treatment using a genome-wide miRNA–PCR array consisting of 1066 annotated miRNAs, and we found that ATRA significantly upregulated a subset of microRNAs in MCF12A cells, among which microRNA-200c-3p (miR-200c) was the most significantly upregulated (Supplementary Figure S5a, >2.5-fold increase compared with the mock treatment, P<0.01). Despite the previous finding that links TET proteins to activated miR-200c expression,15 the regulatory mechanism underlying TET-mediated activation of miR-200c expression is largely unclear.
Therefore, to investigate whether ATRA-RARβ indeed has a role in regulation of TET2-mediated activation of miR-200c, we first analyzed, using promoter analysis (Genomatix MetInspector, Ann Arbor, MI, USA), the transcription factor response elements located within the 2-kb region upstream of the transcription starting site of MiR-200c. We found eight putative RAR response elements (RAR-RE, s1–s8) located within the MiR-200c promoter that had high consensus scores (Supplementary Table S3, Supplementary Figure S5b). To validate the direct association of RAR family proteins with miR-200c, we performed ChIP analysis in MCF12A cells targeting the RAR-RE using antibodies specifically against RARα, RARβ and RARγ. We found that among these RAR family members, RARβ was most strongly associated with the MiR-200c promoter (Supplementary Figure S5c). Specifically, ATRA induced a significant enhancement of RARβ association to the MiR-200c promoter region 8 (r8), which encompassed a putative RAR-RE (s8) right next to a CpG island (Supplementary Figures S5b and c). Interestingly, TET2 also showed a significant association with r8 upon ATRA treatment (Supplementary Figure S5d).
ATRA treatment consistently resulted in transcriptional activation of the luciferase driven by MiR-200c promoter, which was reversed by mutations of the RAR-RE s8 (Supplementary Figures S6a and b). The sequential-ChIP results further revealed that RARβ along with TET2 were indeed bound to the MiR-200c promoter (Supplementary Figure S6c). ATRA significantly increased the association of both RARβ and TET2 with the MiR-200c promoter, where the 5 hmc level was enhanced, whereas the 5-methylcytosine level was reduced (Supplementary Figures S6d and e). However, knocking-down RARβ abolished the association between TET2 and the MiR-200c promoter with a markedly reduced 5 hmc level (Supplementary Figures S6d and e). Together, these data suggest that RARβ is required for recruitment of TET2 in a complex bound to miR-200c promoter region.
Lost nuclear TET2 and deficient miR-200c expression is correlated with ATRA resistance in high tumor grade and aggressive breast cancer
To further strengthen the pathological correlation of RARβ-TET2-miR-200c regulation in human breast cancer, we performed a correlation analysis of RARβ (nuclear vs cytoplasmic), TET2 (nuclear vs cytoplasmic) and miR-200c expression levels in human breast tissue microarrays consisting of a cohort of breast tumor samples. We found that RARβ and TET2 were predominantly expressed in the nucleus of the well-differentiated low tumor grade breast tumors (LG, grade I), where miR-200c was highly expressed (Figures 3a and c, arrowheads indicate positive nuclear staining, n=140, P<0.01). In contrast, the poorly differentiated high tumor grade tumors (HG, grades II–III) exhibited lost expression of total/nuclear RARβ and TET2, along with significantly reduced miR-200c levels (Figures 3a and c, n=140, P<0.01). We next examined the correlation of nuclear TET2 expression level with the CSC sensitivity to ATRA, indicated by the number of tumor spheres under treatment of various concentrations of ATRA (sensitive-S vs resistant-R defined by EC50 of sphere suppression under ATRA treatment with a dose range between 10−8 M and 10−4 M), in a cohort of primary human breast cancer cells (six luminal breast cancer, seven TNBC and one Her2-positive breast cancer Her2+), and non-tumorigenic immortalized mammary epithelial cell lines (MCF12A and MCF10A). Notably, TNBC, a breast cancer subtype deficient in expression of estrogen receptor (ER), progesterone receptor (PR) and HER2, tends to be more aggressive than the other subtypes and manifests high recurrence along with poor survival.3 We found that the nuclear TET2 level and the sensitivity of the spheres in response to ATRA are comparable between the non-tumorigenic mammary epithelial cell lines and the luminal/Her2+ breast cancer cells, where nuclear TET2 was highly expressed, and ATRA-EC50 of sphere suppression was <10−6 M (Figure 3d). However, compared with luminal/Her2+ breast cancer cells, nuclear TET2 expression was uniformly and markedly diminished in all of the primary TNBC cells, as well as in the TNBC-like breast cancer cell lines, where a resistance to ATRA-mediated tumor sphere suppression was manifested (Figure 3d, n=23, r=−0.8, P=0.02). Consistently, 5 hmc and miR-200c levels were significantly reduced in the ATRA-resistant cancer cells compared with the ATRA-sensitive cancer cells (Figures 3e and f, n=23, P<0.05). Together, these data indicate that deficient RARβ-TET2-miR-200c regulation is functionally linked to ATRA resistance and is correlated with a poorly differentiated, TNBC phenotype.
Figure 3.
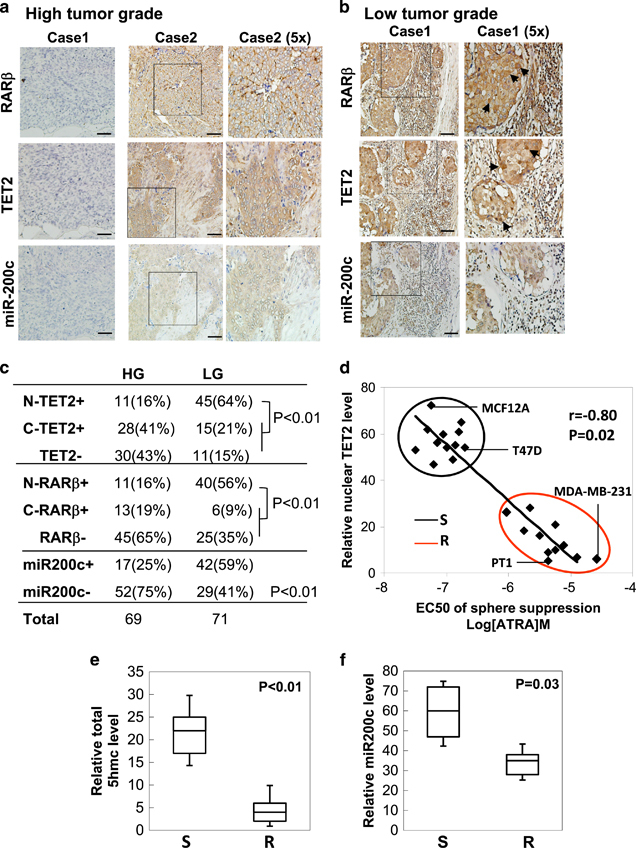
Lost nuclear RARβ/TET2 and deficient miR-200c expression is correlated with ATRA resistance in high-grade, aggressive breast cancer. (a, b) Representative IHC staining images showing expression levels of RARβ, TET2 and miR-200c in 140 human breast tissue specimens, including low-grade tumors (a: tumor grade I, LG) and high-grade tumors (b: tumor grades II–III, HG, arrowhead indicates positive nuclear staining, scale bar: 100 μm). (c) The nuclear/cytoplasmic RARβ, nuclear/cytoplasmic TET2 and total miR-200c levels were scored and their correlation with tumor grade was analyzed by Chi-square analysis. N, nuclear staining; C, cytoplasmic staining; (−), negative-low staining; (+), strong-positive staining. (d) Pearson’s correlation analysis of ATRA sensitivity (S) and resistance (R) defined by ATRA-EC50 for tumor sphere suppression with the relative expression level of nuclear TET2 showing that ATRA-EC50 is negatively correlated with nuclear TET2 level (r=−0.8, P=0.02). ATRA-resistant cells (n=11, grouped in red circle) that include seven triple-negative primary tumor samples (for example, PT1), as well as BT549, MDA-MB-231, BT20, and SUM149PT breast cancer cell lines; ATRA-sensitive cells (n=12, grouped in black circle) that include six luminal and one HER2+ primary tumor samples, as well as MCF7, T47D, ZR75-1 breast cancer cell lines and MCF10A and MCF12A cell lines. Box plots showing (e) the relative 5hmc levels, and (f) the relative miR-200c levels in ATRA-sensitive (S) cells and ATRA-resistant (R) cells shown in d. R, resistant cells with ATRA-EC50 of sphere suppression ⩾10−6M; S, sensitive cells with ATRA-EC50 of sphere suppression <10−6 M. Relative expression levels of nuclear TET2, 5 hmc, and miR-200c levels were determined by expression ratios to normalized total input lysates, total input genomic DNA and RNU6B expression, respectively. Error bars denote ±s.d.
TET2 expression is required for sensitizing breast CSCs to ATRA
Next, we attempted to determine whether ATRA-induced TET2 signaling is involved in regulation of stemness properties, including the EMT phenotype that is known to link to generation of the CSC traits.20 We found that ATRA treatment in T47D cells led to an increase in the expression of the epithelial marker, E-cadherin, which was abolished upon further knock-down of TET2 (Figure 4a). Knockdown of TET2 also substantially increased the expression of the mesenchymal marker, N-cadherin (Figure 4a).
Figure 4.
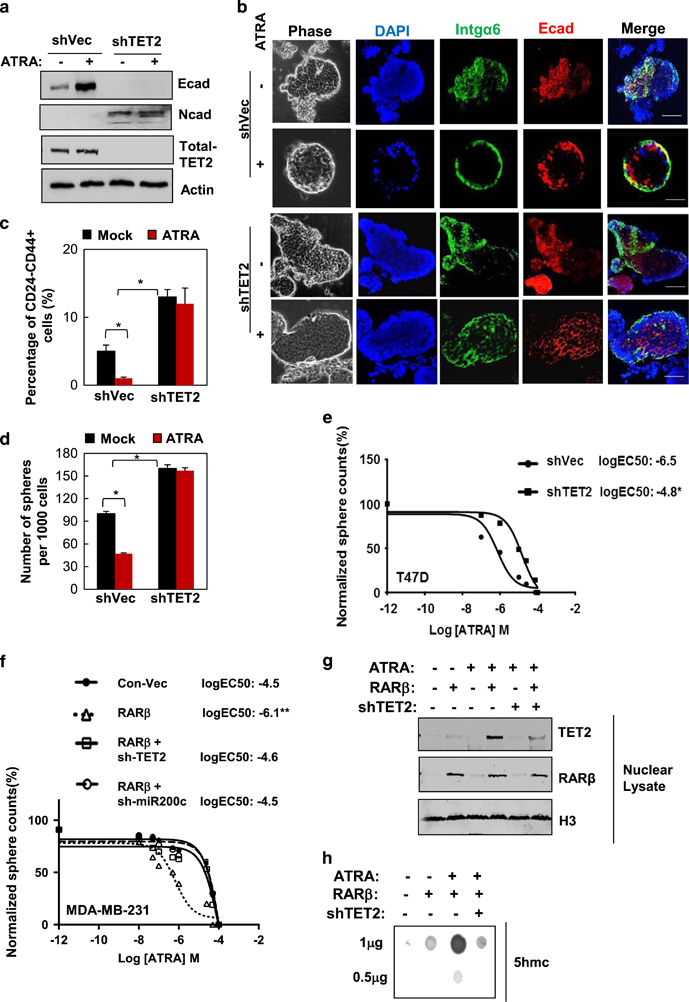
TET2 activation is required for breast CSC sensitivity to ATRA. (a) Immunoblots showing protein expression levels of E-cadherin (epithelial marker) and N-cadherin (mesenchymal marker) in T47D breast cancer cells that stably expressed sh-TET2 or sh-Vec and treated with 10−6 M ATRA or vehicle for 3 days. (b) T47D-sh-TET2 and T47D-sh-Vec single-cell suspensions were subjected to three-dimensional (3D) on-top matrigel culture (20 000 cells per well in 24-well plate) under 10−6 M ATRA or vehicle treatment for 12 days (treatment medium was replenished every 2 days); acini were stained for basal polarity marker (α6-integrin, green), epithelial marker (E-cadherin, red) and DAPI (nucleus, blue), and then subjected to confocal immnunofluorscence microscopic analysis (scale bar: 50 μm, 600 acini were imaged and analyzed from three independent experiments, also see Supplementary Figure S7). (c) The percentage of the CD24−CD44+ population isolated from T47D-sh-TET2 or T47D-sh-Vec cells treated with 10−6 M ATRA or vehicle for 3 days (n=3 independent experiments, asterisk indicates P<0.05). (d) The number of spheres (sphere size >100 μm) per 1000 initially plated T47D-sh-TET2 or T47D-sh-Vec cells treated with 10−6 M ATRA or vehicle for 7 days. (n=3 independent experiments, asterisk indicates P<0.05). (e) The dose response was plotted and EC50 was calculated using GraphPad Prism software (La Jolla, CA, USA), based on the number of the spheres generate from T47D-sh-TET2 or T47D-sh-Vec cells treated with ATRA or vehicle for 7 days with the dose range between 10−12M and 10−4M (where Y axis is the normalized sphere number counts (%) and X axis is the logarithm of (ATRA) concentration, n=3 independent experiments, asterisk indicates P<0.05). (f) Dose-response curve (normalized sphere number (%) vs log (ATRA) M), (g) nuclear TET2 and RARβ2 protein expression levels and (h) total 5hmc levels shown by 5 hmc dot blot using diluted genomic DNA (1 and 0.5 μg) of MDA-MB-231 breast cancer cells that expressed RARβ, RARβ+sh-TET2, RARβ+sh-miR-200c or sh-Vec under ATRA or vehicle treatment (n=3 independent experiments, double asterisk indicates P<0.01). Comparable amount of control vector and vehicle were included as negative control (-) in each group. Error bars denote ±s.d.
We further attempted to understand the role of ATRA-TET2 regulation on mammary cell stemness vs differentiation using a three-dimensional acini culture in matrigel.21 We found that T47D cells exhibited disorganized acinar formation (enlarged acini size with luminal filling), along with disrupted expression of α6-integrin (basal polarity marker) and E-cadherin (epithelia marker) (Figure 4b and Supplementary Figure S7), where ATRA treatment led to retrieval of the normal epithelial-like, well-differentiated acinar structure with polarized expression of α6-integrin and E-cadherin (Figure 4b and Supplementary Figure S7). ATRA treatment consistently resulted in a substantial reduction in the putative stem cell population marked by CD24−CD44+ or enriched by the side population using Hoechst dye (Figure 4c, Supplementary Figures S8a and b), and also suppressed the number of spheres, suggesting a diminished stem cell pool under ATRA treatment (Figure 4d: no significant changes in cell death or cell cycle profile; data not shown). In contrast, knockdown of TET2 enhanced the CD24−CD44+ population and the side population (Figure 4c, Supplementary Figures S8a and b), along with increasing the sphere formation and abolishing ATRA-mediated effects on cell polarization and acinar formation (Figures 4b and d).
We also found that T47D cells were sensitive to ATRA, which blocked 50% of the sphere formation with a half maximal effective concentration (EC50) at 10−6.5 M (Figure 4e). However, knockdown of TET2 significantly diminished the sensitivity of T47D cells to ATRA as indicated by the elevated EC50 (to EC50=10−4.8 M, Figure 4e). In contrast to T47D, sphere formation in MDA-MB-231 cells, which exhibited a deficiency in endogenous RARβ expression and TET2 nuclear localization as shown in Figure 1g, was highly resistant to ATRA treatment (EC50=10−4.5 M, Figure 4f). We found that ectopic expression of RARβ in MDA-MB-231 cells was able to sensitize the cells to ATRA treatment (to EC50=10−6.1 M, Figure 4f), accompanied by the enhanced nuclear TET2 expression and increased 5 hmc level (Figures 4g and h), whereas further knockdown of TET2 diminished 5 hmc expression and reversed ATRA sensitization caused by RARβ re-expression (Figures 4f and h).
Our data in Figure 2 suggest that miR-200c is a major target of ATRA-TET2 in regulation of stemness. Indeed, knockdown of miR-200c in ATRA-sensitive T47D cells promoted sphere formation and tumor-seeding capacity (Supplementary Figures S8c and d), which were previously inhibited upon ATRA treatment. However, ectopic expression of miR-200c sensitized ATRA-resistant cells MDA-MB-231 to ATRA as evidenced by significantly reduced sphere formation and abolished xenograft tumor formation, suggesting a markedly diminished CSC pool (Supplementary Figures S8e and f). Together, these data reveal the critical role that TET2-miR-200c has in regulation of breast CSC sensitivity to ATRA treatment.
ATRA downregulates PKCζ through activation of miR-200c
We next attempted to identify novel gene targets and the associated mechanism regulated by TET2-miR-200c in response to ATRA. Using global gene expression profiling analysis of T47D cells that were under ATRA treatment compared with that of the mock-treated cells, combined with the analysis result of miR-200c seed sequence matching targets from three miRNA-target prediction databases (DIANSmT, miRand and TargetScan), 26 putative miR-200c targets were identified with expression levels that were significantly downregulated by ATRA and also with high context and conservation seed sequence matching scores of miR-200c interaction (Figure 5a, TargetScan 6.0); among these candidates, PRKCζ (encodes protein PKCζ) was the most significantly downregulated gene under ATRA treatment (Figure 5a, 9.7-fold change, n=3, false discovery rate <0.05, P<0.01). PKCζ, a member of the aPKC family, has an evolutionarily conserved role in directing self-renewing AD of mammalian stem cells.5, 6, 7 Notably, PKCζ overexpression is linked to breast cancer invasiveness and metastasis.22
Figure 5.
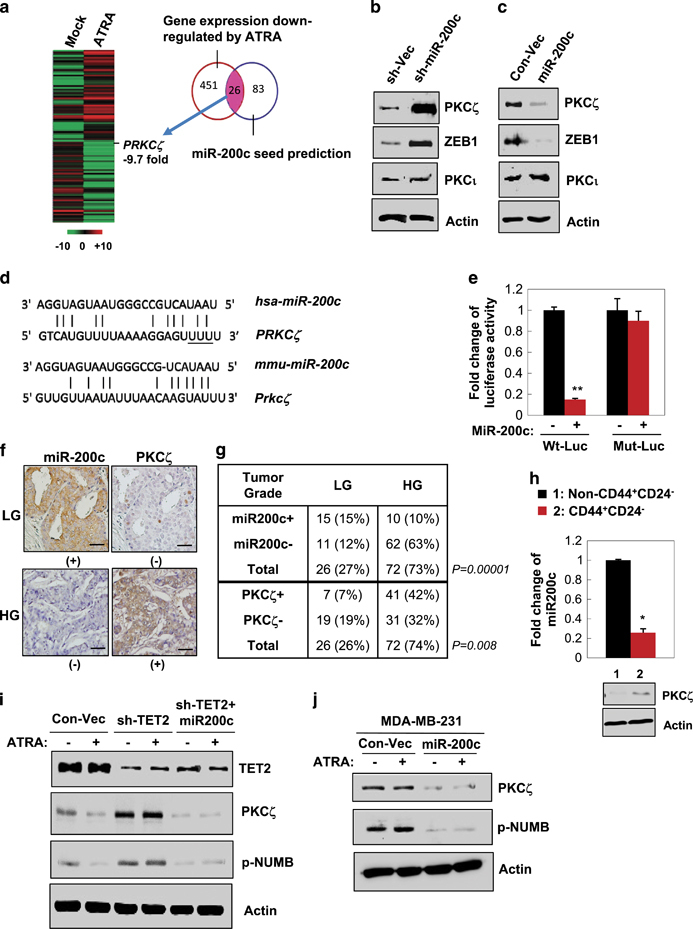
ATRA treatment leads to downregulation of PKCζ, a new miR-200c target. (a) Heat map showing mean expression values of the putative miR-200c target genes with expression levels that were most significantly downregulated in 10−6 M ATRA-treated T47D cells for 24 h (n=3 independent experiments, fold change >2 compared with vehicle-treated cells, P<0.05). Putative miR-200c targets were identified by seed sequence matching from three prediction databases (DIANSmT, miRand and TargetScan). (b) Immunoblots showing PKCζ expression levels in T47D cells that stably expressed sh-miR-200c and the control vector. (c) Immunoblots showing PKCζ expression levels in T47D cells that stably expressed miR-200c and the control vector. (d) Diagram showing the putative miR-200c targeting seed sequence on PKCζ-3′-UTR, which was conserved between human and mouse (underline indicating mutation of UUU to CCC in the mutated PKCζ-3′UTR luciferase reporter, Mut-Luc). (e) Fold change of luciferase activity driven by the wild-type or mutant PKCζ-3′-UTR reporter in T47D cells that stably expressed miR-200c and treated with 10−6 M ATRA or vehicle for 24 h (n=3 independent experiments, double asterisk indicates P<0.01). (f) Representative IHC staining images showing a reverse correlation between expression of miR-200c and PKCζ in human breast tumor sections. (-), negative-low staining; (+), strong-positive staining. (g) Chi-square correlation analysis of the expression levels of PKCζ and miR-200c with the tumor grade (n=98, P<0.01; tumor grades II-III, HG; tumor grade I, LG). (h) miR-200c and PKCζ expression levels in the CD44+CD24− population versus the non-CD44+CD24− population isolated from primary human breast tumor cells (n=3 independent experiments, asterisk indicates P<0.05). (i) Protein expression in T47D cells that stably expressed miR-200c, sh-TET2, or the control vectors and treated with 10−6 M ATRA or vehicle for 24 h. (j) Protein expression in MDA-MB-231 cells that stably expressed miR-200c or the control vector and treated with 10−6 M ATRA or vehicle for 24 h. Comparable amount of control vector and vehicle were included as negative control (-) in each group. Error bars denote ±s.d.
Based on the data shown in Figure 5a, we speculated that PKCζ could be regulated by ATRA as a new miR-200c target. Immunoblotting data first validated that PKCζ protein expression was markedly upregulated by knockdown of miR-200c, while it was downregulated by ectopic expression of miR-200c in T47D cells (Figures 5b and c). To further determine whether PKCζ is directly regulated by miR-200c through interaction of the complementary 3′-UTR region (conserved between human and mouse Figure 5d), a luciferase reporter linked with 3′-UTR of PKCζ was used. Indeed, expression of miR-200c significantly decreased the luciferase activity driven by PKCζ-UTR, whereas a reporter mutated in PKCζ-3′-UTR failed to respond to miR-200c expression (Figure 5e).
Next, to determine whether miR-200c-PKCζ regulation is functionally linked to the cancer phenotype and to the stem cell traits, we first examined PKCζ and miR-200c expression levels in human breast tissue sections consisting of a cohort of normal and malignant breast tissue samples. We found that miR-200c was most abundantly expressed in benign lesions and well-differentiated low-grade (LG, tumor grade⩽I) breast tumors and was negatively correlated with PKCζ expression (Figures 5e and f). In contrast, PKCζ was overexpressed in high-grade (HG, tumor grades II–III) aggressive breast tumors, where miR-200c was most repressed (Figures 5f and g, n=98, P<0.01). We then examined PKCζ and miR-200c expression levels in the CSC-enriched CD24−CD44+ cell population compared with the non-CD24−CD44+ cell population isolated from primary human breast tumor cells. Compared with the non-CD24−CD44+ cell population, PKCζ protein was found to be highly expressed in the CD24−CD44+ cell population, where miR-200c was repressed (Figure 5h).
Furthermore, we found that ATRA treatment significantly decreased PKCζ expression in ATRA-sensitive T47D cells, whereas knockdown of TET2, which was shown to suppress the miR-200c level in Figure 2, markedly increased PKCζ expression. The enhanced PKCζ level along with its substrate phospho-NUMB (p-NUNB) was diminished upon re-expression of miR-200c in T47D cells (Figure 5i), as well as in ATRA-resistant MDA-MB-231 cells (Figure 5j).
ATRA directs breast cancer cell state changes and decreases the CSC pool through miR-200c-mediated suppression of PKCζ
Our data in Figure 5 showed that PKCζ, a critical member of the aPKC family, was a putative target of miR-200c under the regulation of ATRA-TET2 signaling. Therefore, we next examined whether TET2-miR-200c-PKCζ signaling could have a role in regulation of the breast cancer cell state in response to ATRA.
We have previously established a paired cell imaging analysis to examine the polarity of breast CSC division by analyzing the intracellular distribution of the cell fate determinant NUMB along with the breast CSC surface marker CD44 immediately after mitosis by treatment with blebbistatin, a small molecule that arrests cytokinesis to form a binucleated cell.19 Using paired cell analysis of the isolated CD24−CD44high-CSC population from T47D cells, the data revealed that in the dividing CSC that exhibited the AD pattern, PKCζ was localized to the progeny cell expressing the putative stem cell surface marker CD44 (Figures 6a and b, Supplementary Figure S9), whereas NUMB was exclusively polarized to the CD44-negative progeny cell (Figures 6a and b). Moreover, in the dividing CSC that exhibited the SC pattern, NUMB was highly expressed and uniformly distributed in the binucleated cell, where CD44 was lost in both progenies; in the dividing CSC that exhibited the SD pattern, NUMB was repressed while CD44 was highly expressed and uniformly distributed in both progenies of the binucleated cell (Figures 6a and b).
Figure 6.
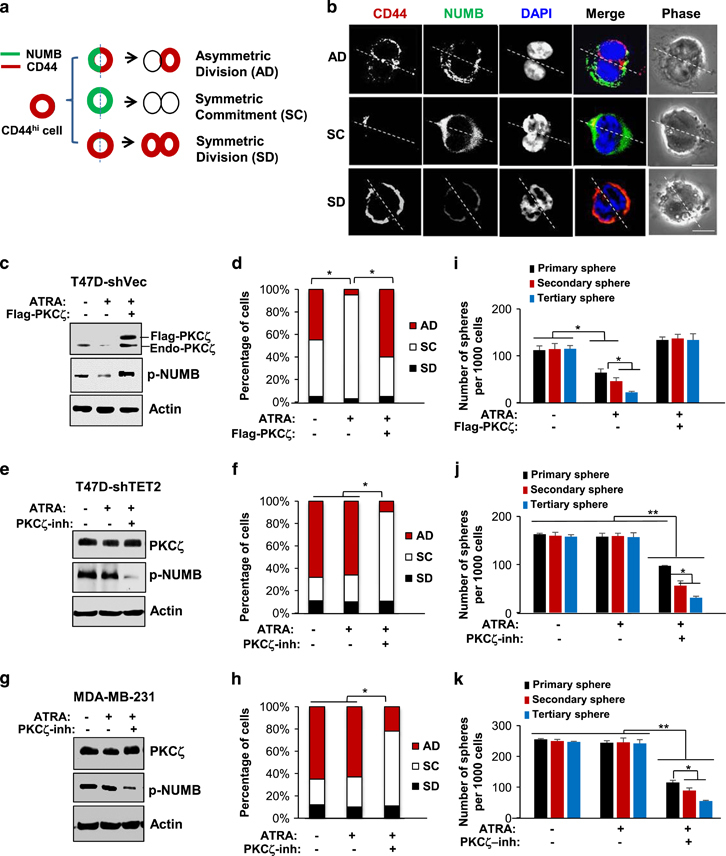
ATRA directs breast cancer cell state changes and decreases breast CSCs through suppression of PKCζ. (a) Schematic showing CSC division patterns: AD, SD or SC patterns. (b) Representative confocal fluorescence images showing intracellular distribution of CD44 (red) and NUMB (green), in the dividing CD24−CD44hi cells isolated from T47D cells using paired cell analysis (scale bar: 10 μm). (c) Protein expression levels of PKCζ and p-NUMB in T47D cells that ectopically expressed PKCζ or the control vector and treated with 10−6 M ATRA or vehicle for 24 h and (d) the percentages of these cells that manifested AD, SC and SD division patterns (n=700–1000 randomly selected cells from three independent experiments, asterisk indicates P<0.05). (e) Protein expression levels of PKCζ and p-NUMB in T47D-sh-TET2 cells that were treated with PKCζ pseudo substrate inhibitor (25μM), ATRA (10−6M), ATRA with PKCζ inhibitor or vehicle for 24 h, and (f) the percentages of these cells that manifested AD, SC and SD division patterns (n=700–1000 randomly selected cells from three independent experiments, asterisk indicates P<0.05). (g) Protein expression levels of PKCζ and p-NUMB in MDA-MB-231 cells that were treated with PKCζ pseudo substrate inhibitor (25 μM), ATRA (10−6M), ATRA with PKCζ inhibitor or vehicle for 24 h, and (h) the percentages of these cells that manifested AD, SC and SD division patterns (n=700–1000 randomly selected cells from three independent experiments, asterisk indicates P<0.05). (i) The number of the serially passaged tumor spheres (sphere size >100 μm) per 1000 initially plated T47D cells that ectopically expressed PKCζ or the control vector and treated with 10−6M ATRA or vehicle for 24 h (n=3 independent experiments, asterisk indicates P<0.05). (j) The number of the serially passaged tumor spheres (sphere size>100 μm) per 1000 initially plated T47D-sh-TET2 cells that were treated with PKCζ pseudo substrate inhibitor (25 μM), ATRA (10−6M), ATRA with PKCζ inhibitor or vehicle for 24 h (n=3 independent experiments, asterisk indicates P<0.05, double asterisk indicates P<0.01). (k) The number of the serially passaged tumor spheres (sphere size>100 μm) per 1000 initially plated MDA-MB-231 cells that were treated with PKCζ pseudo substrate inhibitor (25 μM), ATRA (10−6M), ATRA with PKCζ inhibitor, or vehicle for 24 h (n=3 independent experiments, asterisk indicates P<0.05, double asterisk indicates P<0.01). Comparable amount of control vector and vehicle were included as negative control (−) in each group. Error bars denote ±s.d.
Our data here also showed that ATRA treatment in the ATRA-sensitive T47D cells significantly diminished PKCζ and p-NUMB levels and led to a substantial increase in the frequency of the SC pattern, along with a decrease in the AD pattern of the CSC population (there was no significant change in the SD pattern); however, the ATRA-mediated enhancement of SC pattern was abolished upon ectopic expression of PKCζ (Figures 6c and d). In contrast, knockdown of TET2 cells in T47D cells (T47D-sh-TET2) resulted in resistance to ATRA, whereas treatment of the PKCζ inhibitor along with ATRA led to a markedly increased frequency of the SC pattern along with a reduced frequency of the AD pattern (Figures 6e and f). Similar results were able to be recapitulated in the ATRA-resistant MD-MB-231 cells where knockdown of PKCζ or treatment of the PKCζ inhibitor re-sensitized the CSCs to ATRA-enhanced SC pattern (Figures 6g and h), whereas re-expression of PKCζ reversed the effects caused by knock-down of PKCζ (Supplementary Figure S10).
To determine the role of the TET2-miR-200c-PKCζ signaling axis in regulating the CSC pool, the isolated CD24−CD44high-CSC population from T47D or MDA-MB-231 cells was subjected to a serial sphere formation assay. ATRA treatment in T47D cells substantially diminished sphere formation at each passage (Figure 6i), suggesting that an decreased CSC population; however, the ATRA-diminished CSC pool was able to be fully rescued upon re-expression of PKCζ (Figure 6i). Furthermore, ATRA treatment did not affect sphere formation in both T47D-sh-TET2 cells and MD-MB-231 cells, where the primary spheres were around the same number of the secondary and tertiary spheres (Figures 6j and k), suggesting that a constant CSC population was maintained. But treatment of the PKCζ inhibitor or knockdown of PKCζ led to a continuing diminishment of the spheres at each passage (Figures 6j and k, Supplementary Figure S10), whereas re-expression of PKCζ reversed the effects caused by knockdown of PKCζ (Supplementary Figure S10).
Consistently, downregulation of PKCζ by enforced expression of miR-200c in primary human breast tumor cells manifested ATRA resistance (PT1, Figure 3d) led to a decreased frequency of the AD pattern and significant suppression of the CSC pool, as evidenced by reduced CD24−CD44+ population, diminished tumor sphere formation at each passage for three consecutive passages, and inhibited mammary xenograft tumor formation with a significant reduction in the tumor-seeding CSC frequency (Supplementary Figure S11), whereas re-expression of PKCζ reversed the effects caused by expression of miR-200c (Supplementary Figure S11).
Furthermore, using a three-dimensional acinar differentiation culture in matrigel, the data showed that ATRA-resistant MD-MB-231 cells, which highly expressed the basal/myoepithelial lineage marker α-SMA, failed to form the normal acinar structure even in the presence of ATRA (Supplementary Figure S12), whereas PKCζ inhibitor treatment combined with ATRA was able to reinforce a well-differentiated acinar morphology with predominant expression of the luminal lineage marker CK18, pointing to a commitment to the luminal cell-like state (Supplementary Figure S12). Compared with treatment of ATRA or PKCζ inhibitor alone, in vivo treatment of PKCζ inhibitor along with ATRA treatment significantly suppressed MDA-MB-231 xenograft breast tumor growth and tumor volume (Supplementary Figures S13a and b), and caused the poorly differentiated high-grade adenocarcinoma phenotype to revert to a well-differentiated low-grade cancer phenotype (Supplementary Figure S13c). In addition, PKCζ inhibitor effectively inhibited p-NUMB in the tumor tissues, promoted the luminal cell lineage with a robust expression of CK18 (Supplementary Figure S13c), and also abolished in vitro serial tumor sphere formation of the isolated xenograft tumor cells from the treated mice (Supplementary Figure S13d). Together, these data suggest that ATRA-TET2 has a role in regulation of the breast cancer cell state through suppression of PKCζ expression.
Inhibition of PKCζ suppresses the ATRA-resistant CSC pool and directs CSCs to the luminal cell-like state and re-sensitization to TAM
To further determine the role of PKCζ (encoded by PRKCZ gene) in modulation of the breast cancer cell state and breast tumor progression, PRKCZ-knockout (PRKCZ-KO) MDA-MB-231 cells (Figure 7a) were generated by the CRISPR/Cas9 system.23 We previously showed that inhibition of PKCζ directed MDA-MB-231 CSCs to the luminal cell-like state in vitro and in vivo (re-expression of luminal lineage markers CK18/MUC1, Figure 6 and Supplementary Figure S13), we then asked whether these luminal-like cells also expressed ERα/PR, a major characteristic of luminal subtype breast cancer, and became sensitized to the traditional first-line selective ER modulator treatments for breast cancer, such as tamoxifen (TAM). Indeed, we found that compared with the MDA-MB-231 control cells (TNBC, ER/PR/Her2-negative), the PRKCZ-KO MDA-MB-231 cells exhibited repressed p-NUMB expression along with strong ERα expression (Figures 7a and b). In addition, consistent with the PKCζ inhibitor treatment (Supplementary Figure S13), knockout of PRKCZ markedly inhibited mammary xenograft tumor formation with significant decrease in the tumor-seeding CSC frequency (Figure 7c).
Figure 7.
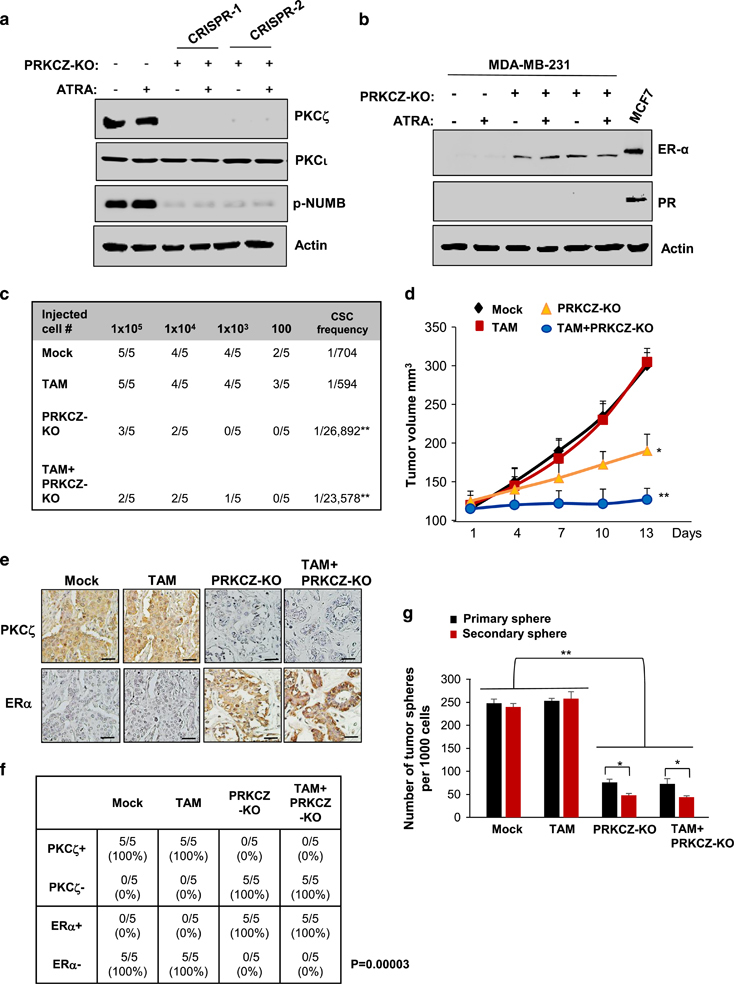
Inhibition of PKCζ directs breast CSCs to the luminal cell-like state and re-sensitization to TAM. (a, b) Protein expression levels of PKCζ, p-NUMB or ERα/PR in PRKCZ-KO or the control MDA-MB-231 breast cancer cells treated with 10−6 M ATRA or vehicle for 24 h. (c) Extreme limiting dilution analysis showing the calculated CSC frequency by the xenograft tumor formation in the mammary fat pads of the 6-week-old female nude mice inoculated with the indicated inoculated number cells pre-treated with ATRA or vehicle for 7 days. (d) Growth curve of mammary xenograft tumors, (e) IHC staining images of PKCζ and ERα expression in the xenograft tumor sections and (f) Fisher’s exact correlation analysis showing a negative correlation of PKCζ with ERα expression levels (n=5 animals/group, P=0.00003, (-), negative-low staining; (+), strong-positive staining), and (g) the number of serially passaged tumor spheres (sphere size>100 μm) per 1000 initially plated cells isolated from the xenograft tumors of the 6-week-old female nude mice that had been inoculated with 105 PRKCZ-KO or control MDA-MB-231 breast cancer cells and then treated with TAM (5 mg/kg i.p. every other day for 2 weeks) or vehicle when the tumors reached 100 mm3 (n=5 animals per group, asterisk indicates P<0.05, double asterisk indicates P<0.05; scale bar: 50 μm). Comparable amount of control vector and vehicle were included as negative control (-) in each group. Error bars denote ±s.d.
To further determine whether knockout of PRKCZ could re-sensitize MDA-MB-231 cells to TAM treatment and block tumor progression in vivo, PRKCZ-KO MDA-MB-231 or the control cells were first injected into mammary fat pads of 6-week-old female nude mice, and when the tumors reached 100 mm3, the animals were given TAM (i.p., 5 mg/kg, every other day, dosing based on Kumar et al.24), or vehicle for 2 weeks (n=5 mice per each group). We found that TAM alone did not have significant effects on MDA-MB-231 xenograft tumor growth, CSC frequency, tumor phenotype or serial tumor sphere formation of the isolated xenograft tumor cells from the treated mice (Figures 7c–g). However, knockout of PRKCZ significantly suppressed tumor growth, abolished serial tumor sphere formation and caused the poorly differentiated high-grade adenocarcinoma phenotype to revert to a well-differentiated low-grade cancer phenotype with a robust expression of ERα (Figures 7c–g). Furthermore, we found that PRKCZ-KO combined with TAM resulted in therapeutic synergism in abrogating tumor growth (Figure 7d). Together, these data suggest that inhibition of PKCζ directs luminal (ER+) differentiation and thereby re-sensitizes TNBC cells to TAM-mediated tumor suppression.
Discussion
Our findings reveal that defective RARβ/TET2 signaling in aggressive tumors promotes an ATRA-resistant breast CSC pool, and miR-200c therapy or strategies that directly inhibit PKCζ likely direct the resistant breast CSCs to the differentiation state, potentially by enhancing SC along with repressing AD of these CSCs (Figures 6d and i), and thereby diminish the CSC pool (Figure 8). It is known that aPKC family members, including PKCζ, can serve as tumor suppressors or tumor promoters among various cancer types.25, 26 Several studies have implied that in breast cancer, PKCζ expression likely promotes cancer cell growth and aggressiveness.27, 28, 29 Furthermore, our data have shown that ATRA specifically downregulates PKCζ, but not the highly homologous isoform PKCι (Supplementary Figure S14a). Downregulation of PKCζ expression by ATRA, knockdown of PKCζ (by si-PKCζ, sh-PKCζ), and knockout of PKCζ (by PKCζ-CRISPR) consistently leads to decreased p-NUMB (Figure 7, Supplementary Figures S10 and S14), whereas knockdown of PKCι does not affect p-NUMB level (Supplementary Figure S14c). Together with the in vivo and in vivo functional data using PKCζ inhibitor along with PKCζ-knock-down and knockout systems (Figures 6 and 7, Supplementary Figures S10 and S13), our evidence suggests that PKCζ is the major aPKC isoform that sustains the breast cancer CSC pool to promote breast tumor progression.
Figure 8.
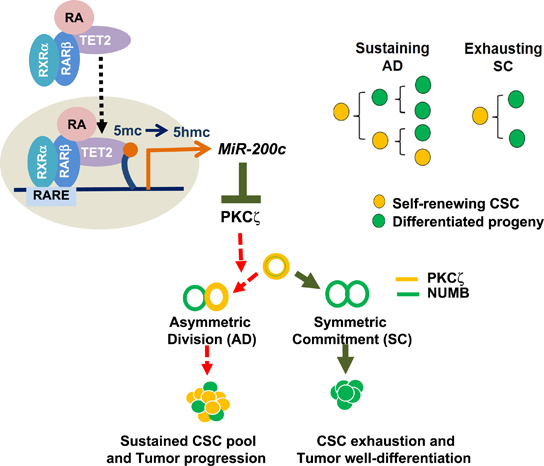
A proposed model illustrating ATRA directs breast cancer cell state changes to diminish the CSC pool through regulation of RARβ-TET2-miR-200c-PKCζ pathway with schematic showing AD or SC cell fate decisions are made over accumulated CSC divisions to sustain or diminish the CSC pool.
Materials and methods
Cell culture and treatment
The immortalized mammary epithelial cell line, MCF12A and MCF10A, and the breast cancer cell lines, including MDA-MB-231, T47D, ZR75-1, BT549 and MCF7, etc., were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Primary breast tumor cells were purchased from Promab Inc (Richmond, CA, USA). All the cell lines were tested for mycoplasma free and authenticated using short tandem repeat profiling by ATCC within 6 months of laboratory use and cultured according to the vendor’s instruction.
TAM treatment in mammary xenograft tumor
Isolation, infection and culture of tumor spheres from tumors were performed as described previously.30 In all, 105 CRISPR-PRKCZ-knockout MDA-MB-231 cells or the control MDA-MB-231 cells were injected into mammary fat pads of 6-week-old female nude mice. When tumors reached the indicated palpable size (~100 mm3), animals were given 100 μl of corn oil (i.p. every 2 days) or TAM (5 mg/kg in 100 μl of corn oil, i.p. every 2 days) for 2 weeks (Sigma, St Louis, MO, USA; regimen based on Kumar et al.24 and manufacturer’s suggestion). For ATRA and PKCζ inhibitor treatment, animals were given vehicle dimethylsulfoxide or ATRA 7.5 mg/kg i.p. daily for 2 weeks (Sigma, regimen based on previous study in Elstner et al.31) and/or PKCζ pseudo substrate inhibitor (Millipore, Billerica, MA, USA; 20 mg/kg i.p. every other day for 2 weeks, regimen according to the manufacturer’s suggestion). Tumor was measured by caliper every other 2 days and tumor volume was calculated using (tumor length × tumor width2)/2. Four weeks after inoculation, tumors were collected for RNA/protein extraction, sphere formation analysis following single-cell isolation, and also fixed in 10% neutral formalin for histological evaluation. The expected frequency of the tumorigenic CSCs was calculated using Extreme Limiting Dilution Analysis.32
Statistical analysis
All data were presented as means±the s.d. of the mean. Statistical calculations were performed with Microsoft Excel analysis tools. Differences between individual groups were analyzed by two-tailed paired t-test. P-values of <0.05 considered statistically significant. Chi-square analysis for tumor samples were performed as described previously.17
Supplementary information
Acknowledgements
We thank the assistance of Dr Chao-Ching Chang as well as Dr Chi-Hong Chao with advice on animal and biochemical experiments, and Mr Chun-Chih Hsu with LC-MS/MS analysis. This study was supported by Department of Defense Breast Cancer Breakthrough Award (W81XWH-15-1-0644 to C-J Chang), V foundation for Cancer Research Grant (V2015-019 to C-J Chang), and P30 CA023168 to the Purdue University Center for Cancer Research in support of the use of facilities.
Author contributions
M-JW and C-JC designed and conceived the study. M-JW and C-JC performed the experiments and wrote the manuscript. MRK and Y-SC helped with the biochemical experiments.
Competing interests
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on the Oncogene website
Change history
3/3/2023
A Correction to this paper has been published: 10.1038/s41388-023-02616-7
References
- 1.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 2.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 3.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 4.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2012;12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- 6.Smith CA, Lau KM, Rahmani Z, Dho SE, Brothers G, She YM. aPKC-mediated phosphorylation regulates asymmetric membrane localization of the cell fate determinant Numb. EMBO J. 2007;26:468–480. doi: 10.1038/sj.emboj.7601495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–860. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garattini E, Gianni M, Terao M. Cytodifferentiation by retinoids, a novel therapeutic option in oncology: rational combinations with other therapeutic agents. Vitam Horm. 2007;75:301–354. doi: 10.1016/S0083-6729(06)75012-9. [DOI] [PubMed] [Google Scholar]
- 9.Cuzick J, DeCensi A, Arun B, Brown PH, Castiglione M, Dunn B. Preventive therapy for breast cancer: a consensus statement. Lancet Oncol. 2011;12:496–503. doi: 10.1016/S1470-2045(11)70030-4. [DOI] [PubMed] [Google Scholar]
- 10.Sutton LM, Warmuth MA, Petros WP, Winer EP. Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: a phase II trial. Cancer Chemother Pharmacol. 1997;40:335–341. doi: 10.1007/s002800050666. [DOI] [PubMed] [Google Scholar]
- 11.Bryan M, Pulte ED, Toomey KC, Pliner L, Pavlick AC, Saunders T. A pilot phase II trial of all-trans retinoic acid (Vesanoid) and paclitaxel (Taxol) in patients with recurrent or metastatic breast cancer. Invest New Drugs. 2011;29:1482–1487. doi: 10.1007/s10637-010-9478-3. [DOI] [PubMed] [Google Scholar]
- 12.Veronesi U, De Palo G, Marubini E, Costa A, Formelli F, Mariani L. Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. J Natl Cancer Inst. 1999;91:1847–1856. doi: 10.1093/jnci/91.21.1847. [DOI] [PubMed] [Google Scholar]
- 13.Tang X-H, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol Mech Dis. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 14.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154:311–324. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang C-J, Chao C-H, Xia W, Yang J-Y, Xiong Y, Li C-W. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao C-H, Chang C-C, Wu M-J, Ko H-W, Wang D, Hung M-C. MicroRNA-205 signaling regulates mammary stem cell fate and tumorigenesis. J Clin Invest. 2014;124:3093–3106. doi: 10.1172/JCI73351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urtreger AJ, Kazanietz MG, Bal de Kier Joffé ED. Contribution of individual PKC isoforms to breast cancer progression. IUBMB Life. 2012;64:18–26. doi: 10.1002/iub.574. [DOI] [PubMed] [Google Scholar]
- 23.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar BNP, Rajput S, Dey KK, Parekh A, Das S, Mazumdar A. Celecoxib alleviates tamoxifen-instigated angiogenic effects by ROS-dependent VEGF/VEGFR2 autocrine signaling. BMC Cancer. 2013;13:273. doi: 10.1186/1471-2407-13-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urtreger AJ, Grossoni VC, Falbo KB, Kazanietz MG, Bal de Kier Joffé ED. Atypical protein kinase C-zeta modulates clonogenicity, motility, and secretion of proteolytic enzymes in murine mammary cells. Mol Carcinogenesis. 2005;42:29–39. doi: 10.1002/mc.20066. [DOI] [PubMed] [Google Scholar]
- 27.Castoria G, Migliaccio A, Di Domenico M, Lombardi M, de Falco A, Varricchio L. Role of atypical protein kinase C in estradiol-triggered G1/S progression of MCF-7 cells. Mol Cell Biol. 2004;24:7643–7653. doi: 10.1128/MCB.24.17.7643-7653.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul A, Danley M, Saha B, Tawfik O, Paul S. PKCζ promotes breast cancer invasion by regulating expression of E-cadherin and Zonula occludens-1 (ZO-1) via NFκB-p65. Sci Rep. 2015;5:12520. doi: 10.1038/srep12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Zhang X, Li M, Chen P, Zhang B, Guo H. mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010;70:9360–9370. doi: 10.1158/0008-5472.CAN-10-0207. [DOI] [PubMed] [Google Scholar]
- 30.Chang C-C, Wu M-J, Yang J-Y, Camarillo IG, Chang C-J. Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 2015;75:2375–2386. doi: 10.1158/0008-5472.CAN-14-3076. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 31.Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA. 1998;95:8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.