

Abstract
Histone phosphorylation influences transcription, chromosome condensation, DNA repair and apoptosis. Previously, we showed that histone H3 Ser10 phosphorylation (pSer10) by the yeast Snf1 kinase regulates INO1 gene activation in part via Gcn5/SAGA complex-mediated Lys14 acetylation (acLys14). How such chromatin modification patterns develop is largely unexplored. Here we examine the mechanisms surrounding pSer10 at INO1, and at GAL1, which herein is identified as a new regulatory target of Snf1/pSer10. Snf1 behaves as a classic coactivator in its recruitment by DNA-bound activators, and in its role in modifying histones and recruiting TATA-binding protein (TBP). However, one important difference in Snf1 function in vivo at these promoters is that SAGA recruitment at INO1 requires histone phosphorylation via Snf1, whereas at GAL1, SAGA recruitment is independent of histone phosphorylation. In addition, the GAL1 activator physically interacts with both Snf1 and SAGA, whereas the INO1 activator interacts only with Snf1. Thus, at INO1, pSer10's role in recruiting SAGA may substitute for recruitment by DNA-bound activator. Our results emphasize that histone modifications share general functions between promoters, but also acquire distinct roles tailored for promoter-specific requirements.
Keywords: acetylation, histone, phosphorylation, SAGA, Snf1
Introduction
Covalent modification of histones is a universal mechanism regulating DNA templated processes, including locus-specific events such as transcription and DNA damage repair. For dynamic transactions during processes that are short-lived, the modifications are generally reversible and include acetylation, phosphorylation, and monoubiquitylation (Berger, 2002).
Phosphorylation occurs in all four core histones that constitute the histone octamer, as well as in some of the histone variants and histone H1 (Iizuka and Smith, 2003; Nowak and Corces, 2004; Peterson and Cote, 2004). Histone H3 Ser10 phosphorylation is involved in chromosome condensation and subsequent segregation during mitosis/meiosis and in transcriptional activation (Nowak and Corces, 2004). H3 phosphorylation during gene induction was discovered in mammalian cells as an immediate early response to mitogenic stimulation, leading to transcription of c-Fos and c-Jun (Mahadevan et al, 1991) mediated by RSK2 and MSK1 (Sassone-Corsi et al, 1999; Thomson et al, 1999; Strelkov and Davie, 2002; Soloaga et al, 2003). Other mammalian gene induction pathways involving pSer10 include upregulation of the IFNβ gene by viral infection (Agalioti et al, 2002) and IKKα kinase-mediated phosphorylation in NFκB pathways (Anest et al, 2003; Yamamoto et al, 2003). In Drosophila melanogaster, H3 pSer10 dramatically increases at heat-induced genes during the heat shock response (Nowak and Corces, 2000).
In Saccharomyces cerevisiae the Snf1 kinase activates transcription in part through histone H3 phosphorylation (Lo et al, 2001; Lin et al, 2003). Snf1 induces many stress-responsive pathways in yeast, including inositol biosynthesis and galactose utilization. In high inositol, transcription of the INO1 gene is strongly repressed (Jackson and Lopes, 1996). In low inositol, the Ino2 and Ino4 basic helix–loop–helix proteins heterodimerize and bind to the INO1 promoter to activate transcription (Ambroziak and Henry, 1994). Numerous cofactors regulate INO1 transcription, including the Swi–Snf ATP-dependent nucleosome remodeling complex, the SAGA acetylation complex, Snf1–Snf4 kinase complexes, and RNA polymerase II holoenzyme itself (reviewed in Arndt et al, 1995; Henry and Patton-Vogt, 1998). We identified Ser10 on histone H3 as a target of Snf1 at the INO1 gene. However, it is unknown whether Snf1 is recruited to the promoter, and, if so, whether recruitment is mediated by the Ino2/Ino4 activator. Indeed, DNA-bound activators recruit both acetylation and Swi/Snf remodeling complexes (Utley et al, 1998; Knoepfler and Eisenman, 1999; Peterson and Workman, 2000), but whether activators also recruit other histone-modifying activities is unknown.
Snf1 also regulates signaling in response to nonfermentable carbon sources. In high glucose, many genes are repressed (reviewed in Carlson, 1999). Low glucose, or alternate carbon sources such as galactose, leads to Snf1 induction of a wide variety of genes, such as galactose-inducible GAL1. The Gal4 activator recruits the SAGA acetylation complex to activate the promoter through Spt3-dependent TATA-binding protein (TBP) recruitment (Larschan and Winston, 2001), Gcn5-mediated histone acetylation (Bhaumik and Green, 2001; Yu et al, 2003) and histone deubiquitylation by the protease Ubp8 (Henry et al, 2003; Kao et al, 2004). Snf1 phosphorylates and thereby dissociates the repressor Mig1, the sole known substrate in the pathway (Treitel et al, 1998).
Histone H3 phosphorylation of Ser10 is linked to acetylation of Lys14. In yeast, Lys14 acetylation at INO1 is partially dependent on phosphorylation (Lo et al, 2001). Biochemical and structural studies show that binding of the Gcn5 HAT domain to the histone H3 tail is potentiated by phosphorylation of Ser10 (Clements et al, 2003). Further, temporal linkage has been established, where pSer10 occurs prior to AcLys14 during immediate early gene activation in mammalian cells (Cheung et al, 2000), in IFNβ (Agalioti et al, 2002; Spilianakis et al, 2003), and in NFκB regulated genes (Yamamoto et al, 2003). In general, linked patterns suggest that combinatorial modifications of histones dictate functional outcomes (Jenuwein and Allis, 2001; Turner, 2002). However, it is not established whether phosphorylation is usually linked to acetylation for gene activation.
Histone modifications are involved in initiation of RNA synthesis and transcriptional elongation. Coactivator recruitment and histone acetylation often occur before TBP binding. Recruitment of SAGA to GAL1 is required for optimal TBP and RNA polymerase II recruitment (Bhaumik and Green, 2001; Larschan and Winston, 2001; Bryant and Ptashne, 2003; Yu et al, 2003; Topalidou et al, 2004). H2B ubiquitylation and deubiquitylation regulate a histone H3 methylation (me) pathway at GAL1, which in turn is linked to other mechanisms of RNA elongation (Santos-Rosa et al, 2002; Morillon et al, 2003; Zhang, 2003). Functional consequences of combinatorial histone modifications are unknown, such as histone phosphorylation/acetylation.
In this study, we investigate Snf1-mediated histone H3 phosphorylation during induction of INO1 and GAL1. We address the mechanisms by which Snf1 is recruited to promoters, how phosphorylation leads to acetylation, and dependence of TBP recruitment on pSer10. Our studies provide evidence for Snf1-mediated histone phosphorylation in transcriptional activation of distinct signaling pathways.
Results
Snf1 is recruited to the INO1 promoter prior to H3 Ser10 phosphorylation
Our previous observations suggest that Snf1 is recruited directly to the promoter during gene activation to mediate pSer10 on histone H3, and further suggest that Snf1, Gcn5, and their modified histone substrates are present at the promoter with similar timing. We used chromatin immunoprecipitation (ChIP) to test these hypotheses. Wild-type (WT) and snf1 null cells were grown in noninducing or inducing (medium lacking inositol) conditions to increase INO1 transcription (Shirra and Arndt, 1999; Lo et al, 2001). We monitored Snf1 binding to the promoter by ChIP, and found that the signal for the INO1 gene increases in the inducing condition by approximately 14-fold (Figure 1A, left). This signal is nearly absent in an snf1 null strain (Figure 1A, right), indicating that the INO1 ChIP signal corresponds to the presence of Snf1.
Figure 1.
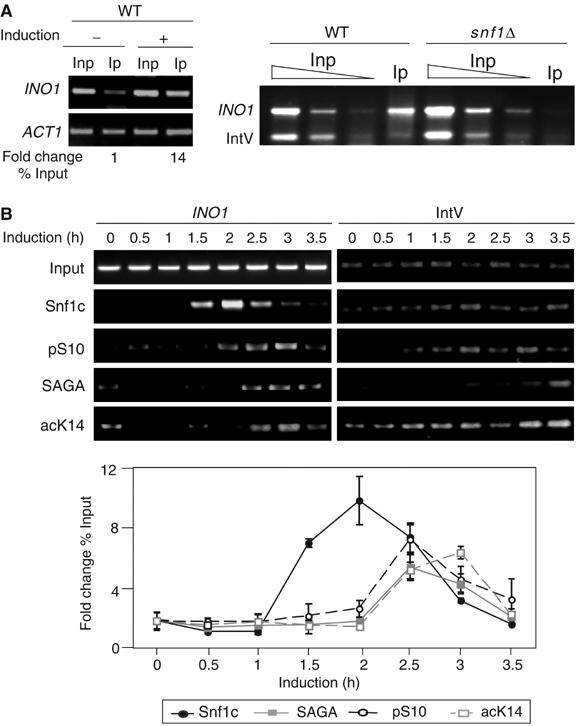
Recruitment of Snf1, SAGA, and cognate histone modifications to the INO1 promoter in vivo. (A) PCR analysis of ChIP with anti-Snf1 antibody (Snf1) on the INO1 promoter. Left panel, PCR results of ChIP-assay-performed WT cells under repressing (−, 10 μM inositol) or inducing conditions (+, inositol starvation). Inp=Input DNA and Ip=immunoprecipitated DNA by anti-Snf1. ACT1 was the internal control. See Materials and methods for quantitation methods. Right panel, PCR results of ChIP assay performed from WT compared to snf1Δ cells. Titration of input DNA (Inp) and immunoprecipitated DNA (Ip) by anti-Snf1 antibody are as indicated. Intergenic region on chromosome V (IntV) was the internal background control. (B) Time-course ChIP analysis of protein complexes and histone modifications on the INO1 promoter during induction. Cells were grown under repressing conditions to 0.8 OD600 (time 0) and then in inducing conditions at the time points indicated. Upper panel: Antibodies were against Snf1 (Snf1c), pS10, Ada2 (SAGA), and acK14. Lower panel: Quantitation of time course and statistical evaluation of multiple ChIP and PCR reactions. IntV was used as a control for ChIP.
A time course of induction was analyzed by ChIP to determine the relative order of appearance of Snf1, pSer10 SAGA, and acK14, at 30-min intervals. The Snf1 signal increases at 1.5 h, and is closely followed by pSer10 (at the next time point) (Figure 1B). SAGA is increased at the same time as pSer10, and acetylation peaks 30 min later (Figure 1B). This pattern of recruitment is consistent with the hypothesis that Snf1/pSer10 and Gcn5/acLys14 have a role in INO1 regulation (Lo et al, 2001).
Snf1 also mediates regulatory Ser10 phosphorylation at GAL1
As introduced above, GAL1 transcription is regulated by Snf1 and by SAGA. We determined whether GAL1 is regulated by H3 pSer10, and, if so, whether this is related to acLys14. We examined the growth of strains deleted for the modification enzymes or bearing substitution mutations in histones on media containing galactose (Figure 2A). Loss of Gcn5 does not impair growth on galactose medium, whereas loss of Snf1 causes poorer growth. The gcn5snf1 double null mutant grows much worse than the snf1 null mutant (Figure 2A). Sas3, a second histone acetyltransferase that, like Gcn5, targets histone H3 but is not known to be required for transcription (Howe et al, 2001), shows no exaggerated sickness when deleted in combination with snf1. The deleterious effects of Snf1 or Gcn5 enzyme deletions are apparently in part due to failure to modify histone substrates, because strains bearing the double mutant histone H3 substitution S10AK14A grow more poorly than either S10A or K14A.
Figure 2.
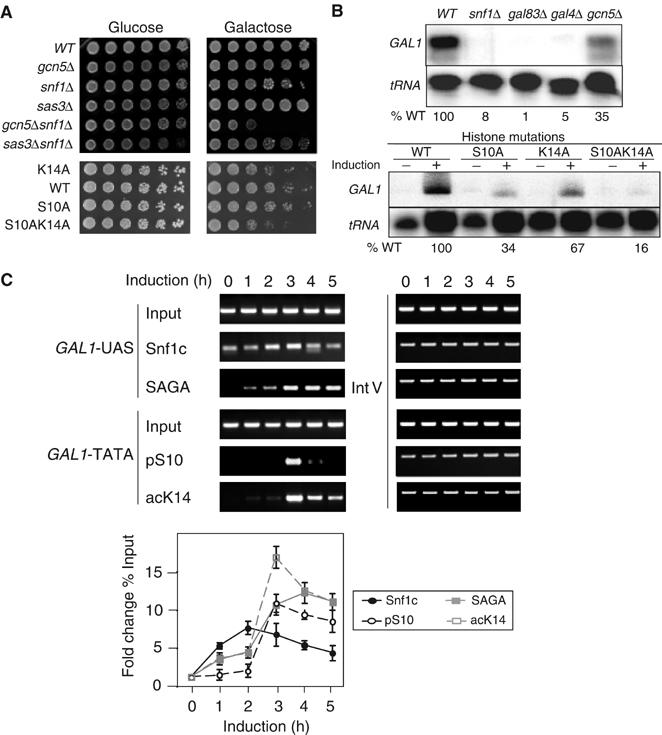
Cooperation of histone H3 phosphorylation and acetylation in galactose utilization and GAL1 transcriptional regulation in vivo. (A) Growth phenotype of enzyme deletions and histone substitutions. Five-fold serial dilutions of strains with the genotypes indicated on the left were spotted on glucose (2% glucose) and galactose (2% galactose) plates. Phenotypes were scored after 3-day growth at 30°C (top panels) and at 39°C (lower panels). (B) S1 nuclease RNA protection assay of enzyme deletions and histone substitutions. GAL1 transcription from WT, deletion strains (snf1, gal83, gal4, gcn5) and histone substitution strains (S10A, K14A, and S10AK14A) under repressing (−, 2% glucose) and inducing conditions (+, 2% galactose; upper panel). The induced level of GAL1 transcription in each strain was normalized to internal tRNA level and then calculated as percentage of WT level. (C) ChIP analysis of histone modifications on the GAL1 promoter during galactose induction time course. Cells were collected every 60 min over a 5 h time course. Antibodies were against Snf1 (Snf1c), pS10, Spt3-myc (SAGA), and acK14. Primer pairs were located in the GAL1 upstream activating sequence (GAL1-UAS) or the core promoter of GAL1 (GAL1-TATA).
We used quantitative S1 nuclease RNA protection analysis to determine whether the growth defects reflect diminished GAL1 transcription (Figure 2B). As expected, GAL1 transcription is very low in the absence of the transcriptional activator Gal4 (upper panel). GAL1 transcription is reduced approximately three-fold in the absence of Gcn5, and is very low in the absence of Snf1, or without Gal83, one targeting subunit of the Snf1 complex (Vincent and Carlson, 1999; Vincent et al, 2001). Analysis of histone substitution mutations (lower panel) shows that GAL1 transcription is lowered modestly in the K14A strain and three-fold in the S10A strain. In parallel with the growth assay, GAL1 RNA is even lower in the S10AK14A double mutant, dropping five-fold compared to WT. These results indicate that Ser10 phosphorylation may have a role in GAL1 transcription, and that genetic interactions between Snf1/Gcn5 and Ser10/Lys14 mutually optimize the effects of histone phosphorylation and acetylation.
ChIP analysis was performed at GAL1 during galactose induction to examine the recruitment of Snf1, pSer10, SAGA, and acLys14 (Figure 2C). Recruitment of enzymes was monitored at the non-nucleosomal GAL1 UAS, where the activator Gal4 binds, and histone modifications were assayed at the nucleosome-associated TATA sequence (Bhaumik and Green, 2001; Larschan and Winston, 2001). The level of Snf1 rises at 1 h and SAGA appears at a similar time as Snf1, but reaches a maximum slightly later. AcLys14 and pSer10 show similar kinetics, rising sharply at the time of SAGA appearance. We noted a greater acLys14 increase at these later times of induction than reported previously at a single 20-min time point (Larschan and Winston, 2001). We examined pSer10 in raffinose compared to galactose to distinguish derepression from activation; pSer10 increases sharply in galactose, but not under derepressing raffinose conditions (M Schwartz and SL Berger, unpublished results). We conclude that Snf1-mediated Ser10 phosphorylation plays a role in GAL1 transcriptional induction as it does for INO1.
The Snf1 protein complex (Snfc) binds to activators in vitro
Previous work established that acetyltransferases are recruited to promoters through interaction with the activation domains (ADs) of DNA-bound activators (Utley et al, 1998). We investigated activator interaction and dependency of Snf1 recruitment. We tested whether the Snf1 complex physically binds to activators. The Snf1 complex (Snf1c) was purified to near homogeneity by affinity chromatography using endogenous Snf1-FLAG. Coomassie staining shows three apparently stoichiometric bands, and mass spectrometry analysis revealed these to be Snf1 (α subunit), Gal83 (β/targeting subunit), and Snf4 (γ/activating subunit) (Figure 3A), as previously shown (Nath et al, 2002). Substoichiometric amounts of Sip2 (an alternate β subunit with Gal83) and very small amounts of Sip1 (the third β subunit) were also identified (Figure 3A and data not shown).
Figure 3.
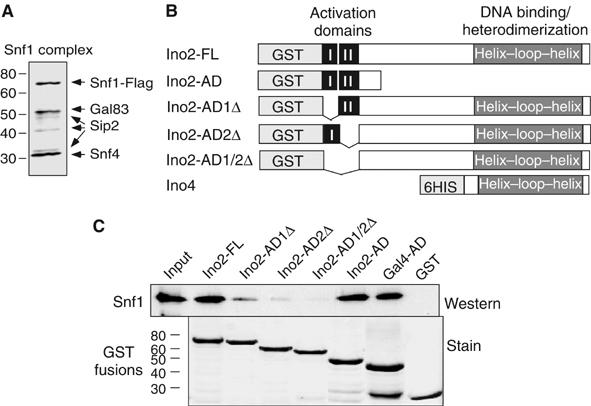
Interaction of Snf1c with transcriptional activators in vitro. (A) Subunit composition of the Snf1c. Purification scheme for the Snf1c from a strain bearing Snf1-FLAG is described in Materials and methods. Purified Snf1c was separated by 4–12% gradient SDS–PAGE and visualized by Coomassie blue staining. Subunits identified by mass spectrometry are indicated, and molecular mass markers (kDa) at left. The bands at 40 and 33 kDa were Sip2 and likely to be degradation products. (B) Description of Ino2 and Ino4 constructs. The diagrams show GST fusion proteins containing full-length Ino2 (Ino2-FL: aa 1–304), acidic AD of Ino2 (Ino2-AD: aa 1–100), deletion of first AD (Ino2-AD1Δ, aa 1–27), deletion of second AD (Ino2-AD2Δ, aa 28–100), or deletion of both (Ino2-AD1/2Δ, aa 1–100). Ino4 (aa 1–151) was purified via a 6His-tag at its N-terminus. The DNA binding and interaction domains (basic helix–loop–helix) of Ino2 (aa 213–304) and Ino4 are indicated. (C) GST pulldown assay of Ino2 or Gal4 activator interaction with Snf1c. Fusion proteins as indicated and GST alone were tested for Snf1c binding. Top panel: Input and bound Snf1c were analyzed on 10% SDS–PAGE and immunoblotted using anti-Snf1antibody. Input represents 50% of amount in binding reaction. Bottom panel: Relative amounts of recombinant fusion proteins used in the assay were visualized by Ponceau S staining on the same membrane.
The INO1 activator is a heterodimer composed of Ino2 and Ino4; Ino2 harbors two tandem and independent ADs (Schwank et al, 1995; Figure 3B). Full-length INO2 was fused to glutathione-S-transferase (GST) and expressed in Escherichia coli (Figure 3C, lower). Snf1c associates with purified GST-Ino2-FL, but not with GST alone (Figure 3C, upper). To evaluate contributions of the Ino2 ADs, either one or both were deleted (Figure 3B). Loss of either AD reduces binding of Snf1c, and deletion of both ADs most strongly lowers binding (Figure 3C, upper). Thus, the ADs are necessary for efficient association of Snf1c with Ino2.
We examined whether the Ino2 ADs (Figure 3B) are sufficient for Snf1c interaction. Snf1c associates with GST-Ino2-AD, and also associates with GST-Gal4-AD (Figure 3C, upper), consistent with the involvement of both Snf1 and pSer10 in GAL1 transcription. Similar amounts of GST fusion proteins were used (Figure 3C, lower) and, since comparable amounts of Snf1c associate with each fusion, there may be a similar efficiency of association between Ino2 or Gal4 ADs and Snf1c.
Ino2/Ino4 activator recruits Snf1, Gcn5, and establishes histone modifications in vivo
The association of Snf1c with Ino2 in vitro suggested that Ino2 may be required to recruit the complex to the INO1 promoter in vivo. We used ChIP assay to investigate Ino2 and Ino4 recruitment of Snf1. The Snf1 signal is greatly reduced in the ino2Δ, ino4Δ or double disruption strain (Figure 4B), consistent with diminution of RNA synthesis in the mutant strains (Figure 4A). Since Snf1 is required for pSer10 and acLys14 at INO1 (Lo et al, 2001), we tested whether loss of activator affects chromatin modification and/or recruitment of SAGA. Deletion of Ino2, Ino4, or both lowers pSer10, SAGA, and acLys14 at INO1 (Figure 4B). Thus, the Ino2/Ino4 heterodimeric activator is required for Snf1association, SAGA association, and corresponding histone modifications in vivo.
Figure 4.
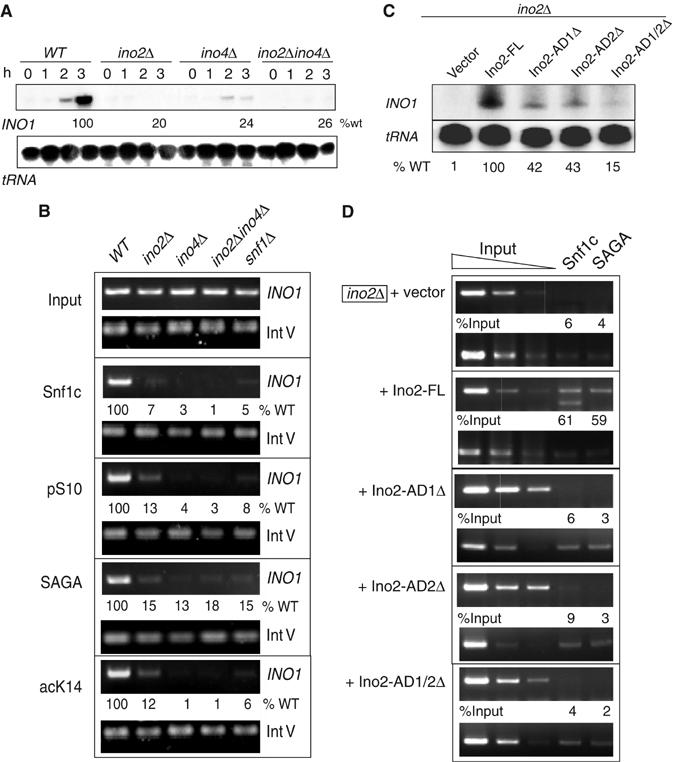
Role of Ino2/Ino4 activator in recruitment of Snf1 and SAGA complexes at the INO1 promoter in vivo. (A, C) S1 nuclease assay of INO1 gene expression in ino2, ino4, and ino2 AD deletion strains. RNA was prepared from indicated deletion strains or ino2D bearing plasmid-borne full-length or deleted Ino2, from repressing conditions (0 time) and inducing conditions (inositol starvation). Plasmid-born Ino2-FL (panel C) conferred 81% of transcription compared to endogenous Ino2 (panel A). (B, D) ChIP analysis of Snf1c, SAGA, and cognate histone modifications of complexes dependant on Ino2 and Ino4 activators at INO1. Same as above, except that Spt3-Myc was used to examine SAGA recruitment.
We extended these assays to determine whether the Ino2 ADs are required for recruitment. As single or both Ino2 ADs were deleted, we found a good correlation between reduction of RNA synthesis (Figure 4C) and reduction of ChIP signal for Snf1 and SAGA (Figure 4D). Recruitment of Snf1 and SAGA complexes correlates with AD-mediated transcriptional activation by Ino2.
Gal4-AD, but not Ino2 ADs, interacts with SAGA in vitro
We investigated why the Ino2/Ino4 activator is required to recruit SAGA in vivo. Two alternative possibilities are that there is direct interaction necessary for activator recruitment of SAGA, or that Ino2 may recruit Snf1c, which in turn recruits SAGA via pSer10, as suggested by our previous epistasis analysis (Lo et al, 2001). We tested direct interaction in vitro by comparing Snf1c and SAGA binding to GST-Ino2-AD. As seen in the previous assay (Figure 3C), Snf1c associates with GST-Ino2-AD; however, SAGA associates poorly (Figure 5A).
Figure 5.
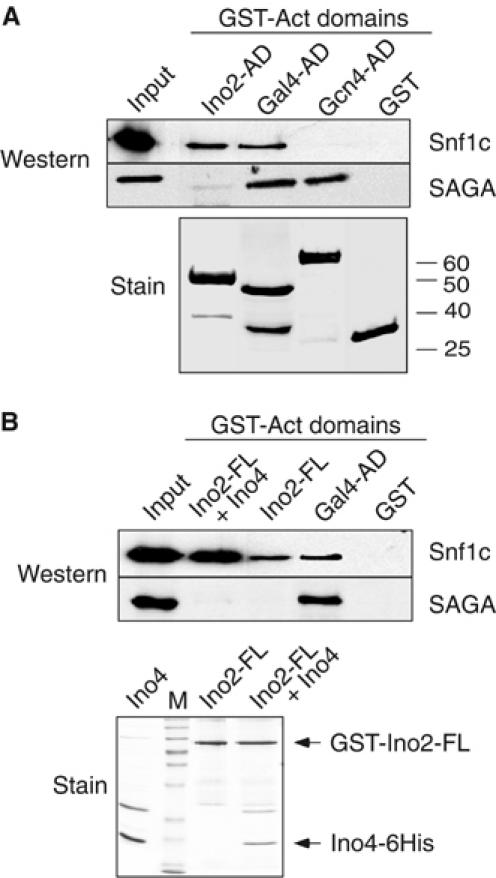
Comparative interaction of SAGA and Snf1c with Ino2/Ino4 and Gal4 activators in vitro. (A, B) GST pulldown between Ino2, Gal4, or Gcn4 ADs, and Snf1c or SAGA. See Figure 3 legend for details. Anti-Ada3 antibody was used to visualize SAGA. In panel B, prior to GST pulldown, 6His-tagged Ino4 protein was incubated with GST-Ino2-FL in high salt buffer (350 mM NaCl) for 30 min at 4°C.
It was surprising that SAGA bound poorly with the Ino2 ADs, as SAGA binds to activators Gal4 and Gcn4 to activate relevant promoters (Drysdale et al, 1998; Larschan and Winston, 2001). In contrast, only Gal4, and not Gcn4, is known to require Snf1 (we confirmed that HIS3 transcription is not reduced in an snf1Δ strain (W-S Lo, L Pillus and SL Berger, unpublished observations)). We then compared SAGA and Snf1c binding to Gal4 and Gcn4. As expected, both complexes associate with GST-Gal4-AD (Figure 5A), whereas SAGA, but not Snf1c, associates with GST-Gcn4-AD (Figure 5A), using similar amounts of each GST fusion protein (Figure 5A, lower). In summary, Snf1c, but not SAGA, binds to Ino2 ADs, both complexes bind to Gal4-AD, and SAGA, but not Snf1c, binds to Gcn4-AD.
We considered an additional, trivial explanation for the inability of SAGA to bind to Ino2 ADs, that is, that SAGA is unable to bind to isolated Ino2 ADs, compared to full-length Ino2 or Ino2/Ino4 heterodimer. This was not the case: GST-Ino2-FL, or GST-Ino2/Ino4–6his (Figure 5B, lower), like GST-Ino2 ADs, interact with Snf1c but not with SAGA (Figure 5B, upper) although GST-Gal4-AD interacts with both complexes (Figure 5B, upper).
These data suggest that SAGA recruitment to INO1 depends upon Snf1c-mediated pSer10 in part because SAGA is not able to bind to the Ino2 activator for direct recruitment. In contrast, both Snf1c and SAGA complexes efficiently interact with the Gal4 activator, suggesting that, although H3 is modified at both INO1 and GAL1, pSer10 is involved in recruitment of SAGA only at INO1.
Snf1 and H3 Ser10 are required for SAGA recruitment at INO1, but not at GAL1 in vivo
We tested the hypothesis that SAGA recruitment at GAL1 is independent of Snf1-mediated pSer10 by determining the effect of S10A substitution on acLys14 at GAL1. The prediction is that acetylation would not be affected by loss of phosphorylation, in contrast to our previous observation at INO1 (Lo et al, 2001). We performed a time course of galactose induction, using ChIP to compare acLys14 and pSer10 at GAL1 in WT and S10A strains. As before (Figure 2C), the levels of both acLys14 and pSer10 increase, reaching a high level at 3 h (Figure 6A). The pattern of acLys14 in the S10A and WT strains is nearly identical here at GAL1 (Figure 6A and B), whereas, at INO1, a significant decrease in acLys14 occurs in the S10A strain (Lo et al, 2001). As expected, there is nearly no pSer10 signal in the S10A strain (Figure 6A). These data indicate that pSer10 is not required for acLys14 at GAL1, in striking contrast to INO1.
Figure 6.
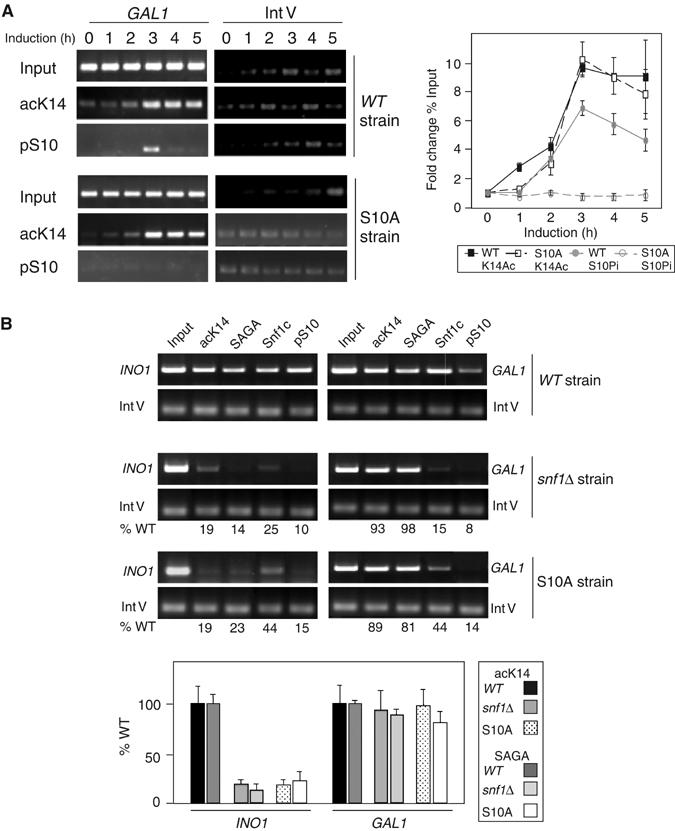
Effect of loss of Snf1 or pSer10 on recruitment of SAGA complex and acK14 to GAL1 and INO1 promoters. (A, B) ChIP assays of snf1Δ or S10A mutant effect on recruitment of SAGA or Snf1c and cognate histone modifications at GAL1 (A, B) and INO1 (B).
Considering these results, we then directly compared recruitment of SAGA to the INO1 and GAL1 promoters. ChIP was done in a WT strain, an snf1Δ strain, and an S10A substitution strain. SAGA recruitment and acLys14 are both strongly reduced at INO1 in either the absence of Snf1 or upon substitution of Ser10 (Figure 6B, left). Thus, it appears that pSer10 is required for SAGA recruitment at INO1. In contrast, neither SAGA recruitment nor acLys14 is reduced at the GAL1 promoter in the snf1Δ or S10A strains (Figure 6B, right). As expected, both Snf1 and pSer10 ChIP signals are low in the snf1Δ and S10A substitution strains at both promoters. Thus, SAGA recruitment is strongly dependent upon pSer10 and Snf1 at the INO1 promoter, but not at the GAL1 promoter.
H3 Ser10 phosphorylation is required for TBP recruitment both at INO1 and GAL1
TBP recruitment is essential for transcriptional initiation at many genes, such as INO1 and GAL (reviewed in Hampsey, 1998). Since pSer10 correlates with optimal INO1 and GAL1 transcription, but SAGA recruitment requires pSer10 at INO1 and not at GAL1, we hypothesized that TBP recruitment might be defective at both promoters. We compared the TBP level to that of trimethylation (me) of Lys4 (meK4) in histone H3 using ChIP analysis. Dimethyl K4 has a high steady-state level in ORFs (Bernstein et al, 2002), while trimethyl K4 increases at the 5′ end of genes during their induction (Briggs et al, 2002; Santos-Rosa et al, 2002), having a likely role in transcriptional elongation (Zhang, 2003).
The level of TBP increases at the TATA box region in the inducing condition for each promoter (Figure 7A). TBP recruitment in the S10A strain is reduced five-fold at INO1 and nearly 20-fold at GAL1 (Figure 7B). In contrast, the level of meLys4 is lowered only marginally at either promoter (Figure 7B). These data indicate that pSer10 is involved in TBP recruitment at both INO1 and GAL1.
Figure 7.
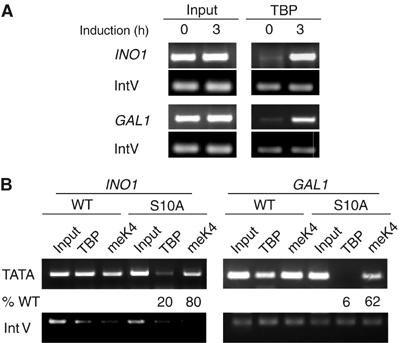
Effect of loss of Snf1 or S10A substitution on recruitment of TBP to INO1 or GAL1 promoters. ChIP assay of TBP and H3 trimethyl Lys4 recruitment. Cells were collected before (A) or after 3 h induction (A, B). In panel B, WT, snf1D, or S10A mutant cells were used. Primer pairs were located in TATA regions.
Discussion
Histone covalent modifications occur in linked patterns. Although a number of patterns have been identified, it is not yet well understood how these patterns develop, nor whether modifications are generally linked to regulate a common mechanism, such as phosphorylation/acetylation in transcriptional activation. In this study, we explore the establishment and function of Snf1-dependent histone H3 phosphorylation in gene regulation. Previously, we reported that INO1 is transcriptionally regulated in part by linked H3 modifications: Snf1-mediated pSer10 and Gcn5-mediated acLys14. Here we describe a pathway at the INO1 gene that establishes pSer10 and then links it to acLys14. We also newly identify GAL1 as a second Snf1-regulated gene that is subject to regulatory H3 phosphorylation. We detect both common and distinguishing features of Snf1-mediated phosphorylation for transcriptional regulation of INO1 and GAL1, supporting the idea that linked modifications may impart exquisite specificity for chromatin-mediated processes.
A pathway linking pSer10 and acLys14 at INO1
In this study, we addressed the question whether Snf1 is recruited to promoters that undergo pSer10 during induction. We showed that Snf1 associates with the promoter regions of INO1 and GAL1 and that an increase in Snf1 precedes the increase of the pSer10 mark. The mechanism of Snf1 association with the promoter is similar to other histone/chromatin regulatory factors which are brought to the promoter via direct activator/repressor interactions with coactivators/corepressors. This has been shown for HAT and HDAC complexes, as well as Swi/Snf (Peterson and Workman, 2000; Sterner and Berger, 2000). We detect in vitro interaction between Snf1c and the ADs of Ino2/Ino4 (or Gal4, see below). The interaction point within Snf1c for activator association may involve the β targeting subunits Gal83, Sip1, and Sip2, since the targeting subunits have distinct functions (Vincent and Carlson, 1999; Schmidt and McCartney, 2000; Vincent et al, 2001; Lin et al, 2003; Vyas et al, 2003).
Consistent with the in vitro interaction, Snf1 recruitment to INO1 and induction of pSer10 in vivo require the heterodimeric activator complex Ino2/Ino4. Thus, in mechanistic parallel to recruitment of HATs (SAGA, NuA4 and CBP/p300), the histone kinase Snf1 appears to be brought to the promoter by direct interaction of a multicomponent Snf1 complex with DNA-bound activator. Recently, Snf1 has been shown to interact with the activator for heat shock genes, HSF (Hahn and Thiele, 2004), suggesting that pSer10 may induce heat shock gene transcription in yeast, as in Drosophila (Nowak and Corces, 2000). In addition, the kinase IKKα is recruited to promoters of certain NFκB-responsive genes for H3 pSer10/acLys14, suggesting a linked pathway as we have demonstrated for INO1. Thus, although the mechanism of recruitment is still unknown for these cofactors, it may be that, similar to HAT recruitment, histone kinases associate with activators as a common mechanism for recruitment to promoters.
Our study also shows that, following Ino2/Ino4 activator recruitment of Snf1c, the pSer10 modification leads to SAGA recruitment to the promoter and acLys14. We and others have shown that histone acetylation by the Gcn5 HAT domain is enzymatically stimulated by pSer10 in vitro (Cheung et al, 2000; Lo et al, 2000), although assay conditions may be critical to detect the stimulation (Shogren-Knaak et al, 2003). In addition, enzyme kinetic and X-ray structural studies show that binding of the Gcn5 HAT domain is strengthened and side-chain interactions with the histone substrate are increased by pSer10 (Clements et al, 2003). Thus, it may be that functionally significant recognition of pSer10 is accomplished by the Gcn5 HAT domain itself. Alternatively, there may be additional modules within SAGA such as the Gcn5 or Spt7 bromodomains or the Ada2 SANT domain, which bind to histone tails (Boyer et al, 2002; Hassan et al, 2002; Sterner et al, 2002b). In this latter case, there may be a transfer or ‘hand-off' of SAGA from pSer10 following initial binding for subsequent Gcn5 HAT domain interaction with pSer10.
Our data here show that SAGA recruitment correlates with pSer10, and thus provide significant evidence to support and extend the previous model that H3 acetylation requires Snf1 and intact Ser10. The timing of recruitment at the promoter (Snf1 associates first, then pSer10, followed closely by SAGA and acLys14) is consistent with functional dependency. Secondly, and importantly, loss of Snf1 or mutation of Ser10 results in loss of both SAGA recruitment and acLys14 to the promoter. Further, there is a striking inability of SAGA to interact with the Ino2/Ino4 activator, in contrast to the ability of Snf1c to interact with Ino2/Ino4. Together, these observations suggest that the inability of Ino2/4 to recruit SAGA leads to an alternate and parallel recruitment strategy through SAGA's association with pSer10.
SAGA recruitment differs between GAL1 and INO1 promoters
We show that GAL1 is an independent promoter regulated by Ser10 phosphorylation. Supporting this observation is that a substitution mutation in histone H3 of Ser10 lowers expression of GAL1 RNA and that Ser10 is phosphorylated in an Snf1-dependant fashion in vivo during GAL1 gene activation. Notably though, in contrast to INO1, where SAGA recruitment depends upon Snf1-mediated pSer10, SAGA recruitment at GAL1 is not dependent on Snf1 or pSer10. These data indicate that, although Ser10 phosphorylation is commonly required for full transcription of both promoters, the role of Ser10 phosphorylation in direct recruitment of SAGA differs between them.
Given the requirement for Snf1-mediated pSer10 in GAL1 transcription, and the similar timing of Snf1/pSer10 and SAGA/acLys14 at the promoter, it is surprising that SAGA recruitment is independent of Snf1 or pSer10 in vivo. Instead, the Gal4 activator interacts with both Snf1c and SAGA in vitro, and can recruit both complexes in vivo. Thus, it appears that recruitment of SAGA at INO1 requires pSer10 because the activator Ino2/Ino4 is not able to directly bind to SAGA, whereas GAL1 is distinct in part because Gal4 is able to associate with both complexes. Some elements of this dissimilar dependency remain to be dissected, yet there are other examples of contrasting patterns of chromatin alterations and recruitment. For example, at the yeast HO promoter, Swi/Snf is recruited before SAGA and is required for SAGA recruitment (Cosma et al, 1999), whereas at the mammalian IFNβ gene it appears that acetylation occurs before Swi/Snf recruitment (Agalioti et al, 2002). In vitro, Swi/Snf utilizes SAGA-dependent acetylation to stably associate with promoters (Hassan et al, 2001), suggesting that acetylation occurs first to recruit Swi/Snf. An example of independence of phosphorylation and acetylation is also found in D. melanogaster, where pSer10 appears to play a major role in regulation of heat-shock-responsive genes, yet may not be linked to acetylation (Labrador and Corces, 2003).
pSer10 is required for TBP recruitment
Ser10 phosphorylation occurs at both INO1 and GAL1. Since pSer10 appears to be required for transcription, but is not required for SAGA recruitment to GAL1, we examined other possible effects of pSer10 loss. Two contrasting events in transcription are TBP recruitment to the TATA box during initiation and histone H3 trimethylation at Lys4, which increases at the 5′ end during the transition from initiation to elongation (Hampsey and Reinberg, 2003). We found that TBP association with either the INO1 or GAL1 promoters was strongly reduced in the S10A strain which is unable to phosphorylate Ser10, yet trimethyl K4 was only modestly lowered. Thus, diminished TBP recruitment is a common feature of these two pSer10-dependent promoters. Perhaps consistent with this commonality is the identification of TBP mutants that cause severely impaired induction of INO1, GAL1, and GAL10 genes (Arndt et al, 1995). It is an attractive possibility that some of these TBP mutants may be defective in recognition of pSer10. A second perhaps relevant observation is that TBP is regulated at INO1 by an unknown substrate of Snf1 that is distinct from the Snf1-phosphorylated repressor Mig1 (Shirra and Arndt, 1999); this substrate could be histone H3.
This apparent requirement for pSer10 in TBP promoter targeting could be caused by several mechanisms. It could be that TBP, or a TBP-associated factor, may directly bind to pSer10 when present at the TATA box. As described above, certain specific domains within chromatin-related proteins associate with covalently modified histones. For example, bromodomains of CBP/p300, Gcn5/PCAF, Taf1, and other proteins associate with acetylated lysines in histones H3 and H4 to regulate gene induction (Marmorstein and Berger, 2001). Following these and other examples, TBP may show a preference for binding to TATA sequence associated with phosphorylated nucleosomes.
A second mechanism is illustrated by the example of INO1, where SAGA recruitment is potentiated by pSer10. SAGA contains proteins that directly interact with and regulate TBP binding at promoters. Spt3 positively regulates TBP at GAL1, and TBP binding is significantly reduced (about 13-fold, which is similar to the reduction of 20-fold in the S10A strain) in an spt3 deletion strain (Larschan and Winston, 2001). As TBP binding in vivo requires Spt3, the requirement for intact Ser10 for TBP binding at INO1 may be due to reduced SAGA association.
A third mechanism is that pSer10 may cause an alteration in SAGA conformation or function to promote TBP recruitment. Spt3, Spt7, and Spt8 each have a role in TBP recruitment or regulation (Sterner et al, 2002a), thus it is possible that their activity is altered in the presence of pSer10.
Model for recruitment and function of Snf1 and pSer10
We observe that Snf1-mediated histone H3 phosphorylation has a significant role in regulating both the INO1 and GAL1 genes. There appear to be common and distinct mechanisms regulating the promoters that relate to the status of pSer10 (Figure 8). There are two common mechanisms. First, the activators for each promoter contribute to recruiting Snf1c for Ser10 phosphorylation (upper panel). Second, pSer10 plays a role in the recruitment of TBP (lower panel). However, the recruitment of SAGA distinguishes the two promoters: whereas the Gal4 activator can apparently directly recruit SAGA to GAL1, in contrast, pSer10 recruits SAGA to INO1 (middle panel). Further examination of the role of histone phosphorylation will continue to define common themes in gene regulation as well as to identify distinct mechanisms used to establish patterns and temporal orders involving this modification.
Figure 8.
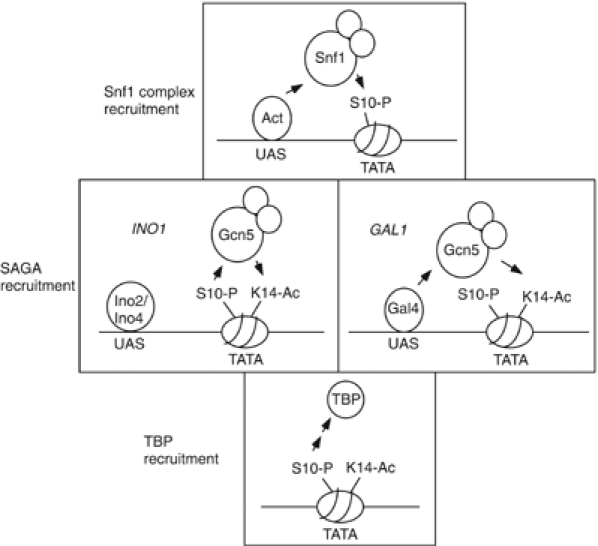
Model for the establishment of pS10, acK14, and recruitment of TBP. The upper panel shows Snf1c recruitment by activators for both INO1 and GAL1 genes to establish pSer10. The middle panels show two putative mechanisms for establishment of acLys14: at INO1 pS10 sets up acK14 via SAGA recruitment, whereas at GAL1 the activator recruits SAGA. Lower panel, TBP recruitment is mediated by pSer10 at both genes.
Materials and methods
Plasmids
Plasmids expressing GST fusion full-length Ino2 (pGST-Ino2FL) and Ino2-AD (pGST-Ino2-AD) were constructed by inserting the sequences encoding amino-acid residues 1–304 (FL) and 1–100 (AD) of Ino2 into pGEX-5X-1 (Pharmacia). Deletions of Ino2 ADs (Ino2-ADIΔ: deletion of aa 1–27: Ino2-ADIIΔ: deletion of aa 28–100; Ino2-ADI & II: deletion of aa 1–100) were generated within pGST-Ino2FL using the QuikChange site-directed mutagenesis kit (Stratagene). Yeast expression plasmids containing Ino2 and its AD deletions were constructed in two steps: first, the INO2 promoter sequence (600 base pairs upstream of the starting codon) was generated by PCR amplification from yeast genomic DNA and inserted into pRS315 (CEN/ARS, LEU2) with three copies of HA tag and an ADH1 terminator cassette at 3′ end of the INO2 promoter (pINO2-HA); second, the full length and AD mutants of INO2 from the series of GST-Ino2 fusion constructs described above were subcloned into pINO2-HA. PCR amplification products and deletions were verified by sequencing.
Yeast strains and antibodies
WT yeast strain SB301 (MATa his3Δ200 leu2Δ1 ura3-52 trp1ΔhisG), gcn5 null mutant strain SB303 (MATα gcn5∷HIS3 his3Δ200 leu2Δ1 ura3-52 trp1Δ hisG, a trp1 derivative of FY1369), FY1370 (Lo et al, 2000, 2001), snf1 null strain LPY7091 (snf1∷TRP1), and ino2 null strain LPY7860 (ino2∷HIS3) were constructed as described previously (Longtine et al, 1998). LPY7097 (MATa gal83∷KanMX), LPY7099 (gal4∷KanMX), LPY7861(ino4∷KanMX), LPY7862, and LPY7589 (sas3∷KanMX) were generated by transformation of SB301with individual PCR-amplified genomic DNA from a 300 base pair region surrounding the target genes disrupted with the KanMX cassette (ResGen). Double mutation strains (snf1gcn5, snf1sas3, and ino2ino4) were generated from mating of haploid single mutation strains followed by sporulation and selection. For mutants with histone mutations, strains were constructed by transforming FY1716 (MATa his3Δ200 leu2Δ1 lys2-128 trp1Δ63 ura3-52 hht1-hhf1∷LEU2, hht2-hhf2∷HIS3, pDM9[HHT1-HHF1-CEN-URA3]) with histone mutation plasmids (HHT2-HHF2-CEN-TRP1). The WT histone plasmid, pDM9, was lost by selection in 5-fluoro-orotic acid (5-FOA) media. Epitope tagging was performed as described previously (Longtine et al, 1998), and was verified by PCR and Western blot analysis.
The Snf1 peptide antibodies were generated for this study and specificity was demonstrated in vitro on peptides and in vivo using an snf1Δ strain. The pSer10 peptide and recombinant Ada2 antibodies were generated previously (Candau and Berger, 1996; Lo et al, 2001). Yeast TBP antibody was a gift from S Buratowski. All other antibodies were purchased from commercial vendors (Upstate or Abcam).
S1 nuclease protection assays were performed as described previously (Lo et al, 2000) (Henry et al, 2003).
Snf1 and SAGA complexes were purified from yeast (WT or Snf1-Flag strain) as described previously (Grant et al, 1997; Lo et al, 2001). Ion trap mass spectrometric sequencing of proteins that cofractionated with Snf1 was done by D Speicher (Wistar Institute). The Mono Q fractions containing the SAGA complex were also confirmed by Ada2 and Ada3 antibodies.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously (Dudley et al, 1999; Kuras et al, 2000). The linearity of PCR reactions was shown by multiple template dilutions of input (Inp) and immunoprecipitated (Ip) DNA and varying the number of PCR cycles. Gene-specific PCR was usually 28–30 PCR cycles. Control PCR for IntV was performed at 35–40 cycles, in order to detect a background signal for normalization. In general, Ip DNA samples were initially diluted 1:100 and inputs were diluted 1:12 500 with ultrapure water. Gene-specific PCR product was normalized by a control PCR product amplified from a noncoding region of chromosome V. PCR products were separated on a 2% agarose gel and the signal was determined by fluorography after ethidium bromide staining, scanning, and quantification using AlhaImager™ 1220 (Alpha Innotech Corp.). Primer pairs used for PCR analysis are available upon request.
Bacterial protein expression, purification, and GST pulldown assay were as previously described (Barlev et al, 1995) with minor modifications.
Acknowledgments
We thank F Winston and CD Allis for yeast histone H3/H4 deletion strains, M Grunstein for histone H3/H4 substitution plasmids, and S Buratowski for TBP antibody. We thank G Moore, KL-C Wang and members of the Berger and Pillus laboratories for discussions and critical reading of the manuscript. W-S Lo was a Special Fellow of the Leukemia and Lymphoma Foundation. The research was supported by grants from the National Institutes of General Medical Sciences (GM56469 to LP and GM 55360 to SLB and from the National Science Foundation to SLB (MCB-78940)).
References
- Agalioti T, Chen G, Thanos D (2002) Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381–392 [DOI] [PubMed] [Google Scholar]
- Ambroziak J, Henry SA (1994) INO2, INO4 gene products, positive regulators of phospholipid biosynthesis in Saccharomyces cerevisiae, form a complex that binds to the INO1 promoter. J Biol Chem 269: 15344–15349 [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423: 659–663 [DOI] [PubMed] [Google Scholar]
- Arndt KM, Ricupero-Hovasse S, Winston F (1995) TBP mutants defective in activated transcription in vivo. EMBO J 14: 1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev NA, Candeau R, Wang L, Darpino P, Silverman N, Berger SL (1995) Characterization of physical interactions of the putative transcriptional adaptor, ADA2, with acidic activation domains and TATA-binding protein. J Biol Chem 270: 19337–19344 [DOI] [PubMed] [Google Scholar]
- Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL (2002) Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA 99: 8695–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik SR, Green MR (2001) SAGA is an essential in vivo target of the yeast acidic activator Gal4p. Genes Dev 15: 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Langer MR, Crowley KA, Tan S, Denu JM, Peterson CL (2002) Essential role for the SANT domain in the functioning of multiple chromatin remodeling enzymes. Mol Cell 10: 935–942 [DOI] [PubMed] [Google Scholar]
- Briggs SD, Xiao T, Sun ZW, Caldwell JA, Shabanowitz J, Hunt DF, Allis CD, Strahl BD (2002) Gene silencing: trans-histone regulatory pathway in chromatin. Nature 418: 498. [DOI] [PubMed] [Google Scholar]
- Bryant GO, Ptashne M (2003) Independent recruitment in vivo by Gal4 of two complexes required for transcription. Mol Cell 11: 1301–1309 [DOI] [PubMed] [Google Scholar]
- Candau R, Berger SL (1996) Structural, functional analysis of yeast putative adaptors. J Biol Chem 271: 5237–5245 [DOI] [PubMed] [Google Scholar]
- Carlson M (1999) Glucose repression in yeast. Curr Opin Microbiol 2: 202–207 [DOI] [PubMed] [Google Scholar]
- Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD (2000) Synergistic coupling of histone H3 phosphorylation, acetylation in response to epidermal growth factor stimulation. Mol Cell 5: 905–915 [DOI] [PubMed] [Google Scholar]
- Clements A, Poux AN, Lo WS, Pillus L, Berger SL, Marmorstein R (2003) Structural basis for histone, phosphohistone binding by the GCN5 histone acetyltransferase. Mol Cell 12: 461–473 [DOI] [PubMed] [Google Scholar]
- Cosma MP, Tanaka T, Nasmyth K (1999) Ordered recruitment of transcription, chromatin remodeling factors to a cell cycle-, developmentally regulated promoter. Cell 97: 299–311 [DOI] [PubMed] [Google Scholar]
- Drysdale CM, Jackson BM, McVeigh R, Klebanow ER, Bai Y, Kokubo T, Swanson M, Nakatani Y, Weil PA, Hinnebusch AG (1998) The Gcn4p activation domain interacts specifically in vitro with RNA polymerase II holoenzyme, TFIID, the Adap-Gcn5p coactivator complex. Mol Cell Biol 18: 1711–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley AM, Rougeulle C, Winston F (1999) The Spt components of SAGA facilitate TBP binding to a promoter at a post-activator-binding step in vivo. Genes Dev 13: 2940–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, Berger SL, Workman JL (1997) Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex, the SAGA (Spt/Ada) complex. Genes Dev 11: 1640–1650 [DOI] [PubMed] [Google Scholar]
- Hahn JS, Thiele DJ (2004) Activation of the Saccharomyces cerevisiae heat shock transcription factor under glucose starvation conditions by Snf1 protein kinase. J Biol Chem 279: 5169–5176 [DOI] [PubMed] [Google Scholar]
- Hampsey M (1998) Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol Mol Biol Rev 62: 465–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampsey M, Reinberg D (2003) Tails of intrigue: phosphorylation of RNA polymerase II mediates histone methylation. Cell 113: 429–432 [DOI] [PubMed] [Google Scholar]
- Hassan AH, Neely KE, Workman JL (2001) Histone acetyltransferase complexes stabilize swi/snf binding to promoter nucleosomes. Cell 104: 817–827 [DOI] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL (2002) Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 111: 369–379 [DOI] [PubMed] [Google Scholar]
- Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev 17: 2648–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry SA, Patton-Vogt JL (1998) Genetic regulation of phospholipid metabolism: yeast as a model eukaryote. Prog Nucleic Acid Res Mol Biol 61: 133–179 [DOI] [PubMed] [Google Scholar]
- Howe L, Auston D, Grant P, John S, Cook RG, Workman JL, Pillus L (2001) Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev 15: 3144–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Smith MM (2003) Functional consequences of histone modifications. Curr Opin Genet Dev 13: 154–160 [DOI] [PubMed] [Google Scholar]
- Jackson JC, Lopes JM (1996) The yeast UME6 gene is required for both negative and positive transcriptional regulation of phospholipid biosynthetic gene expression. Nucleic Acids Res 24: 1322–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Kao CF, Hillyer C, Tsukuda T, Henry K, Berger S, Osley MA (2004) Rad6 plays a role in transcriptional activation through ubiquitylation of histone H2B. Genes Dev 18: 184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Eisenman RN (1999) Sin meets NuRD and other tails of repression. Cell 99: 447–450 [DOI] [PubMed] [Google Scholar]
- Kuras L, Kosa P, Mencia M, Struhl K (2000) TAF-containing and TAF-independent forms of transcriptionally active TBP in vivo. Science 288: 1244–1248 [DOI] [PubMed] [Google Scholar]
- Labrador M, Corces VG (2003) Phosphorylation of histone H3 during transcriptional activation depends on promoter structure. Genes Dev 17: 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Winston F (2001) The S. cerevisiae SAGA complex functions in vivo as a coactivator for transcriptional activation by Gal4. Genes Dev 15: 1946–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SS, Manchester JK, Gordon JI (2003) Sip2, an N-myristoylated beta subunit of Snf1 kinase, regulates aging in Saccharomyces cerevisiae by affecting cellular histone kinase activity, recombination at rDNA loci, and silencing. J Biol Chem 278: 13390–13397 [DOI] [PubMed] [Google Scholar]
- Lo W-S, Duggan L, Emre NCT, Belotserkovskya R, Lane WS, Shiekhattar R, Berger SL (2001) Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293: 1142–1146 [DOI] [PubMed] [Google Scholar]
- Lo W-S, Trievel RC, Rojas JR, Duggan L, Hsu J-Y, Allis CD, Marmorstein R, Berger L (2000) Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell 5: 917–926 [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Mahadevan LC, Willis AC, Barratt MJ (1991) Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell 65: 775–783 [DOI] [PubMed] [Google Scholar]
- Marmorstein R, Berger SL (2001) Structure and function of bromodomains in chromatin-regulating complexes. Gene 272: 1–9 [DOI] [PubMed] [Google Scholar]
- Morillon A, Karabetsou N, O'Sullivan J, Kent N, Proudfoot N, Mellor J (2003) Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell 115: 425–435 [DOI] [PubMed] [Google Scholar]
- Nath N, McCartney RR, Schmidt MC (2002) Purification and characterization of Snf1 kinase complexes containing a defined beta subunit composition. J Biol Chem 277: 50403–50408 [DOI] [PubMed] [Google Scholar]
- Nowak SJ, Corces VG (2000) Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes Dev 14: 3003–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak SJ, Corces VG (2004) Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 20: 214–220 [DOI] [PubMed] [Google Scholar]
- Peterson CL, Cote J (2004) Cellular machineries for chromosomal DNA repair. Genes Dev 18: 602–616 [DOI] [PubMed] [Google Scholar]
- Peterson CL, Workman JL (2000) Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr Opin Genet Dev 10: 187–192 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 285: 886–891 [DOI] [PubMed] [Google Scholar]
- Schmidt MC, McCartney RR (2000) Beta-subunits of Snf1 kinase are required for kinase function and substrate definition. EMBO J 19: 4936–4943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank S, Ebbert R, Rautenstrauss K, Schweizer E, Schuller HJ (1995) Yeast transcriptional activator INO2 interacts as an Ino2p/Ino4p basic helix–loop–helix heteromeric complex with the inositol/choline-responsive element necessary for expression of phospholipid biosynthetic genes in Saccharomyces cerevisiae. Nucleic Acids Res 23: 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirra MK, Arndt KM (1999) Evidence for the involvement of the Glc7-Reg1 phosphatase and the Snf1-Snf4 kinase in the regulation of INO1 transcription in Saccharomyces cerevisiae. Genetics 152: 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak MA, Fry CJ, Peterson CL (2003) A native peptide ligation strategy for deciphering nucleosomal histone modifications. J Biol Chem 278: 15744–15748 [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS (2003) MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 22: 2788–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilianakis C, Kretsovali A, Agalioti T, Makatounakis T, Thanos D, Papamatheakis J (2003) CIITA regulates transcription onset viaSer5-phosphorylation of RNA Pol II. EMBO J 22: 5125–5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Belotserkovskaya R, Berger SL (2002a) SALSA, a variant of yeast SAGA, contains truncated Spt7, which correlates with activated transcription. Proc Natl Acad Sci USA 99: 11622–11627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL (2000) Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64: 435–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Wang X, Bloom MH, Simon GM, Berger SL (2002b) The SANT domain of Ada2 is required for normal acetylation of histones by the yeast SAGA complex. J Biol Chem 277: 8178–8186 [DOI] [PubMed] [Google Scholar]
- Strelkov IS, Davie JR (2002) Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogene-transformed mouse fibroblasts. Cancer Res 62: 75–78 [PubMed] [Google Scholar]
- Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC (1999) The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J 18: 4779–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalidou I, Papamichos-Chronakis M, Thireos G, Tzamarias D (2004) Spt3 and Mot1 cooperate in nucleosome remodeling independently of TBP recruitment. EMBO J 23: 1943–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treitel MA, Kuchin S, Carlson M (1998) Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol Cell Biol 18: 6273–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM (2002) Cellular memory and the histone code. Cell 111: 285–291 [DOI] [PubMed] [Google Scholar]
- Utley RT, Ikeda K, Grant PA, Côt J, Steger DJ, Eberharter A, John S, Workman JL (1998) Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature 394: 498–502 [DOI] [PubMed] [Google Scholar]
- Vincent O, Carlson M (1999) Gal83 mediates the interaction of the Snf1 kinase complex with the transcription activator Sip4. EMBO J 18: 6672–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent O, Townley R, Kuchin S, Carlson M (2001) Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev 15: 1104–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas VK, Kuchin S, Berkey CD, Carlson M (2003) Snf1 kinases with different beta-subunit isoforms play distinct roles in regulating haploid invasive growth. Mol Cell Biol 23: 1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB (2003) Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423: 655–659 [DOI] [PubMed] [Google Scholar]
- Yu Y, Eriksson P, Bhoite LT, Stillman DJ (2003) Regulation of TATA-binding protein binding by the SAGA complex and the Nhp6 high-mobility group protein. Mol Cell Biol 23: 1910–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y (2003) Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev 17: 2733–2740 [DOI] [PubMed] [Google Scholar]